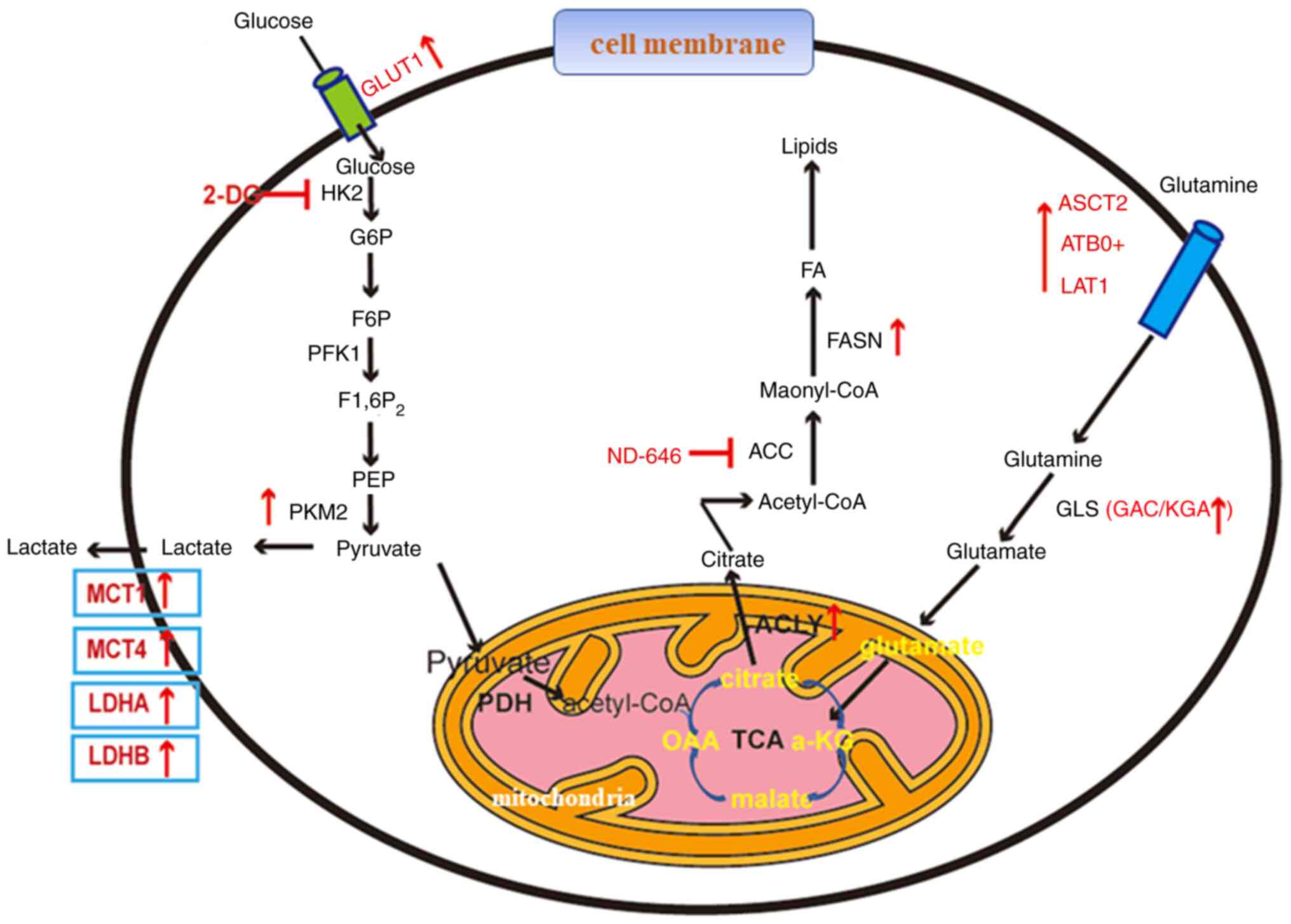
Lung cancer is a disease with high incidence and
mortality rates worldwide. At present, the pathogenic factors of
lung cancer have not been completely clarified. The most common
pathogenic factor of lung cancer is long-term and high-frequency
smoking (1). In addition,
long-term exposure to carcinogens, air pollution, human immune
status, genetic factors and metabolic activities have been
associated with lung cancer (2).
Lung cancer is mainly divided into small cell lung cancer (SCLC)
and non-SCLC (NSCLC). The latter includes lung squamous cell
carcinoma, lung adenocarcinoma (LUAD) and large cell cancer,
accounting for ~85% of all lung cancer cases (3). Although considerable progress has
been achieved in the targeted treatment of lung cancer in recent
years, drug resistance, recurrence and metastasis have brought
great difficulties (4). At
present, surgical resection is still performed for tumors that
limit themselves to the primary location, but 80–85% of patients
are already in the unresectable stage at early diagnosis (5). In addition, the progress of lung
cancer treatment is limited by the adverse reactions of
chemotherapeutic drugs, the high resistance rate of targeted drugs
and the immune tolerance microenvironment of the tumor. Therefore,
novel strategies for lung cancer treatment need to be
developed.
Metabolism is the energy and material basis of life
activities. Under normal circumstances, metabolism occurs in the
body in an orderly manner to ensure physiological functions
(6). However, tumor growth is a
multi-factor and multi-stage dynamic process. Some studies have
shown that the metabolic pattern of tumor cells is different from
that of normal cells (7–9). Metabolic change, also called
metabolic reprogramming, plays an important role in regulating the
occurrence and development of tumors. To meet the needs of rapid
proliferation and growth in a tumor microenvironment with poor
blood vessels and nutrition, tumor cells undergo metabolic
reprogramming to provide energy and raw materials, maintain the
steady state of cell redox and regulate intracellular signal
transduction (10). However, the
mechanisms underlying tumorigenesis and development remain to be
elucidated. Various metabolism-related genes serve as oncogenes and
tumor suppressor genes (11). It
has been shown that oncogenes such as MYC, NF-κB and AKT can
regulate the enzymes in the glycolysis and glutaminolysis pathways
(12–14). MYC increases the transcription rate
of GLUT transporter and hexokinase-2, which enhances glucose uptake
and retention (15).
Therefore, the present review summarizes the
abnormal changes in the metabolism of glucose, fat and amino acids
in lung cancer, and the molecular mechanisms behind them to provide
novel ideas for the prevention, early diagnosis and treatment of
lung cancer (Fig. 1).
