Introduction
Chronic myeloid leukemia (CML) is a hematological
disease, that accounted for ~20% of adult leukemia worldwide in
2009 (1). CML is characterized by
reciprocal translocation between the break-point cluster (BCR) gene
on chromosome 22 and the Abelson leukemia virus oncogene (ABL) on
chromosome 9, also termed the Philadelphia chromosome, which
results in the formation of the BCR-ABL oncogene (2). Constitutive expression of the BCR-ABL1
fusion protein transforms hematopoietic stem cells into CML stem
cells and ultimately leads to myeloproliferative disease (3). The BCR-ABL fusion protein also
constitutively activates tyrosine kinase activity and various
downstream signaling pathways, such as the AKT, JAK/STAT3 and MAPK
pathways, contributing to cell proliferation, and resistance to
apoptosis and disrupting genetic stability (3).
Hepatocyte growth factor (HGF) is a pleiotropic
growth factor, that is secreted by mesenchymal stem cells and
capable of inducing various physiological activities in different
cells, such as proliferation, survival, migration and angiogenesis
(4). The HGF gene is located
on chromosome 7, which is a chromosome that is frequently altered
in various hematological malignancies. HGF is initially produced in
a one-chain inactive form and later cleaved into a two-chain (α, β)
biologically active form by enzymes, such as HGF activator
(4). HGF has been intensively
studied, mainly due to its role in cancer development and
progression. For example, it has been reported that primary CML
cells express high levels of HGF mRNA and protein, as well as its
corresponding tyrosine kinase receptor, MET (5). Furthermore, elevated serum HGF levels
have been associated with poor prognosis in patients with CML
(6). Notably, HGF has been found to
be preferentially generated in CML basophils and basophil-derived
HGF induces endothelial cell migration, and might be used as a
target for CML (7). All these
findings indicate that HGF might play an essential role in the
development of CML.
Tyrosine kinase inhibitors (TKIs) are able to induce
remission and improve survival in patients with CML; however, they
are unable to eliminate leukemia stem cells (LSCs), as these cells
do not depend on the kinase activity of BCR-ABL oncoprotein for
survival (8). VP-16 (etoposide) is a
widely used chemotherapeutic agent for various cancers, including
CML. VP-16 has also been shown to enhance TKI-induced apoptosis in
BCR-ABL-transformed CML cells (9).
More importantly, VP-16 has also been found to effectively repress
the growth of LSCs (10). In the
current study, the effects of HGF on the cytotoxicity of VP-16 in a
K562 CML cell line was investigated. It was found that
overexpression of HGF significantly decreased VP-16-induced
apoptosis. The results from the present study further highlight the
potential value of HGF in the treatment of CML.
Materials and methods
Cells and reagents
The human K562 CML cell line was purchased from the
Cell Bank of the Chinese Academy of Sciences. VP-16 was purchased
from Jiangsu Hengrui Medicine Co., Ltd. (cat. no.10092131). All
other routine chemicals were purchased from MilliporeSigma.
LY294002 (PI3K/Akt-specific inhibitor)
treatment
LY294002 was obtained from MilliporeSigma (cat. no.
L9908). After the cells were cultured in a six-well plate at
4.0×105/well for 24 h, the medium was discarded and the
cells were treated for another 24 h with LY294002 at a final
concentration of 20 µM (dissolved in the fresh culture medium).
Cell culture and transfection
The cell line was cultured in Iscove modified
Dulbecco's Media, with 10% FCS, 100 IU/ml penicillin and 100 IU/ml
streptomycin. The cells were maintained at 37°C in a humidified
atmosphere with 5% CO2. The pVITRO2-HGF plasmid was
constructed using the pVITRO2 vector (Shanghai, GenePharma, Co.
Ltd.). Small interfering (si)RNA against Bcl-2 (si-Bcl-2) and
negative control (si-ctrl) were purchased from Shanghai,
GenePharma, Co. Ltd. The following sequences were used: Bcl-2:
5′-AAGGUGUCUUCCAGAUCCUGA −3′; Ctrl: 5′-AAAUGUGUGUACGUCUCCUCC −3′.
Human HGF cDNA was subcloned into the pVITRO2 vector using the
SalI enzyme. For overexpression of Bax, human Bax cDNA was
subcloned into the pcDNA3.1 vector by Shanghai, GenePharma, Co.
Ltd. The K562 cells were seeded, at a density of 4×105
cells/well in six-well plates and incubated for 24 h. si-Ctrl (200
pmol), si-Bcl-2 (200 pmol), pVITRO2-mcs (mock-transfectant; 3 µg),
pVITRO2-HGF (3 µg) or pcDNA3.1 Bax (3 µg) was transfected using
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.) for
24 h according to the manufacturer's instructions. The cells
without transfection were used as a negative control
(untreated).
