Introduction
Breast cancer is one of the leading cancers in
women, with 1 in 8 women showing a lifetime risk of developing it
(1). Breast cancer is
heterogeneous, and patient demographics show considerable health
disparities (2,3). For example, African-American women
are more likely to develop breast cancer at a younger age and
suffer from an aggressive sub-type called triple-negative breast
cancer more often than their white counterparts (4). Next-generation sequencing
technologies have been used to study cancer genomics to determine
causative mutations leading to the disease. Whole-Genome (WGS) and
Whole-Exome sequencing (WES) technologies are used for this
purpose. Many large consortia made an effort to understand cancer
biology using these technologies. The Cancer Genome Atlas (TCGA) is
one of the most significant multicenter initiatives that use exome
and genome sequencing for all cancers. The data from TCGA showed
that mutations in PIK3C, PTEN, TP53, and CDH1 are
highly enriched in breast cancer samples with an increased risk or
less overall survival (5).
However, the TCGA dataset primarily has samples from white
patients, even with contributions from African-American or Hispanic
patients. Thus, a significant effort is underway to understand the
effect of ancestry/ethnicity on breast cancer (6,7).
Recent studies show that the overall breast cancer
incidence rates are similar among white and black patients.
However, Black patients are more like to be diagnosed with larger
tumor size (>5 cm (12%) and with high-grade (42%) breast cancer
(8). There is also a noticeable
disparity in Triple-Negative Breast Cancer (TNBC), with black women
showing 19% of all breast tumors as TNBC compared to 9% in white
patients. Utilizing exome technologies will be crucial to
understanding the genetic aspects of this health disparity. We
report our analysis on WES of breast tumor biopsies from
African-American and Hispanic patients from South Los Angeles, a
region with significant health disparity. Our work highlights the
overall mutational landscape and specific genetic mutations that
might provide insight into the biological aspects of breast cancer
in minority patients.
Materials and methods
Tumor sample and patient
ethnicity
Breast cancer patient samples from minority patients
were collected (Table I). Patients
belong to the Los Angeles SPA6 (Service Planning Area 6) region,
which traditionally shows significant health disparity. A total of
thirteen African-American and fifteen Hispanic patients were
analyzed for the study with confirmed invasive ductal carcinoma,
except for one African-American sample with a Ductal Carcinoma in
situ (DCIS) diagnosis. We also utilized matched normal
(tumor-adjacent) for eight samples from each ethnicity. Patient
samples were used from our ongoing Breast Cancer Study in the
Division of Cancer Research and Training at the Charles R. Drew
University of Medicine and Science in collaboration with Martin
Luther King Ambulatory Care Center (formerly known as King-Drew
Medical Center, #IRB 00-06-041) and the protocol has been approved
since 1999 and continuing review approved annually (recent
continuing review approval was August 18, 2021. The patient
demographics and breast cancer subtypes are listed in Table I.
 | Table I.Description of the breast cancer
samples used in the present study. |
Table I.
Description of the breast cancer
samples used in the present study.
| Variable | No. (%) |
|---|
| Ethnicity |
|
|
African-American | 13 (44.8) |
|
Hispanic | 15 (55.2) |
| Subtype |
|
|
Luminal | 15 (55.2) |
|
AA | 8 (61.5) |
|
Hisp | 7 (46.7) |
| Her2
enriched | 5 (17.2) |
|
AA | 1 (7.7) |
|
Hisp | 4 (26.6) |
|
TNBC | 6 (17.2) |
|
AA | 3 (23.1) |
|
Hisp | 3 (23.1) |
| Age, years |
|
|
30-50 | 10 (35.7) |
|
AA | 3 (23.1) |
|
Hisp | 7 (46.7) |
|
>50 | 18 (64.3) |
|
AA | 10 (76.9) |
|
Hisp | 8 (53.3) |
Illumina nextera exome library
preparation
Total DNA was isolated from fresh-frozen breast
cancer biopsies and matched normal tissues using the QIAGEN QiaAmp
DNA purification kit and Quantified using Nanodrop (Thermo Fischer
Scientific, USA). 250 to 500 ng DNA was used to prepare the library
using Nextera Flex for Enrichment (Illumina, USA, Cat No 20025524).
Libraries were run on a Bioanalyzer DNA 1000 chip (Agilent, USA) to
assess quality. Library quantitation was done using the qubit 3.0
fluorometer (Thermo Fischer Scientific, USA) using the dsDNA high
sensitivity kit (ThermoFisher, USA, Cat No. Q32851). The exome
capture probes cover about 45 Mb of the primarily protein-coding
region of the human genome (hg19 assembly). The bed file with the
chromosomal coordinates is available at: (https://support.illumina.com/content/dam/illumina-support/documents/downloads/productfiles/nextera/nextera-flex-for-enrichment/TruSeq_Exome_TargetedRegions_v1.2.bed).
Whole exome nextera flex from
enrichment workflow
250 to 500 ng DNA was subjected to library
preparation using the Nextera Flex for Enrichment (Currently, DNA
Prep for Enrichment, Illumina, USA) following
manufacturer-recommended protocol. Briefly, DNA was ‘tagmented’
Tagmentation is the initial step in library prep where genomic DNA
is cleaved and tagged for analysis, cut into small pieces of
300–400 bp length by a transposase, and bead-linked transposes
ligated adaptor for sequencing in the same process. The
tagmentation was followed by captured by biotinylated
oligonucleotides covering approximately 45 Mb of human genomic
exons (sequence version UCSC hg19). Finally, exome library capture
hybridization was performed for 1.5 h. Twelve patient pools were
run on a NextSeq 550 sequencer (Illumina, USA) with paired-end 70
bp reads.
Bioinformatics analysis of exome
sequencing data
Fastq generated by the NextSeq 550 run was mapped to
the hg19 human genome using the Burrows-Wheeler Aligner (BWA, BWA
mem), and variants were identified using the Genome Analysis
Toolkit (GATK). To this end, we utilized Illumina BaseSpace ‘BWA
Enrichment’ pipeline (https://www.illumina.com/products/by-type/informatics-products/basespace-sequence-hub/apps/bwa-enrichment.html).
Mapping was restricted to the chromosomal regions mentioned in
‘TruSeq_Exome_TargetedRegions_v1.2.bed’. This ‘bed’ file specifies
the coordinates of the regions used to generate probes targeting
all known protein-coding genes in the human genome. Variants were
annotated using Illumina Variant Annotator. For the figures shown
below, the variant call format (VCF) file generated by the Illumina
BWA enrichment pipeline was filtered for the common/germline
variants in the QIAGEN QCI software platform using the following
strategy. The following criteria were used to exclude variants from
the cancer exome VCFs. Variants that are present >=1% of Allele
Frequency Community (QIAGEN Database) or >=3% in the following:
the Genome Aggregation Database (gnomAD http://gnomad.broadinstitute.org) or ExAC (https://exac.broadinstitute.org, currently merged
with gnomAD) or of NHLBI GO Exome Sequencing Project (NHLBI ESP
Exomes: http://evs.gs.washington.edu/EVS/) Or 1000 Genomes
project (https://www.internationalgenome.org). Variants were
also excluded if present in the dbSNP database. However, common
germline variants were kept for analysis if established as
pathogenic variants with support from literature published. Matched
normal samples were also used to filter out germline and common
variants. However, variants with known pathogenicity in any disease
were included in the analysis. The QIAGEN Clinical Insight contains
gnomAD (v2.1.1), Exome Variant Server (EVS, vESP6500SI–V2), 1000
Genome Frequency (phase3v5b), Single Nucleotide Polymorphism
Database (dbSNP v154), Combined Annotation Dependent Depletion
(CADD v1.6), Sorting Intolerant from Tolerant (SIFT4G v2016-02-23),
BSIFT (2016-02-23), Polymorphism Phenotyping (PolyPhen-2 v2.2.2)
versions (PhyloP (2009–11). The version information was obtained
from the release notes available at the https://variants.ingenuity.com/qci/website. The
filtered VCF was annotated using Ensemble Variant Effect Predicted
(VEP) (9), which was then
converted to Mutation Annotation Format (MAF) using vcf2maf script
(https://github.com/mskcc/vcf2maf). All
figures were generated using the Maftools R package [R version 4.10
(2021-05-18) and maftools 2.8.0] (10). Mutational signatures were
determined by maftools ‘extractSignature’ and Plot Signature
functions. Finally, mutational profiles were compared with the
current Single base substitutions (SBS) signature from the
Catalogue Of Somatic Mutations In Cancer (COSMIC) database
(https://cancer.sanger.ac.uk/signatures/sbs/). The
overall survival analysis was conducted using maftools mafSurvival
function and comparison between patients with high or low DFS was
carried out with mafCompare function for Fisher's exact test. The
survival analysis utilized Cox proportional hazard function and
correlated gene mutations individually or in groups on the overall
survival of the patients.