Tumor cells produce intermediate metabolites through
glycolysis. These intermediate metabolites provide raw materials
for biosynthetic pathways, such as nucleotides, lipids, amino acids
and reduced nicotinamide adenine dinucleotide phosphate (NADPH), to
meet the needs of rapid cell proliferation (21). These metabolic pathways include the
pentose phosphate pathway for the production of RNA and NADPH, the
hexosamine pathway for protein glycosylation and glycogen
production, and the serine biosynthesis pathway (22–24).
The first step in glycolysis is glucose entering the
cell through the plasma membrane. Glucose transporters (GLUTs) are
important carrier proteins responsible for transporting glucose.
Treatment of A549 lung cancer cells with WZB117, an irreversible
inhibitor of GLUT1, reduces GLUT1 expression and glucose uptake; it
also inhibits the growth of lung cancer cells in cooperation with
cisplatin and paclitaxel. The addition of exogenous ATP alleviates
this inhibitory effect, which indicates that the inhibition of
GLUT1 prevents tumor growth by blocking ATP synthesis (25).
After glucose enters the cell, the first
irreversible reaction is the production of glucose-6-phosphate
catalyzed by hexokinase 2 (HK2). Tumor cells promote glucose
metabolism by increasing glucose uptake and inducing high HK2
expression (26). Targeting HK can
reverse the drug resistance of tumor cells (27). 2-Deoxy-D-glucose (2-DG) is a
small-molecule inhibitor targeting HK. 2-DG combined with
Adriamycin or paclitaxel can significantly delay tumor growth and
prolong the survival time of mice with NSCLC (28). Moreover, inhibition of glycolysis
by 2-DG can improve the sensitivity of NSCLC with T790M secondary
drug resistance mutation to epidermal growth factor receptor
(EGFR)-tyrosine kinase inhibitor (TKI) (29).
In the second step, fructose-1,6-diphosphate is
catalyzed by phosphofructokinase 1 (PFK1). Fascin promotes the
transcription of PFKFB3 by promoting the binding of YAP1 to a
TEAD1/4 binding site, which activates the expression of PFK1 and
mediates glycolysis in lung cancer (30). Not only is PFK expression
upregulated in malignant tumors, platelet-type PFK can also
regulate the glycolysis level of lung cancer and promote cell
proliferation (31).
In the third step, pyruvate kinase catalyzes the
conversion of phosphoenolpyruvic acid and ADP to pyruvate and ATP,
respectively. Low-affinity pyruvate kinase M2 (PKM2) is highly
expressed in lung cancer, and its upregulation is often associated
with the hypomethylation of the PKM2 gene promoter. Silencing PKM2
can improve the sensitivity of lung cancer to the chemotherapy
drugs cisplatin and docetaxel by increasing apoptosis and
inhibiting proliferation. PKM2 is a potential adjuvant therapeutic
target (32,33). However, PKM1 rather than PKM2 shows
tumor-promoting function in pulmonary neuroendocrine tumors,
including SCLC. This finding challenges the view that PKM2 limiting
glucose metabolism is a prerequisite for tumorigenesis and provides
a theoretical basis for PKM1 as a therapeutic target in SCLC
(34).
The attenuation of the last reaction step promotes
the entry of intermediate metabolites into the above biosynthetic
pathway (35). However, the
expression of lactate dehydrogenase (LDH) in lung cancer cells is
upregulated (36), which can
convert pyruvate into lactate. In addition, lactate secretion is
increased, which is characterized by a high expression of
monocarboxylate transporter (MCT), as lactate retained in cells
inhibits the expression of PFK1 (37). Lactate secretion into the
environment can also promote the development of tumors (38). Lactate, as a signal transduction
substance under hypoxia, activates the signal pathway associated
with tumor cell survival (39).
Targeted lactate production is a possible strategy to overcome
tumor drug resistance. Inhibition of LDH activity by small
interfering RNA or oxalate can overcome the drug resistance of
tumor cells to paclitaxel and trastuzumab (40,41).
In mice and patients with lung cancer, lactate in tissues and
circulation provides an equivalent carbon source for the aerobic
oxidation of normal and tumor tissues, and its contribution to
mitochondrial metabolism is no less than that of glucose (38,42).
Faubert et al (42)
injected 13C-labeled lactate into patients with lung
cancer and found that lactate can be used as the carbon source of
the TCA cycle in patients with different lung cancer types, and
that the expression levels of MCT1, MCT4, LDHA and LDHB in tumors
are upregulated. In the transplanted tumor model of human lung
cancer cells in mice, the levels of isotopic-labeled lactic acid
and isotopic-labeled TCA cycle intermediate metabolites at the
tumor site are increased. Knockout of MCT1 in mice could reduce the
uptake of lactic acid by tumors (43).
The upregulated expression of these key glycolytic
enzymes in lung cancer cells is often closely associated with the
abnormally activated oncogenes and cancer-promoting signaling
pathways in cells. Hypoxia inducible factor-1α (HIF-1α) is an
important transcription factor involved in glycolysis regulation in
cells, which can directly transcribe and regulate the expression of
multiple key glycolysis enzymes (44). The expression of aldolase A in the
glycolysis pathway is increased in NSCLC, which consequently
increases lactate activity to inhibit prolyl hydroxylase activity
and further induce HIF-1α (45).
The positive feedback of lactate further promotes the aerobic
glycolysis of tumor cells. MYC is an important oncogene that
promotes the glycolysis of lung cancer cells by transcriptionally
regulating the expression of multiple key glycolytic enzymes
(46).
Amino acid metabolic abnormalities include those in
glutamine, serine and glycine, among others, the most important of
which are the glutamine metabolic abnormalities (47). In addition to glycolysis, numerous
tumor cells also rely on glutamine to meet their bioenergy and
metabolic needs. Although glutamine is an important non-essential
amino acid required for cell proliferation, it is also an essential
amino acid under specific circumstances (48). Glutamine depends on glutamine
metabolism, provides metabolic energy for rapidly proliferating
tumor cells, provides carbon and nitrogen sources for the synthesis
and metabolism of substances, such as nucleotides, amino acids and
fatty acids, and maintains the balance and stability of cellular
reactive oxygen species (49).
Therefore, similar to glucose, glutamine is considered the main
nutrient to promote tumor proliferation.