Cell viability assay
Cell viability was analyzed as previously described
(11). Briefly, the cells
(2.0×103/well) were seeded into 96-well plates. Cells
were transfected with pVITRO2-HFG for 24 h, followed by another 24
h treatment of VP-16 (final concentration of 1 µg/ml). Then cells
were co-incubated with MTT reagent (20 µl/well) for 4 h at 37°C,
after which the supernatant was removed and cells were co-incubated
with 200 µl of DMSO for 20 min at 37°C. Absorbance at 490 nm was
measured using a microplate reader (BioTek, USA).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted using TRIzol®
(Thermo Fisher Scientific, Inc.). cDNA was synthesized from total
RNA (1 µg) using a ThermoScript RT-PCR system (Thermo Fisher
Scientific, Inc.). Reverse transcription reactions was performed
with the following conditions: 37°C for 15 min, 42°C for 50 min and
85°C for 5 min. The reverse transcription products were visualized
using gel electrophoresis. The qPCR reactions were performed using
the ABI7500 system (Applied Biosystems; Thermo Fisher Scientific,
Inc.), and was performed with 20 ng cDNA, 2.5 pmol forward and
reverse primers, 10 µl of SYBR Green Fast qPCR Mix (2X; Takara
Biotechnology Co. Ltd.) and the final volume was adjusted to 20 µl
with RNase-free water. The qPCR reaction was performed at 95°C for
5 min, 60°C for 30 sec, followed by 40 cycles at 95°C for 30 sec
and 58°C for 30 sec. The ΔCt value was calculated by the CT value
of the target gene minus the CT value of endogenous control. The
ΔΔCt value (ΔCt target-ΔCt calibrator) was used to determine the
fold changes in gene levels. The relative gene expressions were
determined by the 2−ΔΔCq method (12). GAPDH was used as an internal control.
The following primers were used: HGF forward,
5′-GGATGGATGGTTAGTTTGAGATACA-3′ and reverse,
5′-CTCTTCCGTGGACATCATGAAT-3′; Bcl-2 forward
5′-GTGGAGGAGCTCTTCAGGGA-3′ and reverse, 5′-AGGCACCCAGGGTGATGCAA-3′;
Bax forward 5′-TTTGCTTCAGGGTTTCATCCA −3′ and reverse,
5′-CTCCATGTTACTGTCCAGTTCGT-3′; and GAPDH forward
5′-GTGAGGAGGGGAGATTCAG-3′ and reverse, 5′-GCATCCTGGGCTACACTG-3′.
GAPDH was used as an internal control. Experiments were performed
independently 3 times.
Western blot analysis
Western blot analysis was performed as previously
described (13). The cells were
lysed with RIPA buffer (Beijing Solarbio Science and Technology
Co., Ltd.). Equal amount (20 µg) of protein lysate was separated
using 12% SDS-PAGE, then transferred onto PVDF membranes (Millipore
Sigma). The PVDF membranes were blocked with 10% skimmed milk in
TBS-Tween-20 (TBST, 0.1% Tween 20) at room temperature for 1 h,
washed with TBST and incubated with a primary antibody in TBST,
containing 5% BSA (cat. no. A1933; MilliporeSigma) overnight at
4°C. The following primary antibodies were used: Caspase-3
(1:1,000; cat. no. 9662; Cell Signaling Technology, Inc.), Cleaved
Caspase-3 (1:1,000; cat. no. 9664; Cell Signaling Technology,
Inc.), Caspase-9 (1:1,000; cat. no. 9502; Cell Signaling
Technology, Inc.), Cleaved Caspase-9 (1:1,000; cat. no. 20750; Cell
Signaling Technology, Inc.), Bcl-2 (1:1,000; cat. no. 4223; Cell
Signaling Technology, Inc.), Bax (1:1,000; cat. no. 5023; Cell
Signaling Technology, Inc.), phosphorylated (p)-PI3K (1:1,000; cat.
no. 17366; Cell Signaling Technology, Inc.), PI3K (1:1,000; cat.
no. 4255; Cell Signaling Technology, Inc.), p-Akt (1:1,000; cat.
no. 4060; Cell Signaling Technology, Inc.), Akt (1:1,000; cat. no.
4691; Cell Signaling Technology, Inc.) and GAPDH (1:2,000; G9545;
MilliporeSigma). The membranes were washed three times with TBST,
then incubated with anti-rabbit HRP-conjugated secondary antibody
(1:4,000; A0545; MilliporeSigma) for 1 h at room temperature.
Signals were visualized using an ECL reagent (Pierce; Thermo Fisher
Scientific, Inc.). Band intensities were quantified using ImageJ
software (https://imagej.niv.gov/) and GAPDH was
used as the loading control.
Apoptosis assay
To measure apoptosis, an Annexin V-FITC Apoptosis
Detection kit (MilliporeSigma) was used according to the
manufacturer's instructions. Briefly, after treatment by VP-16 or
transfection, the cells were washed with PBS and resuspended in
binding buffer, containing Annexin V and PI. The apoptosis rate was
measured using an Attune NxT Flow Cytometer (Thermo Fisher
Scientific, Inc.) within 30 min of staining. The results were
analyzed using the FlowJo v10.0 software (FlowJo, LLC).
Caspase activity assay
The activity level of caspase-3 and −9 was analyzed
using the Caspase-3 and Caspase-9 Activity Assay kit (Fluorometric;
Abcam) according to the manufacturer's instructions. After
treatment by VP-16 or transfection, 100 µl substrate was added to
each well and the samples were incubated for 1 h at room
temperature. Luminescence at 405 nm was measured using a BioTek
312e microplate reader (BioTek Instruments, Inc.).
Statistical analysis
All statistical analyses were conducted using SPSS
v20.0 software (IBM Corp). All measurement data are presented as
the mean ± SD. Multiple groups were analyzed using one-way ANOVA
followed by the Bonferroni post hoc test. P<0.05 was considered
to indicate a statistically significantly difference.
