Introduction
Cholangiocarcinoma (CCA) is a rare and aggressive
type of cancer arising from cholangiocytes of the biliary tract. It
is the second most common primary liver malignancy and its
incidence is increasing worldwide, with the highest CCA incidence
rates reported in countries in Southeast Asia (1–3). CCA
is characterized as asymptomatic until it is diagnosed having
progressed to the advanced stage (4,5).
Despite advances in surgical, medical and molecular-targeted
therapy the 5-year survival rates of patients with CCA have
remained low in recent decades (6). Therefore, it is important to identify
more effective therapeutic strategies against CCA, especially those
that overcome drug resistance and improve clinical outcomes.
All-trans-retinoic acid (ATRA) is a natural
active metabolite of vitamin A, which serves an essential role in
several physiological processes, and aids cell growth and
development, particularly in early embryogenesis (7–9).
ATRA acts as a pan-agonist of retinoic acid receptors (RARs), which
consist of RARA, RARB and RARG subtypes (10). ATRA activates RARs and forms
heterodimers with retinoid-X receptors (RXRs), which bind to the
retinoic acid response elements and initiate the transcription of
retinoic acid-targeted genes. Numerous target genes are associated
with cell differentiation and have particular relevance to the
retinoid-mediated regulation of myelopoiesis (7,8).
Currently, ATRA is clinically used to treat acute
promyelocytic leukemia by promoting terminal differentiation of
hematopoietic progression (11–13).
ATRA has also been studied as a preventative and therapeutic agent
against several types of cancer, including breast cancer,
hepatocellular carcinoma, esophageal cancer and thyroid cancer
(14–17), and as an adjunct medication to
increase chemotherapeutic response (18–21).
Previously, the effects of ATRA on apoptosis, proliferation,
migration and invasion have been reported in CCA (22). In addition, a previous report
demonstrated that upregulating RARB in CCA tissue enhanced
apoptosis in CCA and thus improved CCA chemotherapeutic sensitivity
(23). Therefore, it may be
suggested that, in CCA, RARB has a tumor-suppressive role.
Previous studies have indicated that retinoids and
vitamin A derivatives cause mitochondrial dysfunction, and trigger
reactive oxygen species (ROS) production to induce cellular damage
and apoptosis (24,25). Additionally, ATRA has been reported
to induce ROS production in Sertoli cells and NB4 cells (26,27).
Furthermore, ROS has been shown to be associated with the
activation of apoptosis in CCA cells (28). In CCA, the high expression of
nuclear factor erythroid 2-related factor 2 (NFE2L2 or NRF2), which
is the master regulator of cytoprotective and antioxidant enzymes,
may contribute to promote cancer cell growth, anti-apoptosis and
chemotherapeutic resistance (29,30).
Moreover, it has been suggested that ATRA may act as a potential
inhibitor of NRF2 activation (31).
Based on these aforementioned studies, the
therapeutic potential and underlying mechanisms of ATRA against CCA
should be elucidated. Therefore, the present study aimed to explore
the apoptotic effect and underlying molecular mechanisms of ATRA in
CCA cells. In addition, the potential use of ATRA for enhancing the
sensitivity of anticancer agents was investigated.
Materials and methods
Cell lines and cell culture
In the present study, two human CCA cell lines,
KKU-100 and KKU-213B, were established and generously provided by
Professor Banchob Sripa (Department of Pathology, Faculty of
Medicine, Khon Kaen University, Khon Kaen, Thailand). Cell line
authentication was verified by highly polymorphic short tandem
repeat (STR) analysis; DNA markers of 23 STR loci and the sex
marker (amelogenin) were analyzed using the AmpFLSTR Identifiler
PCR Amplification kit (Applied Biosystems; Thermo Fisher
Scientific, Inc.). Based on the STR analysis, the cell lines used
in the present study, KKU-213B and KKU-100, shared similar markers
and matched well with the partial STR profile of cell line
identity, as described previously (32,33).
Both cell lines were routinely grown in monolayer
cultures in Ham's F12 medium (Gibco; Thermo Fisher Scientific,
Inc.) pH 7.4, supplemented with 10% (v/v) fetal bovine serum
(Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin G and
50 µg/ml gentamicin sulfate. Cells were maintained under an
atmosphere containing 5% CO2 at 37°C and were
sub-passaged every 3 days using 0.5% trypsin-EDTA. Cell numbers
were counted using a hemocytometer.
Sulforhodamine B (SRB) assay
Cell viability was detected using the SRB assay.
Briefly, KKU-100 and KKU-213B cells were treated with 0.00, 1.25,
2.50, 5.00, 10.00 and 20.00 µM ATRA (R2625; MilliporeSigma) in
serum-free Ham's F12 medium for 12, 24, and 48 h under an
atmosphere containing 5% CO2 at 37°C. The cells were
then incubated with 100 µl ice-cold 10% of trichloroacetic acid at
4°C for 1 h and washed with deionized water. Subsequently, the
cells were stained with 50 µl 0.4% SRB (MilliporeSigma) in 1%
acetic acid at room temperature for 30 min. The cells were
solubilized in 10 mM Tris-base (pH 10.5) solution, and the
absorbance was measured at 540 nm using a microplate reader. Cell
cytotoxicity was expressed in terms of a percentage compared with
the untreated control group; half-maximal inhibitory concentration
(IC50) was calculated from the dose-response curves. To
study the effects of ATRA on the sensitivity of anticancer drugs,
cells were treated with ATRA or anticancer drugs or the combined
treatment of ATRA and anticancer drugs for 48 h at 37°C before SRB
assay. The concentrations of drugs were as follows: 2.5 µM ATRA;
30, 100, 300 µM 5-fluorouracil (5-FU); 0.001, 0.010, 0.100 µM
gemcitabine (Gem); 2.5, 5.0, 10.0 µM cisplatin (Cis); and 0.01,
0.10 and 1.00 µM doxorubicin (Doxo). Doxo, Cis and 5-FU were from
Boryung pharmceutical and Gem was from Eli Lily.
Annexin V-PE/7-AAD cell apoptosis
analysis
For the apoptosis assay, KKU-100 and KKU-213B cells
were treated with 0.00, 1.25, and 5.00 µM ATRA in serum-free Ham's
F12 medium for 48 h at 37°C. Subsequently, the cells were
collected, washed with PBS twice and resuspended in 1X Annexin V
binding buffer included in the kit at a concentration of
1×106 cells/ml. The cell suspension was then incubated
with Annexin V-PE and 7-ADD (BD Pharmingen™ PE Annexin V Apoptosis
Detection Kit I; BD Biosciences) for 15 min at room temperature in
the dark, after which flow cytometry was performed using BD FACS
Canto™ II and FACSDiva™ software v6.1.3 (both from BD Biosciences).
To study the role of ROS in apoptosis induction by ATRA, cells were
pre-treated with 2.0 mM NAC (A7250, Sigma Chemical) or 0.5 mM
TEMPOL (176141, Sigma Chemical) for 3 h and then incubated with 0,
1.25 and 2.5 µM ATRA for 48 h at 37°C before flow cytometry
analysis.
Dihydroethidium (DHE) staining
analysis of ROS
For cellular ROS detection, KKU-100 and KKU-213B
cells were treated with 0.00, 1.25 and 2.50 µM ATRA combined with
25 µM DHE (Calbiochem; Merck KGaA) in serum-free Ham's F12 medium;
the cells were incubated in a humidified atmosphere containing 5%
CO2 at 37°C for 90 min. Subsequently, the cells were
measured for the intensity of fluorescent signals of ethidium using
a Gemini XPS fluorescent plate reader (Molecular Devices, LLC.),
with excitation and emission wavelengths of 518 and 605 nm,
respectively. Furthermore, the fluorescence signal of ethidium was
captured under a 4× magnification power florescence microscope with
a G-2A filter.
Caspase activity assays
To assess the protease activities of caspase-3, −8
and −9, KKU-100 and KKU-213B cells were treated with 0.00, 1.25 and
2.50 µM ATRA for 12 h at 37°C. The cells were then trypsinized and
the cell pellets were lysed on ice for 30 min with 50 µl lysis
buffer included in the kit per 1×106 cells.
Subsequently, the supernatant was transferred into a 96-black well
clear-bottom plate. To measure caspase-3 activity, the supernatant
was incubated with the caspase-3 substrate (Z-DEVD-AMC, cat. no.
E13183) according to the manufacturer's instructions (Molecular
Probes; Thermo Fisher Scientific, Inc.). The fluorescent signals
were detected at excitation and emission wavelengths of 340 and 440
nm, respectively. To assess the activities of caspase-8 and −9, the
supernatant was incubated with the caspase-8 substrate (IETD-AFC;
cat. no. ab39534; Abcam) or caspase-9 substrate (Ac-LEHD-AFC, cat.
no. 218765; Calbiochem) according to the manufacturer's
instructions. The fluorescent signals were detected at excitation
and emission wavelengths of 400 and 440 nm, respectively.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Following treatment with 1.25 and 2.50 µM ATRA for
48 h at 37°C, total RNA was isolated using TRIzol® LS
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. For RT, a mixture of total RNA and 5X
iScript™ Reverse Transcription Supermix for RT-qPCR (Bio-Rad
Laboratories Inc.) was mixed with RNase-free water, and the
reaction was performed in a C1000™ Thermal Cycler (Bio-Rad
Laboratories, Inc.). The conditions for cDNA synthesis included
priming for 5 min at 25°C followed by RT for 30 min at 42°C; the
reaction was terminated by incubation for 15 min at 70°C. A mixture
consisting of specific primers, 2X QPCR Green Master Mix
(biotechrabbit GmbH), cDNA and sterile water underwent qPCR using a
Light Cycler® 480 II/384 (Roche Applied Science). The
thermocycling conditions were as follows: 95°C for 3 min, followed
by 40 cycles at 95°C for 15 sec and 60°C for 31 sec, 1 cycle of
melting curve (95°C for 5 s, 72°C for 5 s, and 97°C continuous),
and a cooling cycle (40°C for 10 min). To verify the purity of the
products, a melting curve analysis was performed after each run. To
quantify the relative expression levels of genes, relative
quantitation using the standard curve method was performed
(29). The expression levels of
target mRNA were expressed as a ratio to ACTB mRNA. Primers
used are listed in Table SI.
Western blot analysis
After treatment with 1.25 and 2.50 µM ATRA for 6 h
at 37°C, cells were lysed using RIPA cell lysis buffer (Amresco,
LLC), and protein concentrations were determined using the Bradford
reagent (Bio-Rad Laboratories Inc.) according to the manufacturer's
instructions. Whole-cell protein extracts were separated by
SDS-PAGE on 10% gels using an SE 260 mini-vertical gel
electrophoresis unit (Hoefer, Inc.). The proteins were then
transferred onto a PVDF membrane (Immobilon®-P; cat. no.