In tumor cells, glutamine can be transported into
cells by amino acid transporters as a substrate. Glutamine is then
converted into glutamate in the mitochondria and enters the TCA
cycle. Some amino acid transporters, such as
alanine-serine-cysteine transporter 2, amino acid transporter
B0,+ and Human L-type amino acid transporter 1, are
overexpressed in lung cancer and upregulate the intake of glutamine
by cancer cells (50). Meanwhile,
glutamine and glutamate play important roles in the growth of
pC9/IR-resistant cells. The growth of erlotinib-resistant NSCLC
depends on glutamine (51).
Clinical studies have found high plasma glutamine
levels in patients with malignant tumors (52). Glutamine can be converted into
glutamate under the action of glutaminase (GLS) for the synthesis
of fatty acids and glutathione. Antioxidant glutathione maintains
the balance of redox reaction in tumor cells, helps tumor cells
resist oxidative stress and prolongs tumor cell survival (53).
Lipids contain thousands of different types of
molecules, including glycerophosphingolipids, glycerides, fatty
acids, sphingolipids, sterol lipids, pregnenolone lipids,
glycolipids and polyketones. Lipids are widely distributed in
organelles and are a key component of all membranes (62). Lipid metabolism provides energy for
tumor cells, cell proliferation and signaling molecule generation
(63).
The biosynthesis of fatty acids begins with
acetyl-CoA carboxylase (ACC) carboxylating acetyl CoA in the
cytoplasm to produce malonyl CoA. Silencing ACC accelerates the
growth of lung cancer cells by promoting the redox balance of NADPH
(53). ND-646, an allosteric
inhibitor of ACC, also exerts an antitumor effect on NSCLC cells
(66).
Fatty acid synthase (FASN) catalyzes the assembly of
malonyl CoA or acetyl CoA into fatty acids; it is also one of the
key enzymes in fatty acid anabolism. The increase in fatty acid
synthesis is due to the increase in FASN level, which is closely
associated with a poor prognosis, as confirmed in various tumors,
such as pancreatic and breast cancer (67,68).
Currie et al (69) found a
high expression level of fatty acid synthase in NSCLC. A high
expression level of FASN not only promotes the growth of tumor
cells but also improves the metastatic ability of NSCLC cells and
cisplatin resistance (70). Ali
et al (71) found that the
palmitoylation of EGFR is specifically expressed in mutated EGFR
NSCLC with acquired TKI resistance. Mutated EGFR activates FASN
mediated by sterol regulatory element-binding proteins (SREBPs),
which consequently promote the palmitoylation of EGFR. To produce
TKI resistance, orlistat, a selective inhibitor of FASN, can
inhibit this effect. Therefore, FASN is an attractive therapeutic
target. Most tumor cells rely on the FASN-mediated de novo
synthesis of fatty acids, whereas most non-tumor cells rely on
exogenous fatty acids. The first targeted drug of FASN, TVB-2640,
has entered clinical trials. Combined with paclitaxel, it can
stabilize the disease progression in the medium and long term
(72,73).
Fatty acid synthesis can also be performed through
reductive carboxylation. Glutamine-derived α-ketoglutarate is
catalyzed by isocitrate dehydrogenase (IDH) to form citric acid
(49). IDH mutations are
associated with various tumor types, including gliomas, myeloid
malignancies and myelodysplastic syndormes (74). Genetic hybridization of IDH2 mutant
mice with carcinogenic FLT3 or NRAS alleles can promote leukemia
transformation by inhibiting myeloid cell differentiation (75). The expression levels of FASN, ACC
and ATP-citrate lyase (ACLY) are upregulated in fatty acid
synthesis in various cancer types, including prostate cancer,
breast cancer, colorectal cancer, lung cancer, hepatocellular
carcinoma, pancreatic cancer, gastric cancer and multiple myeloma.
Inhibition of these enzymes prevents tumor growth in vitro
and in vivo (76–78).
ACLY is the most critical enzyme in glucose
catabolism, and cholesterol and fatty acid anabolism. In the
presence of ATP and CoA, it catalyzes citric acid in the cytoplasm
to produce acetyl CoA and oxaloacetate. Acetyl CoA is not only an
important raw material for the de novo synthesis of
cholesterol and fatty acids, but also a substrate for protein
acetylation modification. ACLY knockdown or inhibition with
SB-204990 changes the metabolic pathway to reduce the occurrence of
mouse tumors and the formation of transplanted human tumor cells
(79,80). Acetylation of 540, 546 and 554
lysine residues (3K) of ACLY polypeptide chain promotes lipid
biosynthesis and tumor growth. The acetylation level of ACLY is
significantly increased in lung cancer tissues (81). Further investigations have
confirmed that ACLY 3K is also a ubiquitin modification site, and a
competitive association exists between them. Under high-glucose
conditions, P300/calcium-binding protein-associated factor
acetyltransferase is activated to promote ACLY acetylation, block
ACLY ubiquitination and degradation, improve its stability, and
promote de novo lipid synthesis and tumor growth (81). ACLY is a promising therapeutic
target for lung cancer; its product, acetyl CoA, is not only an
important metabolite, but also a substrate for the acetylation of
proteins and nucleic acids. Therefore, inhibiting its production
affects de novo fatty acid biosynthesis.
Fatty acids entering the bioactive pool must be
activated by acetyl CoA synthetase (ACS) to produce fatty acid CoA.
Bioactive fatty acids contribute to protein palmitoylation, a
post-translational modification that is particularly important in
certain tumors (82). Triacsin C,
a chemical inhibitor of ACS, induces the apoptosis of lung cancer
cells, and the expression of ACS is negatively correlated with the
overall survival of patients with lung cancer (83).
The polyunsaturated fatty acid metabolism and
biotransformation pathways have a great impact on tumor apoptosis
and proliferation. A number of tumor cells highly express
clyclooxygenase (COX), lipoxygenase (LOX) and cytochrome P450.
These enzymes transform ω-6 polyunsaturated fatty acids into highly
active arachidonic acid (AA) to regulate the proliferation and
apoptosis of tumor cells (84).
Various ω-3 metabolites of polyunsaturated fatty acids inhibit
tumorigenic pathways; for example, the eicosapentaenoic acid
metabolite ω-3,17,18-epoxide cascade activates anti-proliferation
and pro-apoptosis pathways (85).
Resolvins produced by LOX metabolites are effective inhibitors of
tumor-derived inflammatory pathways (86). The cytosolic phospholipase
A2-AA-COX-2 pathway is an important signaling pathway of
inflammation, and some key factors of the pathway are associated
with lung cancer. Overexpression of COX-2 initiates and promotes
lung cancer development (87,88).
