Lung cancer is the leading cause of cancer-related
mortality worldwide, with non-small cell lung cancer (NSCLC) being
the most common subtype (1). The
majority of patients with NSCLC are diagnosed at an advanced,
inoperable stage (2) and have an
overall 5-year survival rate of just 5% (3). This poor prognosis may be associated
with tumor heterogeneity, acquisition and intrinsic resistance to
therapeutic agents in NSCLC (4).
If the most appropriate treatment is identified early and drug
resistance is addressed to a satisfactory degree, patient survival
can be significantly improved. Current non-surgical treatments for
NSCLC in clinical practice include systemic chemotherapy,
radiotherapy, targeted therapy and immunotherapy (IO). In recent
years, rapid developments have been made in cellular and molecular
biotechnology, and targeted gene therapy and IO are gradually
gaining traction (5). Molecular
testing is commonly used in NSCLC, and the detection of epidermal
growth factor receptor (EGFR), B-Raf proto-oncogene,
serine/threonine kinase (BRAF) and MET proto-oncogene,
receptor tyrosine kinase (MET) mutations, as well as
anaplastic lymphoma kinase (ALK), ROS proto-oncogene 1,
receptor tyrosine kinase (ROS1), ret proto-oncogene
(RET) and neurotrophic receptor tyrosine kinase 1
(NTRK1) translocations have been incorporated into the
diagnostic criteria for NSCLC, and inhibitors of these kinases are
now routinely used in the clinic (6). An increasing number of signaling
pathways and driver genes are being identified, and therapeutic
drugs for NSCLC are emerging (7,8).
Targeted drugs can bind specifically to the oncogenic site and
induce cancer cell-specific death (9). Targeted drugs have a higher efficacy
and fewer side effects than chemotherapy (10,11).
However, each generation of targeted drugs shows different degrees
of resistance; therefore, identifying new therapeutic targets
following resistance is crucial. Multiple mutations are not
uncommon in clinical practice in recent years, and exploring
serine/threonine kinase 11 (STK11) co-mutations in
ALK-positive NSCLC patients is important.
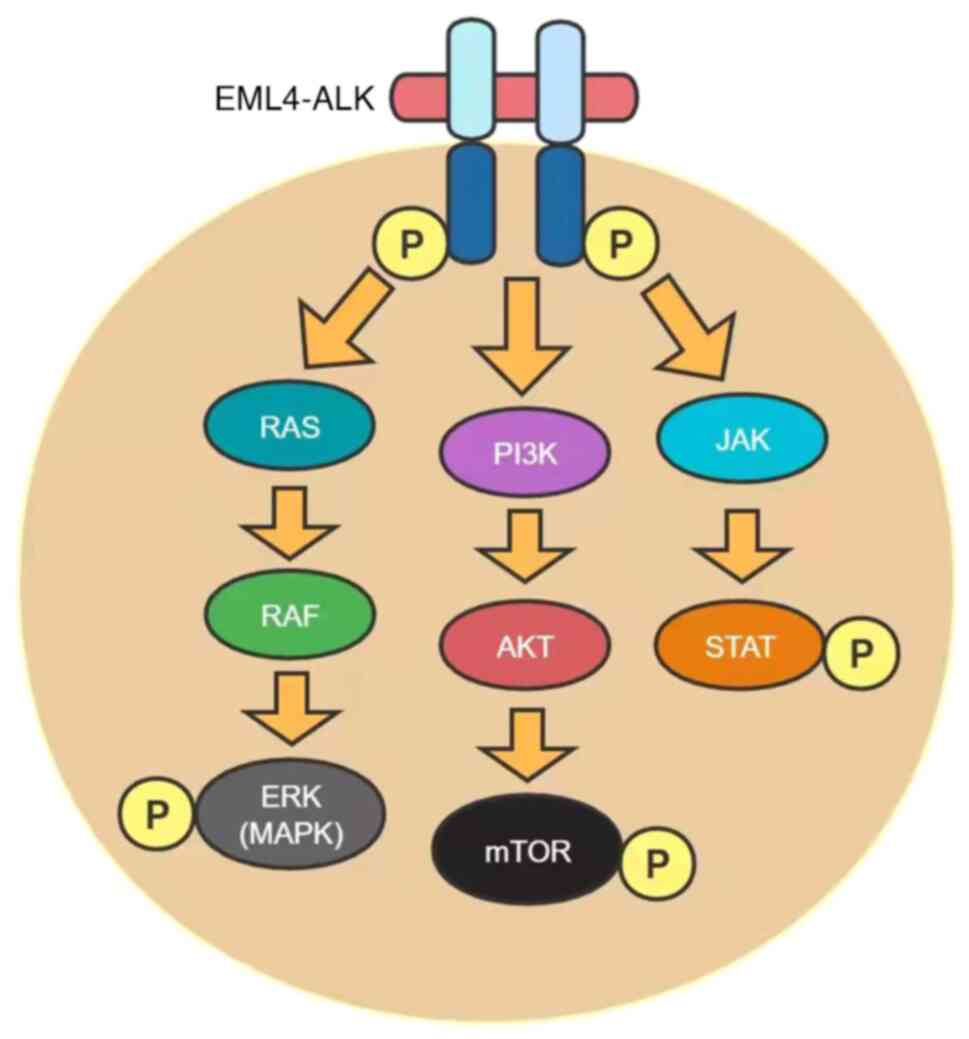
As a receptor tyrosine kinase of the insulin
receptor (IR) subfamily, ALK has been found to play an important
role in various types of cancer, particularly in anaplastic large
cell lymphoma (ALCL), NSCLC and neuroblastoma (21). In 2007, the echinoderm
microtubule-associated protein like protein 4
(EML4)-ALK fusion gene was identified in a group of
NSCLC patients (22). This fusion
is the result of an inversion of the short arm of chromosome 2,
where the human EML4 and ALK genes are present
(23). EML4 contains a
coiled oligomeric structural domain, which mediates the
dimerization and structural activation of ALK. Like in ALCL,
many different ALK fusions have been identified, but
EML4-ALK is the most common variant (24). ALK has been reported to
regulate several different pathways involved in cell proliferation
and survival, such as the phosphatidylinositol-3-kinase
(PI3K)/AKT/mammalian targets of rapamycin (mTOR), RAS/RAF/MAP
kinase-extracellular signal-regulated kinase (ERK) kinase (MEK)/ERK
and JAK/STAT pathways (Fig. 1),
once it is dimerized and activated by autophosphorylation upon
binding to its ligands, pleiotrophin and midkine (25,26).
