Introduction
There are various types of polyposis syndrome
including familial adenomatous polyposis (FAP), serrated polyposis
syndrome, Peutz-Jeghers syndrome, juvenile polyposis syndrome, and
PTEN-hamartoma tumor syndrome (1). Patients with these syndromes are at
higher risk of developing colorectal cancer. Therefore, appropriate
management via genetic testing and endoscopic surveillance is
essential for the treatment and surgical prophylaxis of patients
with colorectal polyposis. FAP is an autosomal dominant colorectal
tumor syndrome caused by an APC pathogenic germline variant,
and is characterized by the formation of numerous adenomatous
polyps throughout the entire colon (2). Since 15–20% of cases are de
novo without clinical or genetic evidence of FAP (3), it is important for clinical diagnosis
to check for more than 100 colorectal adenomas via colonoscopy,
regardless of family history of colorectal adenomatous polyposis
(4). Each adenoma is typically
polypoid in shape in patients with FAP. Therefore, it is difficult
to judge whether a phenotype with vast numbers of an alternate
morphological type of adenoma is associated to FAP.
Although the main colorectal tumorigenesis pathway
is via protruded adenomas through the adenoma-carcinoma sequence,
some colorectal cancers (CRCs) develop from these flat lesions via
a de novo pathway (5,6).
Kudo et al first called these flat, early lesions laterally
spreading tumors (LSTs), and classified the horizontal growth
lesions into two subtypes: the LST-granular type (LST-G), with
granules and nodules on the tumor surface; the LST-nongranular type
(LST-NG), with flat, smooth surfaces (7,8). In
terms of histopathology, most LST-G and LST-NG lesions comprise
tubular or tubulovillous adenomas, although the molecular
characteristics of the two subtypes differ. Hiraoka et al
demonstrated that LST-G lesions have KRAS mutations and are
intermediate with regard to hypermethylation, whereas LST-NG
lesions exhibit little pathogenic variation (9). Sakai et al reported that a
TP53 mutation occurred during the development of cancer in
both LST-NG and LST-G lesions (10,11).
Although the authors reported the molecular characteristics of
tumor progression from LST to CRC, the collected tumor samples were
obtained from patients subjected to various environmental factors,
including different lifestyles and microbiomes, as well as various
genetic germline backgrounds. Therefore, ideal molecular analysis
should be performed using normal colonic mucosa, LSTs, and CRCs
from patients with identical backgrounds.
Herein, we investigate a patient with >150 LST-NG
lesions in the entire large intestine and one adenocarcinoma in the
sigmoid colon, which has not been previously reported. The lesions
appeared to have the FAP phenotype, but otherwise atypical in that
all the adenomatous lesions were LST-NG. The cancer lesion of this
case might have been developed by somatic mutation of FBXW7
and/or TP53, differently with the tumorigenesis by germline
APC mutation followed by acquired APC disfunction in
patients with FAP. We further demonstrate the genomic/epigenomic
difference among the normal colonic mucosa, LST-NGs, and cancer
lesions of the patient as tumor progression occurs via accumulation
of epigenetic alterations as well as pathogenic mutations of tumor
suppressor genes. In this study, for the first time, we report that
the hypermethylation of ZNF625, LONRF2, SDC2, and
WDR17 as well as somatic mutation of FBXW7 and/or
TP53 contribute to tumorigenesis from LST-NG.
Case report
A 72-year-old female presented at our hospital after
a positive fecal occult blood test without any change to her bowel
habits. She had no family history of colorectal polyposis or
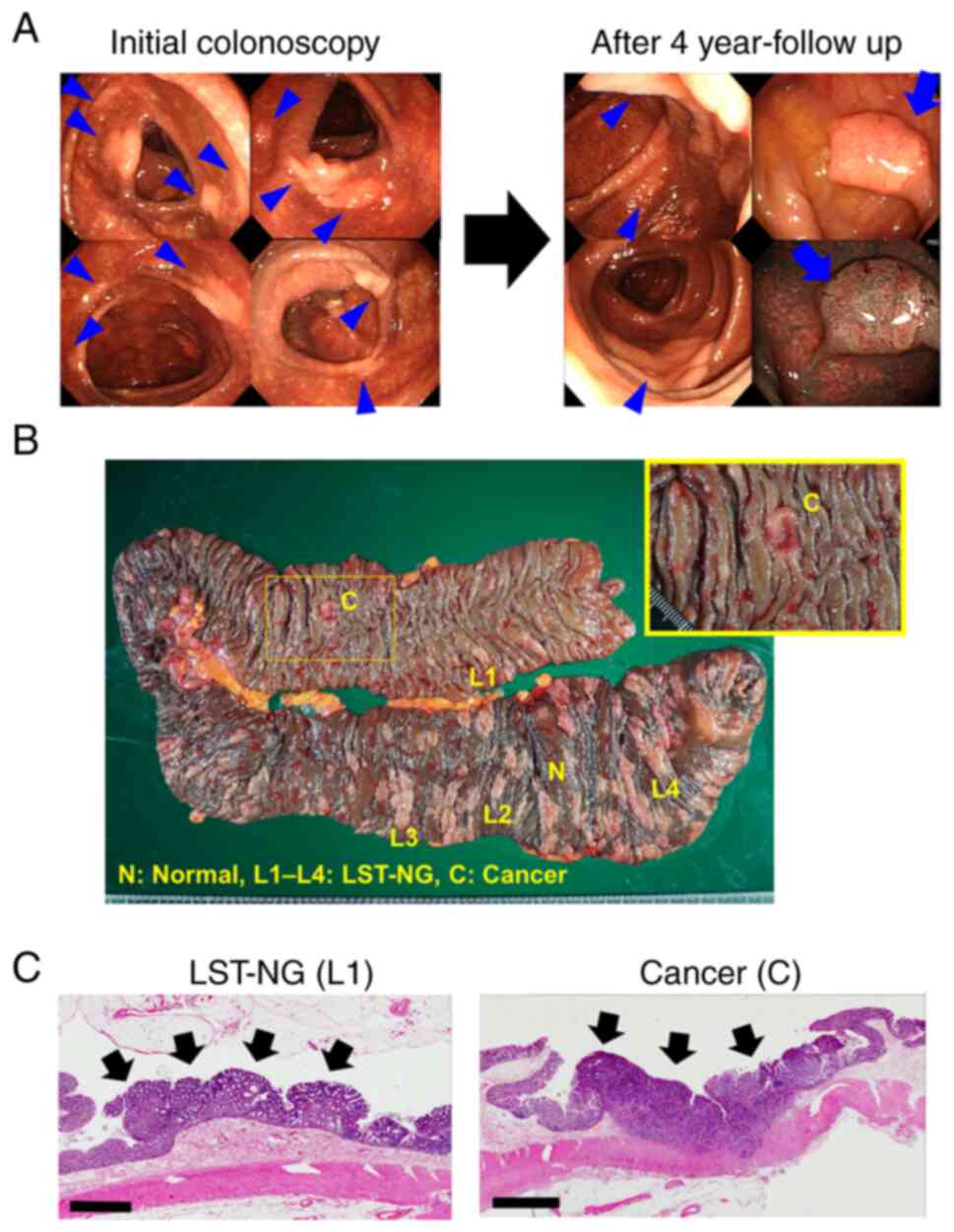
colorectal cancer. A complete colonoscopy revealed more than 150
amelanotic, flat, elevated lesions in the entire large intestine
with melanotic background mucosa owing to prolonged
self-administration of Sennoside A+B calcium (Fig. 1A). All the flat lesions were
LST-NG, and the biopsy samples obtained from the lesions were
pathologically diagnosed as tubular adenomas.