Analysis of significantly mutated
genes in the patient sample
Tumor Mutational Burden (TMB) was calculated as
total nonsynonymous mutations per mb (megabases, log2 transformed,
per mb is calculated from the capture size of the exome capture
baits). The statistical test was performed with Graphpad Prism 9
for the Mann-Whitney U test or maftools for the pairwise t-test.
All differences in variant comparison between groups of the sample
were calculated using 2×2 Fisher's Exact Test in maftools. In all
cases, P-value <=0.05 was considered significant. We have used
MutSig v1.4 to analyze significantly mutated genes in
African-American and Hispanic samples. The genes with a q-value
less than 0.0001 were analyzed for enrichment using the ShinyGO
v.0741 web portal (http://bioinformatics.sdstate.edu/go/) against the
Gene Ontology Biological pathways and the Hallmark Dataset form The
Molecular Signatures Database (MSigDB) (11,12).
For MutSig cancer driver identification, we used variants with the
‘PASS’ criteria attached to the variants of interest in the final
filtered variant list for our samples utilizing the maftools
prepareMutsig function. For the TCGA cohorts, we directly exported
the MutSig Compatible variant list file for analysis from the GDC
mc3 maf file. We did the comparative analysis in the ‘Ingenuity
QCI’ (app.ingenuity.com/). The result includes SIFT (https://sift.bii.a-star.edu.sg/) and Polyphen
(http://genetics.bwh.harvard.edu/pph2/) scores
(13,14). These scores predict the functional
consequences of a variation for a protein. We filter the variants
to predict detrimental ‘damaging’ mutations and ‘activating’
mutations.
Analysis of publicly available COSMIC
and TCGA data
TCGA was accessed via cBioPortal using the general
web interface (cbioportal.org). We also downloaded the publicly
available ‘mc3.v0.2.8.PUBLIC.maf.gz ‘from the https://gdc.cancer.gov/about-data/publications/mc3-2017
website and used clinical data available from cBioPortal for the
TCGA Breast Cancer (TCGA-BRCA) cohort to subset the maf file into
African-American and Hispanic categories. We used the clinical
category ‘Race’ as ‘Black or African-American’ (Total 162
extracted) and the ‘Ethnicity’ category ‘Hispanic’ (total 33
extracted) to create the ethnicity-specific mafs using the subset
Maf function in maftools. We analyzed these ethnic categories for
comparative analysis with our cohort of patients. COSMIC Census
genes were downloaded from Sanger's COSMIC site (https://cancer.sanger.ac.uk/census). Venn diagram
comparison of somatic mutations was conducted using the web portal
http://genevenn.sourceforge.net/vennresults.php.
Statistical analysis
We analyzed variants in total of 13 African-American
and 15 Hispanic samples. We also utilized TCGA breast cancer data
set with 163 African-American and 33 Hispanic breast cancer
samples. The comparative analysis was conducted using an unpaired
Mann-Whitney U test, one-way ANOVA with Tukey's multiple comparison
or Fisher's exact test (comparison between high and low
disease-free survival groups). The P-value generated by MutSig for
a gene is a combination of three P-values for the mutation
abundance, location in the genome and the conservation of genetic
sequence across species. Details of the MutSig algorithm are
available at https://www.broadinstitute.org/cancer/cga/mutsig.
The data used for ANOVA and Mann-Whitney U test are available at
the synapse project page (project ID, syn42137028). GraphPad Prism
version 9, GraphPad Inc., San Diego, USA was used for one-way ANOVA
with Tukey's multiple comparison and Mann-Whitney U test. Fisher
exact test was carried out using maftools in R [Maftools R package
(R version 4.10 (2021-05-18, http://cran.r-project.org/) and maftools 2.8.0,
https://github.com/PoisonAlien/maftools/].
Kaplan-Meier plots were generated with univariate cox proportional
hazard model analysis using maftools. In all cases, P-value
<=0.05 was considered significant.
Results
Significant genetic variants in the
African-American and Hispanic cohorts
Our current study examines patient samples from a
small cohort of ethnic minority groups, with an overall of 13
African-American (AA) and 16 Hispanic/Latinx Tumor samples
(Table I Method section, 15
samples for the Hispanic group analyzed). Overall, our exome
analysis recovered single nucleotide variations (SNVs) and a small
number of insertions and deletions. The average coverage for
African-American and Hispanic tumor samples was ~52X and ~48X,
respectively, excluding one Hispanic sample with low coverage
(excluded from analysis). The median Tumor mutational burden
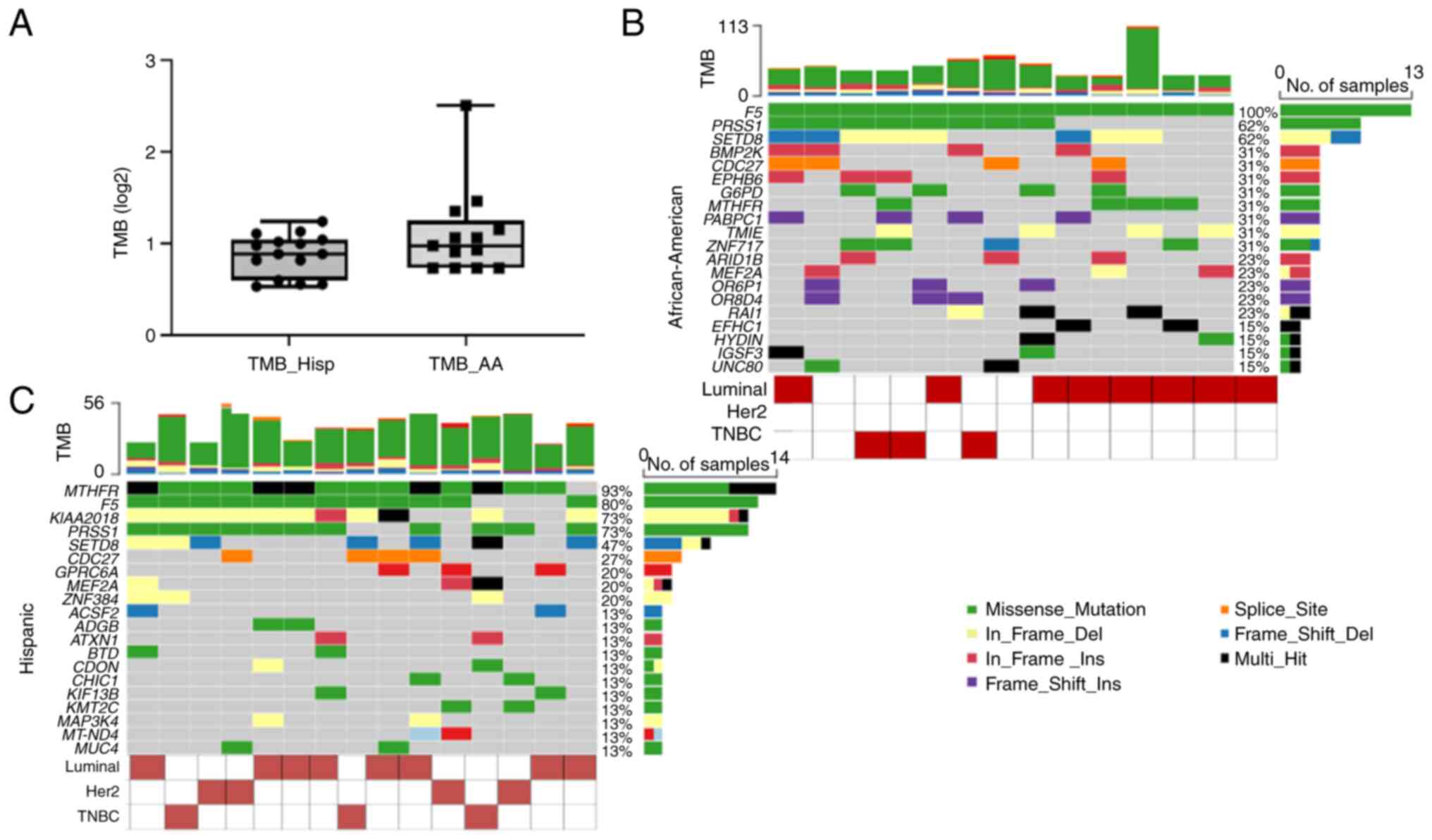
(calculated a log2 (missense mutations/Mb of capture size) were
0.98 and 0.89 in African-American and Hispanic samples,
respectively (Fig. 1). The TMB
values were slightly lower than the TCGA cohorts. However, we did
not observe any statistically significant difference between our
and TCGA samples. After filtering for common variants, the
African-American samples had 647 mutations (SNVs including
insertion and deletion). On the other hand, Hispanic samples had
594 mutations (Table SI: Summary
of mutation after applying common variant filters).
