Introduction
Lung cancer is the second most common cancer in the
world and remains the leading cause of cancer death proven by a
high global diagnosis rate (11.4%) and mortality rate (18%). In
2020, there were 19.3 million new cases of cancer and 10 million
cancer-associated deaths worldwide, and it was estimated that ~1.8
million of these individuals died of lung cancer (1). Among all subtypes of lung cancer,
non-small cell lung cancer (NSCLC) accounts for 80–85%, of which
~40% cases are adenocarcinomas (AC), 25–30% cases are squamous cell
carcinomas (SCCs) and 10–15% cases are large cell carcinomas
(2). In the USA, the 5-year
survival rate for NSCLC is ~26% (3). Since NSCLC is the most common type of
lung cancer, research into the disease is necessary. The treatment
of NSCLC includes traditional surgery, chemotherapy (including
targeted drug therapy), radiotherapy and emerging immunotherapy for
advanced tumors. In addition, some nanodrugs are at the research
stage (4). The option of treatment
mainly depends on, but not necessarily determined by, the stage of
NSCLC (5). Despite these promising
treatment options, the five-year survival rate of advanced NSCLC is
still very low, particularly at stage IIIB, which is only 26%
(6). Therefore, it is necessary to
discover and explore new therapies.
It has been nearly a century since the first study
on tumor metabolism, the Warburg effect, was published in 1924
(7). Research on tumor metabolism
has burgeoned over the past decade. In the sense of exploring tumor
metabolism, it not only elucidates the mechanism of tumorigenesis
and progression, but is also conducive to diagnosis and treatment.
The metabolism of cancer cells is both flexible and plastic.
Through metabolic reprogramming, cancer cells can maintain cell
vitality and growth, even under nutrition-deprived conditions or in
an oxygen-deficient environment (8). Metabolic reprogramming is regarded as
one of the emerging hallmarks of cancer (9). Glucose and glutamine are two major
nutrients supporting survival and biosynthesis in tumor metabolism
(10). The Warburg effect revealed
the process of aerobic glycolysis in tumor cells, which is one of
the most prominent features of tumor metabolism (11). Glutamine is a rich and versatile
nutrient involved in energy generation, redox homeostasis,
macromolecular synthesis and signal transduction (12). Based on the important role of
glutamine in tumorigenesis and progression, it may be a promising
direction of targeting its metabolic process to develop new
clinical therapies.
A previous study indicated that NSCLC cells may use
glutamine as a substrate through metabolic reprogramming to induce
glutamine addiction, which is a promising therapeutic target
(13). However, tumor metabolism is
not a specific metabolic map, presenting challenges for targeting
glutamine metabolism in cancer therapy (14). In this perspective, the present
review combined the glutamine-dependent metabolism of NSCLC with
the therapeutic strategy of targeting glutamine to provide new
approaches for the treatment of NSCLC and to select those patients
who may benefit the most from glutamine metabolism-targeted
therapy, and ultimately to improve the overall survival rate and
the long-term quality of life.
Metabolic reprogramming in NSCLC
It is widely accepted that metabolic programming is
one of the hallmarks of cancer (15). Most normal cells use external
stimuli to activate growth factor signals and to absorb a large
amount of nutrients from the external environment for metabolism,
whereas cancer cells maintain the activation of signal pathways
through gene mutations (16). In
this way, the metabolism of cancer cells is flexible and plastic,
which provides the basis for metabolic reprogramming. The
significance of metabolic reprogramming is to promote the growth
and proliferation of tumor cells by generating energy, synthesizing
necessary precursors and maintaining oxidative balance, especially
under hypoxic and hypo-nutrient conditions (17). Furthermore, metabolic reprogramming
plays an important role in malignant transformation, the
progression of tumors and the resistance to antitumor therapy
(18). In NSCLC cells, reactive
oxygen species (ROS)-mediated metabolic reprogramming leads to
oxidative phosphorylation, which results in cisplatin resistance
(19). It has been shown that
smoking can greatly increase the incidence of lung cancer partly
due to the overexpression of enzymes related to glutamine
metabolism, fatty acid degradation and lactate synthesis, resulting