IPVH00010; MerckMillipore) using Owl™ HEP-1 Semidry Electroblotter
(cat. no. HEP1; Thermo Fisher Scientific, Inc.) and blocked with 5%
(w/v) skimmed milk at room temperature for 1 h. Subsequently, the
membranes were incubated with the following primary antibodies:
Anti-AIF, anti-Bax, anti-cytochrome c, anti-RARA, anti-RARB,
anti-ACTB (all from Santa Cruz Biotechnology, Inc.; antibodies are
described in Table SII) and
anti-RARG (Cell Signaling Technology Inc.). The membranes were then
incubated with appropriate horseradish peroxidase-conjugated
secondary antibodies. The antibodies used are listed in Table SII. The protein bands were
detected using a Luminata™ Forte Western HRP Substrate (Merck
Millipore Corporation) under a ChemiDoc™ MP Imaging system (Bio-Rad
Laboratories, Inc.). To semi-quantify the target protein expression
levels, the intensity of the target protein bands was analyzed
using Image Lab 6.0 software (Bio-Rad, Hercules) and normalized to
that of ACTB.
CRISPR/Cas9-mediated RARγ
knockout
To assess the role of RAR in the response of CCA
cells to ATRA, CRISPR/Cas9-mediated knockout was performed. The
RARG CRISPR cloning vector pLentiCRISPR v2 was purchased from
GenScript® (RARG CRISPR guide RNA 2; Cat. no. SC1805),
sequence of the gRNA is shown in Table SIII. Transfection of RARG CRISPR
into the cells was carried out using Lipofectamine® LTX
with Plus Reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. KKU-213B cells were
transfected with 0.5 µg RARG CRISPR in a liposome complex for 24 h
at 37°C and maintained for cell growth. After that, the transfected
cells were selected using 0.5 µg/ml puromycin followed by single
clone selection. The single clones were assessed for RARG protein
expression using western blot analysis and DNA sequencing was
performed to confirm RARA knockout. PCR amplification was performed
using C1000™ Thermal Cycler (BioRad) and the conditions were as
follows: an initial denaturation at 94°C for 2 min followed by 35
amplification cycles of 30 sec denaturation at 94°C, 30 sec
annealing at 60°C, 45 sec elongation at 72°C and one cycle of final
elongation at 72°C for 5 min. The purified PCR products were
subjected to nucleotide sequencing by Apical Scientific Sdn. Bhd.
The Sanger sequences of KKU-M213B cells with detected
CRISPR-mediated mutation in exon 1 of RARG (NM_000966.6) are
shown in Data S1 and the
sequencing traces of RARG CRISPR is shown in Fig. S1. RARG CRISPR mediated indel which
encodes a truncated protein with a 133 amino acid sequence, as
predicted using web.expasy.org/translate/(Data S1).
Small interfering RNA (siRNA)
siRNAs are a frequently used tool to study the role
of proteins of interest on cell function. siRNA transfection is a
knockdown technique that has a transient effect on the expression
of the targeted protein; by contrast, CRISPR/Cas9 is a knockout
technique that has permanent effect. However, the CRISPR/Cas9
method is a complex and time-consuming process with a high failure
rate. In the present study, knockout cells were developed using the
CRISPR/Cas9 technique for all three RARs (data not shown); however,
only CRISPR/Cas9-mediated RARG knockout cells were successfully
created. Therefore, the siRNA technique was used to evaluate the
role of the other RARs on the response of CCA cells to ATRA.
Notably, the use of different techniques for knockdown/knockout may
be a limitation of the present study.
Since the CRISPR/Cas9 transfection of at least two
different gRNA target sequences for RARA or RARB failed (data not
shown), RARA and RARB siRNA transfection was performed. RARA, RARB
and non-targeting siRNA were purchased from GE Healthcare
Dharmacon, Inc., and a detailed list of siRNA sequences is provided
in Table SIII. Transfection of
siRNA into cells was performed using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. Briefly, KKU-213B cells at a density of
1.5×105 cells/well were seeded into a 6-well plate, and
cells were transfected with 100 pmol RARA siRNA, RARB siRNA or
non-targeting (NT) siRNA. The liposome-siRNA complex was added to
cells in serum-free medium without antibiotics for 6 h at 37°C.
Subsequently, the transfected cells were incubated further in
culture medium for 48 h at 37°C; the control group transfected with
non-targeting siRNA was cultured under the same conditions. The
efficiency of siRNAs was determined by western blotting of RARA and
RARB proteins. siRNA-transfected cells were used to assess effects
of ATRA on cell viability.
Statistical analysis
Data are presented as the mean ± SD from three
independent experiments. Statistical comparisons between the
control and treatment groups were performed by one-way ANOVA
followed by Tukey's post hoc test using GraphPad Prism v8.0
software (GraphPad Software, Inc.). P<0.05 was considered to
indicate a statistically significant difference.
Results
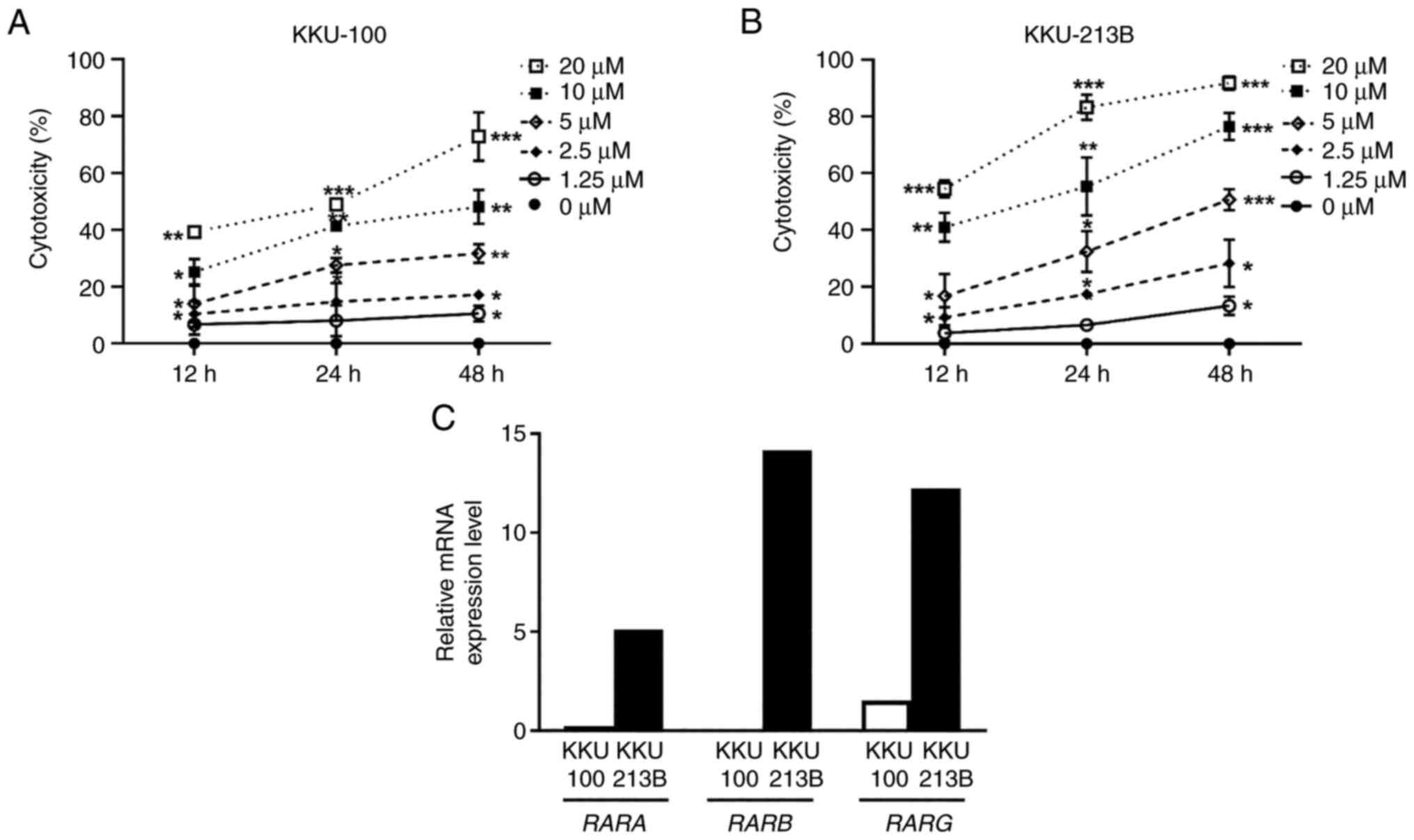
ATRA reduces CCA cell viability
The present study evaluated the effect of ATRA on
the viability of KKU-100 and KKU-213B cells. The SRB assay revealed
that ATRA significantly reduced the viability of both CCA cell
lines in a dose- and time-dependent manner (Fig. 1A and B). The IC50 values
at 48 h were 10.29±3.86 µM for KKU-100 cells and 4.58±1.90 µM for
KKU-213B cells. Subsequently, RT-qPCR analysis was used to assess
the basal mRNA expression levels of RARs; KKU-100 expressed low
levels of RARA, RARB and RARG, whereas KKU-213B cells
expressed higher levels of these receptors (Fig. 1C). These findings indicated that
KKU-213B cells were more sensitive to ATRA cytotoxicity than
KKU-100 cells.