In either case, while including known pathogenic
mutations, the significantly mutated genes were F5
(Coagulation Factor V, p.Q534R) and PRSS1 (Serine Protease
1) in the African-American samples. On the other hand, Both
African-American and Hispanic samples had MTHFR
(methylenetetrahydrofolate reductase, p.E470A, and p.A263V)
variants. Both samples showed variants in histone lysine
methyltransferase gene SETD8. SETD8 had two in-frame
deletions, namely p.A20_A21del and p.L181Hfs*20. One Hispanic and 3
AA samples showed insertion in the ARID1B (p.H1534Q and
p.Q130_Q131dup). PRSS1 mutations were observed in 11
Hispanic and 8 AA samples (p.N29I). The p.N29I polymorphism in
PRSS1 is possibly pathogenic (e.g., ClinVar Accession
RCV000012652.31). Due to the higher frequencies observed in our
samples and associated dbSNP identifiers, these genetic variants
could be due to germline contribution and are predicted to be
benign.
Variants in breast cancer-related and
DNA damage response genes
We examined single nucleotide variants in our
dataset for known breast cancer genes. These genes are reported to
be frequently mutated in breast cancer [Online Mendelian
Inheritance of Man (OMIM) entry for breast carcinoma: https://omim.org/entry/114480 and Human phenotype
ontology (HPO): https://hpo.jax.org/app/browse/term/HP:0003002]
(15). We also compared the gene
list we obtained for each cohort with the COSMIC Cancer Gene Census
(CGC) genes, which in many cases, have experimental evidence as
oncogenes and tumor suppressors (Tier 1) (16).
The cancer genome atlas lists PIK3CA (45%) as
the most frequently mutated gene, followed by MAP3K1, GATA3,
TP53, CDH1, and MAP2K [5]. Although TP53 was the most
frequently mutated gene in African-American breast cancer patients
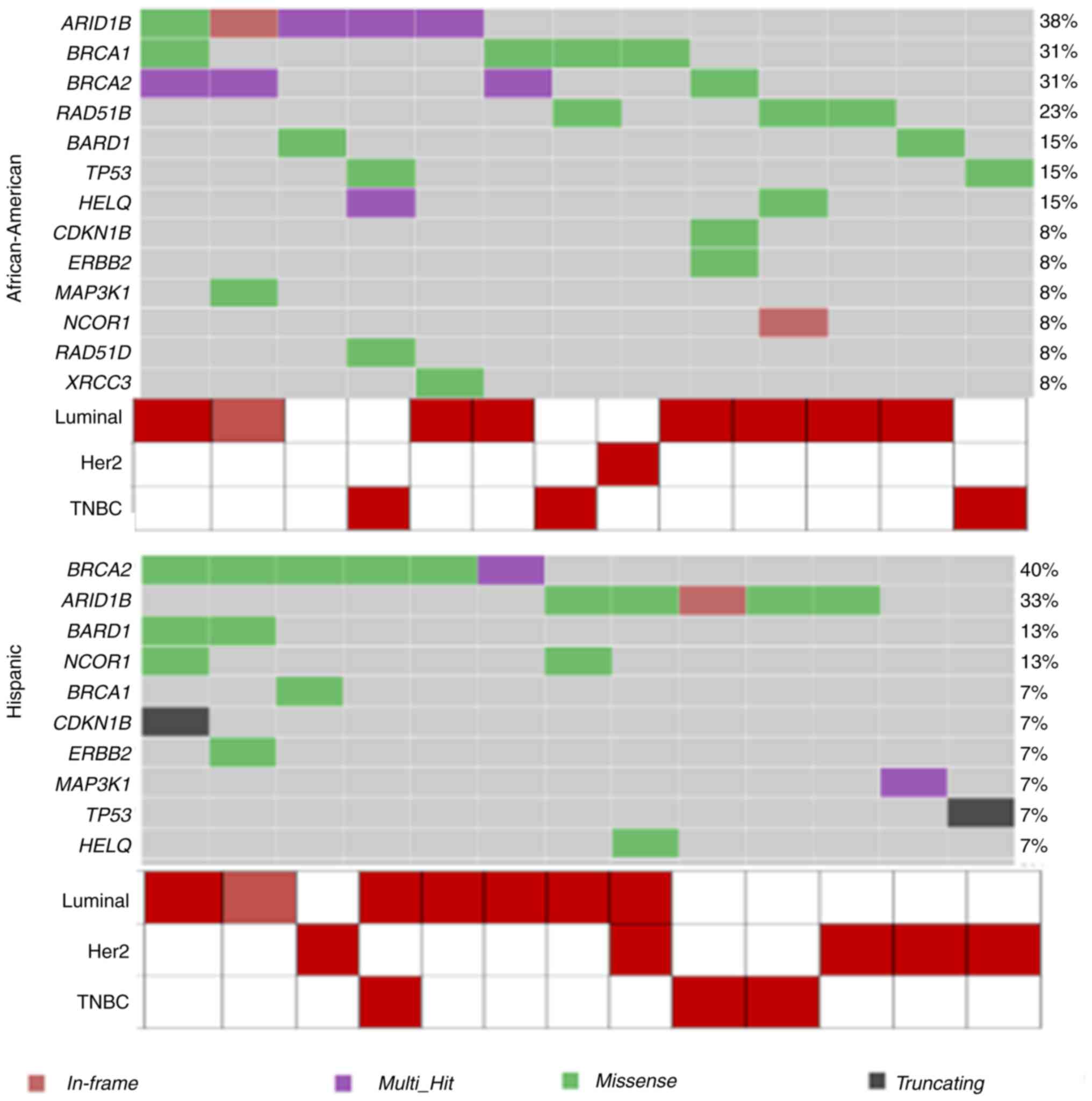
in our cohort, we found missense mutations in ARID1B (5
samples), BRCA1 (4 samples), BRCA2 (4 samples), and
RAD51B (3 samples) (Fig. 2,
Table II). The details of example
genes from the top 50 frequently mutated genes [Online Mendelian
Inheritance of Man (https://omim.org/) and Human
Phenotype Ontology] and the COSMIC Census (updated as of February
2022) are shown in Table II. To
capture genetic variants in these genes, we included variants with
dbSNP ids while comparing the tumor with matched normal (Table III). As a result, BRCA2 (6
samples), ARID1B (AT-rich interactive domain, 5 samples), and
BARD1 (BRCA1 Associated RING Domain 1B, 2 samples) showed
variants in this cohort. In addition, a single sample in each
cohort showed variants in the TP53, ERBB2, and HELQ
(Helicase, POLQ Like, a single-stranded DNA-dependent ATPase, and
DNA helicase) genes. On the other hand, Hispanic patients also
exhibited similar mutational profiles in the top frequently mutated
breast cancer or DNA damage response genes (Fig. 2, Table II).
| Table II.Details of mutations in the
African-American and Hispanic patients. |
Table II.
Details of mutations in the
African-American and Hispanic patients.
| Symbol | Approved name | Mutations | Total samples,
n |
|---|
| KDM6A | Lysine demethylase
6A | c.2703-5dup/del,
intronic | 4 |
| PABPC1 | Poly(A) binding
protein cytoplasmic 1 | p.K254Nfs*24 | 4 |
| ARID1B | AT-rich interaction
domain 1B | p.Q130_Q131dup;
p.Q131dup | 2 |
| MUC16 | Mucin 16, cell
surface associated | p.M2786V;
P13559N | 2 |
| AKT3 | AKT
serine/threonine kinase 3 | p.L208* | 1 |
| ZNF384 | Zinc finger protein
384 | p.Q547del | 3 |
| EIF1AX | Eukaryotic
translation initiation factor 1A X-linked | c.337+1G>C;
C256-3A>C, splice site | 2 |
| KMT2C | Lysine
methyltransferase 2C | p.A30P;
p.M1774T | 2 |
| MUC4 | Mucin 4, cell
surface associated | p.V3635F;
p.G4028S | 2 |
| SGK1 |
Serum/glucocorticoid regulated kinase
1 | c.285+50T>C;
c.362-3230T>G; intron | 2 |
| Table III.Breast cancer and DNA damage
response-related genetic variants. |
Table III.