Direct evidence for the oncogenic potential of
EML4-ALK in lung carcinogenesis has been found in
mice. The transgenic overexpression of EML4-ALK in
type II alveolar cells via the surface activated protein-c or Clara
cell secretory protein promoter leads to the rapid development of
tumors with features of lung adenocarcinoma (27,28).
A total of 3–7% of NSCLC (mainly adenocarcinoma subtypes) cases are
characterized by ALK rearrangements, which occur in a
mutually exclusive manner with KRAS and EGFR
mutations (29,30). Of note, a previous study developed
in vivo induction models by inducing EML4-ALK
rearrangement, which leads to lung carcinogenesis. These models
have shown a sensitivity to ALK inhibition, thus serving as
valuable tools to explore the mechanisms of
EML4-ALK-induced lung cancer and response to
ALK-targeted therapy (31).
Of note, ALK-positive NSCLC patients have a healthy weight,
are non-smokers or are young (32).
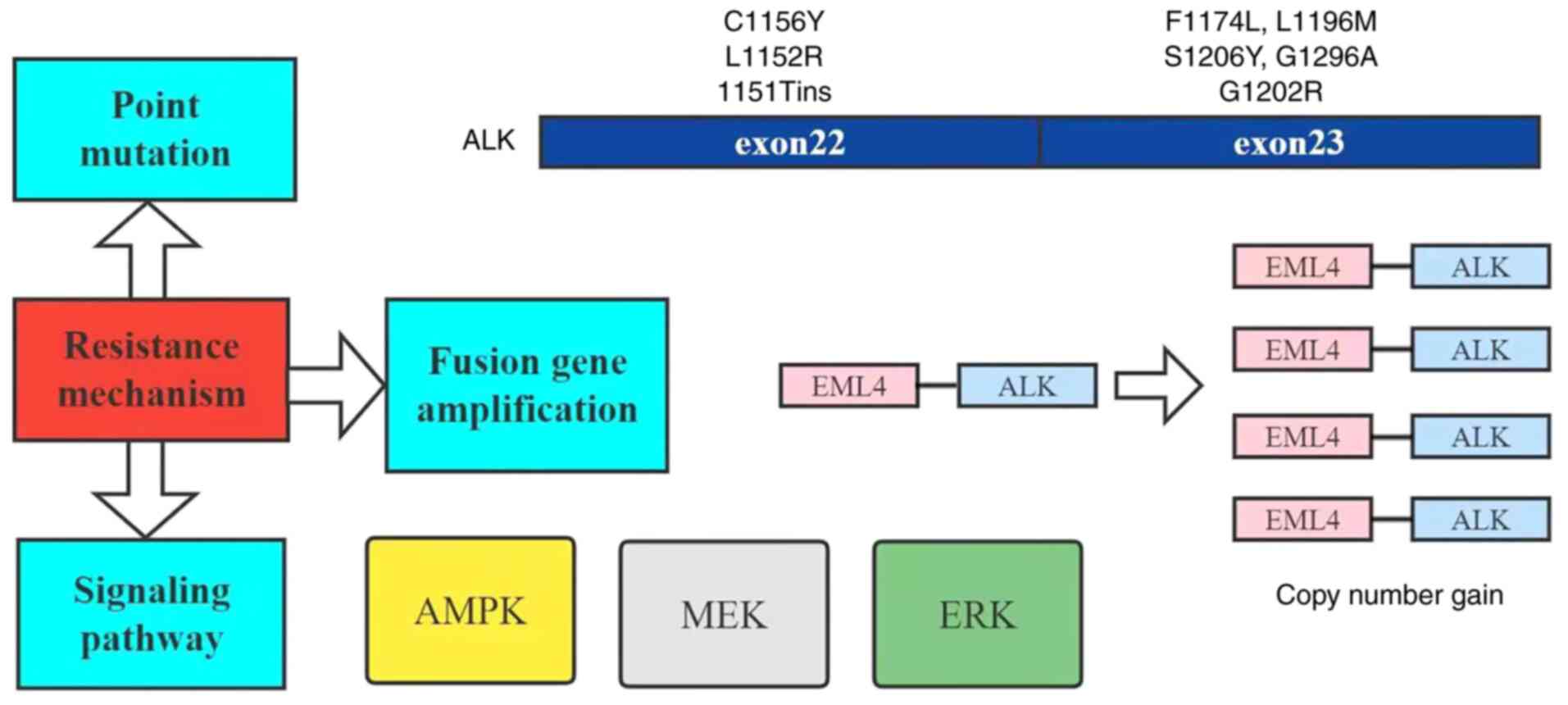
Known mechanisms of resistance include point
mutations, fusion gene amplification and bypass signaling through
the activation of other oncogenes (Fig. 2) (63), and AMPK, which is closely
associated with STK11 mutation and is one of the important
pathways in the mechanism of ALK inhibitor resistance
(30).
Cancer typically evades immune surveillance by
aberrantly expressing immune checkpoints (e.g. PD-1) that isolate
tumor cells from the host immune system. Immune blockade using
monoclonal antibodies against the immune checkpoint PD-1 and its
primary ligand PD-L1 can greatly improve survival in advanced
NSCLC, with the greatest impact in patients with stage III and
first-line stage IV lung cancer (126–128). However, in patients with other
types, the response rate was just 20% (129). In a retrospective cohort study,
data from the Clinico-Genomic Database were used to identify
patients with metastatic NSCLC who received first-line IO (alone or
in combination) or chemotherapy in routine clinical practice. The
results suggested that in NSCLC, patients with STK11
mutation (STK11m) exhibit poorer overall survival (OS) and
PFS compared with patients with STK11 wild-type
(STK11wt) receiving IO or chemotherapy. Survival outcomes
analyzed by treatment line and type showed that OS and PFS were
worse in the IO treatment group for STK11m vs.