Esophagogastroduodenoscopy was also performed and that no fundic
gland polyposis was observed. After 4 years of annual endoscopic
surveillance, one of the LST-NG lesions had developed into an
adenocarcinoma in the sigmoid colon (Fig. 1A). Since LST-NG is a pre-cancerous
condition, a subtotal colectomy by ileorectal anastomosis (IRA) was
performed. The postoperative histopathology results demonstrated
the presence of a tubular adenocarcinoma and multiple tubular
adenomas (Fig. 1B and C). No cases
of both multiple LST-NG lesions and CRC have been previously
reported; we hypothesize that the tumorigenesis might have had a
genetic/epigenetic cause. The patient provided written informed
consent, and the study was approved by the Institutional Review
Board of the Hamamatsu University School of Medicine (approval no.
17-222).
For genetic analysis, we first performed multigene
panel testing using MiSeq sequencer (Illumina) and collected one
normal colonic mucosa (N), four LST-NG lesions (L1-L4), and one
cancer sample (C) from the patient immediately following IRA,
extracted the DNA, and froze the fresh samples for storage. The
extracted DNA was quantified using a Qubit dsDNA BR Assay Kit
(Q32850; Thermo Fisher Scientific) on a Qubit 2.0 Fluorometer
(Q33216, Thermo Fisher Scientific), and was prepared for shearing
according to the SureSelect XT HS Target Enrichment System Manual
(Agilent Technologies). Custom capture probes were designed using
SureDesign (Agilent Technologies) covering the exons and boundary
regions of 96 genes, including APC. For library preparation,
a SureSelect XT HS Reagent Kit (G9702A, Agilent Technologies) was
used according to the manufacturer's instructions. In brief,
pre-enriched adapter-ligated libraries were prepared. Quality and
quantity of libraries were determined by 4150 TapeStation System
(G2992AA, Agilent Technologies) using D1000 ScreenTape (5067-5582,
Agilent Technologies). 3.8 pmol of each library was used for
hybridization. Subsequently, custom capture probes were hybridized
to target sequences to enable sequence enrichment using
streptavidin beads. Post-enrichment, libraries were quantified,
pooled, and sequenced using MiSeq Reagent Kit v3 (MS-102-3001,
Illumina) on a MiSeq sequencer. SureCall v4.0.1.46 (Agilent
Technologies) and VariantStudio software (Illumina) were used for
data analysis and alignment. GRCh37 was used as the reference
genome.
All detected variants were validated using
Integrative Genomics Viewer 2.9.2 (Broad Institute, Cambridge, MA,
USA). We detected no pathogenic variants, including APC, in
the normal mucosa. Of the four LST-NG lesions, only one had a
pathogenic variant of APC (NM_000038.5: c.2396_2397delAT,
NP_000029.2: p.Tyr799CysfsTer3), which was a somatic change because
there was no mutation of APC in the normal colonic mucosa.
In contrast, pathogenic mutations in both TP53 (NM_000546.5:
c.499_500delCA, NP_000537.3: p.Gln167AlafsTer13) and FBXW7
(NM_033632.3: c.1513C>T, NP_361014.1: p.Arg505Cys) were detected
in the cancer (Table I). These
results suggest that the patient had no possibility of developing
FAP because there was no APC mutation in the normal mucosa,
and the somatic mutation of FBXW7 and/or TP53
contributed to tumorigenesis.
 | Table I.Genetic alterations in normal colonic
mucosa, four LST-NG lesions (L1-L4) and one colonic cancer
lesion. |
Table I.