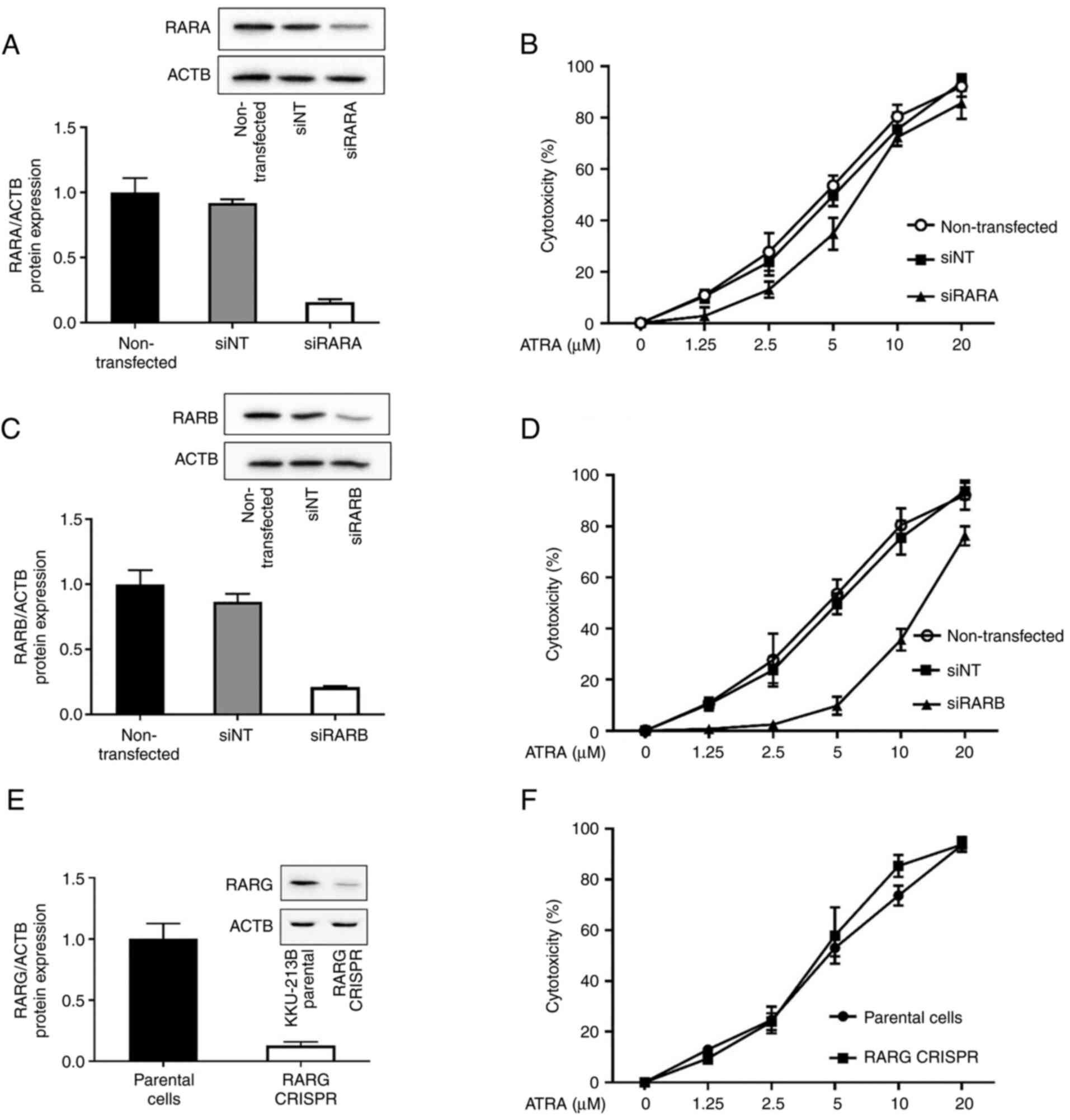
ATRA cytotoxicity in CCA cells is
RARB-dependent
ATRA is a pan-agonist of RARs (RARA, RARB and RARG).
ATRA binding to RARs has been reported to be sufficient for RAR-RXR
heterodimers to confer ligand-dependent activation of target gene
transcription (10), which can
affect cell growth and development, and responses to toxicant
exposure. To examine whether RARA, RARB and RARG contributed to
ATRA cytotoxicity in CCA cells, a loss-of-function approach was
used. KKU-213B cells with high RAR expression were transfected with
siRARA, siRARB, non-targeting control siRNA or RARG CRISPR, and the
expression levels of RARs were validated by western blotting.
Knockdown of RARA and RARB using siRNA efficiently decreased the
protein expression levels of RARA and RARB (Fig. 2A and C). Compared with in parental
KKU-213B cells, CRISPR/Cas9-mediated RARG knockout cells exhibited
a clear loss of RARG expression (Fig.
2E). Both siRNA transfection and CRISPR/Cas9 are very useful
techniques to study the role of the proteins of interest on cell
function. Notably, the effects of siRNA are transient, whereas
those of CRISPR/Cas9 are permanent; however, both techniques
perform a similar function and effectively suppressed the
expression levels of RAR subtypes by >80%.
When the RAR-deficient cells were tested for ATRA
cytotoxicity, the results revealed that the IC50 values
of 48-h ATRA treatment were not altered by siRARA or RARG CRISPR
compared with in cells transfected with the non-targeting control
siRNA or in KKU-213B parental cells, respectively (Fig. 2B and F; Table I). Notably, knockdown of RARβ by
siRNA caused a 3-fold increase in the IC50 value of ATRA
(14.7±2.4 µM) when compared with the IC50 value observed
in cells transfected with the non-targeting control siRNA (4.8±2.5
µM) (Fig. 2D; Table I). These results suggested that
RARβ was, at least in part, required for ATRA cytotoxicity in CCA
cells.
 | Table I.IC50 values of ATRA in
KKU-213B cells following RAR knockdown/knockout. |
Table I.
IC50 values of ATRA in
KKU-213B cells following RAR knockdown/knockout.
| Condition | IC50 of
48-h ATRA, µM |
|---|
| siNT | 4.8±2.5 |
| siRARA | 6.3±4.8 |
| siRARB | 14.7±2.4 |
| Parental cells | 4.6±2.2 |
| RARG CRISPR | 4.1±2.6 |
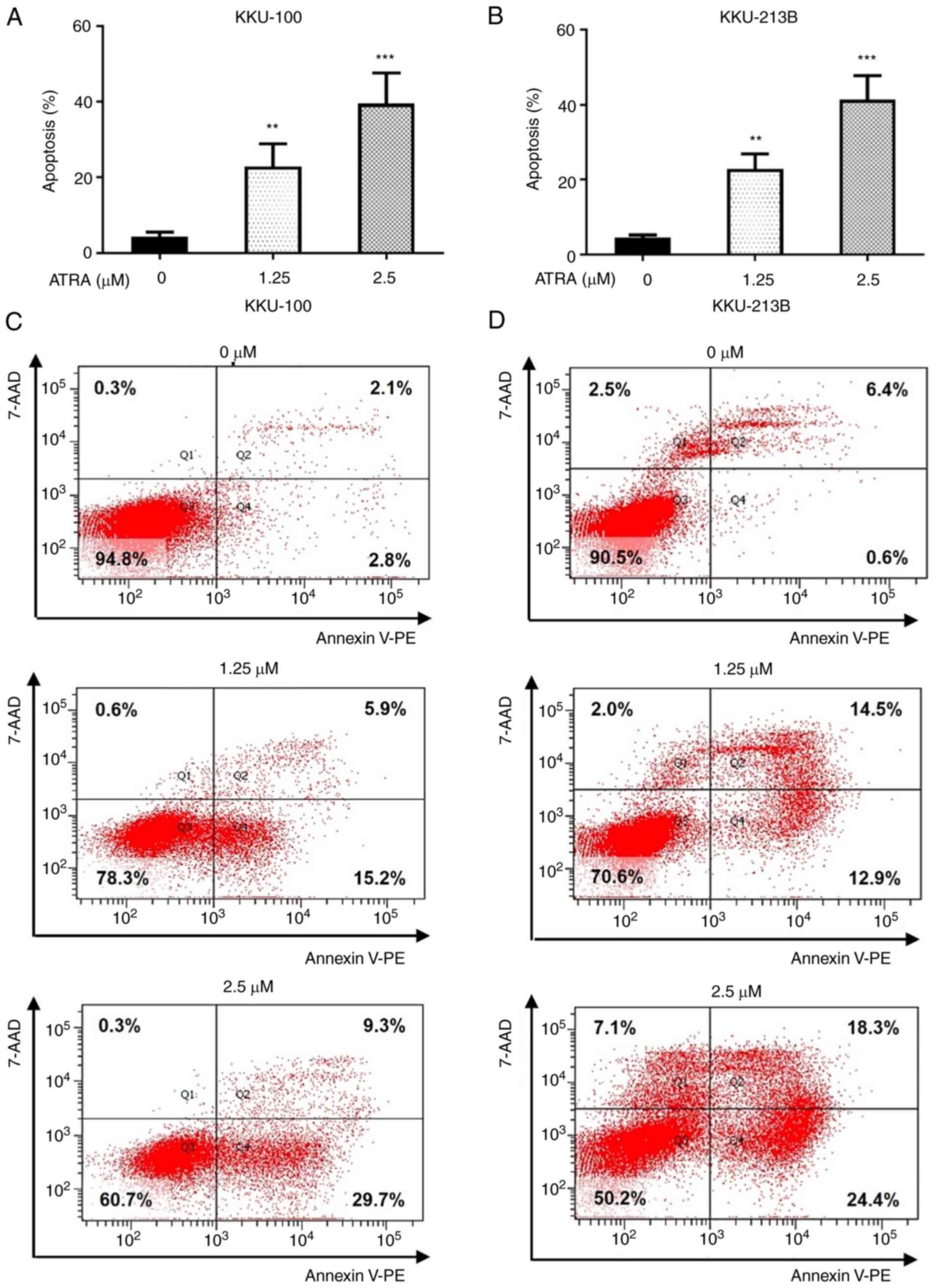
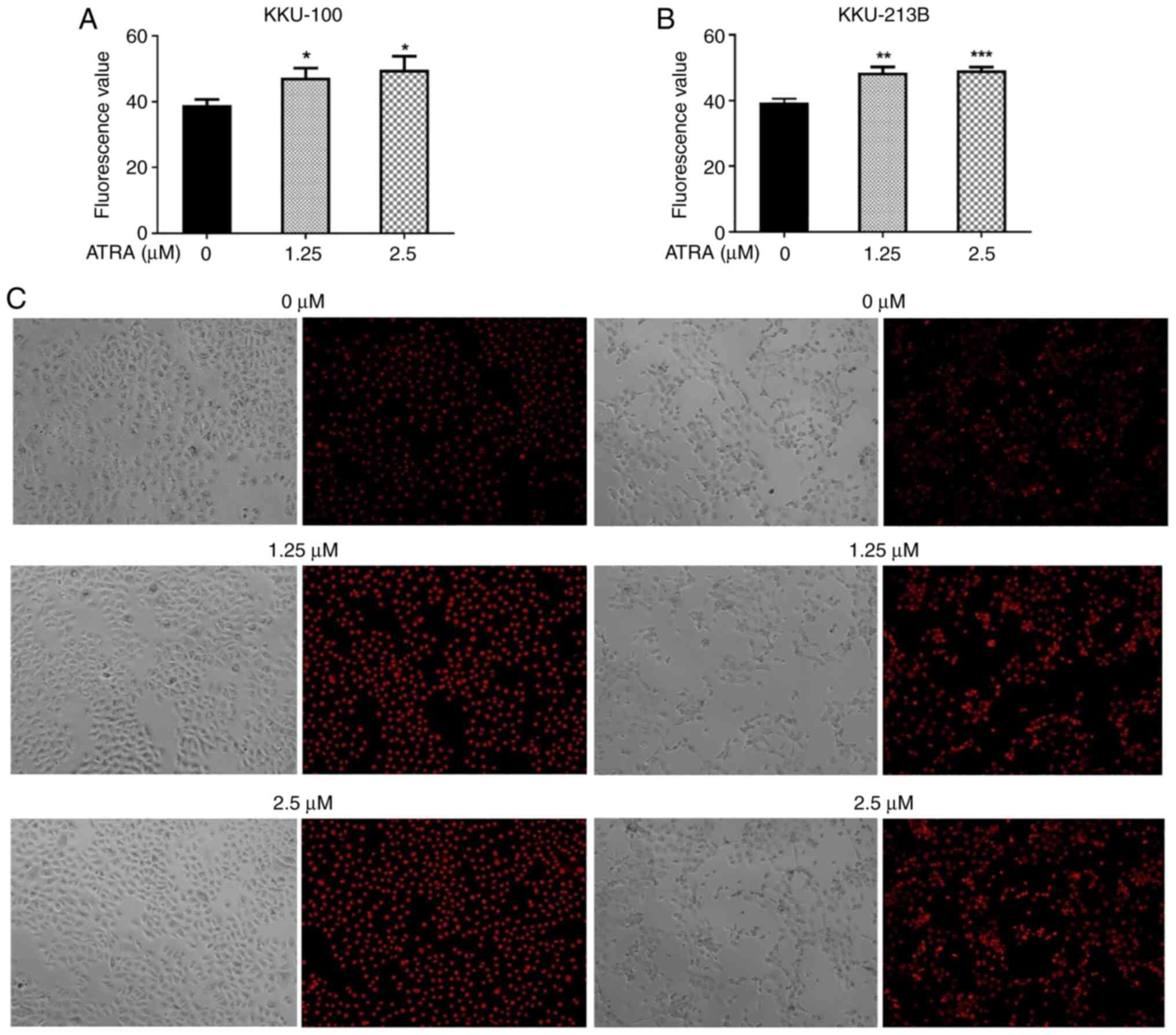
ATRA dose-dependently induces CCA cell
apoptosis
To further examine the cell death mechanism induced
by ATRA treatment in CCA cells, apoptosis was evaluated by flow
cytometry. We selected 1.25 and 2.5 µM ATRA, which induced
cytotoxicity in both CCA cell lines, to explore the time sequencing
of the apoptosis cascade. The results revealed that ATRA
significantly increased the number of Annexin V-PE-positive cells
in Q2 and Q4, which is an index of apoptotic cells, compared with
in the untreated control group (Fig.