Breast cancer and DNA damage
response-related genetic variants.
| A,
African-American |
|---|
|
|---|
| Gene symbol | Protein
variant | dbSNP ID | gnomAD, % | COSMIC ID |
|---|
| BRCA2 | p.Y600H | 75419644 | 0.051 | 7349601 |
| BRCA2 | p.G715G | 112566179 | 0.015 |
|
| BRCA2 | p.L929S | 2227943 | 0.097 |
|
| BRCA2 | p.N987I | 2227944 | 0.096 |
|
| BRCA2 | p.D1902N | 4987048 | 0.195 | 9269275 |
| BRCA2 | p.H2116R | 55953736 | 0.134 | 4985277 |
| BRCA2 | p.R2502C | 55716624 | 0.033 | 6958612 |
| BRCA1 | p.T826K | 28897683 | 0.018 | 7343747 |
| BRCA1 | p.N723D;
p.N676D | 4986845 | 0.058 |
|
| RAD51B | p.S212A; p.S131A;
p.S250A | 33929366 | 0.284 | 9494712 |
| XRCC3 | p.R243H | 77381814 | 0.198 | 8488089 |
| BARD1 | p.I738V | 61754118 | 0.747 | 7349100 |
| BARD1 | p.G184G;
p.G203G | 28997574 | 0.878 | 9494804 |
| RAD52 | p.Q377*;
p.Q300* | 1024866946 | 0.001 |
|
|
| B,
Hispanic |
|
| Gene
symbol | Protein
variant | dbSNP
ID | gnomAD,
% | COSMIC
ID |
|
| BRCA1 | p.I1275V;
p.I1228V | 80357280 | 0.015 |
|
| HELQ | p.L802V; p.L372V;
p.L325V; p.L869V | 1344701424 |
|
|
| PALB2 | p.S524S;
p.S229S | 45472400 | 0.319 | 9494341 |
As mentioned earlier, we compared the somatic
mutation data from our cohort with the COSMIC CGCs, for variants
causally linked with cancer. Overall, 47 genes from the
African-American and 52 in the Hispanic cohorts overlap with the
CGC lists (Table SII: COSMIC
Census genes found in our patient cohort). Most of the genes were
found to be mutated in a single sample. In the African-American
cohort, both KDM6A [Lysine (K)-specific demethylase 6A,
c.2703-5dup/del, Intronic] and PABPC1 [poly (A) binding
protein cytoplasmic 1, p.K254Nfs*24] showed variants in 4 samples.
In the Hispanic group, KMT2C [lysine (K)-specific
methyltransferase 2C, p.A30P; p.M1774T], EIF1AX (eukaryotic
translation initiation factor 1A; X-linked, c.337+1G>C;
C256-3A>C, Splice Site), and SGK1 (serum/glucocorticoid
regulated kinase 1 c.285+50T>C; c.362-3230T>G; Intronic) each
showed variants in 2 samples. ZNF384 (p.Q547del) showed the
same variants in 3 Hispanic samples.
Mutations in significant genes in
breast cancer patients in comparison to the TCGA data
The Cancer Genome Atlas (TCGA, http://www.cancer.gov/tcga.) provides a large dataset
on various cancer types to querying for mutations and gene
expression with samples from multiple ethnicities. We queried the
TCGA data set via cBioPortal (cbioportal.org) (17). We utilized the TCGA Pan-Can Atlas
2018 dataset to this end as a maf (mutation annotation format) file
from the NCI Genomic Data Common (GDC). We compared the mutational
profile of African-American (Race Category) and Hispanic patients
(Ethnicity Category) with our cohorts. According to the mutations
listed in the TCGA data, the most frequently mutated gene is
TP53 (accessed via cBioPortal). TP53 was the most
significantly mutated gene in African-American samples (42.7%, 72
patients out of 182 on cBioPortal), followed by FBXW7 (F-box
and WD repeat domain containing 7) at 8%. This finding is in
concordance with other reported studies, which found TP53
mutation to be highly enriched in African-American patients
(18). The GDC maf file also
listed PIK3CA, TTN, GATA3, and KMT2C, MAP3K1 as
frequently mutated in the Black or African-American cohort. TP53
mutations were found in our patient set in two African-American and
only one Hispanic sample. A somatic FBXW7 variant was not observed
in our cohorts (Filtered) except for one Hispanic patient
(p.I394*). In white patients, on the other hand, the mutation level
of TP53 was 29.23% (216 patients out of 752). PIK3CA
was the most frequently mutated gene in white patients (34.64%
compared to 19.66% in African-American patients).
The genetic variants in the TCGA cohort are in
well-known tumor suppressors or oncogenes. However, excluding
variants in databases such as gnomAD or dbSNP and applying the
‘PASS’ filter to the vcf files, the variants in genes such as TP53
and FBXW7 were filtered out from most of our samples. We only
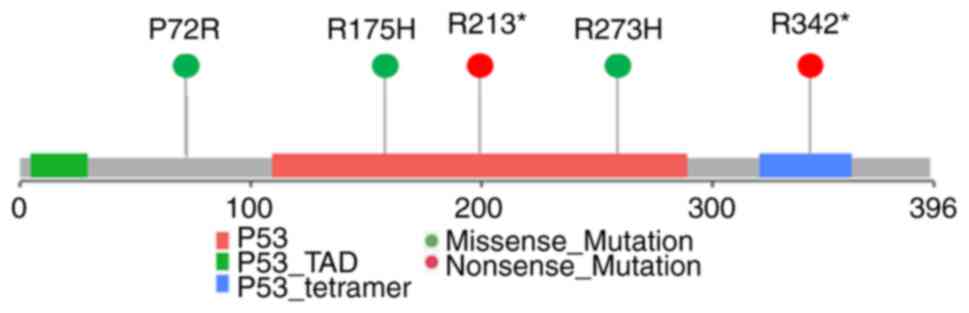
observed TP53 p.R175H in one African-American and p.R213* in
one Hispanic patient. Therefore, we examined the tumor and normal
samples for known variants in those genes to capture the possible
contribution of tumor matched-normal tissues for TP53
variants and other gene mutations in our samples. In our patient
samples, TP53 mutations that were most frequent were p.P72R
(COSMIC id: COSV52666208), p.R273H (COSMIC id: COSV52660980),
p.R342* (stop codon, COSMIC id: COSV52665487). The Hispanic group
only showed the P72R mutations (Fig.
3). All of these TP53 variants are implicated in cancer
(19).
On the other hand, one additional FBXW7
mutation was found in 2 African-American patients. The FBWX7
p.P160L missense mutations might be a loss-of-function implicated
in cancer (COSMIC ID: COSV55920521). In addition, the Hispanic
breast cancer cohort also showed p.D600N mutation, which might be a
somatic loss of function of the protein (COSMIC ID:
COSV55951044).