Results
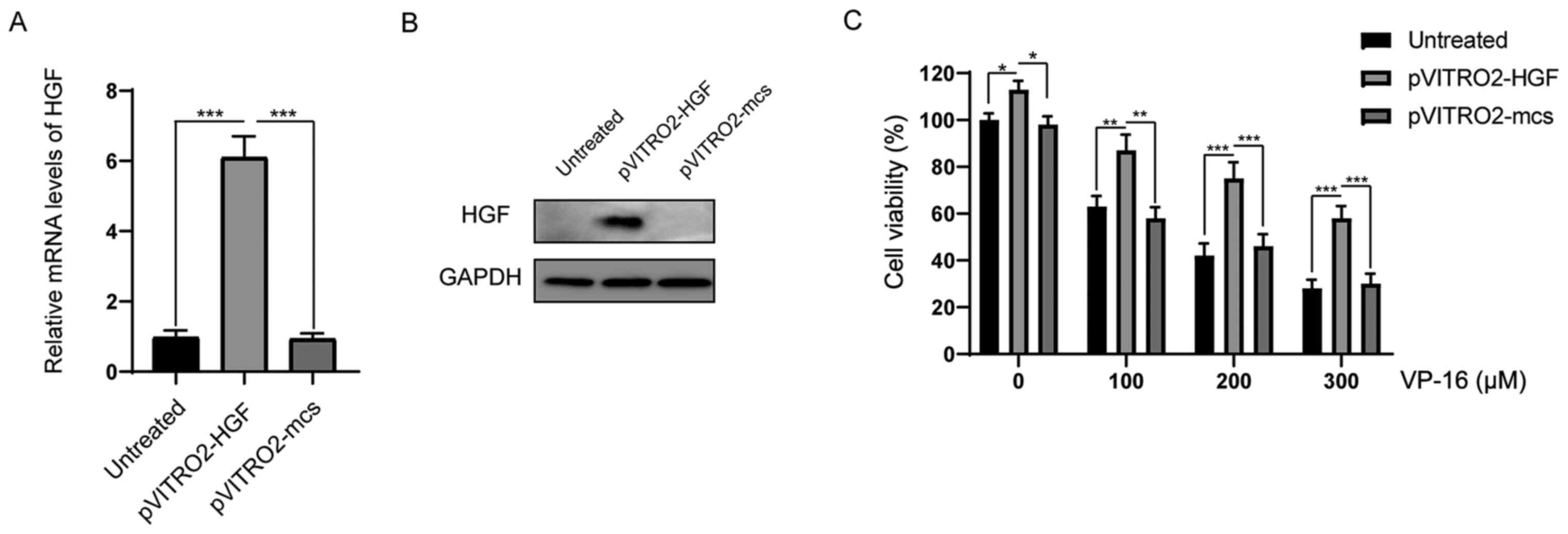
Overexpression of HGF protects the
K562 cells from cytotoxicity induced by VP-16
To investigate the protective role of HGF, the K562
cells were transfected with pVITRO2-HGF or pVITRO2-mcs (control).
As indicated in Fig. 1A, after
transfection for 24 h, the mRNA expression level of HGF was
successfully upregulated in the pVITRO2-HGF transfection group
compared with that in the pVITRO2-mcs and untreated groups. Western
blot assays confirmed that the protein expression level of HGF was
markedly increased following transfection with pVITRO2-HGF
(Fig. 1B). Next, it was investigated
whether the increase in HGF expression could affect the response of
the K562 cells to VP-16. The K562 cells were treated with different
concentrations of VP-16 (0, 100, 200 and 300 µM) for 48 h, then the
cell viabilities were analyzed. As shown in Fig. 1C, overexpression of HGF significantly
enhanced cell survival following treatment with VP-16.