Cholesterol homeostasis is strictly regulated by a
complex protein grid, including its intake, synthesis, efflux,
metabolism and esterification. High levels of cholesterol are found
in prostate, breast, liver, gastric, colorectal and lung cancer
(89,90).
3-Hydroxy-3-methyl glutaryl CoA reductase is the
rate-limiting enzyme of cholesterol biosynthesis; it is upregulated
in lung cancer and the target of statins regulating plasma
cholesterol (91).
In a number of tissues and organs, cholesterol can
be converted to 27-hydroxycholersterol (27HC) through sterol 27
hydroxylase (CYP27A1). 27HC is the most abundant hydroxyl
cholesterol substance in the circulating blood; its role in cancer
mainly depends on the properties of endogenous SREBP and the
function of liver X receptor (LXR) regulator.
In normal cells, the rate of cholesterol production
is low, occurring through the transcriptional regulation of key
genes involved in lipid biosynthesis. Tumor cells can regulate and
activate SREBP through the PI3K/Akt/mTOR, MAPK/ERK1/2, HIF-1α, p53
and SHH pathways to increase the signal activity of growth factors
or steroid hormone receptors (92). In vivo, 27HC inhibits
cholesterol synthesis by regulating SREBP (93).
LXRs are members of the nuclear receptor family of
ligand-dependent transcription factors. Activation of LXRs affects
cancer progression in various ways. Specifically, LXRs inhibit the
proliferation, migration and invasion of breast, prostate, ovarian,
lung, skin and colorectal cancer cells (94,95).
27HC is a risk factor for a number of cancer types; it triggers a
tumor response to endocrine therapy and reduces the activation of
estrogen receptor (ER) to promote angiogenesis, proliferation,
invasion and migration (96). By
contrast, 27HC inhibits cell viability, proliferation, invasion and
migration by activating LXRs in EGFR-mutated malignant tumors, such
as lung cancer and colon cancer (97,98).
Lung cancer tissues are rich in 27HC, and the expression of 27HC
synthase CYP27A1 in lung cancer cells is also higher than that in
normal lung cells. 27HC may be the main source of lung cancer
occurrence and development (99).
Existing research results show that 27HC promotes lung cancer cells
proliferation in an ERβ-dependent manner. The role of 27HC is not
affected by the membrane-bound ER G protein-coupled receptor 30.
Conversely, the role of 27HC is not associated with the activation
of EGFR or MAPK, but is mediated by the PI3K/Akt signaling pathway.
In other respects, 27HC promotes the production of osteoclasts in
the microenvironment of LUAD by inhibiting the expression of
miR-139 and activating the STAT3/c-Fos/NFATc1 pathway to accelerate
bone metastasis (100). A
high-cholesterol diet upregulates the level of 27HC in vivo,
whereas 27HC downregulates the cholesterol level of ER-negative
NSCLC A549 cells by activating the LXR signaling pathway, and
inhibits the activity and proliferation of A549 cells.
Cholesterol is also involved in the drug resistance
of tumor cells. For example, Chen et al (101) observed that the cholesterol level
is significantly higher in the lipid raft of gefitinib-resistant
cells than in that of gefitinib-sensitive cells in NSCLC. After
depletion of lipid raft cholesterol, the cells recover their
sensitivity to gefitinib. Colenemine and betulin improve the
sensitivity of patients with NSCLC to gefitinib by inhibiting
SREBP/SCAP or SREBP (102).
Carcinogenesis is a complex process involving
multiple genes and steps. In cell carcinogenesis, the imbalance in
cell metabolism is not only an important biochemical basis for
maintaining various malignant phenotypes, such as tumor cell
growth, proliferation and apoptosis, but also a result of the
imbalance of the intracellular regulation grid; it involves the
activation of oncogenes, the inactivation of tumor suppressor genes
and gene mutations. With the continuous development of
metabolomics, genomics, proteomics and other associated disciplines
and technologies, great breakthroughs have been achieved in the
research into the metabolic reprogramming of lung cancer in recent
years, and the molecular mechanisms underlying metabolic
reprogramming have been gradually understood. However, tumor
metabolic reprogramming includes different metabolic pathways that
usually involve several regulatory genes, metabolic enzymes and
signaling pathways. The research progress associated with lung
cancer metabolism is far less than expected, and each metabolic
pathway and its specific regulatory mechanism need to be further
studied. The identification of the specific metabolic enzymes or
metabolites involved in the occurrence and development of lung
cancer, and the elucidation of their roles and mechanisms in
tumorigenesis warrant further investigation.
Not applicable.
This research was supported by a grant from the Scientific
Research Program of Tianjin Education Commission (no.
2020KJ151).
Not applicable.
XL was responsible for the conception of the
manuscript, and ML was responsible for consulting relevant
literature and manuscript drafting. JC and HL are responsible for
designing and overseeing the study, and reviewing the manuscript.
All authors read and approved the final manuscript. Data
authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Tammemagi MC, Berg CD, Riley TL,
Cunningham CR and Taylor KL: Impact of lung cancer screening
results on smoking cessation. J Natl Cancer Inst. 106:dju0842014.
View Article : Google Scholar
|
|
2
|
Sun S, Schiller JH and Gazdar AF: Lung
cancer in never smokers-a different disease. Nat Rev Cancer.
7:778–790. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Relli V, Trerotola M, Guerra E and Alberti
S: Abandoning the notion of non-small cell lung cancer. Trends Mol
Med. 25:585–594. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zheng H, Zhan Y, Liu S, Lu J, Luo J, Feng
J and Fan S: The roles of tumor-derived exosomes in non-small cell
lung cancer and their clinical implications. J Exp Clin Cancer Res.
37:2262018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fernandez Y, Viesca M and Arvanitakis M:
Early diagnosis and management of malignant distal biliary
obstruction: A review on current recommendations and guidelines.
Clin Exp Gastroenterol. 12:415–432. 2019. View Article : Google Scholar
|
|
6
|
Murphy RM, Watt MJ and Febbraio MA:
Metabolic communication during exercise. Nat Metab. 2:805–816.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gururaja Rao S: Mitochondrial changes in
cancer. Handb Exp Pharmacol. 240:211–227. 2017. View Article : Google Scholar
|
|
8
|
Chen Y, Chen Z, Feng JH, Chen YB, Liao NS,
Su Y and Zou CY: Metabolic profiling of normal hepatocyte and
hepatocellular carcinoma cells via 1H nuclear magnetic
resonance spectroscopy. Cell Biol Int. 42:425–434. 2018. View Article : Google Scholar
|
|
9
|
Guppy M: The hypoxic core: A possible
answer to the cancer paradox. Biochem Biophys Res Commun.