STK11wt (Table II)
(130). The results of the study
were not optimistic, which further suggested that STK11
mutations reduce the survival rate of patients with NSCLC. Most
importantly, it is unclear whether STK11 can be used as a
predictive biomarker to guide treatment selection, and prospective
evaluation is still lacking. Therefore, immunotherapy should not be
administered to patients with STK11-mutated tumors at the
present time (131). At the same
time, it has also been reported that LKB1 encoded by the
STK11 gene may be associated with radioresistance in
patients, and several previous studies have shown the role of
LKB1 expression in regulating the response to radiotherapy,
based on preclinical experiments (132–134). However, to the best of our
knowledge, there are no clinical trials on STK11 mutations,
so a comprehensive evaluation of patients with STK11
mutations in NSCLC could not be performed.
Not applicable.
The present study was supported by the Hubei Province Health and
Family Planning Scientific Research Project (grant nos. WJ2019-21)
to NL.
All data generated or analyzed during this study
are included in this published article.
XCP and JC contributed to the design of this
review. WZ, MW, MXM and ZQY reviewed the references. WZ, JC and XCP
wrote the manuscript. WZ, LDY, YYC and NL designed and produced the
tables and figures. YYC and NL revised the manuscript critically
for important intellectual content. NL acquired the funding. YHY
analyzed the data and designed the figures. LDY put a lot of effort
into revising the manuscript and is listed as co-first author. Data
authentication is not applicable. All authors read and approved the
manuscript for publication.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Rotow J and Bivona TG: Understanding and
targeting resistance mechanisms in NSCLC. Nat Rev Cancer.
17:637–658. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ye Z, Huang Y, Ke J, Zhu X, Leng S and Luo
H: Breakthrough in targeted therapy for non-small cell lung cancer.
Biomed Pharmacother. 133:1110792021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Molina JR, Yang P, Cassivi SD, Schild SE
and Adjei AA: Non-small cell lung cancer: Epidemiology, risk
factors, treatment, and survivorship. Mayo Clin Proc. 83:584–594.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang L, Li N, Wang M, Zhang YH, Yan LD,
Zhou W, Yu ZQ, Peng XC and Cai J: Tumorigenic effect of TERT and
its potential therapeutic target in NSCLC (Review). Oncol Rep.
46:1822021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brueckl WM, Ficker JH and Zeitler G:
Clinically relevant prognostic and predictive markers for
immune-checkpoint-inhibitor (ICI) therapy in non-small cell lung
cancer (NSCLC). BMC Cancer. 20:11852020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Imyanitov EN, Iyevleva AG and Levchenko
EV: Molecular testing and targeted therapy for non-small cell lung
cancer: Current status and perspectives. Crit Rev Oncol Hematol.
157:1031942021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu J, Li D, Luo H and Zhu X: Circular
RNAs: The star molecules in cancer. Mol Aspects Med. 70:141–152.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guo B, Li D, Du L and Zhu X: piRNAs:
Biogenesis and their potential roles in cancer. Cancer Metastasis
Rev. 39:567–575. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gerlinger M: Targeted drugs ramp up cancer
mutability. Science. 366:1452–1453. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liang G, Fan W, Luo H and Zhu X: The
emerging roles of artificial intelligence in cancer drug
development and precision therapy. Biomed Pharmacother.
128:1102552020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li S, Zhang Z, Lai WF, Cui L and Zhu X:
How to overcome the side effects of tumor immunotherapy. Biomed
Pharmacother. 130:1106392020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ceccon M, Mologni L, Bisson W, Scapozza L
and Gambacorti-Passerini C: Crizotinib-resistant NPM-ALK mutants
confer differential sensitivity to unrelated Alk inhibitors. Mol
Cancer Res. 11:122–132. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Toyokawa G, Hirai F, Inamasu E, Yoshida T,
Nosaki K, Takenaka T, Yamaguchi M, Seto T, Takenoyama M and
Ichinose Y: Secondary mutations at I1171 in the ALK gene confer
resistance to both Crizotinib and Alectinib. J Thorac Oncol.
9:e86–e87. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gainor JF, Dardaei L, Yoda S, Friboulet L,
Leshchiner I, Katayama R, Dagogo-Jack I, Gadgeel S, Schultz K,
Singh M, et al: Molecular mechanisms of resistance to first- and
second-generation ALK Inhibitors in ALK-rearranged lung cancer.
Cancer Discov. 6:1118–1133. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Toyokawa G and Seto T: Updated evidence on
the mechanisms of resistance to ALKInhibitors and strategies to
overcome such resistance: Clinical and preclinical data. Oncol Res
Treat. 38:291–298. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Costa DB: Clinical development and
approval of second generation ALK inhibitors for ALKrearranged lung
cancer. Transl Lung Cancer Res. 3:373–375. 2014.PubMed/NCBI
|
|
17
|
Roskoski R Jr: ROS1 protein-tyrosine
kinase inhibitors in the treatment of ROS1 fusion protein-driven
non-small cell lung cancers. Pharmacol Res. 121:202–212. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shaw AT, Kim DW, Mehra R, Tan DS, Felip E,
Chow LQ, Camidge DR, Vansteenkiste J, Sharma S, De Pas T, et al:
Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J
Med. 370:1189–1197. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rothschild SI: New treatment options for
ALK+ advanced non-small-cell lung cancer: Critical appraisal of
ceritinib. Ther Clin Risk Manag. 12:735–741. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rossi A: Alectinib for ALK-positive
non-small-cell lung cancer. Expert Rev Clin Pharmacol. 9:1005–1013.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kong X, Pan P, Sun H, Xia H, Wang X, Li Y
and Hou T: Drug discovery targeting anaplastic lymphoma kinase
(ALK). J Med Chem. 62:10927–10954. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Qian M, Zhu B, Wang X and Liebman M: Drug
resistance in ALK-positiveNon-small cell lungcancer patients. Semin
Cell Dev Biol. 64:150–157. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Soda M, Choi YL, Enomoto M, Takada S,
Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K,
Hatanaka H, et al: Identification of the transforming EML4-ALK
fusion gene in non-small-cell lung cancer. Nature. 448:561–566.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Katayama R, Lovly CM and Shaw AT:
Therapeutic targeting of anaplastic lymphoma kinase in lung cancer:
A paradigm for precision cancer medicine. Clin Cancer Res.
21:2227–2235. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Morales La Madrid A, Campbell N, Smith S,
Cohn SL and Salgia R: Targeting ALK: A promising strategy for the
treatment of non-small cell lung cancer, non-Hodgkin's lymphoma,
and neuroblastoma. Target Oncol. 7:199–210. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shaw AT and Solomon B: Targeting
anaplastic lymphoma kinase in lung cancer. Clin Cancer Res.