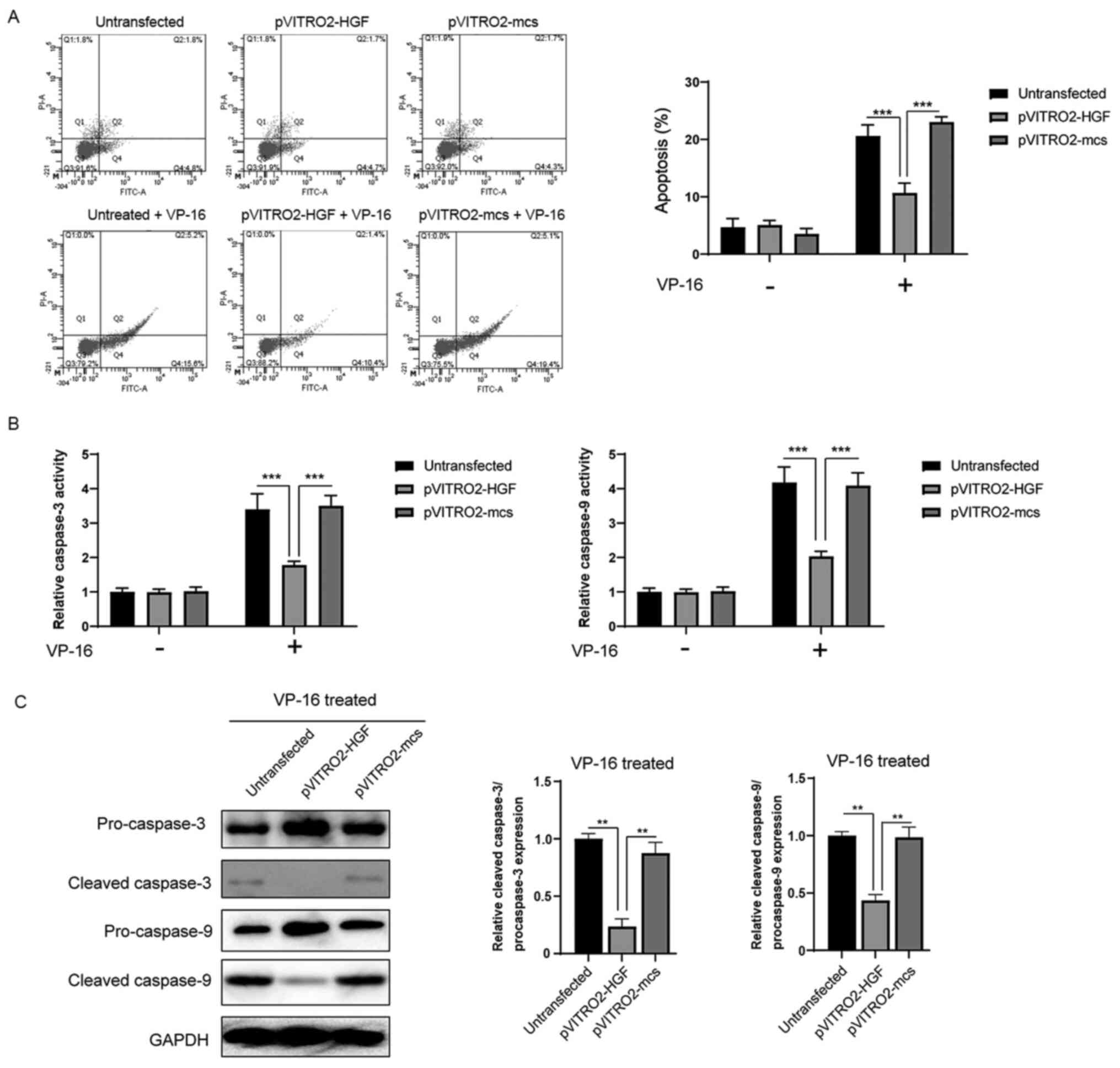
Overexpression of HGF reduces
apoptosis induced by VP-16 in the K562 cells
Then, it was investigated whether the protective
role of HGF against VP-16 developed by affecting apoptosis in the
K562 cells. Annexin V staining revealed that in the presence of
VP-16 (300 µM) for 24 h, the control group (without transfection)
or cells transfected with pVITRO2-mcs had apoptosis rates of 20.8±3
and 24.6±1.9%, respectively. Furthermore, overexpression of HGF
significantly decreased apoptosis induced by VP-16 to 12.0±1.4%
(Fig. 2A). Consistent with the
results from the apoptosis assay, the caspase activities also
revealed that the activation of caspase-3/-9 induced by VP-16 was
significantly inhibited following overexpression of HGF (Fig. 2B). In addition, western blot analysis
showed that the cleavage of caspase-3/-9 was significantly
inhibited by the overexpression of HGF (Fig. 2C). Taken together, these results
indicate that HGF confers protection against VP-16-induced
apoptosis via the regulation of caspase-3/9 activation.
Overexpression of HGF affects the
expression level of Bcl-2 proteins
Since apoptosis can be regulated by the Bcl-2
protein family, it was investigated whether HGF could affect the
expression level of the Bcl-2 protein family. First, the mRNA
expression level of Bcl-2 and Bax was analyzed using RT-qPCR. The
results revealed that VP-16 treatment led to the decrease of Bcl-2
mRNA expression level and the increase in the mRNA expression level
of Bax in the pVITRO2-mcs and untreated groups compared with that
in the pVITRO2-HGF transfection group (Fig. 3A). Notably, overexpression of HGF
reversed the effects of VP-16 on the mRNA expression levels of both
Bcl-2 and Bax (Fig. 3A). Western
blot analysis demonstrated similar effects (Fig. 3B). To further investigate the role of
the Bcl-2 proteins in the protective role of HGF, siRNA was used to
knockdown the expression level of Bcl-2 (Fig. 3C). According to the results, the
apoptosis induced by VP-16 was significantly enhanced in
pVITRO2-HGF + si-Bcl-2 group compared with pVITRO2-HGF or
pVITRO2-HGF + si-Ctrl group (Fig.
3D). At the same time, Bax was overexpressed in the K562 cells
following transfection with pcDNA3.1 Bax plasmid (Fig. 3E). Compared with pVITRO2-HGF group or
pVITRO2-HGF + pcDNA3.1 group, the VP-16-induced apoptosis was
further enhanced in case of high expression of Bax in K562 cells
(Fig. 3F). Taken together, these
findings revealed that HGF exerts its protective effects at least
partly via the regulation of Bcl-2 and Bax.