Genetic alterations in normal colonic
mucosa, four LST-NG lesions (L1-L4) and one colonic cancer
lesion.
|
|
|
|
|
| Variant allele
frequency (%) |
|---|
| Gene | Clinically relevant
variants | Type of
mutation | COSMIC legacy
identifier | COSMIC
significance |
|
|---|
| N | L1 | L2 | L3 | L4 | C |
|---|
| TP53 | NM_000546.5 | Frameshift | 44275 | N/A | 0 | 0 | 0 | 0 | 0 | 19 |
|
| c.499_500delCA |
|
|
|
|
|
|
|
|
|
|
| p.Gln167Alafs |
|
|
|
|
|
|
|
|
|
|
| Ter13 |
|
|
|
|
|
|
|
|
|
| FBXW7 | NM_033632.3 | Missense | 22975 | Pathogenic | 0 | 0 | 0 | 0 | 0 | 35 |
|
| c.1513C>T |
|
|
|
|
|
|
|
|
|
|
| p.Arg505Cys |
|
|
|
|
|
|
|
|
|
| APC | NM_000038.5 | Frameshift | 4167217 | N/A | 0 | 0 | 0 | 0 | 20 | 0 |
|
|
c.2396_2397delAT |
|
|
|
|
|
|
|
|
|
|
|
p.Tyr799CysfsTer3 |
|
|
|
|
|
|
|
|
|
As tumor progression generally occurs by
accumulation of epigenetic alterations as well as pathogenic
mutations of tumor suppressor genes, it is important to understand
the role of DNA methylation in tumorigenesis. Therefore, we next
conducted a comprehensive genome-wide analysis using an Infinium
MethylationEPIC BeadChip Kit (Illumina) according to the
manufacturer's recommendations. Briefly, bisulfite-treated DNA was
subjected to whole-genome amplification before fragmentation and
precipitation. The resuspended DNA was subsequently hybridized to
probes attached to the BeadChips (Illumina), which contained
>850,000 CpG sites, and the nonhybridized DNA was removed. The
attached probes were then subjected to single-base extension and
stained. The BeadChips were scanned using the iScan™
system (Illumina) according to the manufacturer's recommendations.
The red and green signals from the iScan™ system were
converted into unmethylated and methylated signals. For each CpG
site in the CpG island gene region, a DiffScore value of >100
between the normal mucosa and the cancer was defined as the
absolute DMS value of the sample and calculated using GenomeStudio
FrameWork v2011.1 software (Illumina) and the R statistical
environment (version 3.1.3). Among the 766 detected DMSs, we
selected one methylated CpG site from each of the cancer and normal
mucosa samples, and performed clustering analysis of the normal
mucosa sample, four LST-NG lesions, and cancer sample, as shown in
Fig. 2A. In most DMSs, the
methylation levels of the LST-NG lesions were the same as those in
the normal mucosa (Group a), as previously demonstrated using sets
of patients with LST (9–11). We next focused on the minority
group (Group b) in which the DMS levels of the LST-NG lesions were
as high as those in the cancer sample (Fig. 2B). DMS occurred at the CpG islands
of the following nine genes: ZNF625, LONRF2, MSC, OPLAH,
PCDHGA4, GSG1L, BEND5, SDC2, and WDR17. We further
checked the methylation values at the CpG islands including the DMS
site detected in Group b (Fig. 3).
Among the regions, the CpG sites of ZNF625, LONRF2, MSC, and
OPLAH were methylated in all four LST-NG lesions as in the
cancer sample, and SDC2 and WDR17 were methylated in
three of the LST-NG lesions. In the CpG islands in GSG1L,
BEND5, and PCDHGA4, the CpG sites were not methylated as
in the cancer lesions. When we observed on the gene regions of
these CpG sites, we noticed that ZNF625, LONRF2, SDC2, and
WDR17 might have been methylated at the promoter regions in
both the LST-NG lesions and the cancer sample, because these
methylated CpG sites were located 200 bases upstream of the
transcriptional start site, or 5′ untranslated region. This
suggests that methylation-silenced ZNF625, LONRF2, SDC2, and
WDR17 play roles in tumorigenesis as early as the LST-NG
phase.