3). Following treatment with 1.25 and 2.5 µM ATRA for 48 h, the
apoptotic rates of KKU-100 cells were 22 and 38%, whereas the
apoptotic rates of KKU-213B cells were 22 and 41%, respectively
(Fig. 3A and B). The
representative images from one experiment of flow cytometry were
shown in Fig. 3C for KKU-100 and
Fig. 3D for KKU-213B cells. These
results indicated that ATRA induced cytotoxicity in CCA cells via
the induction of apoptosis.
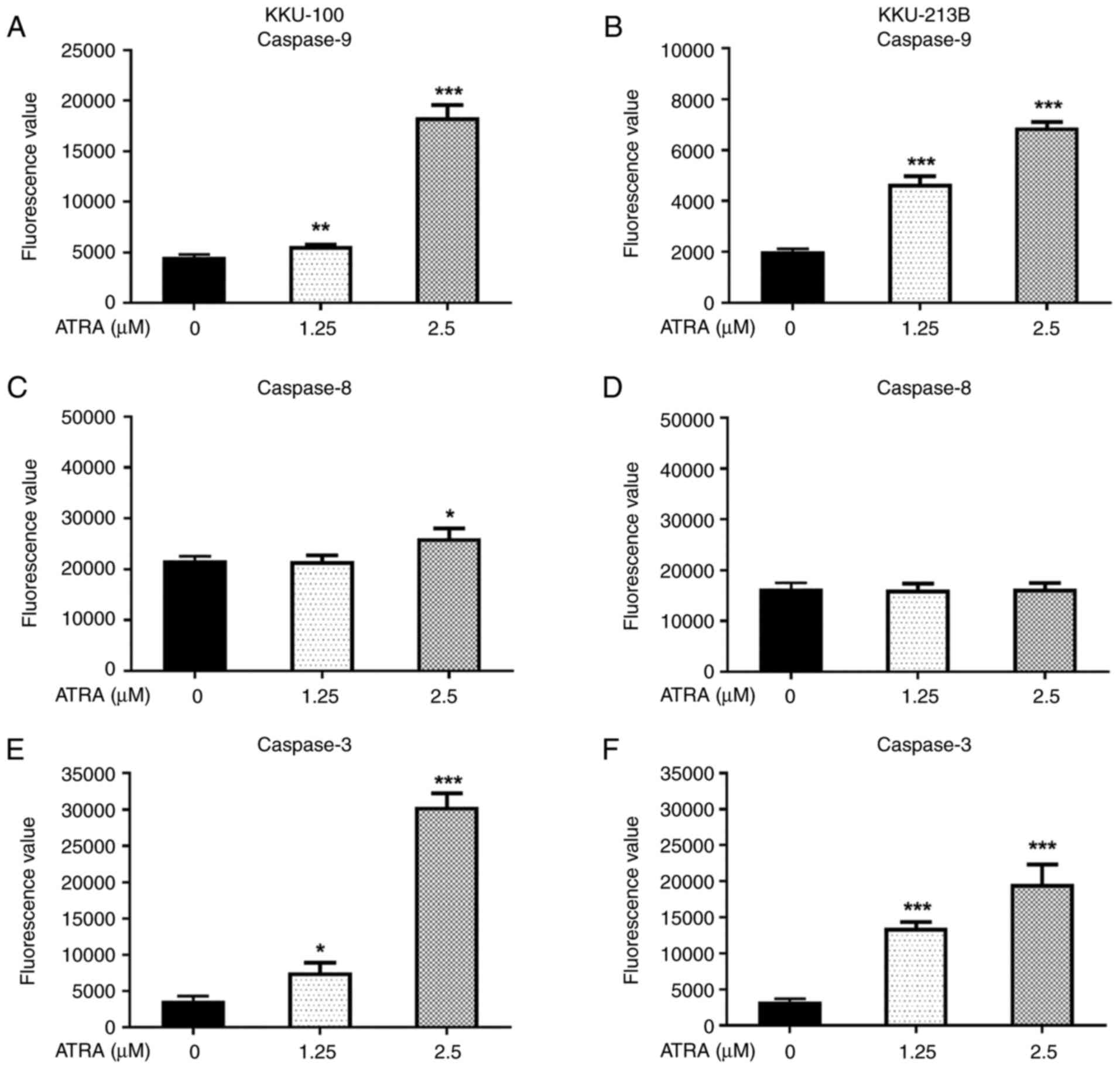
ATRA-induced CCA cell apoptosis is
mediated through activation of caspase-3 and −9
Since ATRA was revealed to induce apoptosis, the
present study further explored the mechanisms underlying its
effects. The pathways of apoptotic induction following ATRA
treatment were examined using enzymatic caspase activity assays.
Following treatment with 1.25 and 2.5 µM ATRA for 12 h, there was
an increase in caspase-9 and −3 enzyme activity in both CCA cell
lines compared with that in the untreated control cells (P<0.05;
Fig. 4A, B, E and F). These
results indicated that the mitochondrial apoptotic pathway was
activated. Notably, ATRA at a concentration of 2.5 µM significantly
increased caspase-8 activity in KKU-100 cells (Fig. 4C), but not in KKU-213B (Fig. 4D); suggesting ATRA-induced
activation of the extrinsic pathway may be dependent on
concentration and cell type.
ATRA increases the expression levels
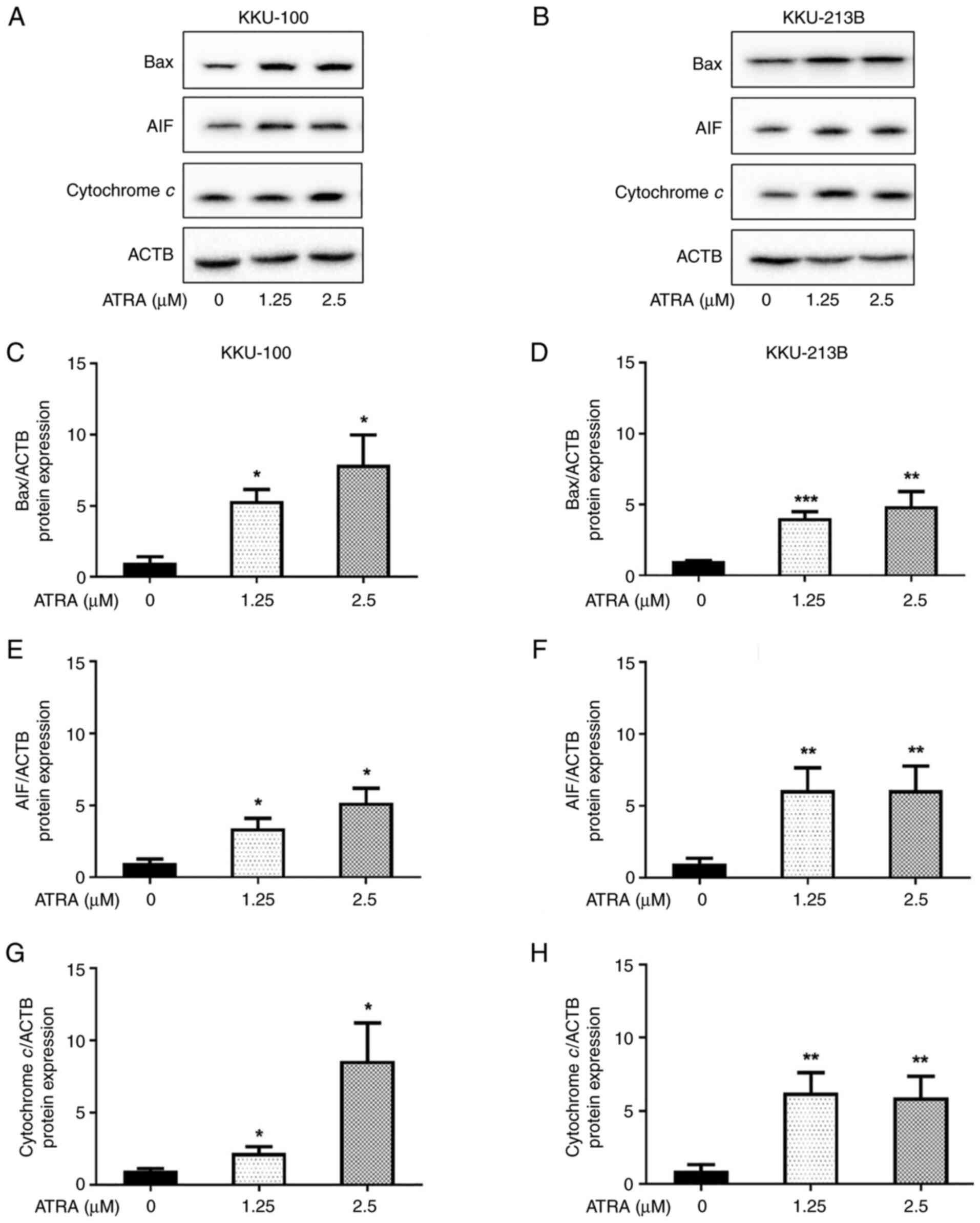
of apoptosis-associated proteins, Bax, AIF and cytochrome c, in CCA
cells
The mechanism underlying ATRA-induced apoptosis was
further investigated. The effect of ATRA on the expression levels
of pro-apoptotic proteins, including Bax, cytochrome c and
AIF, were determined by western blot analysis. Following treatment
with ATRA (1.25 and 2.5 µM) for 6 h, the protein expression levels
of Bax, cytochrome c and AIF were significantly increased in
KKU-100 (Fig. 5A, C, E and G) and
KKU-213B (Fig. 5B, D, F and -H)
cells. These results revealed that ATRA upregulated the expression
levels of apoptosis-inducing proteins in CCA cells, resulting in
the initiation of apoptosis.