Mutational signatures in minority
breast cancer patients
In recent years, it has become apparent that certain
mutational processes are causative for a specific type of single
nucleotide variations in the genome. These processes involving the
APOBEC3 (Apolipoprotein B Editing Complex) enzyme or DNA damage
response protein will leave a ‘signature’ behind, which can be
revealed by WES or WGS (20). This
mutational signature is displayed using a 96-mutational profile
classification. The signature is calculated using the substitution
class (A>G, T>C) and three nucleotides 3′ downstream or 5′
upstream to the mutated base. We applied the signature algorithms
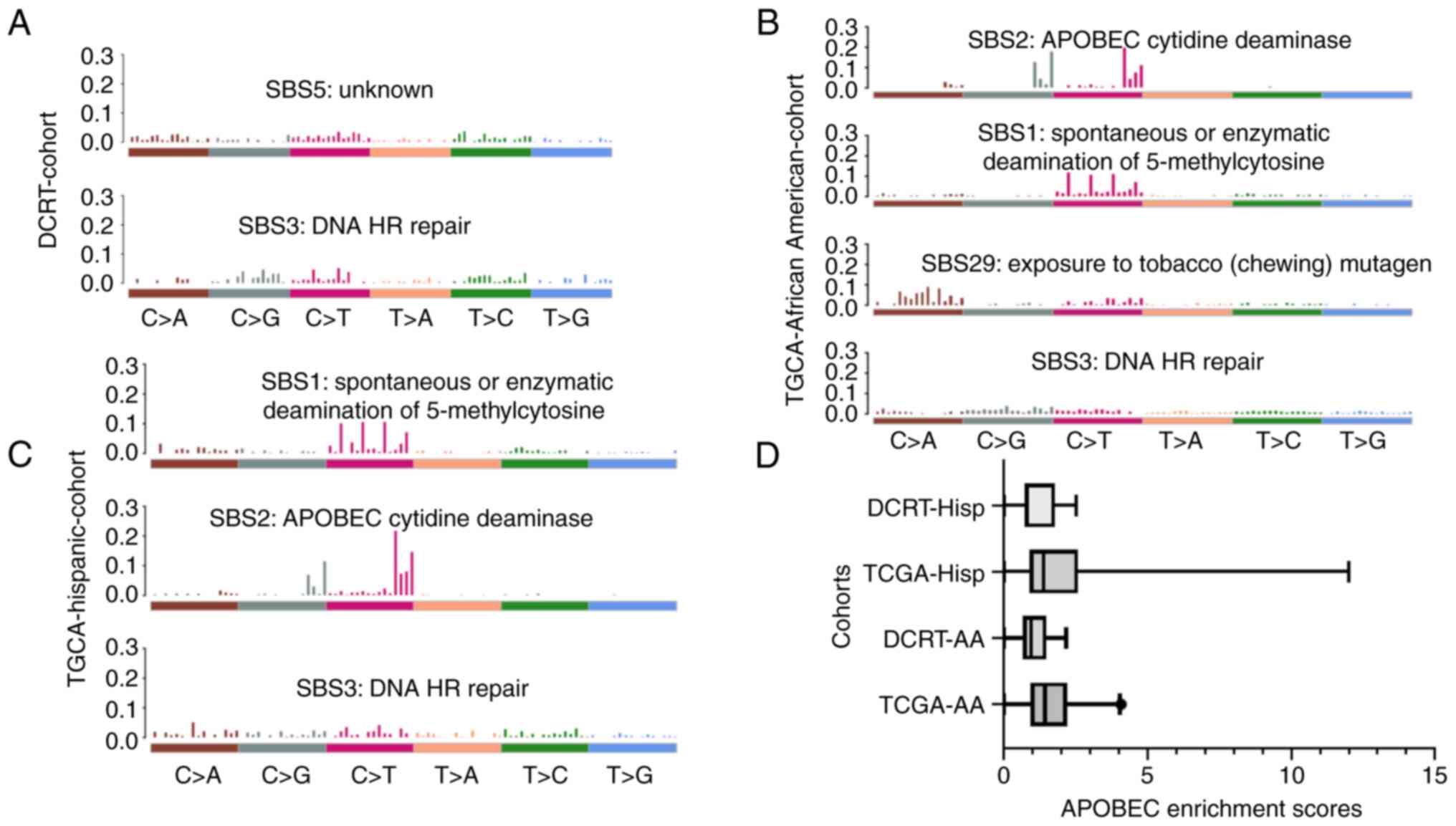
on the exome profile of our patient data. We did not find APOBEC
mutational pattern enrichment in our patient samples. However, all
breast cancer patients in our cohort showed similar mutational
profiles. Furthermore, we discovered that Signatures 3 and 5 are
enriched in African-American and Hispanic samples (Fig. 4). Signature 3 is associated with
DNA double-strand Homologous Recombination repair. We compared the
signature profile with TCGA African-American and Hispanic cohorts.
Signature 3 was found to be over-represented in all samples. The
TCGA cohorts showed Spontaneous or Enzymatic deamination of
5-methylcytosine (SBS1) and APOBEC Cytidine Deaminase (SBS2). The
cytosine deaminase APOBEC3 mediated C to T mutation is prevalent in
breast cancer, and generally, multiple cancer shows mutational
patterns indicative of APOBEC activity (20,21).
However, we did not observe these signatures in our datasets. Only
one African-American patient and three Hispanic patients had APOBEC
enrichment (>2) in our cohort (Table SIII: APOBEC Enrichment scores for
patient samples). APOBEC enrichment score profiles are shown as a
boxplot in Fig. 4D. The TCGA-AA
group has a higher APOBEC value than our cohort (DCRT-AA)
(Mann-Whitney U test, Median TCGA-AA=1.439 and DCRT-0.954,
two-tailed P=0.039). However, all other comparisons between AA and
Hispanic categories did not significantly differ.
Oncogenic pathways in African-American
and Hispanic samples
We also examined somatic variants in the signaling
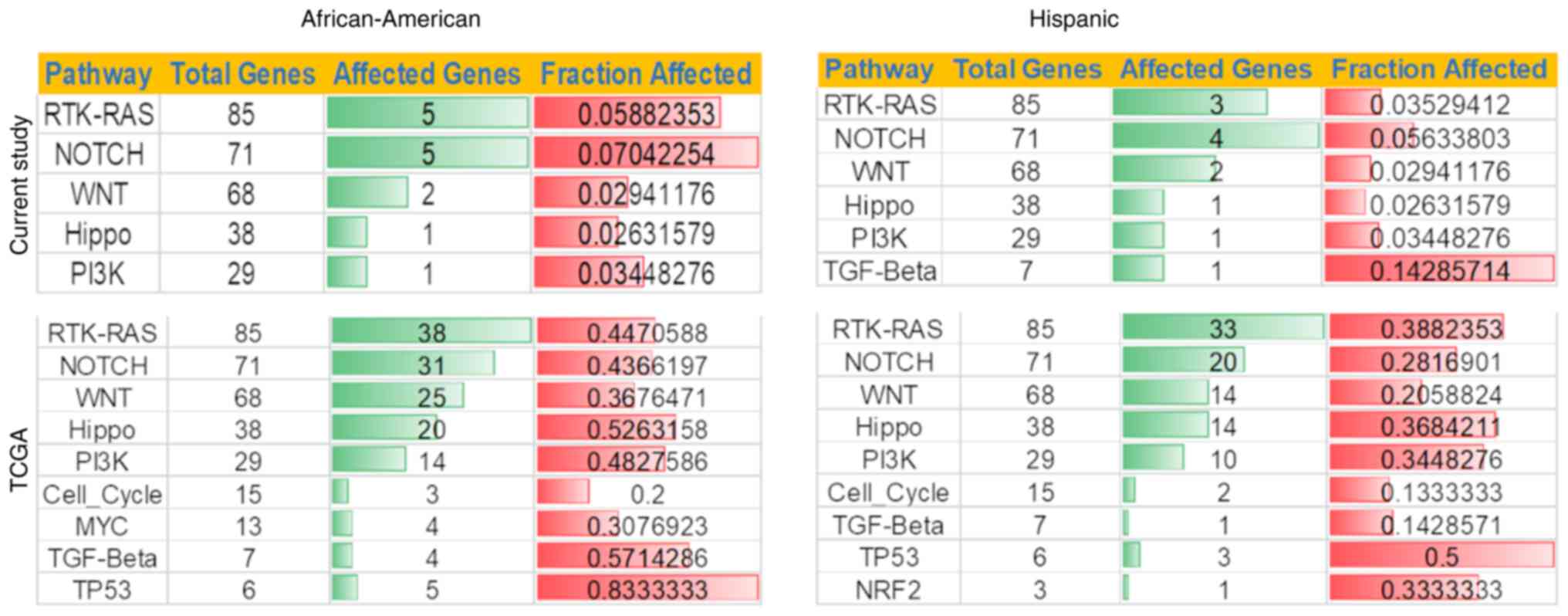
pathways associated with cancer. Ten most affected pathways were
examined using maftools. The pathways are derived from Sanchez-Vega
et al (22). In analyzing
African-American and Hispanic tumor samples, we found that both
have similar profiles regarding affected genes in the pathways. For
example, African-American and Hispanic tumors have RTK-RAS, NOTCH,
and WNT as the top three pathways, although each category's number
of affected genes varies. For example, the RTK-RAS pathway showed 5
and 3 genes mutated for African-American and Hispanic patients,
respectively (Fig. 5). Hispanic
patients had more variants in The TGFβ signaling pathway-associated
genes than African-American patients. Similarly, the Notch pathway
showed variants in 5 and 4 genes out of 71 in African-American and
Hispanic patients. The TCGA data for African-American and Hispanic
cohorts showed similar patterns in the oncogenic pathways. Hippo
and PI3K pathways showed similar profiles in all cohorts
analyzed.