299:676–680. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kalyanaraman B: Teaching the basics of
cancer metabolism: Developing antitumor strategies by exploiting
the differences between normal and cancer cell metabolism. Redox
Biol. 12:833–842. 2017. View Article : Google Scholar
|
|
11
|
Levine AJ and Puzio-Kuter AM: The control
of the metabolic switch in cancers by oncogenes and tumor
suppressor genes. Science. 330:1340–1344. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stine ZE, Walton ZE, Altman BJ, Hsieh AL
and Dang CV: MYC, metabolism, and cancer. Cancer Discov.
5:1024–1039. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Moretti M, Bennett J, Tornatore L,
Thotakura AK and Franzoso G: Cancer: NF-κB regulates energy
metabolism. Int J Biochem Cell Biol. 44:2238–2243. 2012. View Article : Google Scholar
|
|
14
|
Courtnay R, Ngo DC, Malik N, Ververis K,
Tortorella SM and Karagiannis TC: Cancer metabolism and the Warburg
effect: The role of HIF-1 and PI3K. Mol Biol Rep. 42:841–851. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Christofk HR, Vander Heiden MG, Harris MH,
Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL and
Cantley LC: The M2 splice isoform of pyruvate kinase is important
for cancer metabolism and tumour growth. Nature. 452:230–233. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ward PS and Thompson CB: Metabolic
reprogramming: A cancer hallmark even warburg did not anticipate.
Cancer Cell. 21:297–308. 2012. View Article : Google Scholar
|
|
17
|
Pavlova NN and Thompson CB: The emerging
hallmarks of cancer metabolism. Cell Metab. 23:27–47. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liberti MV and Locasale JW: The warburg
effect: How does it benefit cancer cells? Trends Biochem Sci.
41:211–218. 2016. View Article : Google Scholar
|
|
19
|
Nagao A, Kobayashi M, Koyasu S, Chow CCT
and Harada H: HIF-1-dependent reprogramming of glucose metabolic
pathway of cancer cells and its therapeutic significance. Int J Mol
Sci. 20:2382019. View Article : Google Scholar
|
|
20
|
Musharraf SG, Mazhar S, Choudhary MI, Rizi
N and Atta-ur-Rahman: Plasma metabolite profiling and chemometric
analyses of lung cancer along with three controls through gas
chromatography-mass spectrometry. Sci Rep. 5:86072015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Patra KC and Hay N: The pentose phosphate
pathway and cancer. Trends Biochem Sci. 39:347–354. 2014.
View Article : Google Scholar
|
|
23
|
Lam C, Low JY, Tran PT and Wang H: The
hexosamine biosynthetic pathway and cancer: Current knowledge and
future therapeutic strategies. Cancer Lett. 503:11–18. 2021.
View Article : Google Scholar
|
|
24
|
Amelio I, Cutruzzola F, Antonov A,
Agostini M and Melino G: Serine and glycine metabolism in cancer.
Trends Biochem Sci. 39:191–198. 2014. View Article : Google Scholar
|
|
25
|
Liu Y, Cao Y, Zhang W, Bergmeier S, Qian
Y, Akbar H, Colvin R, Ding J, Tong L, Wu S, et al: A small-molecule
inhibitor of glucose transporter 1 downregulates glycolysis,
induces cell-cycle arrest, and inhibits cancer cell growth in vitro
and in vivo. Mol Cancer Ther. 11:1672–1682. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu S and Herschman HR: A tumor agnostic
therapeutic strategy for hexokinase 1-Null/Hexokinase 2-positive
cancers. Cancer Res. 79:5907–5914. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang XY, Zhang M, Cong Q, Zhang MX, Zhang
MY, Lu YY and Xu CJ: Hexokinase 2 confers resistance to cisplatin
in ovarian cancer cells by enhancing cisplatin-induced autophagy.
Int J Biochem Cell Biol. 95:9–16. 2018. View Article : Google Scholar
|
|
28
|
Maschek G, Savaraj N, Priebe W,
Braunschweiger P, Hamilton K, Tidmarsh GF, De Young LR and Lampidis
TJ: 2-deoxy-D-glucose increases the efficacy of adriamycin and
paclitaxel in human osteosarcoma and non-small cell lung cancers in
vivo. Cancer Res. 64:31–34. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim SM, Yun MR, Hong YK, Solca F, Kim JH,
Kim HJ and Cho BC: Glycolysis inhibition sensitizes non-small cell
lung cancer with T790M mutation to irreversible EGFR inhibitors via
translational suppression of Mcl-1 by AMPK activation. Mol Cancer
Ther. 12:2145–2156. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lin S, Li Y, Wang D, Huang C, Marino D,
Bollt O, Wu C, Taylor MD, Li W, DeNicola GM, et al: Fascin promotes
lung cancer growth and metastasis by enhancing glycolysis and
PFKFB3 expression. Cancer Lett. 518:230–242. 2021. View Article : Google Scholar
|
|
31
|
Shen J, Jin Z, Lv H, Jin K, Jonas K, Zhu C
and Chen B: PFKP is highly expressed in lung cancer and regulates
glucose metabolism. Cell Oncol (Dordr). 43:617–629. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guo W, Zhang Y, Chen T, Wang Y, Xue J,
Zhang Y, Xiao W, Mo X and Lu Y: Efficacy of RNAi targeting of
pyruvate kinase M2 combined with cisplatin in a lung cancer model.