ATRA increases the cellular content of
ROS in CCA cells
Several studies have reported that increased ROS are
associated with the initiation and activation of apoptosis in
various types of cancer, including CCA (28,34,35).
To investigate the effects of ATRA on intracellular ROS content, a
DHE assay was performed. Following treatment with ATRA (1.25 and
2.5 µM) for 90 min, the intracellular ROS levels were significantly
increased in KKU-100 (Fig. 6A) and
KKU-213B cells (Fig. 6B).
Phase-contrast microscopy revealed that ATRA-treated and untreated
control cells maintained their original morphology and cell density
and the representative images of ethidium staining in the nucleus
are shown in Fig. 6C.
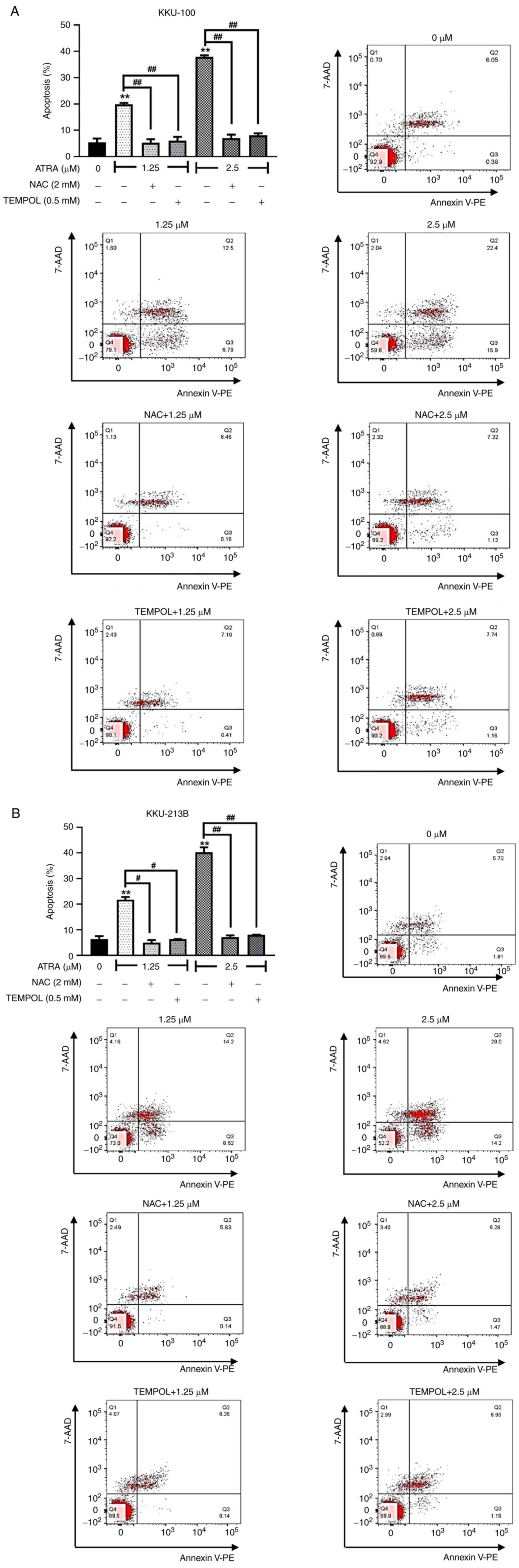
ATRA-induced cellular ROS accumulation
contributes to CCA cell apoptosis
Previous observations revealed that ATRA increased
cellular ROS levels in CCA cells. Subsequently, the causal
relationship between ROS and apoptosis induction by ATRA was
further investigated. KKU-100 and KKU-213B cells were pre-treated
with ROS scavengers NAC and TEMPOL, and were then exposed to 1.25
and 2.5 µM ATRA prior to apoptosis analysis by flow cytometry. When
cells were pre-treated with NAC or TEMPOL, these ROS scavengers
completely blocked the apoptotic effect of ATRA (KKU-100, Fig. 7A; KKU-213B, Fig. 7B). These results supported that
ATRA-induced ROS accumulation was essential for the apoptosis of
CCA cells. Moreover, in cells treated with a high dose of ATRA (5
µM), NAC and TEM also partially suppressed ATRA-induced apoptosis
(Figs. S2 and S3). NAC or TEMPOL partly reduce the
percentage of cell death by high dose of ATRA, suggesting non-ROS
mechanisms involving in ATRA cytotoxicity. These findings indicated
that ATRA induced the apoptosis of CCA cells by both ROS-dependent
and ROS-independent mechanisms.
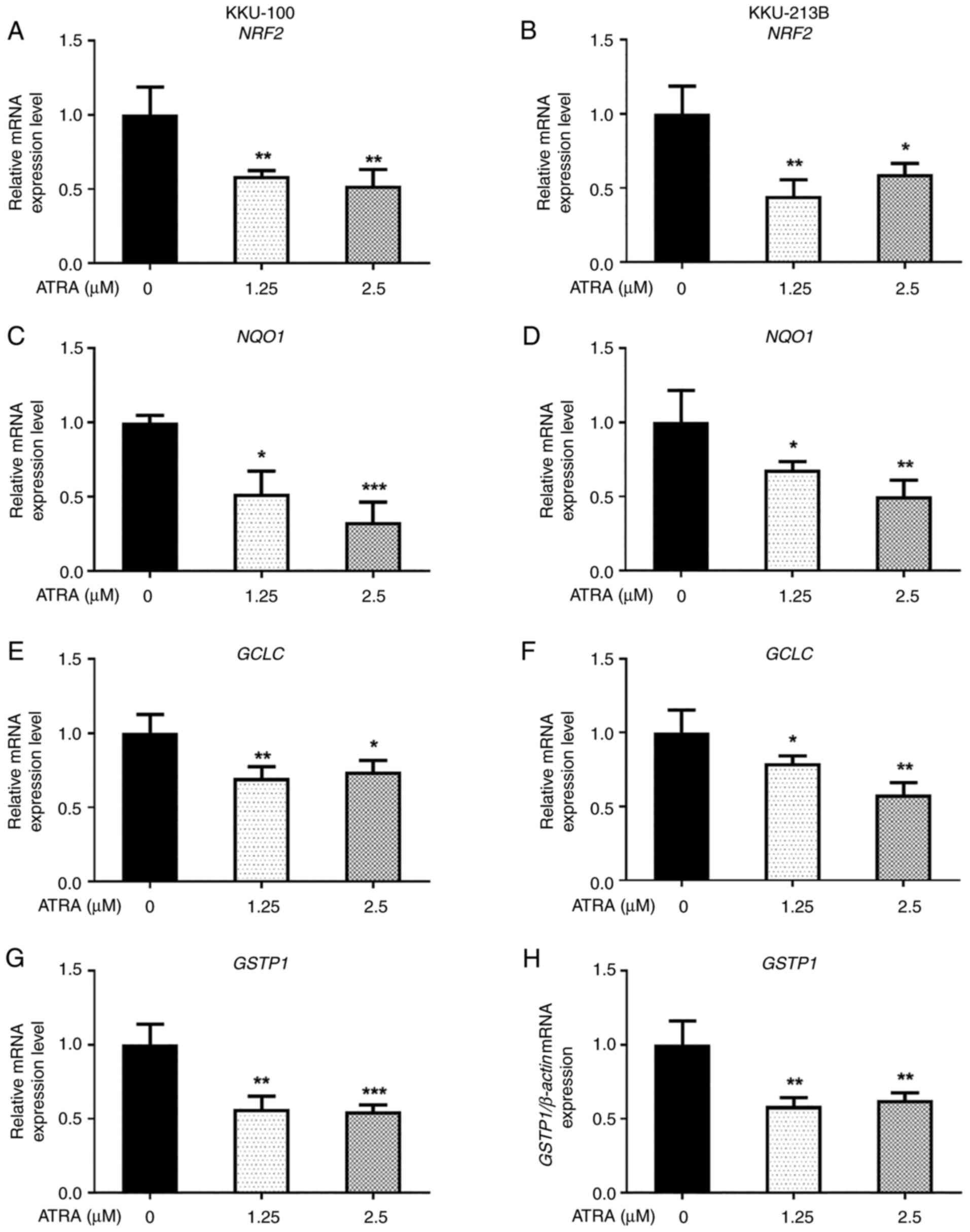
ATRA downregulates the expression
levels of NRF2 and NRF2 target antioxidant genes in CCA cells
Since NRF2 signaling is the primary regulator for
the balance of cellular ROS levels and ROS accumulation was
essential for the ATRA-induced apoptosis of CCA cells, the present
study investigated the effects of ATRA on the expression levels of
NRF2 and NRF2 target genes, which encode for cellular
antioxidant proteins. The results revealed that the expression
levels of NRF2 and NRF2 target genes, including
NQO1, GCLC and GSTP1, were decreased in both KKU-100
(Fig. 8A, C, E and G) and KKU-213B
(Fig. 8B, D, F and H) cells
following ATRA treatment. These results suggested that ROS
accumulation after ATRA treatment may be caused by downregulation
of the NRF2 pathway, leading to the induction of CCA cell
apoptosis.
ATRA enhances the cytotoxicity of
anticancer drugs partly through NRF2 downregulation
To evaluate whether ATRA could enhance the cytotoxic
effect of anticancer drugs, an SRB assay was performed. Cells were
treated with 2.5 µM ATRA or anticancer drugs, or a combination of
ATRA and anticancer drugs, including 5-fluorouracil, gemcitabine,
cisplatin and doxorubicin, for 48 h. In KKU-100 cells, ATRA
significantly increased the cytotoxicity of 10 µM cisplatin
(Fig. 9E); however, it had no
effects on the cytotoxicity of other drugs used in the present
study (Fig. 9A, B and D). In
KKU-213B cells, ATRA significantly enhanced the cytotoxicity of 100
and 300 µM 5-fluorouracil and 2.5, 5, 10 µM cisplatin (Fig. 9B and F). ATRA treatment resulted in
markedly improved cisplatin sensitivity in KKU-213B cells compared
with in KKU-100 cells. ATRA did not change the sensitivity of
KKU-213B cells to gemcitabine and doxorubicin (Fig. 9D and H).