However, individual subtype-based analysis shows
other genes and the genes described above. For example, we compared
the tumor of three African-American patients with three Hispanic
patients with triple-negative (TNBC) subtype in QCI interpretation.
In this case, we observed enrichment for variants in IGSF3
(p.R456C, p.D254N), ZNF717 (p.E370Q; p.Y499*, p.K622fs*79,
p.K790*, p.S861fs*?) KIR3DL3 (p.V324A) and KMT2C (p.W858L,
p.P860S) while selecting pathogenic or likely pathogenic variants.
A G6PD p.V68M (known loss of function) was found in one
African-American sample. However, this variant has a high
prevalence in the African-American population (gnomAD 11.64% in
Africans, Table SIV: Details of
all genetic variants).
Effect of genetic variants on overall
survival
The challenge with survival analysis is the low
frequency of somatic mutations, significantly reducing the number
of patients carrying a mutation. Therefore, we conducted the
overall survival analysis on the TCGA cohorts due to the higher
available number. In addition, we chose the genes that are
frequently mutated in our cohorts. In our survival analysis for the
TCGA African-American cohort, we observed a statically significant
(P<0.05) reduction in the probability of overall survival with a
mutation in F5 (Median Survival 174.5 days vs. 414.5 days in the
WT, HR=2.96) or BRCA2 (HR=5.2). We also conducted survival analysis
with gene sets instead of individual genes. We found that patients
with mutations in at least two genes among XRCC3, TP53, BRCA1 and
BRCA2 have reduced overall survival (Median survival 176 days vs.
413 in patients with Wt alleles, HR=2.37).
Additionally, patients with at least one mutation in
either SETD8, PRSS1, ARID1B, F5, or CDC27 genes showed reduced
survival to 176 days compared to 426 days in Wt patients (Wt
patients) HR=3.06) (Fig. S1).
Additionally, we also utilized the Disease-Free Survival (DFS) data
(Table SV) for our patients
(median=38 months) and compared the genetic variants in patients
with Low DFS with patients who had higher DFS (Median Split). Even
though not statistically significant, African-American patients
with lower DFS had more PRSS1 and CDC27 frequently mutated. On the
other hand, Hispanic patients had F5 and MTHFR more frequently
mutated along with SETD8 and PRSS1 (Table SVI).
Novel driver gene mutation and
differential mutations in African American and Hispanic
samples
We wanted to understand the differentially mutated
genes in African-American and Hispanic breast cancer patient
samples. To this end, we utilized MutSig v1.4 to determine the
novel driver genes in the patients in both African-American and
Hispanic samples (23). MutSig
algorithm frequently determines mutated genes while considering
gene expression and chromatin state and has been tested to find
novel driver mutations across 21 different tumor types (24). The somatic mutation rate is
calculated with respect to a background mutation rate. Among the
frequently mutated somatic genes, 97 genes are shared between
African-American and Hispanic samples among the top 250 genes as
determined by MutSig [Table SVII:
MutSig scores for genes in the African-American and Hispanic sample
and Table SVIII: Top mutation
significance (MutSig) genes across various ethnicities]. After
Filtering the dataset with P-values (<0.05) in African-American
samples, the top ten mutated genes are SETD8, PRSS1, TMIE,
PABPC1, OR8D4, OR6P1, EPHB6, BMP2K, MEF2A, and TPP1.
However, in the TCGA African-American cohort, GATA3, TP53,
PIK3CA, CDH1, PTEN, MAP3K1, MAP2K4, MUC4, RUNX1, and FBXW7 were
the top ten candidate genes. We also conducted GO (Gene Ontology)
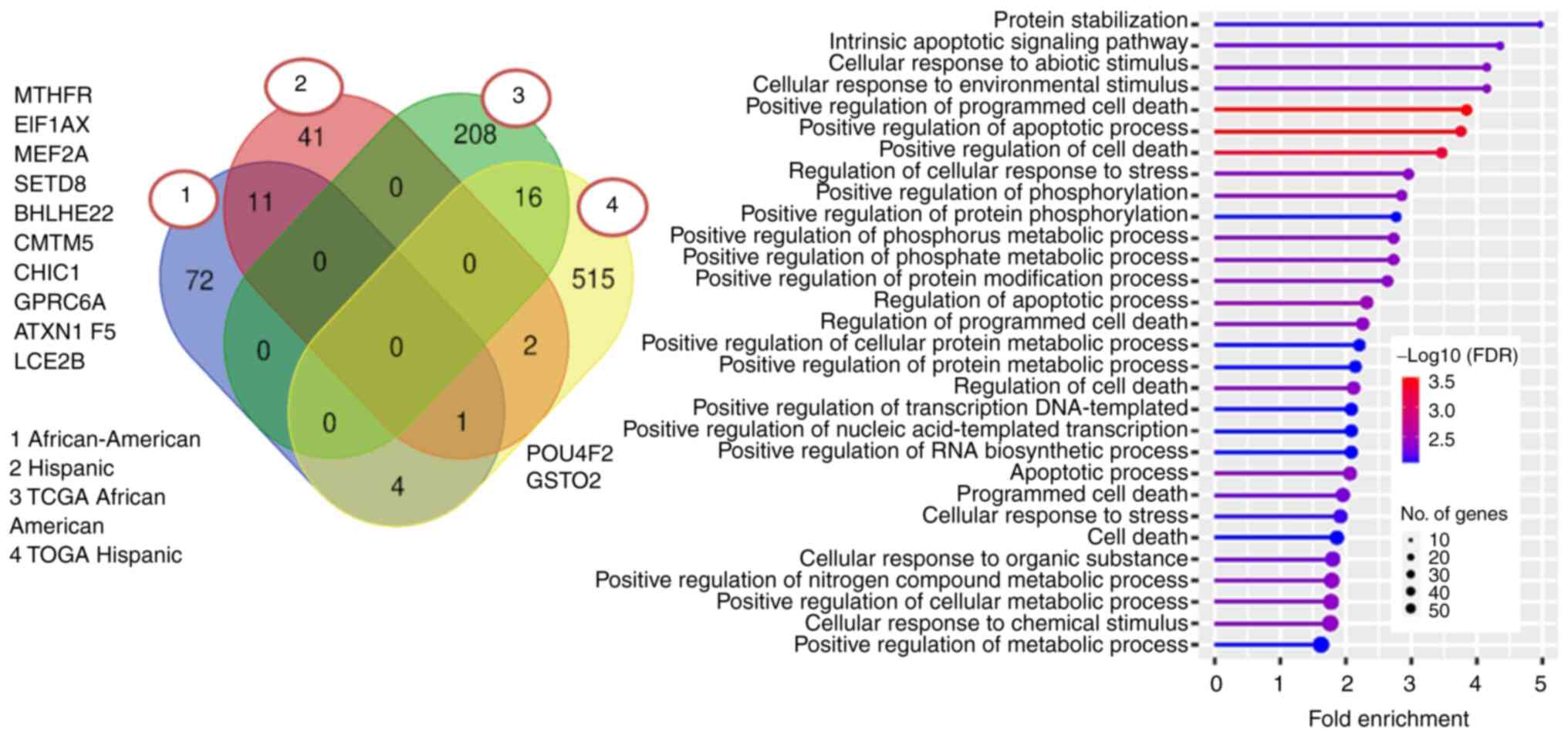
analysis on the significant driver genes for both our and TCGA
cohorts. Our data set did not show enrichment in any particular
category. The TCGA African-American cohorts showed enrichment in
the protein stabilization and intrinsic apoptotic signaling
pathways (Fig. 6). Hallmark MSigDB
Hypoxia, P53 Pathway, and E2F targets were also enriched in the
candidate driver gene profiles from the TCGA African-American
cohort set.
In the TCGA Hispanic group, the top ten genes found
by MutSig were TP53, PIK3CA, GATA3, MAP3K1, TYW3, KHDC1,
CEACAM8, TCP10L2, GPS2, and SNAP29. However, GO ontology
analysis failed to show enrichment in any specific category. Our
Hispanic patient cohort showed a similar candidate driver gene
profile, with the top ten being SETD8, MTHFR, PRSS1, KIAA2018,
CDC27, ZNF384, MEF2A, F5, and NUDT15. As a candidate
driver gene, we observed SETD18, a histone lysine
methyltransferase. SETD8 is known to play a role in breast
cancer metabolism by stabilizing hypoxia-inducible factor 1α
(HIF1α) and is also implicated in DNA damage response maintaining
genomic integrity (25,26).