17:2081–2086. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen Z, Sasaki T, Tan X, Carretero J,
Shimamura T, Li D, Xu C, Wang Y, Adelmant GO, Capelletti M, et al:
Inhibition of ALK, PI3K/MEK, and HSP90 in murine lung
adenocarcinoma induced by EML4-ALK fusion oncogene. Cancer Res.
70:9827–9836. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pyo KH, Lim SM, Kim HR, Sung YH, Yun MR,
Kim SM, Kim H, Kang HN, Lee JM, Kim SG, et al: Establishment of a
conditional transgenic mouse model recapitulating EML4-ALK-positive
human non-small cell lung cancer. J Thorac Oncol. 12:491–500. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Takeuchi K, Choi YL, Soda M, Inamura K,
Togashi Y, Hatano S, Enomoto M, Takada S, Yamashita Y, Satoh Y, et
al: Multiplex reverse transcription-PCR screening for
EML4-ALKfusion transcripts. Clin Cancer Res. 14:6618–6624. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sharma GG, Mota I, Mologni L, Patrucco E,
Gambacorti-Passerini C and Chiarle R: Tumor resistance against
ALKTargeted therapy-where it comes from and where it goes. Cancers
(Basel). 10:622018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Maddalo D, Manchado E, Concepcion CP,
Bonetti C, Vidigal JA, Han YC, Ogrodowski P, Crippa A, Rekhtman N,
de Stanchina E, et al: In vivo engineering of oncogenic chromosomal
rearrangements with the CRISPR/Cas9 system. Nature. 516:423–427.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rodig SJ, Mino-Kenudson M, Dacic S, Yeap
BY, Shaw A, Barletta JA, Stubbs H, Law K, Lindeman N, Mark E, et
al: Unique clinicopathologic features characterize ALK-rearranged
lung adenocarcinoma in the western population. Clin Cancer Res.
15:5216–5223. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gristina V, La Mantia M, Iacono F, Galvano
A, Russo A and Bazan V: The emerging therapeutic landscape of ALK
inhibitors in non-small cell lung cancer. Pharmaceuticals (Basel).
13:4742020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Du X, Shao Y, Qin HF, Tai YH and Gao HJ:
ALK-rearrangement in non-small-cell lung cancer (NSCLC). Thorac
Cancer. 9:423–430. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Camidge DR, Bang YJ, Kwak EL, Iafrate AJ,
Varella-Garcia M, Fox SB, Riely GJ, Solomon B, Ou SH, Kim DW, et
al: Activity and safety of crizotinib in patients with ALK-positive
non-small-cell lung cancer: Updated results from a phase 1 study.
Lancet Oncol. 13:1011–1019. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Blackhall F, Ross Camidge D, Shaw AT,
Soria JC, Solomon BJ, Mok T, Hirsh V, Jänne PA, Shi Y, Yang PC, et
al: Final results of the large-scale multinational trial PROFILE
1005: Efficacy and safety of crizotinib in previously treated
patients with advanced/metastatic ALK-positive non-small-cell lung
cancer. ESMO Open. 2:e0002192017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nishio M, Kim DW, Wu YL, Nakagawa K,
Solomon BJ, Shaw AT, Hashigaki S, Ohki E, Usari T, Paolini J, et
al: Crizotinib versus chemotherapy in Asian patients with
ALK-positive advanced non-small cell lung cancer. Cancer Res Treat.
50:691–700. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Solomon BJ, Kim DW, Wu YL, Nakagawa K,
Mekhail T, Felip E, Cappuzzo F, Paolini J, Usari T, Tang Y, et al:
Final overall survival analysis from a study comparing first-line
crizotinib versus chemotherapy in ALK-mutation-positive
non-small-cell lung cancer. J Clin Oncol. 36:2251–2258. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim DW, Mehra R, Tan DSW, Felip E, Chow
LQM, Camidge DR, Vansteenkiste J, Sharma S, De Pas T, Riely GJ, et
al: Activity and safety of ceritinib in patients with
ALK-rearranged non-small-cell lung cancer (ASCEND-1): Updated
results from the multicentre, open-label, phase 1 trial. Lancet
Oncol. 17:452–463. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Crinò L, Ahn MJ, De Marinis F, Groen HJ,
Wakelee H, Hida T, Mok T, Spigel D, Felip E, Nishio M, et al:
Multicenter phase ii study of whole-body and intracranial activity
with ceritinib in patients With ALK-rearranged non-small-cell lung
cancer previously treated with chemotherapy and crizotinib: Results
from ASCEND-2. J Clin Oncol. 34:2866–2873. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Soria JC, Tan DSW, Chiari R, Wu YL,
Paz-Ares L, Wolf J, Geater SL, Orlov S, Cortinovis D, Yu CJ, et al:
First-line ceritinib versus platinum-based chemotherapy in advanced
ALK-rearranged non-small-cell lung cancer (ASCEND-4): A randomised,
open-label, phase 3 study. Lancet. 389:917–929. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hida T, Nokihara H, Kondo M, Kim YH, Azuma
K, Seto T, Takiguchi Y, Nishio M, Yoshioka H, Imamura F, et al:
Alectinib versus crizotinib in patients with ALK-positive
non-small-cell lung cancer (J-ALEX): An open-label, randomised
phase 3 trial. Lancet. 390:29–39. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pérol M, Pavlakis N, Levchenko E, Platania
M, Oliveira J, Novello S, Chiari R, Moran T, Mitry E, Nüesch E, et
al: Patient-reported outcomes from the randomized phase III ALEX
study of alectinib versus crizotinib in patients with ALK-positive
non-small-cell lung cancer. Lung Cancer. 138:79–87. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Camidge DR, Dziadziuszko R, Peters S, Mok
T, Noe J, Nowicka M, Gadgeel SM, Cheema P, Pavlakis N, de Marinis
F, et al: Updated efficacy and safety data and impact of the
EML4-ALK fusion variant on the efficacy of alectinib in untreated
ALK-positive advanced non-small cell lung cancer in the global
phase III ALEX study. J Thorac Oncol. 14:1233–1243. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Camidge DR, Kim HR, Ahn MJ, Yang JCH, Han
JY, Hochmair MJ, Lee KH, Delmonte A, García Campelo MR, Kim DW, et
al: Brigatinib versus crizotinib in advanced ALK inhibitor-naive
ALK-positive non-small cell lung cancer: Second interim analysis of
the phase III ALTA-1L Trial. J Clin Oncol. 38:3592–3603. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Solomon BJ, Besse B, Bauer TM, Felip E,
Soo RA, Camidge DR, Chiari R, Bearz A, Lin CC, Gadgeel SM, et al:
Lorlatinib in patients with ALK-positive non-small-cell lung
cancer: Results from a global phase 2 Study. Lancet Oncol.