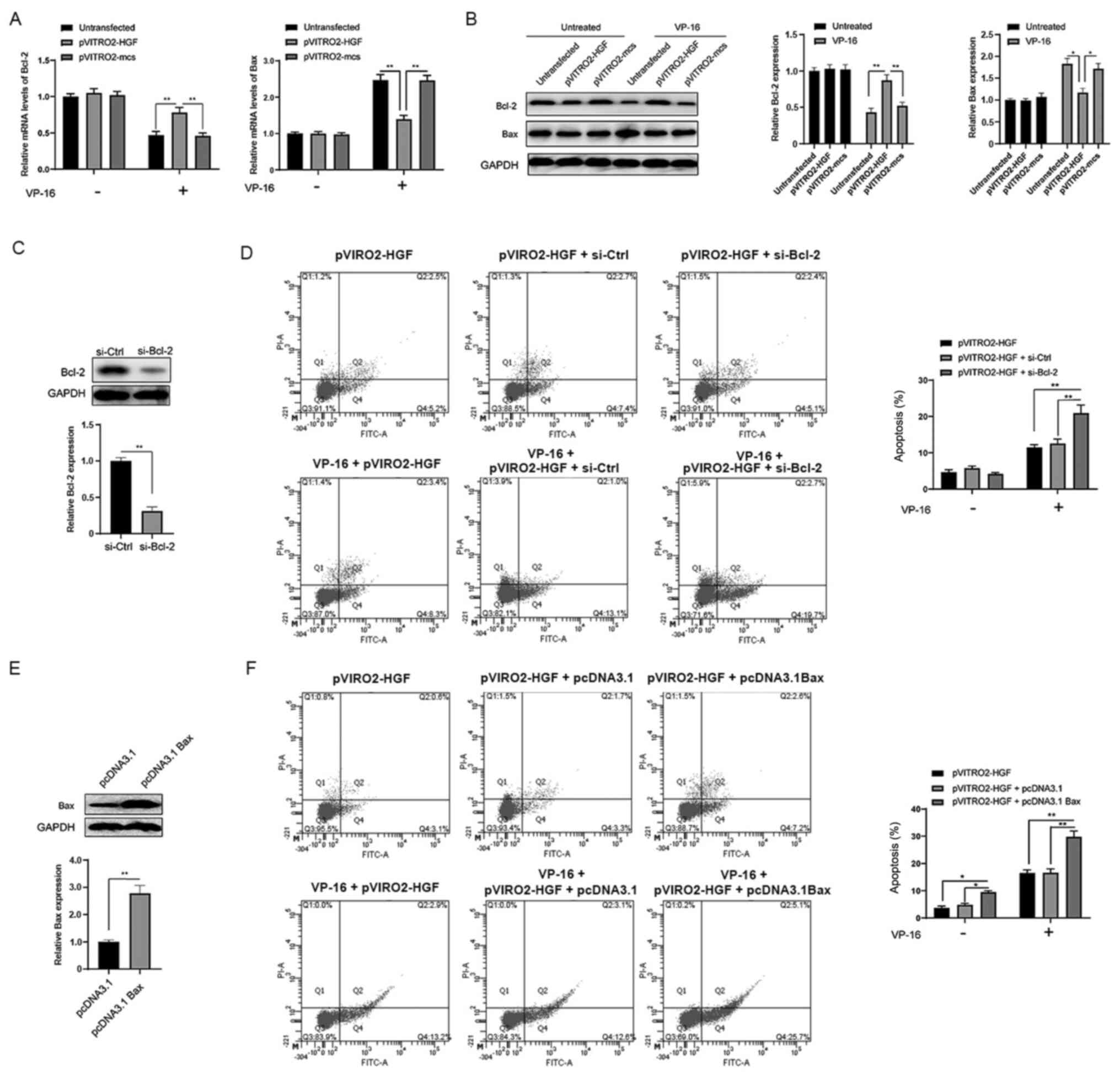 | Figure 3.Overexpression of HGF affects the
expression level of Bcl-2 proteins in the K562 cells. The K562
cells were treated as indicated for 24 h, then the (A) mRNA and (B)
protein expression levels of Bcl-2 and Bax were analyzed using
reverse transcription-quantitative PCR. (C) The K562 cells were
transfected with siRNA targeting Bcl-2 for 24 h, then treated with
or without VP-16 for another 24 h. The protein expression levels of
Bcl-2 and rate of apoptosis was analyzed. (D) The K562 cells were
transfected as indicated for 24 h, then treated with or without
VP-16 for another 24 h. The protein expression levels of Bax and
the rate of apoptosis were analyzed. The data are shown as the mean
and SD from three independent experiments, performed in triplicate.
*P<0.05, **P<0.01. HGF, hepatocyte growth factor; mcs, mock,
si, small interfering; ctrl, control. |
Overexpression of HGF leads to
activation of the PI3K/Akt signaling pathway
To further investigate the possible mechanisms
involved in the protective role of HGF, it was investigated whether
HGF affects the activation of the PI3K/Akt pathway. In the presence
of VP-16, overexpression of HGF resulted in the activation of the
PI3K/Akt signaling pathway compared with that in cells transfected
with pVITRO2-mcs (Fig. 4A). LY294002
was used to further investigate the role of the PI3K/Akt signaling
pathway (Fig. 4B). As indicated in
Fig. 4C, inhibition of the pathway
abrogated the protective effects of HGF against VP-16. Furthermore,
inhibition of PI3K/Akt signaling enhanced the cleavage of
caspase-3/-9 (Fig. 4D). The effects
of HGF overexpression on the protein expression level of Bcl-2 and
Bax were also reversed following inhibition of the PI3K/Akt
signaling pathway (Fig. 4E). Taken
together, these findings indicate that overexpression of HGF leads
to the activation of the PI3K/Akt signaling pathway, which is
essential for the protective role of HGF against VP-16-induced
apoptosis in the K562 cells.