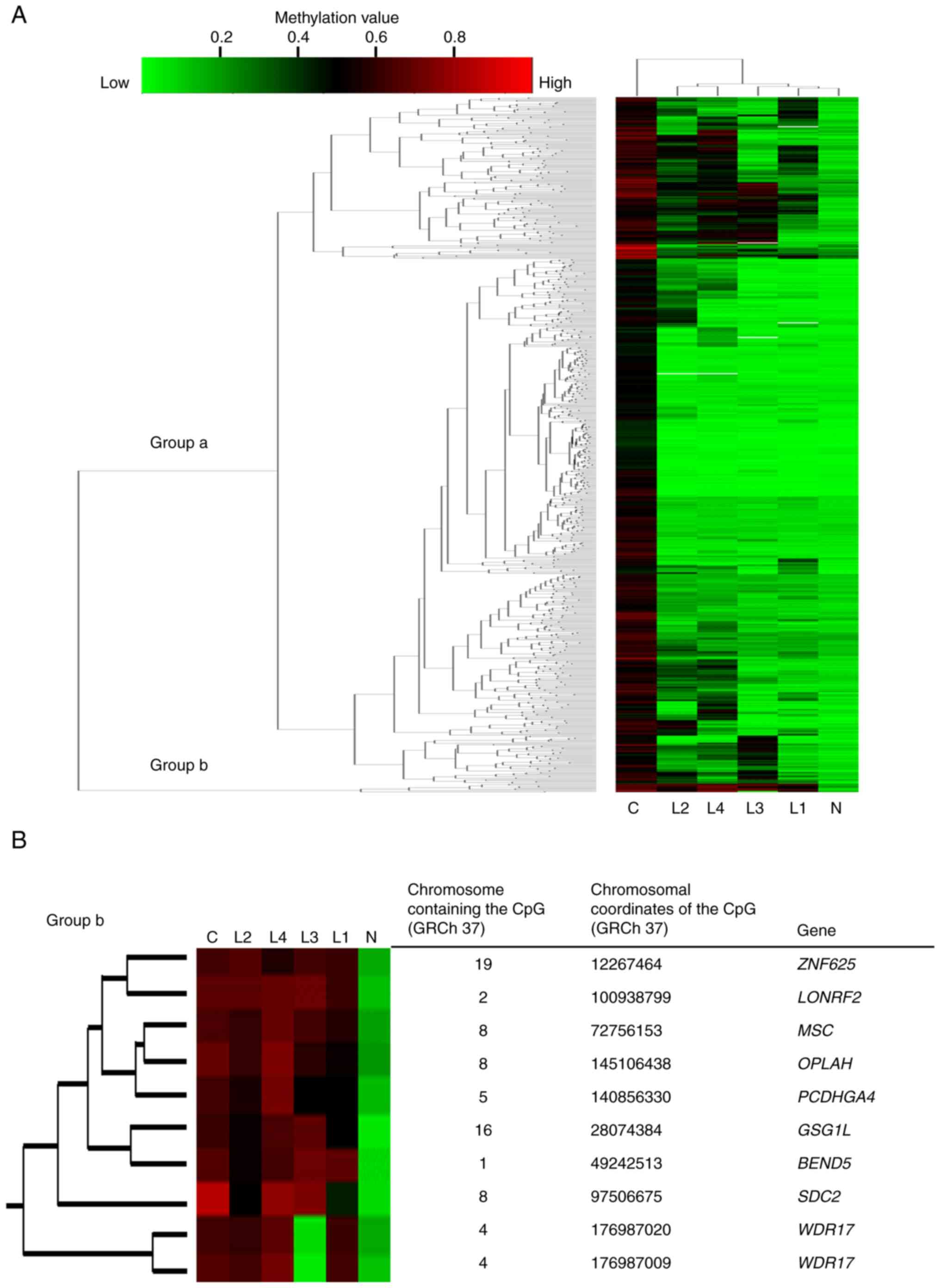 | Figure 2.Methylation profile focused on DMSs
in the analyzed patient. (A) Clustered heatmap composed of 766
DMSs. For each CpG site in the CpG islands of the gene region, a
DiffScore value of >100 between the normal mucosa and the cancer
was defined as the absolute DMS value of the sample. In the
majority of DMSs, the methylation levels of the LST-NG lesions were
the same as those of the normal mucosa (Group a). However, there
were small clusters with methylation levels as high as those of the
cancer lesion (Group b). (B) Heatmap focused on Group b with
information about each DMS. DMSs, differentially methylated sites;
ZNF625, zinc finger protein 625; LONRF2, LON
peptidase N-terminal domain and ring finger 2; MSC,
musculin; OPLAH, 5-oxoprolinase, ATP-hydrolysing;
PCDHGA4, protocadherin gamma subfamily A, 4; GSG1L,
GSG1 like; BEND5, BEN domain containing 5; SDC2,
syndecan 2; WDR17, WD repeat domain 17. |
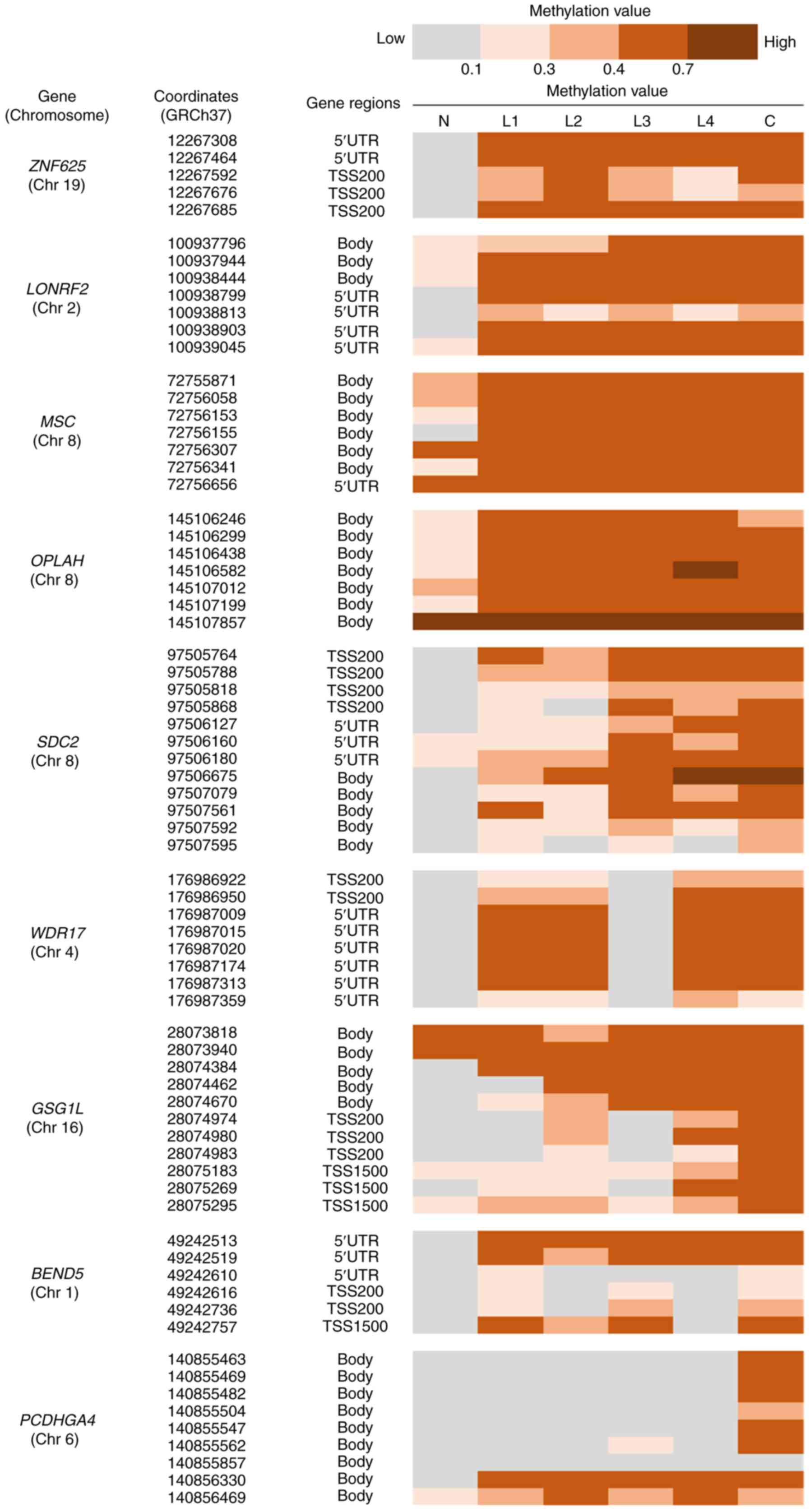 | Figure 3.Methylation profile focused on CpG
island regions including DMSs categorized in group b. The
methylation values of the CpG island regions around the DMS site in
Group b were determined. In the cancer lesions, all the CpG island
regions, including each DMS, were methylated in all nine genes, and
the CpG sites of ZNF625, LONRF2, MSC and OPLAH were
methylated in all four LST-NG lesions. SDC2 and WDR17
were methylated in three LST-NG lesions. In the CpG islands in
GSG1L, BEND5 and PCDHGA4, the CpG sites were not
methylated as cancer lesions. TSS200, 200 bases upstream of the
TSS; TSS500, 500 bases upstream of the TSS; TSS1500, 1,500 bases
upstream of the TSS; 5′UTR, within the 5′ untranslated region,
between the TSS and the ATG start site; body, between the ATG and
stop codon. TSS, transcriptional start site; DMSs, differentially
methylated sites; LST-NG, laterally spreading tumor-nongranular
type lesions; ZNF625, zinc finger protein 625;
LONRF2, LON peptidase N-terminal domain and ring finger 2;
MSC, musculin; OPLAH, 5-oxoprolinase,
ATP-hydrolysing; PCDHGA4, protocadherin gamma subfamily A,
4; GSG1L, GSG1 like; BEND5, BEN domain containing 5;
SDC2, syndecan 2; WDR17, WD repeat domain 17. |
Discussion
The diagnosis and management of patients with
polyposis syndromes is constantly evolving, owing to new scientific
and technological advances with respect to the identification of
causative genes, and the increased sophistication of endoscopic
treatments for polyps. However, we were uncertain as to how to
categorize the patient in the present study who had numerous LST-NG
lesions, among the various colorectal polyposis syndromes, as the
present work is the first report of a patient with >150 LST-NG
lesions that developed a CRC during endoscopic surveillance. Our
genomic and epigenomic analyses showed that: (1) no germline APC pathogenic
variant was detectable via the multigene panel testing; (2) there was only one somatic APC
frameshift mutant site in one of the four LST-NG lesions; (3) the somatic mutations of TP53
and FBXW7 were only present in the cancer sample; (4) there was methylation of the promoter
CpG islands in ZNF625, LONRF2, SDC2, and WDR17 in
most of the LST-NG lesions as well as the cancer lesion.
FAP is clinically diagnosed when approximately ≥100
adenomatous polyposis are detected in the large intestine,
irrespective of the presence or absence of a family history of FAP,
as 15–20% of FAP cases are de novo (3,4,12).
Therefore, we first suspected the indexed patient had sporadic FAP
since there were >100 adenomatous lesions throughout her large
intestine and she had no family history of FAP-associated
lesions.