19:1654–1667. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Friboulet L, Li N, Katayama R, Lee CC,
Gainor JF, Crystal AS, Michellys PY, Awad MM, Yanagitani N, Kim S,
et al: The ALK inhibitor ceritinib overcomes crizotinib resistance
in non-small cell lung cancer. Cancer Discov. 4:662–673. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xia B, Nagasaka M, Zhu VW, Ou SI and Soo
RA: How to select the best upfront therapy for metastatic disease?
Focus on ALK-rearranged non-small cell lung cancer (NSCLC). Transl
Lung Cancer Res. 9:2521–2534. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Werner MT, Zhao C, Zhang Q and Wasik MA:
Nucleophosmin-anaplastic lymphoma kinase: The ultimate oncogene and
therapeutic target. Blood. 129:823–831. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tse BC, Said BI, Fan ZJ, Hueniken K, Patel
D, Gill G, Liang M, Razooqi M, Brown MC, Sacher AG, et al:
Longitudinal health utilities, symptoms and toxicities in patients
with ALK-rearranged lung cancer treated with tyrosine kinase
inhibitors: A prospective real-world assessment. Curr Oncol.
27:e552–e559. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Castellanos EH and Horn L: Re-Evaluating
progression in an era of progress: A review of first- and
second-line treatment options in anaplastic lymphoma
kinase-positive non-small cell lung cancer. Oncologist. 21:755–761.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Herbst RS, Giaccone G, de Marinis F,
Reinmuth N, Vergnenegre A, Barrios CH, Morise M, Felip E, Andric Z,
Geater S, et al: Atezolizumab for first-line treatment of
PD-L1-selected patients with NSCLC. N Engl J Med. 383:1328–1339.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hallberg B and Palmer RH: The role of the
ALK receptor in cancer biology. Ann Oncol. 27 (Suppl 3):iii4–iii15.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Choi YL, Soda M, Yamashita Y, Ueno T,
Takashima J, Nakajima T, Yatabe Y, Takeuchi K, Hamada T, Haruta H,
et al: EML4-ALK mutations in lung cancer that confer resistance to
ALK inhibitors. N Engl J Med. 363:1734–1739. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Azam M, Seeliger MA, Gray NS, Kuriyan J
and Daley GQ: Activation of tyrosine kinases by mutation of the
gatekeeper threonine. Nat Struct Mol Biol. 15:1109–1118. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Heuckmann JM, Hölzel M, Sos ML, Heynck S,
Balke-Want H, Koker M, Peifer M, Weiss J, Lovly CM, Grütter C, et
al: ALK mutations conferring differential resistance to
structurally diverse ALK inhibitors. Clin Cancer Res. 17:7394–7401.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sasaki T, Koivunen J, Ogino A, Yanagita M,
Nikiforow S, Zheng W, Lathan C, Marcoux JP, Du J, Okuda K, et al: A
novel ALK secondary mutation and EGFR signaling cause resistance to
ALK kinase inhibitors. Cancer Res. 71:6051–6060. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Katayama R, Friboulet L, Koike S,
Lockerman EL, Khan TM, Gainor JF, Iafrate AJ, Takeuchi K, Taiji M,
Okuno Y, et al: Two novel ALK mutations mediate acquired resistance
to the next-generation ALK inhibitor alectinib. Clin Cancer Res.
20:5686–5696. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Isozaki H, Hotta K, Ichihara E, Takigawa
N, Ohashi K, Kubo T, Ninomiya T, Ninomiya K, Oda N, Yoshioka H, et
al: Protocol design for the bench to bed trial in
alectinib-refractory non-small-cell lung cancer patients harboring
the EML4-ALK fusion gene (ALRIGHT/OLCSG1405). Clin Lung Cancer.
17:602–605. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Sequist LV, Waltman BA, Dias-Santagata D,
Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger
S, Cosper AK, et al: Genotypic and histological evolution of lung
cancers acquiring resistance to EGFR inhibitors. Sci Transl Med.
3:75ra262011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zou HY, Friboulet L, Kodack DP, Engstrom
LD, Li Q, West M, Tang RW, Wang H, Tsaparikos K, Wang J, et al:
PF-06463922, an ALK/ROS1 inhibitor, overcomes resistance to first
and second generation ALK inhibitors in preclinical models. Cancer
Cell. 28:70–81. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Shaw AT, Friboulet L, Leshchiner I, Gainor
JF, Bergqvist S, Brooun A, Burke BJ, Deng YL, Liu W, Dardaei L, et
al: Resensitization to crizotinib by the lorlatinib ALK resistance
mutation L1198F. N Engl J Med. 374:54–61. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wu YL, Lu S, Lu Y, Zhou J, Shi YK,
Sriuranpong V, Ho JCM, Ong CK, Tsai CM, Chung CH, et al: Results of
PROFILE 1029, a phase iii comparison of first-line crizotinib
versus chemotherapy in East Asian patients with ALK-positive
advanced non-small cell lung cancer. J Thorac Oncol. 13:1539–1548.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Krishnamurthy N, Goodman AM, Barkauskas DA
and Kurzrock R: STK11 alterations in the pan-cancer setting:
Prognostic and therapeutic implications. Eur J Cancer. 148:215–229.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wohlhieter CA, Richards AL, Uddin F,
Hulton CH, Quintanal-Villalonga À, Martin A, de Stanchina E, Bhanot
U, Asher M, Shah NS, et al: Concurrent mutations in STK11 and KEAP1
promote ferroptosis protection and SCD1 dependence in lung cancer.
Cell Rep. 33:1084442020. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Skoulidis F, Goldberg ME, Greenawalt DM,
Hellmann MD, Awad MM, Gainor JF, Schrock AB, Hartmaier RJ, Trabucco
SE, Gay L, et al: STK11/LKB1 mutations and PD-1 inhibitor
resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov.