J Cancer Res Clin Oncol. 137:65–72. 2011. View Article : Google Scholar
|
|
33
|
Shi HS, Li D, Zhang J, Wang YS, Yang L,
Zhang HL, Wang XH, Mu B, Wang W, Ma Y, et al: Silencing of pkm2
increases the efficacy of docetaxel in human lung cancer xenografts
in mice. Cancer Sci. 101:1447–1453. 2010. View Article : Google Scholar
|
|
34
|
Morita M, Sato T, Nomura M, Sakamoto Y,
Inoue Y, Tanaka R, Ito S, Kurosawa K, Yamaguchi K, Sugiura Y, et
al: PKM1 confers metabolic advantages and promotes cell-autonomous
tumor cell growth. Cancer Cell. 33:355–367. e72018. View Article : Google Scholar
|
|
35
|
Israelsen WJ and Vander Heiden MG:
Pyruvate kinase: Function, regulation and role in cancer. Semin
Cell Dev Biol. 43:43–51. 2015. View Article : Google Scholar
|
|
36
|
Zhang X, Guo M, Fan J, Lv Z, Huang Q, Han
J, Wu F, Hu G, Xu J and Jin Y: Prognostic significance of serum LDH
in small cell lung cancer: A systematic review with meta-analysis.
Cancer Biomark. 16:415–423. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Costa Leite T, Da Silva D, Guimaraes
Coelho R, Zancan P and Sola-Penna M: Lactate favours the
dissociation of skeletal muscle 6-phosphofructo-1-kinase tetramers
down-regulating the enzyme and muscle glycolysis. Biochem J.
408:123–130. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hui S, Ghergurovich JM, Morscher RJ, Jang
C, Teng X, Lu W, Esparza LA, Reya T, Le Zhan, Yanxiang Guo J, et
al: Glucose feeds the TCA cycle via circulating lactate. Nature.
551:115–118. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Colegio OR, Chu NQ, Szabo AL, Chu T,
Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC,
Phillips GM, et al: Functional polarization of tumour-associated
macrophages by tumour-derived lactic acid. Nature. 513:559–563.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhao Y, Liu H, Liu Z, Ding Y, Ledoux SP,
Wilson GL, Voellmy R, Lin Y, Lin W, Nahta R, et al: Overcoming
trastuzumab resistance in breast cancer by targeting dysregulated
glucose metabolism. Cancer Res. 71:4585–4597. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou M, Zhao Y, Ding Y, Liu H, Liu Z,
Fodstad O, Riker AI, Kamarajugadda S, Lu J, Owen LB, et al: Warburg
effect in chemosensitivity: Targeting lactate dehydrogenase-A
re-sensitizes taxol-resistant cancer cells to taxol. Mol Cancer.
9:332010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Faubert B, Li KY, Cai L, Hensley CT, Kim
J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, et al:
Lactate metabolism in human lung tumors. Cell. 171:358–371. e92017.
View Article : Google Scholar
|
|
43
|
Hensley CT, Faubert B, Yuan Q, Lev-Cohain
N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, et al:
Metabolic heterogeneity in human lung tumors. Cell. 164:681–694.
2016. View Article : Google Scholar
|
|
44
|
Xie N, Tan Z, Banerjee S, Cui H, Ge J, Liu
RM, Bernard K, Thannickal VJ and Liu G: Glycolytic reprogramming in
myofibroblast differentiation and lung fibrosis. Am J Respir Crit
Care Med. 192:1462–1474. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chang YC, Chan YC, Chang WM, Lin YF, Yang
CJ, Su CY, Huang MS, Wu ATH and Hsiao M: Feedback regulation of
ALDOA activates the HIF-1α/MMP9 axis to promote lung cancer
progression. Cancer Lett. 403:28–36. 2017. View Article : Google Scholar
|
|
46
|
Romero OA, Torres-Diz M, Pros E, Savola S,
Gomez A, Moran S, Saez C, Iwakawa R, Villanueva A, Montuenga LM, et
al: MAX inactivation in small cell lung cancer disrupts MYC-SWI/SNF
programs and is synthetic lethal with BRG1. Cancer Discov.
4:292–303. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
de Koning TJ: Amino acid synthesis
deficiencies. J Inherit Metab Dis. 40:609–620. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cluntun AA, Lukey MJ, Cerione RA and
Locasale JW: Glutamine metabolism in cancer: Understanding the
heterogeneity. Trends Cancer. 3:169–180. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Altman BJ, Stine ZE and Dang CV: From
Krebs to clinic: Glutamine metabolism to cancer therapy. Nat Rev
Cancer. 16:7732016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Scalise M, Pochini L, Galluccio M, Console
L and Indiveri C: Glutamine transport and mitochondrial metabolism
in cancer cell growth. Front Oncol. 7:3062017. View Article : Google Scholar
|
|
51
|
Serizawa M, Kusuhara M, Zangiacomi V,
Urakami K, Watanabe M, Takahashi T, Yamaguchi K, Yamamoto N and Koh
Y: Identification of metabolic signatures associated with erlotinib
resistance of non-small cell lung cancer cells. Anticancer Res.
34:2779–2787. 2014.PubMed/NCBI
|
|
52
|
Dunphy MPS, Harding JJ, Venneti S, Zhang
H, Burnazi EM, Bromberg J, Omuro AM, Hsieh JJ, Mellinghoff IK,
Staton K, et al: In vivo PET assay of tumor glutamine flux and
metabolism: In-Human Trial of
18F-(2S,4R)-4-Fluoroglutamine. Radiology. 287:667–675.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Jeon SM, Chandel NS and Hay N: AMPK
regulates NADPH homeostasis to promote tumour cell survival during
energy stress. Nature. 485:661–665. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yu Y, Newman H, Shen L, Sharma D, Hu G,
Mirando AJ, Zhang H, Knudsen E, Zhang GF, Hilton MJ and Karner CM:
Glutamine metabolism regulates proliferation and lineage allocation
in skeletal stem cells. Cell Metab. 29:966–978. e42019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Simon J, Nunez-Garcia M, Fernandez-Tussy
P, Barbier-Torres L, Fernández-Ramos D, Gómez-Santos B, Buqué X,
Lopitz-Otsoa F, Goikoetxea-Usandizaga N, Serrano-Macia M, et al:
Targeting hepatic glutaminase 1 ameliorates non-alcoholic
steatohepatitis by restoring very-low-density lipoprotein
triglyceride assembly. Cell Metab. 31:605–622. e102020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Mates JM, Campos-Sandoval JA and Marquez
J: Glutaminase isoenzymes in the metabolic therapy of cancer.