As with all pathways, the mutated genes in
African-American samples were similar to Hispanic samples. 11 genes
(including ATXN1, CMTM5, EIF1AX, GPRC6A, MEF2A, PRSS1, and
SETD8) were found to be candidate drivers in both
categories. However, some genes were only enriched in one or the
other sample (72 in African-American, 42 in the Hispanic cohort,
Fig. 6 Venn Diagram). There were
only two genes common between our Hispanic and TCGA cohorts. Our
analysis with the Hispanic samples observed significant mutations
in genes such as CDC27 (cell division cycle 27), making it a
candidate driver gene. CDC27 protein levels and polymorphism are
associated with breast cancer mortality and risk (27,28).
Discussion
Efforts are underway which utilize various omics
approaches to understand cancer health disparity. Next-generation
sequencing like whole genome and exome technologies are paving the
way to understanding the mutational burden and discovering driver
mutations in various tumor types. With technologies like WES, it is
possible to elucidate the biological aspects of health disparity.
The current study shows WES results on African-American and
Hispanic patients, two minority demographics not significantly
represented in large databases like TCGA. We wanted to determine
somatic mutations in our cohort with a tumor-normal comparison and
explore the variants in genes implicated in breast cancer from
publicly available somatic and germline variants databases such as
COSMIC and OMIM (Table SIV:
Details of all variants in our patient samples). However, we
observed multiple breast cancer-related genetic variants in the
germline after relaxing filtering criteria to include dbSNP
variants or variants with known pathogenicity, which might be due
to tumor heterogeneity or purity.
The DNA Damage Response (DDR) pathways are essential
for affecting critical biological processes. The proteins involved
in these pathways can result in mutations in DNA sequences due to
error-prone repair. In addition, multiple proteins affecting this
pathway are implicated in cancer (29). Thus, we examined the DNA damage
response pathway genes and signatures in our cohort of patients and
examined potential driver mutations. We also utilized the TCGA
database to explore the mutational landscaper in African-American
and Hispanic cohorts and did a comparative analysis with our
cohort.
In African-American and Hispanic samples from TCGA,
we overserved that known breast cancer-related genes, including
TP53, PIK3CA, GATA3, and MAP3K1, were significantly
more variants. Even though samples from both ethnic categories of
patients had variants in the genes mentioned, they showed different
frequencies with which these genes were mutated. For example,
TP53 is the most frequently mutated in the African-American
TCGA sample as opposed to PIK3CA in Hispanic samples. Our
cohorts found TP53 variants in a few selected tumor samples,
including tumor-adjacent normal tissue. The variants (Fig. 3) were also found in the TCGA cohort
except for TP53 p.P72R. Among genes that showed a
higher frequency in our cohort were F5 and MTHFR,
with possible germline contributions. However, F5 is a potential
candidate gene for breast cancer and a marker for immune cell
infiltration in breast cancer (30,31).
PRSS1, SETD8, and CDC27 were frequently mutated in
African-American and Hispanic samples.
We observed that the DDR pathway genes are mutated
in the African-American and Hispanic samples. These findings
explain the observed mutational signatures (Current SBS), namely
‘Signature 3 and 5’. Signature 3. These signatures are associated
with DNA Homologous Recombination repair. However, we did not
observe any SBS1/2 associated with APOBEC activity in our breast
cancer samples compared to the TCGA cohorts. Instead, we observed
Signature 5, which may be due to environmental exposure or other
unknown factors.
DNA damage signature can also modify the tumor
microenvironment and affect immune gene expression (32). A ‘DNA damage response-deficient’
subtype shows up-regulation of Programmed Death-Ligand 1 (PD-L1) in
a cyclic GMP-AMP synthase and signaling effector stimulator of
interferon genes (cGAS-STING) dependent manner. The cGAS-STING
pathway is a foreign DNA sensing mechanism associated with multiple
inflammatory responses (33).
Thus, minority breast cancer patients might benefit from checkpoint
inhibitor therapy when multiple genes in the DNA damage response
and homologous recombination pathway have variants with functional
implications (Table II). Other
genomic stability pathways, such as Microsatellite Instability
(MSI), are prevalent in all cancers, with variability across cancer
types (34). It will be
interesting to study the effect of MSI on breast cancer
susceptibility and its occurrence in African-American and Hispanic
patients to understand the contribution of mismatch repair in
breast cancer.
Primarily, the somatic and germline variants show
similarities with some differences between two minority breast
cancer populations that can be further studied in a
sub-type-specific manner. Further studies could help understand
this disparity in our minority breast cancer patients with more
extensive cohort studies. Our exome analysis found variants in the
polymorphic genes in our patient samples, particularly
CDC27. These genes potentially involve cell division and
adipocyte metabolisms (35). In
addition, low CDC27 expression and CDC27
polymorphisms are associated with worse breast cancer outcomes
(27,28). Thus, despite being polymorphic in
the general population, the variants in these genes could also have
functional implications for cancer.
Our findings can have clinical implications in
determining therapy in patients with specific genetic mutations.
For example, MTHFR is associated with drug metabolism and can
affect the patient's ability to respond to chemotherapy. In colon
and breast cancers, 5-Fluorouracila sensitivity is associated with
variants in the MTHFR gene (36,37).
F5 (Coagulation factor V) is an estrogen response gene associated
with CD8+ T cell in cancer immunity (30,38).
These genes are also associated with the TGF-β pathway. Thus, using
inhibitors when the pathway is activated because of mutations can
be a potential therapeutic option. SETD8 variants can affect
epigenetic pathways due to their role as lysine methyltransferases.
SETD8 is also involved in DNA damage repair, thus making it a
potential target via small-molecule inhibition (38). Additionally, PRSS1 is associated
with drug resistance in cancer and higher cancer risk along with
SETD8 and ARID1B (39–41). Thus, our results on genetic
variants can potentially be used as predictors of cancer risk in
minority women.
In our study, we conducted WES on African-American
and Hispanic breast cancer samples to elucidate the genetic makeup
of breast cancer in these patients. We found overlapping genetic
variants in both ethnicities that are potentially causative such as
PRSS1 and SETD8. However, there are significant
differences in the specific genetic variants that belong to DNA
damage response to transcription factors such as BRCA1/2, XRCC3,
HELQ, and ARID1B. In our study, variants shown to be
potentially damaging will need to be further studied to understand
their molecular mechanisms concerning cancer initiation or
progression. In addition, it will be beneficial to validate our
findings in larger cohorts, which could lead to biomarker discovery
towards the goal of alleviating health disparity. Overall, WES and
other next-generation sequencing technologies will be crucial in
our efforts to understand breast cancer health disparity.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The research was funded by NIH/NCI/NIMHD (grant nos. 1U54CA14393
and U54MD007598). Research reported in this publication was
supported by the National Institute on Minority Health and Health
Disparities of the National Institutes of Health (grant no. S21
MD000103).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the Synapse.org
repository, https://doi.org/10.7303/syn42137028 (project ID,
syn42137028).
Authors' contributions
PD was responsible for the conceptualization,
methodology, formal analysis, data curation and original draft
preparation. MYK was involved in the methodology and formal
analysis. YW was involved in obtaining resources, study design,
analysis, writing, review and editing. JVV was responsible for the
conceptualization, supervision, review and editing, and funding
acquisition. PD and JVV confirm the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The study was conducted according to the guidelines
of the Declaration of Helsinki and approved by the Institutional
Review Board of Charles R. Drew University of Medicine and Science
(#IRB 00-06-041; Los Angeles, USA), and the protocol has been
approved since 1999 and is reviewed annually for continuation
(recent continuing review approval was August 18, 2021). Written
informed consent for participation was obtained from all subjects
involved in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Howlader N, Noone AM, Krapcho M, Miller D,
Brest A, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, et al:
SEER cancer statistics review, 1975–2017. National Cancer
Institute; Bethesda, MD: 2020, https://seer.cancer.gov/csr/1975_2017/
|
|
2
|
Polyak K: Heterogeneity in breast cancer.