8:822–835. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Gowans GJ, Hawley SA, Ross FA and Hardie
DG: AMP is a true physiological regulator of AMP-activated protein
kinase by both allosteric activation and enhancing net
phosphorylation. Cell Metab. 18:556–566. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Hemminki A, Markie D, Tomlinson I,
Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M,
Höglund P, et al: A serine/threonine kinase gene defective in
Peutz-Jeghers syndrome. Nature. 391:184–187. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Mahoney CL, Choudhury B, Davies H, Edkins
S, Greenman C, Haaften G, Mironenko T, Santarius T, Stevens C,
Stratton MR and Futreal PA: LKB1/KRAS mutant lung cancers
constitute a genetic subset of NSCLC with increased sensitivity to
MAPK and mTOR signalling inhibition. Br J Cancer. 100:370–375.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Parachoniak CA, Rankin A, Gaffney B,
Hartmaier R, Spritz D, Erlich RL, Miller VA, Morosini D, Stephens
P, Ross JS, et al: Exceptional durable response to everolimus in a
patient with biphenotypic breast cancer harboring an STK11variant.
Cold Spring Harb Mol Case Stud. 3:a0007782017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Sanchez-Cespedes M, Parrella P, Esteller
M, Nomoto S, Trink B, Engles JM, Westra WH, Herman JG and Sidransky
D: Inactivation of LKB1/STK11 is a common event in adenocarcinomas
of the lung. Cancer Res. 62:3659–3662. 2002.PubMed/NCBI
|
|
72
|
Hezel AF, Gurumurthy S, Granot Z, Swisa A,
Chu GC, Bailey G, Dor Y, Bardeesy N and Depinho RA: Pancreatic LKB1
deletion leads to acinar polarity defects and cystic neoplasms. Mol
Cell Biol. 28:2414–2425. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wingo SN, Gallardo TD, Akbay EA, Liang MC,
Contreras CM, Boren T, Shimamura T, Miller DS, Sharpless NE,
Bardeesy N, et al: Somatic LKB1 mutations promote cervical cancer
progression. PLoS One. 4:e51372009. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Gill RK, Yang SH, Meerzaman D, Mechanic
LE, Bowman ED, Jeon HS, Roy Chowdhuri S, Shakoori A, Dracheva T,
Hong KM, et al: Frequent homozygous deletion of the LKB1/STK11gene
in non-small cell lung cancer. Oncogene. 30:3784–3791. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Lee SM, Choi JE, Na YK, Lee EJ, Lee WK,
Choi YY, Yoon GS, Jeon HS, Kim DS and Park JY: Genetic and
epigenetic alterations of the LKB1 gene and their associations with
mutations in TP53 and EGFR pathway genes in Korean non-small cell
lung cancers. Lung Cancer. 81:194–199. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Tanwar PS, Mohapatra G, Chiang S, Engler
DA, Zhang L, Kaneko-Tarui T, Ohguchi Y, Birrer MJ and Teixeira JM:
Loss of LKB1 and PTEN tumor suppressor genes in the ovarian surface
epithelium induces papillary serous ovarian cancer. Carcinogenesis.
35:546–553. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Li J, Liu J, Li P, Mao X, Li W, Yang J and
Liu P: Loss of LKB1 disrupts breast epithelial cell polarity and
promotes breast cancer metastasis and invasion. J Exp Clin Cancer
Res. 33:702014. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Yang JY, Jiang SH, Liu DJ, Yang XM, Huo
YM, Li J, Hua R, Zhang ZG and Sun YW: Decreased LKB1 predicts poor
prognosis in pancreatic ductal adenocarcinoma. Sci Rep.
5:105752015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zhang W, Yin L, Song G, Han X, Yin Z and
Luo D: LKB1 loss cooperating with BRAFV600E promotes melanoma cell
invasion and migration by up-regulation MMP-2 via PI3K/Akt/mTOR
pathway. Oncotarget. 8:113847–113857. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Matsumoto S, Iwakawa R, Takahashi K, Kohno
T, Nakanishi Y, Matsuno Y, Suzuki K, Nakamoto M, Shimizu E, Minna
JD and Yokota J: Prevalence and specificity of LKB1 genetic
alterations in lung cancers. Oncogene. 26:5911–5918. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Fang R, Zheng C, Sun Y, Han X, Gao B, Li
C, Liu H, Wong KK, Liu XY, Chen H and Ji H: Integrative genomic
analysis reveals a high frequency of LKB1 genetic alteration in
Chinese lung adenocarcinomas. J Thorac Oncol. 9:254–258. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Ji H, Ramsey MR, Hayes DN, Fan C, McNamara
K, Kozlowski P, Torrice C, Wu MC, Shimamura T, Perera SA, et al:
LKB1 modulates lung cancer differentiation and metastasis. Nature.
448:807–810. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Liang J and Mills GB: AMPK: A contextual
oncogene or tumor suppressor? Cancer Res. 73:2929–2935. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Cancer Genome Atlas Research Network, .
Comprehensive molecular profiling of lung adenocarcinoma. Nature.
511:543–550. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Imielinski M, Berger AH, Hammerman PS,
Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M,
Sivachenko A, et al: Mapping the hallmarks of lung adenocarcinoma
with massively parallel sequencing. Cell. 150:1107–1120. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Baas AF, Smit L and Clevers H: LKB1 tumor
suppressor protein: PARtaker in cell polarity. Trends Cell Biol.
14:312–319. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Forcet C, Etienne-Manneville S, Gaude H,
Fournier L, Debilly S, Salmi M, Baas A, Olschwang S, Clevers H and
Billaud M: Functional analysis of Peutz-Jeghers mutations reveals
that the LKB1 C-terminal region exerts a crucial role in regulating
both the AMPK pathway and the cell polarity. Hum Mol Genet.
14:1283–1292. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Galan-Cobo A, Sitthideatphaiboon P, Qu X,
Poteete A, Pisegna MA, Tong P, Chen PH, Boroughs LK, Rodriguez MLM,
Zhang W, et al: LKB1 and KEAP1/NRF2 pathways cooperatively promote
metabolic reprogramming with enhanced glutamine dependence in
KRAS-mutant lung adenocarcinoma. Cancer Res. 79:3251–3267. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Koyama S, Akbay EA, Li YY, Aref AR,
Skoulidis F, Herter-Sprie GS, Buczkowski KA, Liu Y, Awad MM,
Denning WL, et al: STK11/LKB1 deficiency promotes neutrophil
recruitment and proinflammatory cytokine production to suppress
T-cell activity in the lung tumor microenvironment. Cancer Res.