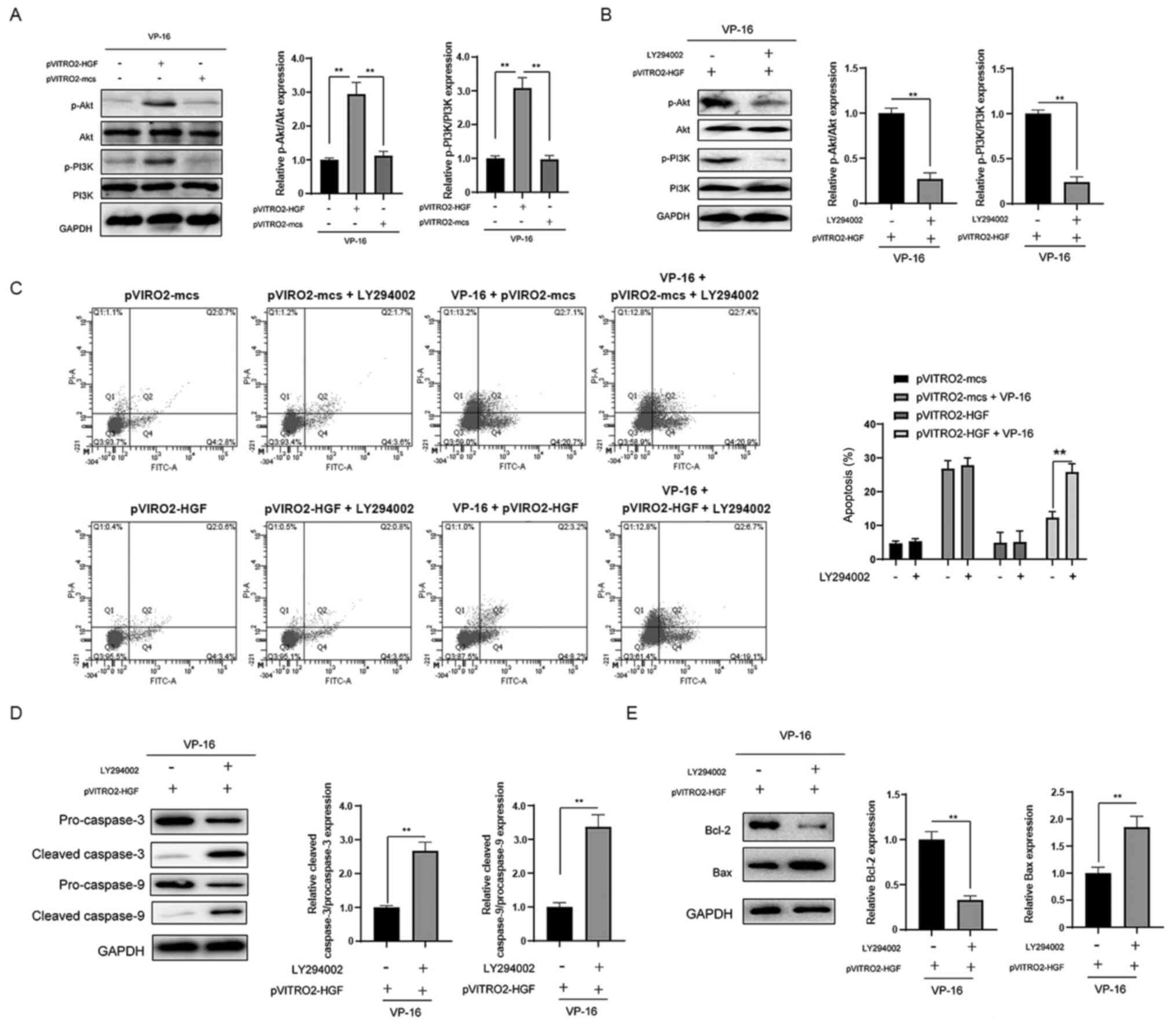 | Figure 4.Overexpression of HGF leads to the
activation of the PI3K/Akt signaling pathway in K562 cells. (A) The
cells were transfected as indicated for 24 h, then treated with
VP-16 for another 24 h. The expression level of proteins in the
PI3K/Akt signaling pathway was analyzed using western blot
analysis. (B) The cells were transfected with pVITRO2-HGF for 24 h,
then treated with or without LY294002. The cells were treated with
VP-16 for another 24 h, then the expression level of proteins in
the PI3K/Akt signaling pathway was analyzed using western blot
analysis. (C) The cells were transfected as indicated for 24 h with
or without LY294002, then treated with or without VP-16 for another
24 h. Apoptosis was subsequently analyzed. The cells were
transfected with pVITRO2-HGF for 24 h, treated with or without
LY294002, treated with VP-16 for another 24 h, then the expression
level of (D) caspase-3/-9 and (E) Bcl-2 and Bax was analyzed using
western blot analysis. The data are shown as the mean and SD from
three independent experiments, performed in triplicate.
**P<0.01. HGF, hepatocyte growth factor; p, phosphorylated; mcs,
mock. |
Discussion
CML is a malignant myeloproliferative disease, that
occurs in pluripotent hematopoietic stem cells and is the third
most common type of leukemia worldwide (14). Significant progress has been made
over the past few decades; however, there is still a lack of
satisfactory treatment for CML.