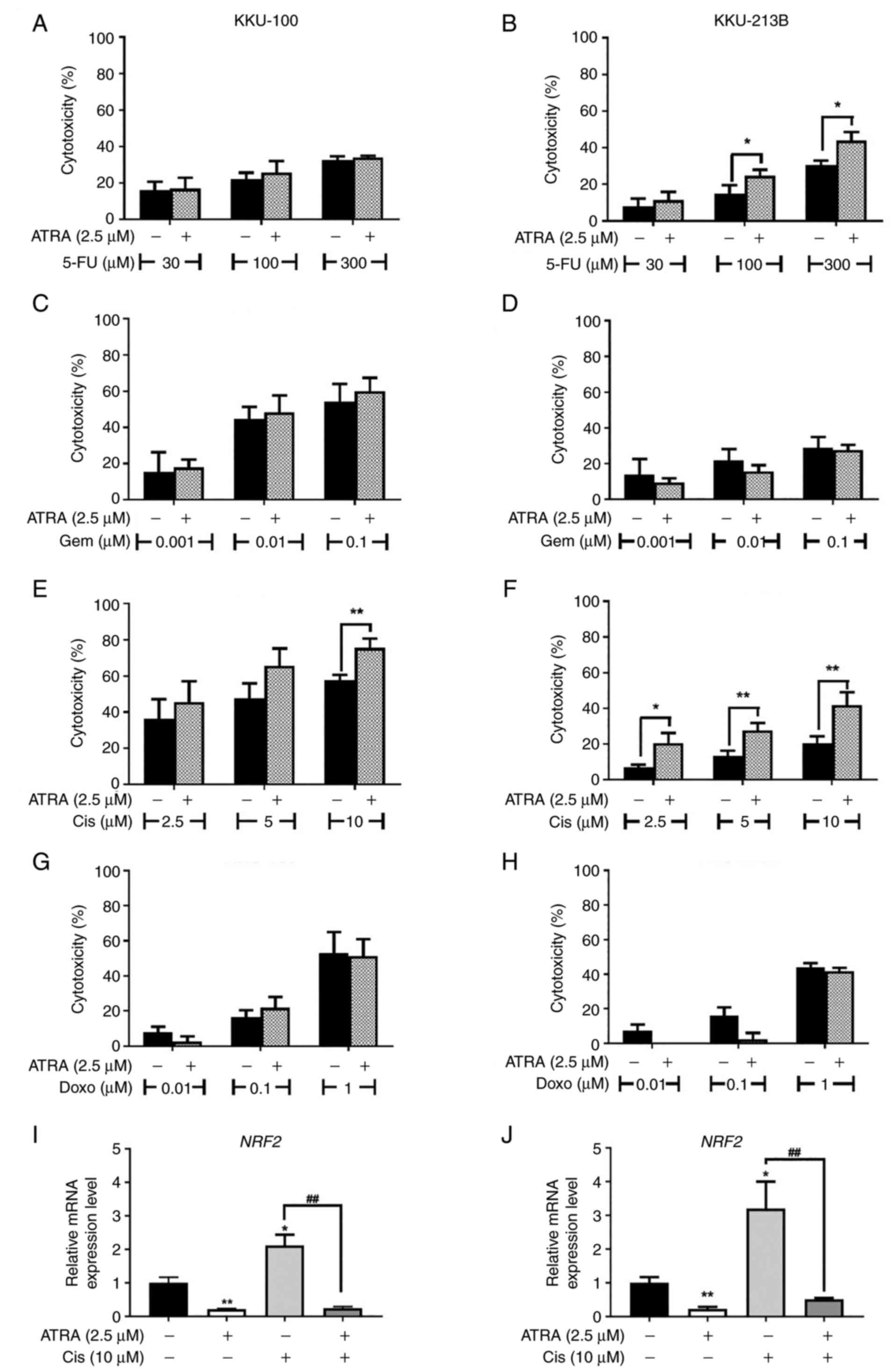 | Figure 9.Cytotoxicity of anticancer drugs is
enhanced by ATRA co-treatment. (A, C, F and G) KKU-100 and (B, D, E
and H) KKU-213B cells were treated with ATRA or anticancer drugs,
or were co-treated with ATRA and (A and B) 5-FU, (C and D) Gem, (E
and F) Cis and (G and H) Doxo anticancer drugs. Data are presented
as the mean ± SD from three independent experiments. *P<0.05 and
**P<0.01. mRNA expression levels of NRF2 in (I) KKU-100
and (J) KKU-213B cells were determined by reverse
transcription-quantitative PCR and normalized to ACTB. Data
are presented as the mean ± SD from three independent experiments.
*P<0.05 and **P<0.01 compared with the untreated control;
##P<0.01 compared with Cis alone. 5-FU,
5-fluorouracil; ATRA, all-trans-retinoic acid; Cis,
cisplatin; Doxo, doxorubicin; Gem, gemcitabine; NRF2,
nuclear factor erythroid 2-related factor 2. |
In CCA, cisplatin has been reported to induce the
expression of NRF2 target antioxidant genes to promote
chemoresistance (30). Since NRF2
is the primary regulator of cellular antioxidant defense, the
present study explored whether ATRA enhanced cisplatin cytotoxicity
in CCA cells by suppressing NRF2. Firstly, it was confirmed
that NRF2 expression was upregulated following cisplatin
treatment, whereas ATRA alone significantly reduced the mRNA
expression levels of NRF2 in KKU-100 and KKU-213B cells
compared with untreated control cells (Fig. 9I and J). Both CCA cell lines,
co-treatment with ATRA and cisplatin showed decreased mRNA
expression levels of NRF2 compared with cells treated with
cisplatin alone (Fig. 9I, J).
These results suggested that ATRA suppressed cisplatin-induced
NRF2, which may increase the cisplatin sensitivity of CCA
cells.
Discussion
The present study assessed the effects of ATRA
treatment on CCA cell apoptosis at different timepoints, and on the
underlying mechanism and cascade. ROS are important factors that
induce intracellular stress and trigger apoptosis; notably, several
studies have suggested that 90 min is sufficient to monitor the
changes in intracellular ROS following exposure to stimuli
(29,30). Similarly, in the present study,
ATRA-induced ROS production was observed at 90 min and alterations
in the expression levels of pro-apoptotic proteins were detected 6
h after ATRA-induced ROS production. According to apoptotic
signaling cascades, the increase in pro-apoptotic proteins can
trigger caspase activation; in the present study, the activity of
caspases was elevated at 12 h. Following ATRA-induced caspase
activation, it was further confirmed that apoptosis was induced at
48 h by flow cytometry. Previously, decreases in NRF2 and
NRF2 target genes have been reported to be associated with
apoptosis induction (29,30); therefore, the effect of ATRA on the
expression levels of NRF2 and NRF2 target genes were
assessed at 48 h, which is the time at which apoptosis induction
was detected.
The results of the present study revealed that
ATRA-induced cytotoxicity in CCA cells, at least in part, depended
on the specific receptor RARB. Treatment with ATRA promoted
apoptosis in CCA cells by activating the intrinsic pathway via
induction of Bax, AIF, cytochrome c and caspase-9 enzyme.
ATRA caused an increase in intracellular ROS content, leading to
the observed cytotoxicity of ATRA. By contrast, pre-treatment with
ROS scavengers NAC and TEMPOL diminished the apoptosis-inducing
effect of ATRA; however, NAC and TEMPOL also have other
anti-apoptotic mechanisms and can act as direct antioxidants to
scavenge radical molecules or as indirect antioxidants by restoring
redox cycling system in cells (29). NAC and TEMPOL also control the
redox balance of redox-sensitive proteins, such as PI3K, NRF2 and
p53, thus they may exert anti-apoptotic effects via regulation of
these proteins (36–38).
ATRA could suppress the expression of NRF2
and NRF2 target antioxidant genes; ATRA also caused cellular
ROS accumulation. In addition, ATRA significantly enhanced the
sensitivity of CCA cells to cisplatin. Notably, the improvement of
the cisplatin response in ATRA-treated CCA cells may be related to
the ATRA-downregulated NRF2 gene, which is considered the
master regulator for cellular antioxidant defense.
Previous studies reported the cytotoxic effects of
ATRA on several types of cancer cells, such as breast cancer,
non-small cell lung carcinoma, gastric cancer and CCA (22,39–41).
The present study confirmed that ATRA had a potent cytotoxic effect
on CCA with IC50 values at 48 h as 4.58 µM in KKU-213B
and 10.29 µM in KKU-100 cells. It is well known that ATRA acts as a
pan-agonist of the RARs to regulate several physiological
processes, and control growth and development. Two CCA cell lines
with different expression levels of RARs were used in the present
study. KKU-213B cells possessed high expression levels of RARs,
whereas KKU-100 cells exhibited lower RAR expression. In the
current study, the IC50 of ATRA in KKU-213B cells was
2-fold lower than that in KKU-100, thus suggesting that cells with
higher RAR expression were more sensitive to ATRA cytotoxicity. To
prove that ATRA sensitivity was dependent on RARs, cells deficient
in different types of RAR were created by transfecting KKU-213B
cells with siRARA, siRARB, non-targeting control siRNA or RARG
CRISPR. Knockdown/knockout of RARA or RARG had no effect on ATRA
sensitivity compared with in the control groups. Notably, cells
with a loss in RARB expression via siRNA were less sensitive to
ATRA, suggesting that ATRA cytotoxicity was partly mediated through
RARB. Previous reports have identified RARB-mediated cell
cytotoxicity through alterations in histone acetyltransferase,
apoptosis-associated proteins and cell cycle-associated proteins in
oral cancer and breast cancer cells (42,43).
Furthermore, the association between altered expression of RARs or
dysfunctional RARs with the malignant transformation of human cells
has previously been presented. In CCA, Ren et al (23) proposed the role of RARB as a tumor
suppressor and reported that upregulation of RARβ reversed drug
resistance by enhancing apoptotic susceptibility. Similarly, the
present study demonstrated that RARB expression was one of the
required factors for ATRA sensitivity, supporting a
tumor-suppressor role of RARB in CCA.
The present study demonstrated that ATRA induced
cell death by apoptosis induction in CCA cells. Previous studies
have reported that ATRA can promote the apoptosis of several types
of cancer cells, such as pancreatic cancer, breast cancer and
medulloblastoma (15–20). Apoptotic cell death occurs when
cells have a loss of mitochondria membrane potential, upregulation
of caspase-3 and an increase in DNA damage (44–46).
It is well accepted that ROS accumulation is one of the initial
events that trigger apoptosis in cancer cells, including in CCA
(28,34,35).