The patient's adenomatous lesions were LST-NG, which
are histologically the same as in protruding adenomatous polyposes
(tubular adenomas) but differ morphologically to the naked eye.
Moreover, no cases have been reported of FAP with multiple LST-NG
lesions. Therefore, we explored the germline pathogenic variants
using the normal colonic mucosa and customized multigene panel test
and found no pathogenic variants, including APC, suggesting
that the patient had no genetic evidence of FAP. The limitation of
our analysis was that we could not completely exclude hereditary
polyposis syndrome since the multigene panel did not include
polyposis-related genes such as MUTYH and BMPR1A.
Therefore, it is necessary to conduct whole exome/genome sequencing
to detect any unknown germline genetic alterations to confirm the
presence of new types of hereditary polyposis syndrome.
The pathogenicity of somatic variants of cancer
including CRC is assessed by examining general population data,
functional data, predictive data, cancer hotspots, and
computational evidence (13).
Therefore, numerous patients with CRC and healthy controls have
been registered to establish reliable evidence. However, the
registered CRC patients' own environmental factors and gut
microbial compositions considerably differ. The potential role of
epigenetic alterations has been reported in links between obesity,
gut microbiota, and colorectal cancer. For colorectal cancer
progression, high-fat diet-induced obesity leads to epigenetic
remodeling of the acetylation landscape based on the gut
microbiota, promotes changes in DNA methylation, and enhances
production of deoxycholic acid, a secondary bile acid that is
produced solely by Gram-positive gut bacteria and known to cause
DNA damage through reactive oxygen species production (14). Therefore, pure genetic/epigenetic
factors for colorectal tumorigenesis should be detected under the
same environmental and microbiological conditions if possible. One
way of accomplishing that is to compare genetic/epigenetic profiles
among the normal mucosa, pre-cancerous lesions, and cancer lesions
obtained from patients with identical backgrounds at the same time.
In the present study, we analyzed the normal colonic mucosa, LST-NG
lesions, and sigmoid cancer lesion of the same patient obtained
immediately following colorectal resection.
In the present study, genetic analysis using
customized multigene panels revealed only one APC frameshift
variant in one LST-NG lesion, while the remaining three LST-NG
lesions had no pathogenic variants. Metz et al reported that
more than 90% of the LST lesions examined exhibited an APC
mutation, but it did not exhibit the mutation frequency of an
LST-NG lesion (15). Sugimoto
et al detected loss of heterozygosity (LOH) at the
APC locus in 60% of the LST-NG lesions they examined,
whereas only 28% LST-G lesions harbored the mutation (16). In contrast, precise analysis by
Sugai et al showed that null APC variants (i.e.,
nonsense and frameshift type pathogenic variants) were numerous in
LST-G lesions compared with LST-NG lesions (17). These previous reports suggest that
somatic pathogenic APC variants play a role in the
occurrence of LST-NG lesions but are not the main contributors to
tumorigenesis. In the present study, we detected somatic pathogenic
alterations in TP53 and FBXW7, both of which variants
were only present in the cancer lesion. Previous studies have
demonstrated that the synergistic contributions of wild type FBXW7
and TP53 proteins contribute to the suppression of gastrointestinal
cancer (18,19), and most FBXW7 mutations in
cancers, including CRC, exhibit a TP53 mutation (20–22).