In the present study, to the best of our knowledge,
for the first time the role of HGF in the drug resistance of the
K562 cells was analyzed, and the potential mechanisms were also
investigated. It was found that overexpression of HGF significantly
inhibited the cytotoxicity of VP-16 in the K562 cells. Further
analysis into the molecular mechanism revealed that overexpression
of HGF inhibited VP-16-induced apoptosis. Overexpression of HGF
prevented the activation of caspase-3 and −9. The results from the
present study are consistent with previous studies, that also found
that HGF inhibited caspase activation in hepatocytes and human
proximal tubular epithelial cells (15,16).
Notably, another study reported that HGF was able to activate the
apoptosis signaling pathway by increasing caspase-3 activity in
sarcoma cells (17). This
discrepancy might be due to the different cell types and/or
treatments used, and further investigation is required to evaluate
the role of HGF in the progression of apoptosis.
It is well-known that there are two apoptotic
pathways, namely, the extrinsic and intrinsic pathways (11). The extrinsic and intrinsic pathways
are initiated by caspase-8 and −9, respectively, with both
ultimately leading to the activation of caspase-3 (11). As caspase-3/-9 activation was
analyzed in the present study, the intrinsic pathway was triggered.
It is also well-known that the intrinsic apoptosis pathway is
subjected to the regulation of the Bcl-2 family of proteins
(18). Therefore, it was
investigated whether HGF could affect the Bcl-2 proteins. It was
found that overexpression of HGF led to the increase in protein
expression level of Bcl-2 and the decrease in Bax protein
expression level. The results from the present study are consistent
with previous studies that reported that HGF enhanced the protein
and mRNA expression levels of Bcl-2 and inhibited the activation of
Bax (15,19). Bcl-2 proteins are promising targets
for the treatment of hematologic malignancies, and numerous small
molecule inhibitors have been designed to target the Bcl-2 proteins
(20).
The PI3K/Akt pathway is activated in a wide variety
of hematological malignancies, such as CML, acute myeloid leukemia,
diffuse large B-cell lymphoma and chronic lymphoblastic leukemia
(21). By promoting proliferation
and/or inhibiting apoptosis, the PI3K/Akt pathway is considered
vital for tumorigenesis (21), and
several studies have suggested that targeting the PI3K/Akt pathway
could overcome chemoresistance in CML cells (22,23). It
was found that the resistant CML cell line K562/ADM presented
higher PI3K/Akt activity compared with that in a sensitive cell
line (23). In the present study, to
understand why HGF overexpression attenuated VP-16-induced
apoptosis, the expression level of proteins in the PI3K/Akt
signaling pathway was further investigated. Notably, overexpression
of HGF activated the PI3K/Akt signaling pathway. This finding is in
accordance with previous studies, indicating that HGF could induce
the activation of the PI3K/Akt signaling pathway (24,25).
These results indicate that the HGF-mediated K562 cell response to
VP-16 is, at least in part, PI3K/Akt dependent.
In conclusion, it was found that overexpression of
HGF conferred resistance to VP-16 in the K562 cells. The inhibition
of apoptosis caused by HGF overexpression was associated with
suppression of caspase activation. Furthermore, the association
between HGF and the PI3K/Akt signaling pathway were further
elucidated, with the conclusion that overexpression of HGF could
activate this pathway. These initial findings are promising;
however, further comprehensive investigations are still required,
such as animal models. Overall, targeting HGF could be a potential
therapeutic target to overcome chemoresistance in the human CML
K562 cells.
Acknowledgements
Not applicable.
Funding
The present study was supported by Ningbo Natural Science
Foundation Project (grant no. 2021J026) and Zhejiang Medical
Science and Technology Project (grant no. 2022520748).
Availability of data and materials
The datasets used and/or analyzed are available from
the corresponding author upon reasonable request.
Authors' contributions
DC designed the experiments and revised the
manuscript. XZ performed the experiments and wrote the manuscript.
SH, HZ and ZG contributed to acquisition and analysis of data. XZ
and DC confirmed the authenticity of all the raw data. All authors
reviewed and approved the final manuscript.
Ethics approval and patients consent
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Quintás-Cardama A and Cortes J: Molecular
biology of bcr-abl1-positive chronic myeloid leukemia. Blood.
113:1619–1630. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shimada A: Hematological malignancies and
molecular targeting therapy. Eur J Pharmacol. 862:1726412019.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Holyoake TL and Vetrie D: The chronic
myeloid leukemia stem cell: Stemming the tide of persistence.