The present study revealed that ATRA enhanced ROS production,
consequently inducing intracellular stress and upregulating the
expression levels of pro-apoptotic proteins. Upregulated Bax
protein may form pores in mitochondria leading to loss of membrane
potential and leakage of apoptotic inducer components, such as AIF
and cytochrome c, into cytosol. When translocated into the
nucleus, AIF can trigger chromatin condensation and DNA
fragmentation, whereas cytochrome c forms the apoptosome to
activate caspase-9 and −3 activities leading to apoptosis (28). Although the extrinsic (caspase-8
mediated) pathway was unlikely affected by ATRA, the present study
revealed that, at a high dose, ATRA could increase the extrinsic
pathway in KKU-100 cells by increasing caspase-8 enzymatic activity
(Fig. 4C). Dhandapani et al
(47) previously showed that ATRA
sensitized cancer cells to TRAIL-induced apoptosis by upregulating
the expression of TRAIL-R1. However, in the present study, whether
ATRA increased caspase-8 activity and induced the extrinsic pathway
through the TRAIL mechanism in CCA has not been confirmed. In
addition, the ATRA-induced extrinsic pathway was only observed in
KKU-100 cells; thus, ATRA-induced activation of the extrinsic
pathway may be concentration-dependent and cell type-specific.
The present study demonstrated that ATRA-induced ROS
production mediated CCA cell apoptosis. Human cells use NRF2
signaling as the primary regulator for controlling the balance of
cellular ROS levels. Loss or dysfunction of NRF2 signaling can lead
to abnormal cell survival and death (29,30).
The present study identified a suppressive effect of ATRA on the
expression levels of NRF2 and NRF2 target genes in
CCA cells, which may explain the increasing cellular ROS content
induced by ATRA treatment.
CCA is a type of cancer that can exhibit
chemoresistance, thus identifying more effective therapeutics
against CCA resistance is essential to research. Previous studies
have revealed that ATRA can enhance cisplatin sensitivity in liver
cancer cells, 5-fluorouracil sensitivity in breast cancer cells and
gemcitabine sensitivity in pancreatic cancer cells (18–20).
In the present study, the effects of ATRA on the sensitivity of the
common four chemotherapeutic agents used in CCA treatment,
5-fluorouracil, gemcitabine, cisplatin and doxorubicin, were
tested. The results revealed that ATRA could increase the
sensitivity of cisplatin in both CCA cell lines and 5-FU in
KKU-213B cells. Notably, the improved sensitivity of cisplatin and
5-fluorouracil was more marked in KKU-213B cells than in KKU-100
cells. Cisplatin sensitivity may depend on the type of cancer
cells; differences in genetic background between KKU-213B and
KKU-100 CCA cells may be the cause of the different anticancer
response (30). Therefore, RARs
may not be the only factor driving cisplatin sensitivity in CCA
cells, and it was hypothesized that the cytotoxic effect of ATRA in
CCA cells may be both RAR-dependent in KKU-213B cells and
RAR-independent in KKU-100 cells. There was no significant effect
of ATRA on doxorubicin sensitivity in both CCA cells. A previous
report demonstrated that ATRA reduced doxorubicin cytotoxicity
through suppressing ROS generation by restoring the mRNA and
protein expression levels of phase II detoxifying enzyme and via
ERK2 activation in cardiomyocytes, without compromising doxorubicin
cytotoxicity in gastric cancer cells (48). However, the combination effect of
ATRA and doxorubicin in CCA treatment need to be confirmed in a
further study.
In most types of cancer, drug resistance after
long-term chemotherapy is an important event associated with poor
clinical outcomes. Impaired anticancer drug response can be caused
by increasing cellular protection and antioxidant defense via NRF2
activation. Increased NRF2 and NRF2 target genes
after anticancer drug treatment has been presented in several types
of cancer. Furthermore, suppressing NRF2 has been proposed as a
strategy to improve the sensitivity of chemotherapeutic agents in
CCA (29,30). In the present study, ATRA
suppressed the expression levels of NRF2 and NRF2
target genes, and prevented cisplatin-induced NRF2
expression; therefore, the effects of ATRA on enhancing the
sensitivity of CCA cells to anticancer agents may be partly
mediated via suppressing NRF2 signaling. The expression levels of
NRF2 and NRF2-related genes were detected at 48 h
after ATRA treatment; therefore, the differences in these
expression levels may not be a direct cause of ROS production at 90
min. However, the downregulation of NRF2 and
NRF2-related genes, which may be associated with increased
cellular oxidative stress, and decreased protection and survival of
cells, could be one important factor driving cell death and
impaired cellular defensive mechanism against noxious stimuli and
anticancer drug toxicity (29,30).
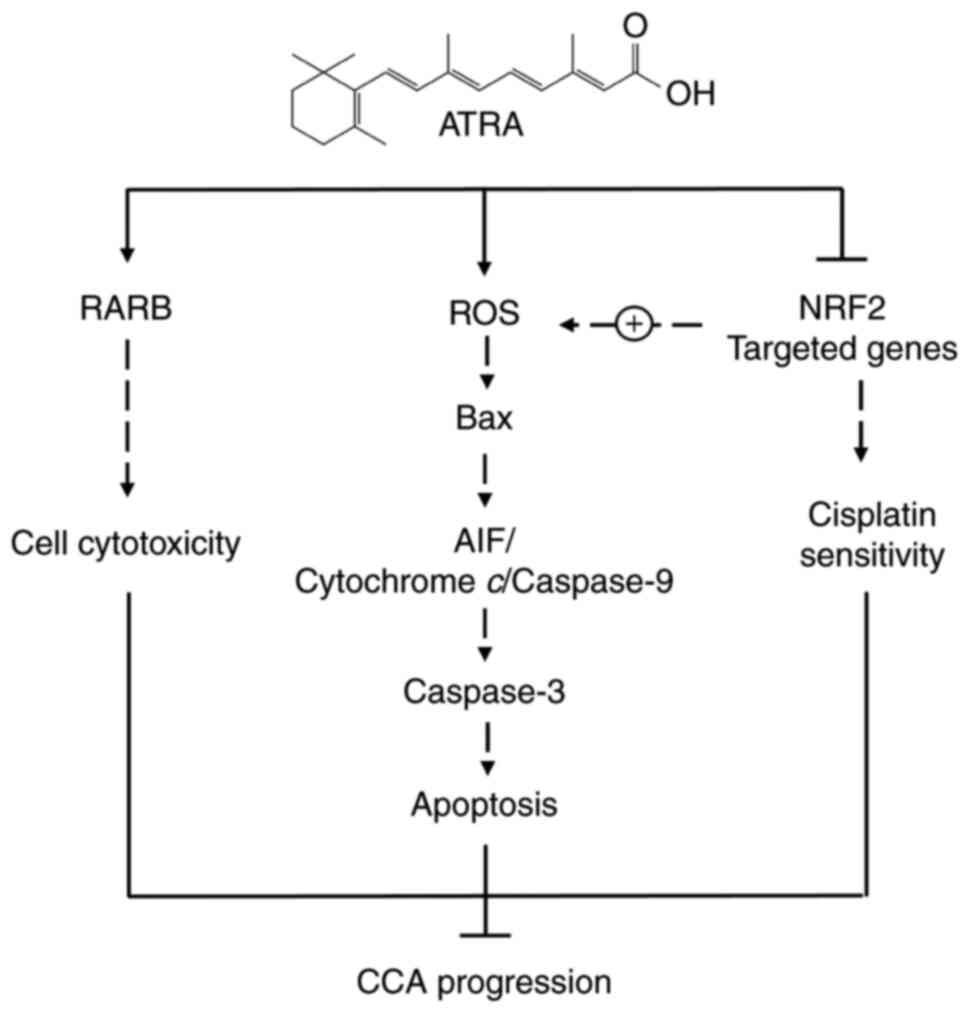
The possible mechanism of action of ATRA in the induction of
apoptosis and enhancement of chemosensitivity in CCA cells has been
summarized in Fig. 10.
In conclusion, the present study demonstrated that
ATRA had an anticancer effect on CCA cells. ATRA promoted
cytotoxicity partly via RARB, and induced the intrinsic pathway of
apoptosis by enhancing ROS accumulation. Furthermore, ATRA
suppressed NRF2 signaling, which in turn may cause impaired ROS
balance and enhance the sensitivity of anticancer drugs. Moreover,
ROS induction may be mediated via RARB; however, the results of the
present study did not prove this concept and further studies are
required. Notably, the present study primarily evaluated the effect
of ATRA on the induction of apoptosis, its potential use for
improvements in the response to anticancer drugs, and explored the
anticancer actions and underlying mechanisms of ATRA in CCA cells.
However, as the present study used a cell culture model to
demonstrate the effect of ATRA on CCA, this may be a limitation of
this study and the findings may differ from the real-world
situation. Therefore, using in vivo animal models and
patient-derived materials is required to assess the role of ATRA.
Notably, the results of the present study indicated that ATRA may
be of use in CCA therapy; however, further in vivo studies
are warranted to approve the potential use of ATRA for CCA
therapy.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank Mr. Kevin McCracken
(Publication Clinic, Khon Kaen University) for editing the
manuscript and Dr Kanha Muisuk (Department of Pathology, Faculty of
Medicine, Khon Kaen University) for technical support and STR
analysis of cell lines.
Funding
This research was supported by the Cholangiocarcinoma Research
Institute, Khon Kaen University (grant no. 6200011002). Siriwoot
Butsri was supported by a scholarship for Postgraduate Study
Support of Faculty of Medicine, Khon Kaen University, Program Year
2016.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SB, AP, VK, LS and SK designed the study. SB and AP
performed the experiments. SB, AP, VK, LS and SK analyzed the data
and interpreted the results. AP, SB and SK wrote the manuscript. SB
and AP confirm the authenticity of all the raw data. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Anderson CD, Pinson CW, Berlin J and Chari
RS: Diagnosis and treatment of cholangiocarcinoma. Oncologist.
9:43–57. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Braconi C and Patel T: Cholangiocarcinoma:
New insights into disease pathogenesis and biology. Infect Dis Clin
North Am. 24871–884. (vii)2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Global Burden of Disease Cancer and
Collaboration, . Fitzmaurice C, Dicker D, Pain A, Hamavid H,
Moradi-Lakeh M, MacIntyre MF, Allen C, Hansen G, Woodbrook R, et
al: The global burden of cancer 2013. JAMA Oncol. 1:505–527. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Banales JM, Cardinale V, Carpino G,
Marzioni M, Andersen JB, Invernizzi P, Lind GE, Folseraas T, Forbes
SJ, Fouassier L, et al: Expert consensus document:
Cholangiocarcinoma: Current knowledge and future perspectives
consensus statement from the European Network for the Study of
Cholangiocarcinoma (ENS-CCA). Nat Rev Gastroenterol Hepatol.
13:261–280. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Blechacz B: Cholangiocarcinoma: Current
knowledge and new developments. Gut Liver. 11:13–26. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Valle JW, Furuse J, Jitlal M, Beare S,
Mizuno N, Wasan H, Bridgewater J and Okusaka T: Cisplatin and
gemcitabine for advanced biliary tract cancer: A meta-analysis of
two randomised trials. Ann Oncol. 25:391–398. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rhinn M and Dolle P: Retinoic acid
signalling during development. Development. 139:843–858. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Blomhoff R and Blomhoff HK: Overview of
retinoid metabolism and function. J Neurobiol. 66:606–630. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Barnard JH, Collings JC, Whiting A,
Przyborski SA and Marder TB: Synthetic retinoids:
Structure-activity relationships. Chemistry. 15:11430–11442. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Idres N, Marill J, Flexor MA and Chabot
GG: Activation of retinoic acid receptor-dependent transcription by
all-trans-retinoic acid metabolites and isomers. J Biol Chem.