Biochim Biophys Acta Rev Cancer. 1870:158–164. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Daemen A, Liu B, Song K, Kwong M, Gao M,
Hong R, Nannini M, Peterson D, Liederer BM, de la Cruz C, et al:
Pan-Cancer metabolic signature predicts co-dependency on
glutaminase and de novo glutathione synthesis linked to a
high-mesenchymal cell state. Cell Metab. 28:383–399. e3892018.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Han T, Zhan W, Gan M, Liu F, Yu B, Chin YE
and Wang JB: Phosphorylation of glutaminase by PKCε is essential
for its enzymatic activity and critically contributes to
tumorigenesis. Cell Res. 28:655–669. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Jacque N, Ronchetti AM, Larrue C, Meunier
G, Birsen R, Willems L, Saland E, Decroocq J, Maciel TT, Lambert M,
et al: Targeting glutaminolysis has antileukemic activity in acute
myeloid leukemia and synergizes with BCL-2 inhibition. Blood.
126:1346–1356. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
van den Heuvel AP, Jing J, Wooster RF and
Bachman KE: Analysis of glutamine dependency in non-small cell lung
cancer: GLS1 splice variant GAC is essential for cancer cell
growth. Cancer Biol Ther. 13:1185–1194. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kim J, Lee HM, Cai F, Ko B, Yang C, Lieu
EL, Muhammad N, Rhyne S, Li K, Haloul M, et al: The hexosamine
biosynthesis pathway is a targetable liability in KRAS/LKB1 mutant
lung cancer. Nat Metab. 2:1401–1412. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sezgin E, Levental I, Mayor S and Eggeling
C: The mystery of membrane organization: Composition, regulation
and roles of lipid rafts. Nat Rev Mol Cell Biol. 18:361–374. 2017.
View Article : Google Scholar
|
|
63
|
Snaebjornsson MT, Janaki-Raman S and
Schulze A: Greasing the wheels of the cancer machine: The role of
lipid metabolism in cancer. Cell Metab. 31:62–76. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Menendez JA and Lupu R: Fatty acid
synthase and the lipogenic phenotype in cancer pathogenesis. Nat
Rev Cancer. 7:763–777. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Corbet C and Feron O: Cancer cell
metabolism and mitochondria: Nutrient plasticity for TCA cycle
fueling. Biochim Biophys Acta Rev Cancer. 1868:7–15. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Svensson RU, Parker SJ, Eichner LJ, Kolar
MJ, Wallace M, Brun SN, Lombardo PS, Van Nostrand JL, Hutchins A,
Vera L, et al: Inhibition of acetyl-CoA carboxylase suppresses
fatty acid synthesis and tumor growth of non-small-cell lung cancer
in preclinical models. Nat Med. 22:1108–1119. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Tadros S, Shukla SK, King RJ, Gunda V,
Vernucci E, Abrego J, Chaika NV, Yu F, Lazenby AJ, Berim L, et al:
De novo lipid synthesis facilitates gemcitabine resistance through
endoplasmic reticulum stress in pancreatic cancer. Cancer Res.
77:5503–5517. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Buckley D, Duke G, Heuer TS, O'Farrell M,
Wagman AS, McCulloch W and Kemble G: Fatty acid synthase-Modern
tumor cell biology insights into a classical oncology target.
Pharmacol Ther. 177:23–31. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Currie E, Schulze A, Zechner R, Walther TC
and Farese RV Jr: Cellular fatty acid metabolism and cancer. Cell
Metab. 18:153–161. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Shen M, Tsai Y, Zhu R, Keng PC, Chen Y,
Chen Y and Lee SO: FASN-TGF-β1-PD-L1 axis contributes to the
development of resistance to NK cell cytotoxicity of
cisplatin-resistant lung cancer cells. Biochim Biophys Acta Mol
Cell Biol Lipids. 1863:313–322. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Ali A, Levantini E, Teo JT, Goggi J,
Clohessy JG, Wu CS, Chen L, Yang H, Krishnan I, Kocher O, et al:
Fatty acid synthase mediates EGFR palmitoylation in EGFR mutated
non-small cell lung cancer. EMBO Mol Med. 10:e83132018. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Falchook G, Infante J, Arkenau HT, Patel
MR, Dean E, Borazanci E, Brenner A, Cook N, Lopez J, Pant S, et al:
First-in-human study of the safety, pharmacokinetics, and
pharmacodynamics of first-in-class fatty acid synthase inhibitor
TVB-2640 alone and with a taxane in advanced tumors.
EClinicalMedicine. 34:1007972021. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Jones SF and Infante JR: Molecular
pathways: Fatty acid synthase. Clin Cancer Res. 21:5434–5438. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Dang L, Yen K and Attar EC: IDH mutations
in cancer and progress toward development of targeted therapeutics.
Ann Oncol. 27:599–608. 2016. View Article : Google Scholar
|
|
75
|
Chen C, Liu Y, Lu C, Cross JR, Morris JP
IV, Shroff AS, Ward PS, Bradner JE, Thompson C and Lowe SW:
Cancer-associated IDH2 mutants drive an acute myeloid leukemia that
is susceptible to Brd4 inhibition. Genes Dev. 27:1974–1985. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Migita T, Narita T, Nomura K, Miyagi E,
Inazuka F, Matsuura M, Ushijima M, Mashima T, Seimiya H, Satoh Y,
et al: ATP citrate lyase: Activation and therapeutic implications
in non-small cell lung cancer. Cancer Res. 68:8547–8554. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Chajes V, Cambot M, Moreau K, Lenoir GM
and Joulin V: Acetyl-CoA carboxylase alpha is essential to breast
cancer cell survival. Cancer Res. 66:5287–5294. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Carrer A, Trefely S, Zhao S, Campbell SL,
Norgard RJ, Schultz KC, Sidoli S, Parris JLD, Affronti HC, Sivanand
S, et al: Acetyl-CoA metabolism supports multistep pancreatic
tumorigenesis. Cancer Discov. 9:416–435. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zaidi N, Swinnen JV and Smans K:
ATP-citrate lyase: A key player in cancer metabolism. Cancer Res.
72:3709–3714. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Hatzivassiliou G, Zhao F, Bauer DE,
Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA and
Thompson CB: ATP citrate lyase inhibition can suppress tumor cell
growth. Cancer Cell. 8:311–321. 2005. View Article : Google Scholar
|
|
81
|
Lin R, Tao R, Gao X, Li T, Zhou X, Guan
KL, Xiong Y and Lei QY: Acetylation stabilizes ATP-citrate lyase to
promote lipid biosynthesis and tumor growth. Mol Cell. 51:506–518.