Therefore, we speculated that our study patient had a cancer lesion
that simultaneously lost the two tumor-suppressors that usually
cooperate in the inhibition of tumorigenesis. In addition, Sakai
et al have suggested that the TP53 mutation is more
closely involved at an earlier stage in LST-NG lesions than in
LST-G lesions during cancer development (11). In the same manner, the TP53
mutation might occur in an earlier phase of the patient's cancer
lesions than the phase in which LST-NG lesions appear, and the
mutation may continuously influence sigmoid tumor progression to
the advanced level.
We further performed epigenome-wide analysis to
determine whether there were any pathogenic epigenetic alterations
that cause tumorigenesis after the LST-NG phase. Of the 766 DMSs
identified, 756 were hypermethylated only in the cancer lesion, and
the methylation levels in the LST-NG lesions were as low as in the
normal colonic mucosa (Group a). This result was expected since
Sakai et al were able to cluster LSTs into two epigenotypes
in 108 LST samples (51 LST-G and 57 LST-NG lesions), i.e.,
intermediate- and low methylation-groups. The authors found that
the intermediate methylation epigenotype was associated with LST-G
lesion morphology, while the low methylation LSTs mostly reflected
LST-NG lesion morphology (10,11).
When we assessed the remaining 10 DMSs, all of which were
categorized in the same cluster group (Group b), we noticed that
the methylation levels of all 10 DMSs were as high as those in the
cancer lesions. Moreover, all 10 DMSs were located in the CpG
island region, indicating that the genes where the 10 DMS were
located play roles in tumorigenesis by silencing the pre-cancerous
phase of the LST-NG lesions. We further determined whether the CpG
island regions, including the 10 DMSs, were methylated to the same
extent in the LST-NG lesions as in the cancer lesion and found that
ZNF625, LONRF2, SDC2, and WDR17 may have been
methylated at the promoter region in both the LST-NG lesions and
the cancer lesion. Among those four genes, SDC2 has been
investigated most extensively regarding methylation in colorectal
neoplasms. SDC2 has the chromosomal locus 8q22.1 and encodes
syndecan-2 protein. The syndecan-2 protein participates in cell
proliferation, cell migration, and cell-matrix interactions via its
extracellular matrix proteins receptor (23–27),
and altered syndecan-2 expression has been detected in several
different tumor types (28–30).
As reported, overexpressed SDC2 has tumor activity in CRC
(31–33), and it apparently exerts its
oncogenic character when activated. Oh et al first reported
the hypermethylation of SDC2 in colorectal adenoma as well
as in CRC, indicating its contribution to tumorigenesis (34). Their results have been corroborated
by other groups, and various useful evaluation methods using stool
samples, blood, and urine have been demonstrated (35–40).
In contrast to many previous reports concerning SDC2 in
colorectal adenomas and CRC, little is known about alterations in
ZNF625, LONRF2, and WDR17 concerning CRC
tumorigenesis. ZNF625 has the chromosomal locus 19p13.2 and
encodes zinc finger protein 625, which is predicted to enable
DNA-binding transcription factor activity, but has not yet been
completely analyzed. Lin et al reported that among 228
hypermethylated promoter-associated CpG islands, ZNF625 is
one of the most frequently hypermethylated genes in colorectal
cancer (41). LONRF2 has
the chromosomal locus 2q11.2 and encodes LON peptidase N-terminal
domain and ring finger 2. The gene is conserved in various species
including chimpanzees, mice, dogs, chickens, zebrafish, and frogs.
Hua et al reported LONRF2 hypermethylation in the
rectal adenocarcinomas from 171 patients (42). WDR17 has the chromosomal
locus 4q34.2, encodes WD repeat-containing protein 17, and is
abundantly expressed in the retina and testes. It has been
suggested as a candidate gene for retinal disease (43). In anal cancer, WDR17 is
hypermethylated, regardless of HIV infection status (44), but to date there has been no report
on WDR17 methylation in CRC. Considering the findings of
this study along with those of previous studies, we suggest that
SDC2 hypermethylation contributes to colorectal
tumorigenesis at the adenoma stage, and is not limited to LST-NG
lesions. Although little is known about the methylation of
ZNF625, LONRF2, and WDR17 concerning CRC
tumorigenesis, it is possible that methylation of the CpG island
promoters at ZNF625, LONRF2, and WDR17 plays a unique
key role in the tumorigenesis of LST-NG lesions.
However, the present study has some limitations: i)
the genetic/epigenetic analysis was performed using a different
number of samples in each group, that is, only one cancer lesion,
four LST-NG, and one normal mucosa; ii) only one patient was
analyzed.
In conclusion, we successfully demonstrated the
acquired genomic/epigenomic status of pre-cancerous and cancerous
phases under identical germline and environmental conditions by
analyzing a patient with multiple LST-NG lesions and sigmoid colon
cancer. We detected four genes methylated at the CpG island
promoters during the LST-NG lesion phase. Although rare, patients
with both pre-cancer and cancer lesions should be further
investigated to elucidate the contribution made by pure somatic
genomic/epigenomic alterations to tumorigenesis.
Acknowledgements
Not applicable.
Funding
This study was supported by the Japan Society for the Promotion
of Science (JSPS) (KAKENHI grant no. 22K08053).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MI conceived the study. MI, TT, and MM analyzed and
interpreted the data. KK, SO, KS, HS and SB interpreted
clinicopathological features. MI and MM drafted the manuscript and
critically revised it for important intellectual content. MI and MM
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Hamamatsu University School of Medicine (approval no.
17-222).