J Clin Invest. 121:3786–3788. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yedjou CG, Sims JN, Miele L, Noubissi F,
Lowe L, Fonseca DD, Alo RA, Payton M and Tchounwou PB: Health and
racial disparity in breast cancer. Adv Exp Med Biol. 1152:31–49.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chlebowski RT, Chen Z, Anderson GL, Rohan
T, Aragaki A, Lane D, Dolan NC, Paskett ED, McTiernan A, Hubbell
FA, et al: Ethnicity and breast cancer: Factors influencing
differences in incidence and outcome. J Natl Cancer Inst.
97:439–448. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Koboldt DC, Fulton RS, McLellan MD,
Schmidt H, Kalicki-Veizer J, McMichael JF, Fulton LL, Dooling DJ,
Ding J, Mardis ER, et al: Comprehensive molecular portraits of
human breast tumours. Nature. 490:61–70. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Carrot-Zhang J, Chambwe N, Damrauer JS,
Knijnenburg TA, Robertson AG, Yau C, Zhou W, Berger AC, Huang KL,
Newberg JY, et al: Comprehensive analysis of genetic ancestry and
its molecular correlates in cancer. Cancer Cell. 37:639–654.e6.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huo D, Hu H, Rhie SK, Gamazon ER,
Cherniack AD, Liu J, Yoshimatsu TF, Pitt JJ, Hoadley KA, Troester
M, et al: Comparison of breast cancer molecular features and
survival by african and european ancestry in the cancer genome
atlas. JAMA Oncol. 3:1654–1662. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
DeSantis CE, Ma J, Gaudet MM, Newman LA,
Miller KD, Goding Sauer A, Jemal A and Siegel RL: Breast cancer
statistics, 2019. CA Cancer J Clin. 69:438–451. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
McLaren W, Gil L, Hunt SE, Riat HS,
Ritchie GR, Thormann A, Flicek P and Cunningham F: The ensembl
variant effect predictor. Genome Biol. 17:1222016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mayakonda A, Lin DC, Assenov Y, Plass C
and Koeffler HP: Maftools: Efficient and comprehensive analysis of
somatic variants in cancer. Genome Res. 28:1747–1756. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ge SX, Jung D and Yao R: ShinyGO: A
graphical gene-set enrichment tool for animals and plants.
Bioinformatics. 36:2628–2629. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liberzon A, Subramanian A, Pinchback R,
Thorvaldsdóttir H, Tamayo P and Mesirov JP: Molecular signatures
database (MSigDB) 3.0. Bioinformatics. 27:1739–1740. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vaser R, Adusumalli S, Leng SN, Sikic M
and Ng PC: SIFT missense predictions for genomes. Nat Protoc.
11:1–9. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Köhler S, Gargano M, Matentzoglu N,
Carmody LC, Lewis-Smith D, Vasilevsky NA, Danis D, Balagura G,
Baynam G, Brower AM, et al: The human phenotype ontology in 2021.
Nucleic Acids Res. 49(D1): D1207–D1217. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sondka Z, Bamford S, Cole CG, Ward SA,
Dunham I and Forbes SA: The COSMIC cancer gene census: Describing
genetic dysfunction across all human cancers. Nat Rev Cancer.
18:696–705. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: An open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Keenan T, Moy B, Mroz EA, Ross K,
Niemierko A, Rocco JW, Isakoff S, Ellisen LW and Bardia A:
Comparison of the genomic landscape between primary breast cancer
in African American versus white women and the association of
racial differences with tumor recurrence. J Clin Oncol.
33:3621–3627. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Olivier M, Hollstein M and Hainaut P: TP53
mutations in human cancers: Origins, consequences, and clinical
use. Cold Spring Harb Perspect Biol. 2:a0010082010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Roberts SA, Lawrence MS, Klimczak LJ,
Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL,
Saksena G, et al: An APOBEC cytidine deaminase mutagenesis pattern
is widespread in human cancers. Nat Genet. 45:970–976. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Burns MB, Lackey L, Carpenter MA, Rathore
A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N,
Nikas JB, et al: APOBEC3B is an enzymatic source of mutation in
breast cancer. Nature. 494:366–370. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sanchez-Vega F, Mina M, Armenia J, Chatila
WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia
S, et al: Oncogenic signaling pathways in the cancer genome atlas.
Cell. 173:321–337.e10. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lawrence MS, Stojanov P, Polak P, Kryukov
GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH,
Roberts SA, et al: Mutational heterogeneity in cancer and the
search for new cancer-associated genes. Nature. 499:214–218. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lawrence MS, Stojanov P, Mermel CH,
Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander
ES and Getz G: Discovery and saturation analysis of cancer genes
across 21 tumour types. Nature. 505:495–501. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang R, Yu Y, Zong X, Li X, Ma L and
Zheng Q: Monomethyltransferase SETD8 regulates breast cancer
metabolism via stabilizing hypoxia-inducible factor 1α. Cancer
Lett. 390:1–10. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Paulsen RD, Soni DV, Wollman R, Hahn AT,
Yee MC, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero
DE, et al: A genome-wide siRNA screen reveals diverse cellular
processes and pathways that mediate genome stability. Mol Cell.
35:228–239. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guo H, Chen W, Ming J, Zhong R, Yi P, Zhu
B, Miao X and Huang T: Association between polymorphisms in cdc27
and breast cancer in a Chinese population. Tumour Biol.
36:5299–5304. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Talvinen K, Karra H, Pitkänen R, Ahonen I,
Nykänen M, Lintunen M, Söderström M, Kuopio T and Kronqvist P: Low
cdc27 and high securin expression predict short survival for breast
cancer patients. APMIS. 121:945–953. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jackson SP and Bartek J: The DNA-damage
response in human biology and disease. Nature. 461:1071–1078. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Andresen MS, Sletten M, Sandset PM,
Iversen N, Stavik B and Tinholt M: Coagulation factor V (F5) is an
estrogen-responsive gene in breast cancer cells. Thromb Haemost.
122:1288–1295. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tinholt M, Stavik B, Tekpli X, Garred Ø,
Borgen E, Kristensen V, Sahlberg KK, Sandset PM and Iversen N:
Coagulation factor V is a marker of tumor-infiltrating immune cells
in breast cancer. Oncoimmunology. 9:18246442020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Parkes EE, Walker SM, Taggart LE, McCabe
N, Knight LA, Wilkinson R, McCloskey KD, Buckley NE, Savage KI,
Salto-Tellez M, et al: Activation of STING-dependent innate immune
signaling by S-phase-specific DNA damage in breast cancer. J Natl
Cancer Inst. 109:djw1992016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Motwani M, Pesiridis S and Fitzgerald KA:
DNA sensing by the cGAS-STING pathway in health and disease. Nat
Rev Genet. 20:657–674. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cortes-Ciriano I, Lee S, Park WY, Kim TM
and Park PJ: A molecular portrait of microsatellite instability
across multiple cancers. Nat Commun. 8:151802017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vernochet C, Peres SB, Davis KE, McDonald
ME, Qiang L, Wang H, Scherer PE and Farmer SR: C/EBPalpha and the
corepressors CtBP1 and CtBP2 regulate repression of select visceral
white adipose genes during induction of the brown phenotype in
white adipocytes by peroxisome proliferator-activated receptor
gamma agonists. Mol Cell Biol. 29:4714–4728. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kim YI: Role of the MTHFR polymorphisms in
cancer risk modification and treatment. Future Oncol. 5:523–542.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sohn KJ, Croxford R, Yates Z, Lucock M and
Kim YI: Effect of the methylenetetrahydrofolate reductase C677T
polymorphism on chemosensitivity of colon and breast cancer cells
to 5-fluorouracil and methotrexate. J Natl Cancer Inst. 96:134–144.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Guan Y, Xu B, Sui Y, Chen Z, Luan Y, Jiang
Y, Wei L, Long W, Zhao S, Han L, et al: Pan-cancer analysis and
validation reveals that D-dimer-related genes are prognostic and
downregulate CD8+ T cells via TGF-Beta signaling in
gastric cancer. Front Mol Biosci. 9:7907062022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tan Z, Gao L and Wang Y, Xu J and Wang Y:
PRSS contributes to cetuximab resistance in colorectal cancer. Sci
Adv. 6:eaax55762020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Weiss FU: Pancreatic cancer risk in
hereditary pancreatitis. Front Physiol. 5:702014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Milite C, Feoli A, Viviano M, Rescigno D,
Cianciulli A, Balzano AL, Mai A, Castellano S and Sbardella G: The
emerging role of lysine methyltransferase SETD8 in human diseases.
Clin Epigenetics. 8:1022016. View Article : Google Scholar : PubMed/NCBI
|