2013. View Article : Google Scholar
|
|
82
|
Niu J, Sun Y, Chen B, Zheng B, Jarugumilli
GK, Walker SR, Hata AN, Mino-Kenudson M, Frank DA and Wu X: Fatty
acids and cancer-amplified ZDHHC19 promote STAT3 activation through
S-palmitoylation. Nature. 573:139–143. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Mashima T, Oh-hara T, Sato S, Mochizuki M,
Sugimoto Y, Yamazaki K, Hamada J, Tada M, Moriuchi T, Ishikawa Y,
et al: p53-defective tumors with a functional apoptosome-mediated
pathway: A new therapeutic target. J Natl Cancer Inst. 97:765–777.
2005. View Article : Google Scholar
|
|
84
|
Yarla NS, Bishayee A, Sethi G, Reddanna P,
Kalle AM, Dhananjaya BL, Dowluru KS, Chintala R and Duddukuri GR:
Targeting arachidonic acid pathway by natural products for cancer
prevention and therapy. Semin Cancer Biol. 40–41:48–81. 2016.
View Article : Google Scholar
|
|
85
|
Murray M, Hraiki A, Bebawy M, Pazderka C
and Rawling T: Anti-tumor activities of lipids and lipid analogues
and their development as potential anticancer drugs. Pharmacol
Ther. 150:109–128. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Vannitamby A, Saad MI, Aloe C, Wang H,
Kumar B, Vlahos R, Selemidis S, Irving L, Steinfort D, Jenkins BJ
and Bozinovski S: Aspirin-triggered resolvin D1 reduces
proliferation and the neutrophil to lymphocyte ratio in a mutant
KRAS-driven lung adenocarcinoma model. Cancers (Basel).
13:32242021. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Riedl K, Krysan K, Pold M, Dalwadi H,
Heuze-Vourc'h N, Dohadwala M, Liu M, Cui X, Figlin R, Mao JT, et
al: Multifaceted roles of cyclooxygenase-2 in lung cancer. Drug
Resist Updat. 7:169–184. 2004. View Article : Google Scholar
|
|
88
|
Xin C, Chu L, Zhang L, Geng D, Wang Y, Sun
D, Sui P, Zhao X, Gong Z, Sui M and Zhang W: Expression of
cytosolic phospholipase A2 (cPLA2)-arachidonic acid
(AA)-Cyclooxygenase-2 (COX-2) pathway factors in lung cancer
patients and its implication in lung cancer early detection and
prognosis. Med Sci Monit. 25:5543–5551. 2019. View Article : Google Scholar
|
|
89
|
Yue S, Li J, Lee SY, Lee HJ, Shao T, Song
B, Cheng L, Masterson TA, Liu X, Ratliff TL and Cheng JX:
Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT
activation underlies human prostate cancer aggressiveness. Cell
Metab. 19:393–406. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Kopecka J, Trouillas P, Gasparovic AC,
Gazzano E, Assaraf YG and Riganti C: Phospholipids and cholesterol:
Inducers of cancer multidrug resistance and therapeutic targets.
Drug Resist Updat. 49:1006702020. View Article : Google Scholar
|
|
91
|
Yeganeh B, Wiechec E, Ande SR, Sharma P,
Moghadam AR, Post M, Freed DH, Hashemi M, Shojaei S, Zeki AA and
Ghavami S: Targeting the mevalonate cascade as a new therapeutic
approach in heart disease, cancer and pulmonary disease. Pharmacol
Ther. 143:87–110. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Gorin A, Gabitova L and Astsaturov I:
Regulation of cholesterol biosynthesis and cancer signaling. Curr
Opin Pharmacol. 12:710–716. 2012. View Article : Google Scholar
|
|
93
|
Li D, Long W, Huang R, Chen Y and Xia M:
27-Hydroxycholesterol inhibits sterol regulatory element-binding
protein 1 activation and hepatic lipid accumulation in mice.
Obesity (Silver Spring). 26:713–722. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Nelson ER, Wardell SE, Jasper JS, Park S,
Suchindran S, Howe MK, Carver NJ, Pillai RV, Sullivan PM, Sondhi V,
et al: 27-Hydroxycholesterol links hypercholesterolemia and breast
cancer pathophysiology. Science. 342:1094–1098. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Wang B and Tontonoz P: Liver X receptors
in lipid signalling and membrane homeostasis. Nat Rev Endocrinol.
14:452–463. 2018. View Article : Google Scholar
|
|
96
|
Gibson DA, Collins F, Cousins FL, Esnal
Zufiaurre A and Saunders PTK: The impact of 27-hydroxycholesterol
on endometrial cancer proliferation. Endocr Relat Cancer.
25:381–391. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Wu Y, Yu DD, Hu Y, Cao HX, Yu SR, Liu SW
and Feng JF: LXR ligands sensitize EGFR-TKI-resistant human lung
cancer cells in vitro by inhibiting Akt activation. Biochem Biophys
Res Commun. 467:900–905. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Lo Sasso G, Bovenga F, Murzilli S,
Salvatore L, Di Tullio G, Martelli N, D'Orazio A, Rainaldi S, Vacca
M, Mangia A, et al: Liver X receptors inhibit proliferation of
human colorectal cancer cells and growth of intestinal tumors in
mice. Gastroenterology. 144:1497–1507. 1507e1–13. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Hiramitsu S, Ishikawa T, Lee WR, Khan T,
Crumbley C, Khwaja N, Zamanian F, Asghari A, Sen M, Zhang Y, et al:
Estrogen receptor beta-mediated modulation of lung cancer cell
proliferation by 27-hydroxycholesterol. Front Endocrinol
(Lausanne). 9:4702018. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Zhang L, Liu M, Liu J, Li X, Yang M, Su B
and Lin Y: 27-Hydroxycholesterol enhanced osteoclastogenesis in
lung adenocarcinoma microenvironment. J Cell Physiol.
234:12692–12700. 2019. View Article : Google Scholar
|
|
101
|
Chen Q, Pan Z, Zhao M, Wang Q, Qiao C,
Miao L and Ding X: High cholesterol in lipid rafts reduces the
sensitivity to EGFR-TKI therapy in non-small cell lung cancer. J
Cell Physiol. 233:6722–6732. 2018. View Article : Google Scholar
|
|
102
|
Li J, Yan H, Zhao L, Jia W, Yang H, Liu L,
Zhou X, Miao P, Sun X, Song S, et al: Inhibition of SREBP increases
gefitinib sensitivity in non-small cell lung cancer cells.
Oncotarget. 7:52392–52403. 2016. View Article : Google Scholar
|