Patient consent for publication
The patient provided written informed consent for
the participation and publication of her data and images.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
FAP
|
familial adenomatous polyposis
|
|
LST
|
laterally spreading tumor
|
|
LST-G
|
LST-granular type
|
|
LST-NG
|
LST-non-granular type
|
|
IRA
|
ileorectal anastomosis
|
|
DMS
|
differentially methylated site
|
References
|
1
|
Patel R and Hyer W: Practical management
of polyposis syndromes. Frontline Gastroenterol. 10:379–387. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bisgaard ML, Fenger K, Bülow S, Niebuhr E
and Mohr J: Familial adenomatous polyposis (FAP): Frequency,
penetrance, and mutation rate. Hum Mutat. 3:121–125. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Aretz S, Uhlhaas S, Caspari R, Mangold E,
Pagenstecher C, Propping P and Friedl W: Frequency and parental
origin of de novo APC mutations in familial adenomatous polyposis.
Eur J Hum Genet. 12:52–58. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vasen HFA, Möslein G, Alonso A, Aretz S,
Bernstein I, Bertario L, Blanco I, Bülow S, Burn J, Capella G, et
al: Guidelines for the clinical management of familial adenomatous
polyposis (FAP). Gut. 57:704–713. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kudo S, Kashida H, Nakajima T, Tamura S
and Nakajo K: Endoscopic diagnosis and treatment of early
colorectal cancer. World J Surg. 21:694–701. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Minamoto T, Sawaguchi K, Mai M, Yamashita
N, Sugimura T and Esumi H: Infrequent K-ras activation in
superficial-type (flat) colorectal adenomas and adenocarcinomas.
Cancer Res. 54:2841–2844. 1994.PubMed/NCBI
|
|
7
|
Kudo S: Endoscopic mucosal resection of
flat and depressed types of early colorectal cancer. Endoscopy.
25:455–461. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kudo SE, Takemura O and Ohtsuka K: Flat
and depressed types of early colorectal cancers: From East to West.
Gastrointest Endosc Clin N Am. 18581–593. (xi)2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hiraoka S, Kato J, Tatsukawa M, Harada K,
Fujita H, Morikawa T, Shiraha H and Shiratori Y: Laterally
spreading type of colorectal adenoma exhibits a unique methylation
phenotype and K-ras mutations. Gastroenterology. 131:379–389. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sakai E, Ohata K, Chiba H, Matsuhashi N,
Doi N, Fukushima J, Endo H, Takahashi H, Tsuji S, Yagi K, et al:
Methylation epigenotypes and genetic features in colorectal
laterally spreading tumors. Int J Cancer. 135:1586–1595. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sakai E, Fukuyo M, Matsusaka K, Ohata K,
Doi N, Takane K, Matsuhashi N, Fukushima J, Nakajima A and Kaneda
A: TP53 mutation at early stage of colorectal cancer progression
from two types of laterally spreading tumors. Cancer Sci.
107:820–827. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tomita N, Ishida H, Tanakaya K, Yamaguchi
T, Kumamoto K, Tanaka T, Hinoi T, Miyakura Y, Hasegawa H, Takayama
T, et al: Japanese society for cancer of the colon and rectum
(JSCCR) guidelines 2020 for the clinical practice of hereditary
colorectal cancer. Int J Clin Oncol. 26:1353–1419. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Horak P, Griffith M, Danos AM, Pitel BA,
Madhavan S, Liu X, Chow C, Williams H, Carmody L, Barrow-Laing L,
et al: Standards for the classification of pathogenicity of somatic
variants in cancer (oncogenicity): Joint recommendations of
clinical genome resource (ClinGen), cancer genomics consortium
(CGC), and variant interpretation for cancer consortium (VICC).
Genet Med. 24:986–998. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Song M and Chan AT: Environmental factors,
gut microbiota, and colorectal cancer prevention. Clin
Gastroenterol Hepatol. 17:275–289. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Metz AJ, Bourke MJ, Moss A, Dower A,
Zarzour P, Hawkins NJ, Ward RL and Hesson LB: A correlation of the
endoscopic characteristics of colonic laterally spreading tumours
with genetic alterations. Eur J Gastroenterol Hepatol. 25:319–326.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sugimoto T, Ohta M, Ikenoue T, Yamada A,
Tada M, Fujishiro M, Ogura K, Yamaji Y, Okamoto M, Kanai F, et al:
Macroscopic morphologic subtypes of laterally spreading colorectal
tumors showing distinct molecular alterations. Int J Cancer.
127:1562–1569. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sugai T, Habano W, Takagi R, Yamano H,
Eizuka M, Arakawa N, Takahashi Y, Yamamoto E, Kawasaki K, Yanaiet
S, et al: Analysis of molecular alterations in laterally spreading
tumors of the colorectum. J Gastroenterol. 52:715–723. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Grim JE, Knoblaugh SE, Guthrie KA, Hagar
A, Swanger J, Hespelt J, Delrow JJ, Small T, Grady WM, Nakayama KI
and Clurman BE: Fbw7 and p53 cooperatively suppress advanced and
chromosomally unstable intestinal cancer. Mol Cell Biol.
32:2160–2167. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yokobori T, Mimori K, Iwatsuki M, Ishii H,
Onoyama I, Fukagawa T, Kuwano H, Nakayama KI and Mori M:
p53-Altered FBXW7 expression determines poor prognosis in gastric
cancer cases. Cancer Res. 69:3788–3794. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wood LD, Parsons DW, Jones S, Lin J,
Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al: The
genomic landscapes of human breast and colorectal cancers. Science.