76:999–1008. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Shackelford DB and Shaw RJ: The LKB1-AMPK
pathway: Metabolism and growth control in tumour suppression. Nat
Rev Cancer. 9:563–575. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Shen Z, Wen XF, Lan F, Shen ZZ and Shao
ZM: The tumor suppressor gene LKB1 is associated with prognosis in
human breast carcinoma. Clin Cancer Res. 8:2085–2090.
2002.PubMed/NCBI
|
|
92
|
Liu W, Monahan KB, Pfefferle AD, Shimamura
T, Sorrentino J, Chan KT, Roadcap DW, Ollila DW, Thomas NE,
Castrillon DH, et al: LKB1/STK11 inactivation leads to expansion of
a prometastatic tumor subpopulation in melanoma. Cancer Cell.
21:751–764. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Sanchez-Cespedes M: A role for LKB1 gene
in human cancer beyond the Peutz-Jeghers syndrome. Oncogene.
26:7825–7832. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Schabath MB, Welsh EA, Fulp WJ, Chen L,
Teer JK, Thompson ZJ, Engel BE, Xie M, Berglund AE, Creelan BC, et
al: Differential association of STK11 and TP53 with
KRASmutation-associated gene expression, proliferation and immune
surveillance in lung adenocarcinoma. Oncogene. 35:3209–3216. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Lamberti G, Sisi M, Andrini E, Palladini
A, Giunchi F, Lollini PL, Ardizzoni A and Gelsomino F: The
mechanisms of PD-L1 regulation in non-small-cell lung cancer
(NSCLC): Which are the involved players? Cancers (Basel).
12:31292020. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Jordan EJ, Kim HR, Arcila ME, Barron D,
Chakravarty D, Gao J, Chang MT, Ni A, Kundra R, Jonsson P, et al:
Prospective comprehensive molecular characterization of lung
adenocarcinomas for efficient patient matching to approved and
emerging therapies. Cancer Discov. 7:596–609. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Roosan MR, Mambetsariev I, Pharaon R,
Fricke J, Husain H, Reckamp KL, Koczywas M, Massarelli E, Bild AH
and Salgia R: Usefulness of circulating tumor DNA in identifying
somatic mutations and tracking tumor evolution in patients with
non-small cell lung cancer. Chest. 160:1095–1107. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Dahmani R, Just PA, Delay A, Canal F,
Finzi L, Prip-Buus C, Lambert M, Sujobert P, Buchet-Poyau K, Miller
E, et al: A novel LKB1 isoform enhances AMPK metabolic activity and
displays oncogenic properties. Oncogene. 34:2337–2346. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Bouchekioua-Bouzaghou K, Poulard C,
Rambaud J, Lavergne E, Hussein N, Billaud M, Bachelot T, Chabaud S,
Mader S, Dayan G, et al: LKB1 when associated with methylatedERα is
a marker of bad prognosis in breast cancer. Int J Cancer.
135:1307–1318. 2014.PubMed/NCBI
|
|
100
|
Koivunen JP, Kim J, Lee J, Rogers AM, Park
JO, Zhao X, Naoki K, Okamoto I, Nakagawa K, Yeap BY, et al:
Mutations in the LKB1 tumour suppressor are frequently detected in
tumours from Caucasian but not Asian lung cancer patients. Br J
Cancer. 99:245–252. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Roy BC, Kohno T, Iwakawa R, Moriguchi T,
Kiyono T, Morishita K, Sanchez-Cespedes M, Akiyama T and Yokota J:
Involvement of LKB1 in epithelial-mesenchymal transition (EMT) of
human lung cancer cells. Lung Cancer. 70:136–145. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Yao YH, Cui Y, Qiu XN, Zhang LZ, Zhang W,
Li H and Yu JM: Attenuated LKB1-SIK1 signaling promotes
epithelial-mesenchymal transition and radioresistance of non-small
cell lung cancer cells. Chin J Cancer. 35:502016. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Momcilovic M and Shackelford DB: Targeting
LKB1 in cancer-exposing and exploiting vulnerabilities. Br J
Cancer. 113:574–584. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Shackelford DB, Abt E, Gerken L, Vasquez
DS, Seki A, Leblanc M, Wei L, Fishbein MC, Czernin J, Mischel PS,
et al: LKB1 inactivation dictates therapeutic response of non-small
cell lung cancer to the metabolism drug phenformin. Cancer Cell.
23:143–158. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Dobashi Y, Watanabe Y, Miwa C, Suzuki S
and Koyama S: Mammalian target of rapamycin: A central node of
complex signaling cascades. Int J Clin Exp Pathol. 4:476–495.
2011.PubMed/NCBI
|
|
106
|
Saxton RA and Sabatini DM: mTOR signaling
in growth, metabolism, and disease. Cell. 168:960–976. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Engelman JA: Targeting PI3K signalling in
cancer: Opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Chen B, Tan Z, Gao J, Wu W, Liu L, Jin W,
Cao Y, Zhao S, Zhang W, Qiu Z, et al: Hyperphosphorylation of
ribosomal protein S6 predicts unfavorable clinical survival in
non-small cell lung cancer. J Exp Clin Cancer Res. 34:1262015.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Krencz I, Sebestyén A, Fábián K, Márk Á,
Moldvay J, Khoor A, Kopper L and Pápay J: Expression of
mTORC1/2-related proteins in primary and brain metastatic lung
adenocarcinoma. Hum Pathol. 62:66–73. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Seki N, Takasu T, Mandai K, Nakata M,
Saeki H, Heike Y, Takata I, Segawa Y, Hanafusa T and Eguchi K:
Expression of eukaryotic initiation factor 4E in atypical
adenomatous hyperplasia and adenocarcinoma of the human peripheral
lung. Clin Cancer Res. 8:3046–3053. 2002.PubMed/NCBI
|
|
111
|
Yoshizawa A, Fukuoka J, Shimizu S, Shilo
K, Franks TJ, Hewitt SM, Fujii T, Cordon-Cardo C, Jen J and Travis
WD: Overexpression of phospho-eIF4E is associated with survival
through AKT pathway in non-small cell lung cancer. Clin Cancer Res.