Blood. 129:1595–1606. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jiang W, Hiscox S, Matsumoto K and
Nakamura T: Hepatocyte growth factor/scatter factor, its molecular,
cellular and clinical implications in cancer. Crit Rev Oncol
Hematol. 29:209–248. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pons E, Uphoff CC and Drexler HG:
Expression of hepatocyte growth factor and its receptor c-met in
human leukemia-lymphoma cell lines. Leuk Res. 22:797–804. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim JG, Sohn SK, Kim DH, Baek JH, Lee NY,
Suh JS, Chae SC, Lee KS and Lee KB: Clinical implications of
angiogenic factors in patients with acute or chronic leukemia:
Hepatocyte growth factor levels have prognostic impact, especially
in patients with acute myeloid leukemia. Leuk Lymphoma. 46:885–891.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cerny-Reiterer S, Ghanim V, Hoermann G,
Aichberger KJ, Herrmann H, Muellauer L, Repa A, Sillaber C, Walls
AF, Mayerhofer M and Valent P: Identification of basophils as a
major source of hepatocyte growth factor in chronic myeloid
leukemia: A novel mechanism of BCR-ABL1-independent disease
progression. Neoplasia. 14:572–584. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Corbin AS, Agarwal A, Loriaux M, Cortes J,
Deininger MW and Druker BJ: Human chronic myeloid leukemia stem
cells are insensitive to imatinib despite inhibition of BCR-ABL
activity. J Clin Invest. 121:396–409. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Crowley LC, Elzinga BM, O'Sullivan GC and
McKenna SL: Autophagy induction by Bcr-Abl-expressing cells
facilitates their recovery from a targeted or nontargeted
treatment. Am J Hematol. 86:38–47. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu MY, Wang WZ, Liao FF, Wu QQ, Lin XH,
Chen YH, Cheng L, Jin XB and Zhu JY: Selective and effective
targeting of chronic myeloid leukemia stem cells by topoisomerase
II inhibitor etoposide in combination with imatinib mesylate in
vitro. Cell Biol Int. 41:16–23. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yu R, Yu BX, Chen JF, Lv XY, Yan ZJ, Cheng
Y and Ma Q: Anti-tumor effects of Atractylenolide I on bladder
cancer cells. J Exp Clin Cancer Res. 35:402016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu R, Yao J and Ren Y: A novel circRNA,
circNUP98, a potential biomarker, acted as an oncogene via the
miR-567/PRDX3 axis in renal cell carcinoma. J Cell Mol Med.
24:10177–10188. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Noh H, Park MS, Kim SH, Oh SJ, Zang DY,
Park HL, Cho DJ, Kim DW and Lee JI: Optimization of radotinib doses
for the treatment of Asian patients with chronic myelogenous
leukemia based on dose-response relationship analyses. Leuk
Lymphoma. 57:1856–1864. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mizui M, Isaka Y, Takabatake Y, Mizuno S,
Nakamura T, Ito T, Imai E and Hori M: Electroporation-mediated HGF
gene transfer ameliorated cyclosporine nephrotoxicity. Kidney Int.
65:2041–2053. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Suzuki A, Hayashida M, Kawano H, Sugimoto
K, Nakano T and Shiraki K: Hepatocyte growth factor promotes cell
survival from fas-mediated cell death in hepatocellular carcinoma
cells via Akt activation and Fas-death-inducing signaling complex
suppression. Hepatology. 32:796–802. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Arakaki N, Kazi JA, Kazihara T, Ohnishi T
and Daikuhara Y: Hepatocyte growth factor/scatter factor activates
the apoptosis signaling pathway by increasing caspase-3 activity in
sarcoma 180 cells. Biochem Biophys Res Commun. 245:211–215. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schenk RL, Strasser A and Dewson G: BCL-2:
Long and winding path from discovery to therapeutic target. Biochem
Biophys Res Commun. 482:459–469. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Konturek PC, Konturek SJ, Sulekova Z,
Meixner H, Bielanski W, Starzynska T, Karczewska E, Marlicz K,
Stachura J and Hahn EG: Expression of hepatocyte growth factor,
transforming growth factor alpha, apoptosis related proteins Bax
and Bcl-2, and gastrin in human gastric cancer. Aliment Pharmacol
Ther. 15:989–999. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yalniz FF and Wierda WG: Targeting BCL2 in
chronic lymphocytic leukemia and other hematologic malignancies.
Drugs. 79:1287–1304. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Neri LM, Cani A, Martelli AM, Simioni C,
Junghanss C, Tabellini G, Ricci F, Tazzari PL, Pagliaro P, McCubrey
JA and Capitani S: Targeting the PI3K/Akt/mTOR signaling pathway in
B-precursor acute lymphoblastic leukemia and its therapeutic
potential. Leukemia. 28:739–748. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bertacchini J, Heidari N, Mediani L,
Capitani S, Shahjahani M, Ahmadzadeh A and Saki N: Targeting
PI3K/AKT/mTOR network for treatment of leukemia. Cell Mol Life Sci.
72:2337–2347. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen JR, Jia XH, Wang H, Yi YJ, Wang JY
and Li YJ: Timosaponin A-III reverses multi-drug resistance in
human chronic myelogenous leukemia K562/ADM cells via
downregulation of MDR1 and MRP1 expression by inhibiting PI3K/Akt
signaling pathway. Int J Oncol. 48:2063–2070. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ding X, Xi W, Ji J, Cai Q, Jiang J, Shi M,
Yu Y, Zhu Z and Zhang J: HGF derived from cancerassociated
fibroblasts promotes vascularization in gastric cancer via PI3K/AKT
and ERK1/2 signaling. Oncol Rep. 40:1185–1195. 2018.PubMed/NCBI
|
|
25
|
Kuang W, Deng Q, Deng C, Li W, Shu S and
Zhou M: Hepatocyte growth factor induces breast cancer cell
invasion via the PI3K/Akt and p38 MAPK signaling pathways to
up-regulate the expression of COX2. Am J Transl Res. 9:3816–3826.
2017.PubMed/NCBI
|