277:31491–31498. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tallman MS, Andersen JW, Schiffer CA,
Appelbaum FR, Feusner JH, Ogden A, Shepherd L, Willman C,
Bloomfield CD, Rowe JM and Wiernik PH: All-trans-retinoic acid in
acute promyelocytic leukemia. N Engl J Med. 337:1021–1028. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tosi P, Visani G, Gibellini D, Zauli G,
Ottaviani E, Cenacchi A, Gamberi B, Manfroi S, Marchisio M and Tura
S: All-trans retinoic acid and induction of apoptosis in acute
promyelocytic leukemia cells. Leuk Lymphoma. 14:503–507. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fang J, Chen SJ, Tong JH, Wang ZG, Chen GQ
and Chen Z: Treatment of acute promyelocytic leukemia with ATRA and
As2O3: A model of molecular target-based cancer therapy. Cancer
Biol Ther. 1:614–620. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Brigger D, Schlafli AM, Garattini E and
Tschan MP: Activation of RARα induces autophagy in SKBR3 breast
cancer cells and depletion of key autophagy genes enhances ATRA
toxicity. Cell Death Dis. 6:e18612015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cui J, Gong M, He Y, Li Q, He T and Bi Y:
All-trans retinoic acid inhibits proliferation, migration, invasion
and induces differentiation of hepa1-6 cells through reversing EMT
in vitro. Int J Oncol. 48:349–357. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li N, Lu Y, Li D, Zheng X, Lian J, Li S,
Cui H, Zhang L, Sang L, Wang Y, et al: All-trans retinoic acid
suppresses the angiopoietin-Tie2 pathway and inhibits angiogenesis
and metastasis in esophageal squamous cell carcinoma. PLoS One.
12:e01745552017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mei D, Lv B, Chen B, Xiao S, Jiang J, Xie
Y and Jiang L: All-trans retinoic acid suppresses malignant
characteristics of CD133-positive thyroid cancer stem cells and
induces apoptosis. PLoS One. 12:e01828352017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kuroda H, Tachikawa M, Uchida Y, Inoue K,
Ohtsuka H, Ohtsuki S, Unno M and Terasaki T: All-trans retinoic
acid enhances gemcitabine cytotoxicity in human pancreatic cancer
cell line AsPC-1 by up-regulating protein expression of
deoxycytidine kinase. Eur J Pharm Sci. 103:116–121. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Y, Guan DX, Shi J, Gao H, Li JJ,
Zhao JS, Qiu L, Liu J, Li N, Guo WX, et al: All-trans retinoic acid
potentiates the chemotherapeutic effect of cisplatin by inducing
differentiation of tumor initiating cells in liver cancer. J
Hepatol. 59:1255–1263. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yan Y, Li Z, Xu X, Chen C, Wei W, Fan M,
Chen X, Li JJ, Wang Y and Huang J: All-trans retinoic acids induce
differentiation and sensitize a radioresistant breast cancer cells
to chemotherapy. BMC Complement Altern Med. 16:1132016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Najafzadeh N, Mazani M, Abbasi A,
Farassati F and Amani M: Low-dose all-trans retinoic acid enhances
cytotoxicity of cisplatin and 5-fluorouracil on CD44(+) cancer stem
cells. Biomed Pharmacother. 74:243–251. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chung KD, Jeong YI, Chung CW, Kim DH and
Kang DH: Anti-tumor activity of all-trans retinoic
acid-incorporated glycol chitosan nanoparticles against HuCC-T1
human cholangiocarcinoma cells. Int J Pharm. 422:454–461. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ren HY, Chen B, Huang GL, Liu Y and Shen
DY: Upregulation of retinoic acid receptor-beta reverses drug
resistance in cholangiocarcinoma cells by enhancing susceptibility
to apoptosis. Mol Med Rep. 14:3602–3608. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
de Oliveira MR: Vitamin A and retinoids as
mitochondrial toxicants. Oxid Med Cell Longev. 2015:1402672015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Oliveira MR: The neurotoxic effects of
vitamin A and retinoids. An Acad Bras Cienc. 87 (2
Suppl):S1361–S1373. 2015. View Article : Google Scholar
|
|
26
|
Conte da Frota ML Jr, Gomes da Silva E,
Behr GA, Roberto de Oliveira M, Dal-Pizzol F, Klamt F and Moreira
JC: All-trans retinoic acid induces free radical generation and
modulate antioxidant enzyme activities in rat sertoli cells. Mol
Cell Biochem. 285:173–179. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Miyoshi T, Arai T, Yamashita K, Sasada M
and Uchiyama T: NB4 cells treated with all-trans retinoic acid
generate toxic reactive oxygen species that cause endothelial
hyperpermeability. Leuk Res. 34:373–378. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kittiratphatthana N, Kukongviriyapan V,
Prawan A and Senggunprai L: Luteolin induces cholangiocarcinoma
cell apoptosis through the mitochondrial-dependent pathway mediated
by reactive oxygen species. J Pharm Pharmacol. 68:1184–1192. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sompakdee V, Prawan A, Senggunprai L,
Kukongviriyapan U, Samathiwat P, Wandee J and Kukongviriyapan V:
Suppression of Nrf2 confers chemosensitizing effect through
enhanced oxidant-mediated mitochondrial dysfunction. Biomed
Pharmacother. 101:627–634. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Samatiwat P, Prawan A, Senggunprai L,
Kukongviriyapan U and Kukongviriyapan V: Nrf2 inhibition sensitizes
cholangiocarcinoma cells to cytotoxic and antiproliferative
activities of chemotherapeutic agents. Tumour Biol. 37:11495–11507.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang XJ, Hayes JD, Henderson CJ and Wolf
CR: Identification of retinoic acid as an inhibitor of
transcription factor Nrf2 through activation of retinoic acid
receptor alpha. Proc Natl Acad Sci USA. 104:19589–19594. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sripa B, Leungwattanawanit S, Nitta T,
Wongkham C, Bhudhisawasdi V, Puapairoj A, Sripa C and Miwa M:
Establishment and characterization of an opisthorchiasis-associated
cholangiocarcinoma cell line (KKU-100). World J Gastroenterol.
11:3392–3397. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sripa B, Seubwai W, Vaeteewoottacharn K,
Sawanyawisuth K, Silsirivanit A, Kaewkong W, Muisuk K, Dana P,
Phoomak C, Lert-Itthiporn W, et al: Functional and genetic
characterization of three cell lines derived from a single tumor of
an Opisthorchis viverrini-associated cholangiocarcinoma patient.
Hum Cell. 33:695–708. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liang W, Cui J, Zhang K, Xi H, Cai A, Li
J, Gao Y, Hu C, Liu Y, Lu Y, et al: Shikonin induces ROS-based
mitochondria-mediated apoptosis in colon cancer. Oncotarget.
8:109094–109106. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang R, Humphreys I, Sahu RP, Shi Y and
Srivastava SK: In vitro and in vivo induction of apoptosis by
capsaicin in pancreatic cancer cells is mediated through ROS
generation and mitochondrial death pathway. Apoptosis.
13:1465–1478. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang G, Wang Q, Zhou Q, Wang R, Xu M,
Wang H, Wang L, Wilcox CS, Liu R and Lai EY: Protective effect of
tempol on acute kidney injury through PI3K/Akt/Nrf2 signaling
pathway. Kidney Blood Press Res. 41:129–138. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang L, Zhu Z, Liu J, Zhu Z and Hu Z:
Protective effect of N-acetylcysteine (NAC) on renal
ischemia/reperfusion injury through Nrf2 signaling pathway. J
Recept Signal Transduct Res. 34:396–400. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sablina AA, Budanov AV, Ilyinskaya GV,
Agapova LS, Kravchenko JE and Chumakov PM: The antioxidant function
of the p53 tumor suppressor. Nat Med. 11:1306–1313. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Choi EJ, Whang YM, Kim SJ, Kim HJ and Kim
YH: Combinational treatment with retinoic acid derivatives in
non-small cell lung carcinoma in vitro. J Korean Med Sci. 22
(Suppl):S52–S60. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Flamini MI, Gauna GV, Sottile ML, Nadin
BS, Sanchez AM and Vargas-Roig LM: Retinoic acid reduces migration
of human breast cancer cells: Role of retinoic acid receptor beta.
J Cell Mol Med. 18:1113–1123. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Patrad E, Niapour A, Farassati F and Amani
M: Combination treatment of all-trans retinoic acid (ATRA) and
gamma-secretase inhibitor (DAPT) cause growth inhibition and
apoptosis induction in the human gastric cancer cell line.
Cytotechnology. 70:865–877. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hayashi K, Yokozaki H, Naka K, Yasui W,
Lotan R and Tahara E: Overexpression of retinoic acid receptor beta
induces growth arrest and apoptosis in oral cancer cell lines. Jpn
J Cancer Res. 92:42–50. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen H, Zhang H, Lee J, Liang X, Wu X, Zhu
T, Lo PK, Zhang X and Sukumar S: HOXA5 acts directly downstream of
retinoic acid receptor beta and contributes to retinoic
acid-induced apoptosis and growth inhibition. Cancer Res.
67:8007–8013. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
el-Metwally TH, Hussein MR, Pour PM,
Kuszynski CA and Adrian TE: Natural retinoids inhibit proliferation
and induce apoptosis in pancreatic cancer cells previously reported
to be retinoid resistant. Cancer Biol Ther. 4:474–483. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gumireddy K, Sutton LN, Phillips PC and
Reddy CD: All-trans-retinoic acid-induced apoptosis in human
medulloblastoma: Activation of caspase-3/poly(ADP-ribose)
polymerase 1 pathway. Clin Cancer Res. 9:4052–4059. 2003.PubMed/NCBI
|
|
46
|
Mangiarotti R, Danova M, Alberici R and
Pellicciari C: All-trans retinoic acid (ATRA)-induced apoptosis is
preceded by G1 arrest in human MCF-7 breast cancer cells. Br J
Cancer. 77:186–191. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dhandapani L, Yue P, Ramalingam SS, Khuri
FR and Sun SY: Retinoic acid enhances TRAIL-induced apoptosis in
cancer cells by upregulating TRAIL receptor 1 expression. Cancer
Res. 71:5245–5254. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yang L, Luo C, Chen C, Wang X, Shi W and
Liu J: All-trans retinoic acid protects against doxorubicin-induced
cardiotoxicity by activating the ERK2 signalling pathway. Br J
Pharmacol. 173:357–371. 2016. View Article : Google Scholar : PubMed/NCBI
|