318:1108–1113. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sjöblom T, Jones S, Wood LD, Parsons DW,
Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al:
The consensus coding sequences of human breast and colorectal
cancers. Science. 314:268–274. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mao JH, Perez-Losada J, Wu D, Delrosario
R, Tsunematsu R, Nakayama KI, Brown K, Bryson S and Balmain A:
Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor
gene. Nature. 432:775–779. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Choi Y, Kim H, Chung H, Hwang JS, Shin JA,
Han IO and Oh ES: Syndecan-2 regulates cell migration in colon
cancer cells through Tiam1-mediated Rac activation. Biochem Biophys
Res Commun. 391:921–925. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Park H, Kim Y, Lim Y, Han I and Oh ES:
Syndecan-2 mediates adhesion and proliferation of colon carcinoma
cells. J Biol Chem. 277:29730–29736. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang JW and Chuang NN: Shift syndecan-2
from RACK1 to caveolin-2 upon transformation with oncogenic ras.
Biochem Biophys Res Commun. 350:227–232. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Orosco A, Fromigué O, Haÿ E, Marie PJ and
Modrowski D: Dual involvement of protein kinase C delta in
apoptosis induced by syndecan-2 in osteoblasts. J Cell Biochem.
98:838–850. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Munesue S, Yoshitomi Y, Kusano Y, Koyama
Y, Nishiyama A, Nakanishi H, Miyazaki K, Ishimaru T, Miyaura S,
Okayama M and Oguri K: A novel function of syndecan-2, suppression
of matrix metalloproteinase-2 activation, which causes suppression
of metastasis. J Biol Chem. 282:28164–28174. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang X, Xiao DW, Xu LY, Zhong HJ, Liao
LD, Xie ZF and Li EM: Prognostic significance of altered expression
of SDC2 and CYR61 in esophageal squamous cell carcinoma. Oncol Rep.
21:1123–1129. 2009.PubMed/NCBI
|
|
29
|
Marzioni D, Lorenzi T, Mazzucchelli R,
Capparuccia L, Morroni M, Fiorini R, Bracalenti C, Catalano A,
David G, Castellucci M, et al: Expression of basic fibroblast
growth factor, its receptors and syndecans in bladder cancer. Int J
Immunopathol Pharmacol. 22:627–638. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Popović A, Demirović A, Spajić B, Stimac
G, Kruslin B and Tomas D: Expression and prognostic role of
syndecan-2 in prostate cancer. Prostate Cancer Prostatic Dis.
13:78–82. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim Y, Park H, Lim Y, Han I, Kwon HJ,
Woods A and Oh ES: Decreased syndecan-2 expression correlates with
trichostatin-A induced-morphological changes and reduced
tumorigenic activity in colon carcinoma cells. Oncogene.
22:826–830. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Han I, Park H and Oh ES: New insights into
syndecan-2 expression and tumourigenic activity in colon carcinoma
cells. J Mol Histol. 35:319–326. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Choi S, Kim Y, Park H, Han IO, Chung E,
Lee SY, Kim YB, Lee JW, Oh ES and Yi JY: Syndecan-2 overexpression
regulates adhesion and migration through cooperation with integrin
alpha2. Biochem Biophys Res Commun. 384:231–235. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Oh T, Kim N, Moon Y, Kim MS, Hoehn BD,
Park CH, Kim TS, Kim NK, Chung HC and An S: Genome-wide
identification and validation of a novel methylation biomarker,
SDC2, for blood-based detection of colorectal cancer. J Mol Diagn.
15:498–507. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ma L, Qin G, Gai F, Jiang Y, Huang Z, Yang
H, Yao S, Du S and Cao Y: A novel method for early detection of
colorectal cancer based on detection of methylation of two
fragments of syndecan-2 (SDC2) in stool DNA. BMC Gastroenterol.
22:1912022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bach S, Paulis I, Sluiter NR, Tibbesma M,
Martin I, van de Wiel MA, Tuynman JB, Bahce I, Kazemier G and
Steenbergen RDM: Detection of colorectal cancer in urine using DNA
methylation analysis. Sci Rep. 11:23632021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhao G, Ma Y, Li H, Li S, Zhu Y, Liu X,
Xiong S, Liu Y, Miao J, Fei S, et al: A novel plasma based early
colorectal cancer screening assay base on methylated SDC2 and
SFRP2. Clin Chim Acta. 503:84–89. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang J, Liu S, Wang H, Zheng L, Zhou C, Li
G, Huang R, Wang H, Li C, Fan X, et al: Robust performance of a
novel stool DNA test of methylated SDC2 for colorectal cancer
detection: A multicenter clinical study. Clin Epigenetics.
12:1622020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Han YD, Oh TJ, Chung TH, Jang HW, Kim YN,
An S and Kim NK: Early detection of colorectal cancer based on
presence of methylated syndecan-2 (SDC2) in stool DNA. Clin
Epigenetics. 11:512019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Oh TJ, Oh HI, Seo YY, Jeong D, Kim C, Kang
HW, Han YD, Chung HC, Kim NK and An S: Feasibility of quantifying
SDC2 methylation in stool DNA for early detection of colorectal
cancer. Clin Epigenetics. 9:1262017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lin PC, Lin JK, Lin CH, Lin HH, Yang SH,
Jiang JK, Chen WS, Chou CC, Tsai SF and Chang SC: Clinical
relevance of plasma DNA methylation in colorectal cancer patients
identified by using a genome-wide high-resolution array. Ann Surg
Oncol. 22 (Suppl 3):S1419–S1427. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hua Y, Ma X, Liu X, Yuan X, Qin H and
Zhang X: Abnormal expression of mRNA, microRNA alteration and
aberrant DNA methylation patterns in rectal adenocarcinoma. PLoS
One. 12:e01744612017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Stöhr H, Mohr N, Fröhlich S, Mehdi SQ,
Bhattacharya SS and Weber BHF: Cloning and characterization of
WDR17, a novel WD repeat-containing gene on chromosome 4q34.
Biochim Biophys Acta. 1579:18–25. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
van der Zee RP, van Noesel CJM, Martin I,
Ter Braak TJ, Heideman DAM, de Vries HJC, Prins JM and Steenbergen
RDM: DNA methylation markers have universal prognostic value for
anal cancer risk in HIV-negative and HIV-positive individuals. Mol
Oncol. 15:3024–3036. 2021. View Article : Google Scholar : PubMed/NCBI
|