16:240–248. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Jeon SM, Chandel NS and Hay N: AMPK
regulates NADPH homeostasis to promote tumour cell survival during
energy stress. Nature. 485:661–665. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Joo MS, Kim WD, Lee KY, Kim JH, Koo JH and
Kim SG: AMPK facilitates nuclear accumulation of Nrf2 by
phosphorylating at serine 550. Mol Cell Biol. 36:1931–1942. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Ciccarese F, Zulato E and Indraccolo S:
LKB1/AMPK pathway and drug response in cancer: A therapeutic
perspective. Oxid Med Cell Longev. 2019:87308162019. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Singh A, Daemen A, Nickles D, Jeon SM,
Foreman O, Sudini K, Gnad F, Lajoie S, Gour N, Mitzner W, et al:
NRF2 activation promotes aggressive lung cancer and associates with
poor clinical outcomes. Clin Cancer Res. 27:877–888. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Trapp EK, Majunke L, Zill B, Sommer H,
Andergassen U, Koch J, Harbeck N, Mahner S, Friedl TWP, Janni W, et
al: LKB1 pro-oncogenic activity triggers cell survival in
circulating tumor cells. Mol Oncol. 11:1508–1526. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Shaw RJ, Kosmatka M, Bardeesy N, Hurley
RL, Witters LA, DePinho RA and Cantley LC: The tumor suppressor
LKB1 kinase directly activates AMP-activated kinase and regulates
apoptosis in response to energy stress. Proc Natl Acad Sci USA.
101:3329–3335. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Deng L, Yao P, Li L, Ji F, Zhao S, Xu C,
Lan X and Jiang P: p53-mediated control of aspartate-asparagine
homeostasis dictates LKB1 activity and modulates cell survival. Nat
Commun. 11:17552020. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Goodman AM, Kato S, Bazhenova L, Patel SP,
Frampton GM, Miller V, Stephens PJ, Daniels GA and Kurzrock R:
Tumor mutational burden as an independent predictor of response to
immunotherapy in diverse cancers. Mol Cancer Ther. 16:2598–2608.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Goodman AM, Piccioni D, Kato S, Boichard
A, Wang HY, Frampton G, Lippman SM, Connelly C, Fabrizio D, Miller
V, et al: Prevalence of PDL1 amplification and preliminary response
to immune checkpoint blockade in solid Tumors. JAMA Oncol.
4:1237–1244. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Frampton GM, Fichtenholtz A, Otto GA, Wang
K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, et
al: Development and validation of a clinical cancer genomic
profiling test based on massively parallel DNA sequencing. Nat
Biotechnol. 31:1023–1031. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Kobayashi A, Kang MI, Okawa H, Ohtsuji M,
Zenke Y, Chiba T, Igarashi K and Yamamoto M: Oxidative stress
sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to
regulate proteasomal degradation of Nrf2. Mol Cell Biol.
24:7130–7139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Donnelly LL, Hogan TC, Lenahan SM,
Nandagopal G, Eaton JG, Lebeau MA, McCann CL, Sarausky HM, Hampel
KJ, Armstrong JD, et al: Functional assessment of somatic
STK11variants identified in primary human non-small cell lung
cancers. Carcinogenesis. 42:1428–1438. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Gill CM, Loewenstern J, Rutland JW, Arib
H, Pain M, Umphlett M, Kinoshita Y, McBride RB, Bederson J, Donovan
M, et al: STK11 mutation status is associated with decreased
survival in meningiomas. Neurol Sci. 41:2585–2589. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Facchinetti F, Bluthgen MV,
Tergemina-Clain G, Faivre L, Pignon JP, Planchard D, Remon J, Soria
JC, Lacroix L and Besse B: LKB1/STK11 mutations in non-small cell
lung cancer patients: Descriptive analysis and prognostic value.
Lung Cancer. 112:62–68. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Borghaei H, Paz-Ares L, Horn L, Spigel DR,
Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, et al:
Nivolumab versus docetaxel in advanced nonsquamous non-small-cell
lung cancer. N Engl J Med. 373:1627–1639. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Rittmeyer A, Barlesi F, Waterkamp D, Park
K, Ciardiello F, von Pawel J, Gadgeel SM, Hida T, Kowalski DM, Dols
MC, et al: Atezolizumab versus docetaxel in patients with
previously treated non-small-cell lung cancer (OAK): A phase 3,
open-label, multicentre randomised controlled trial. Lancet.
389:255–265. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Herbst RS, Baas P, Perez-Gracia JL, Felip
E, Kim DW, Han JY, Molina JR, Kim JH, Dubos Arvis C, Ahn MJ, et al:
Use of archival versus newly collected tumor samples for assessing
PD-L1 expression and overall survival: An updated analysis of
KEYNOTE-010 trial. Ann Oncol. 30:281–289. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Zugazagoitia J, Molina-Pinelo S,
Lopez-Rios F and Paz-Ares L: Biological therapies in nonsmall cell
lung cancer. Eur Respir J. 49:16015202017. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Shire NJ, Klein AB, Golozar A, Collins JM,
Fraeman KH, Nordstrom BL, McEwen R, Hembrough T and Rizvi NA: STK11
(LKB1) mutations in metastatic NSCLC: Prognostic value in the real
world. PLoS One. 15:e02383582020. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Mograbi B, Heeke S and Hofman P: The
Importance of STK11/LKB1 assessment in non-small cell lung
carcinomas. Diagnostics (Basel). 11:1962021. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Herter-Sprie GS, Korideck H, Christensen
CL, Herter JM, Rhee K, Berbeco RI, Bennett DG, Akbay EA, Kozono D,
Mak RH, et al: Image-guided radiotherapy platform using single
nodule conditional lung cancer mouse models. Nat Commun.
5:58702014. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
He Q, Li J, Dong F, Cai C and Zou X: LKB1
promotes radioresistance in esophageal cancer cells exposed to
radiation, by suppression of apoptosis and activation of autophagy
via the AMPK pathway. Mol Med Rep. 16:2205–2210. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Wang Y, Li N, Jiang W, Deng W, Ye R, Xu C,
Qiao Y, Sharma A, Zhang M, Hung MC, et al: Mutant LKB1 confers
enhanced radiosensitization in combination with trametinib in
KRAS-mutant non-small cell lung cancer. Clin Cancer Res.
24:5744–5756. 2018. View Article : Google Scholar : PubMed/NCBI
|