in mitochondrial metabolic reprogramming of NSCLC cells (20). Research on the metabolic
reprogramming of cancer cells can be traced back to last century
when the Warburg effect appeared. It has been shown that even in an
aerobic environment, glucose is involved in the glycolytic pathway
rather than in aerobic oxidation, revealing the metabolic
reprogramming of glucose in cancer cells (11). The mechanism of metabolic
reprogramming remains to be explored. Fundamentally, metabolic
reprogramming is the consequence of oncogene or tumor suppressor
gene mutation, including direct and indirect effects (21). KRAS mutations are among the most
common mutations leading in NSCLC (22). NSCLC cells can become
glutamine-dependent through a variety of metabolic reprogramming
processes (23). For example, it
activates autophagy to uptake glutamine through the degradation of
cellular proteins (24). In
addition, the increase in macropinocytosis also reflects the
dependence on glutamine, which provides cells with a supply of
amino acids, mainly glutamine (25). However, the process of KRAS-driven
glutamine metabolism in NSCLC cells is different in vivo and
in vitro (26). The LKB1
gene is also associated with altered metabolism in cells. LKB1
deletion promotes the expression of hypoxia-inducible factor
(HIF)-1α and stabilizes it by regulating mTOR activity in NSCLC
cells. This metabolic reprogramming increases the uptake and
utilization of both glucose and glutamine, enhancing aerobic
glycolysis and glutamine catabolism to promote cell growth
(27,28). A previous study has shown that
KRAS-mutant NSCLC cells lacking LBK1 usually also have Kelch-like
ECH-associated protein 1 (KEAP1) gene mutations; these are known as
KLK NSCLC cells. These three genes work corporately to promote
metabolic reprogramming, making KLK NSCLC cells glutamine dependent
and sensitive to glutamine inhibitors (29). Glutamine addiction occurs in c-Myc
mutant NSCLC, where c-Myc is an oncogene that drives metabolic
reprogramming. c-Myc can increase the expression of glutaminase
(GLS) by inhibiting microRNA (miR)-23a/b to promote intracellular
glutamine metabolism (30).
However, metabolic reprogramming is not only determined by genes,
but also by tissue specificity (14,31).
In addition, long non-coding RNAs (lncRNAs) are inseparable from
metabolic reprogramming. For example, the oncogenic lncRNA,
Al355338, activates the EGFR/AKT signaling pathway by preventing
enolase 1 from ubiquitination and degradation, making it
stabilized, leading to metabolic reprogramming and increasing
aerobic glycolysis (32). In NSCLC
cells, lncRNA-AC020978 carries out metabolic reprogramming by
regulating the pyruvate kinase M2/HIF-1α positive-feedback axis
under the condition of hypoxia or glucose deficiency, which adapts
the cells to the hypoxic environment and promotes glycolytic
metabolism (33).
Glutamine metabolism in NSCLC
The Warburg effect demonstrated that cancer cells
use glycolysis in an aerobic environment for rapid proliferation
(11); since this discovery, the
metabolic flexibility of tumors has attracted the interest of
researchers. A number of studies have discussed the characteristics
of tumor glucose metabolism and further verified the Warburg effect
from different aspects. Glucose from the extracellular environment
is not sufficient for tumor growth and proliferation; therefore,
tumor cells use other sources of nutrients as well, glutamine being
the most prominent (10).
Characteristics of metabolism in NSCLC include increased glucose
consumption and lactate production, as well as glutamine addiction
(34).
Glutamine metabolism
Glucose and glutamine are the two basic nutrients
used by cancer cells (10).
Although glutamine is a non-essential amino acid, it can accumulate
from de novo synthesis and serve an indispensable role in
cancer cell growth, signal transduction and maintaining redox
homeostasis (35). Glutamine can be
used as a nitrogen source to synthesize amino acids and nucleotides
to promote cell growth, or as a carbon source to replenish the
tricarboxylic acid (TCA) cycle when glucose levels are deficient
(36). This process originates from
the conversion of glutamine into glutamate through the catalysis of
GLS, followed by the conversion of glutamate into α-ketoglutarate
(α-KG) through catalysis by glutamine dehydrogenase (GDH) or
transaminase (Fig. 1) (37). However, under hypoxia or
mitochondrial dysfunction, α-KG is carbonylated to citrate, the
substrate for fatty acid synthesis, indicating that glutamine
actively participates in lipid synthesis (38). The enzymes involved in fatty acid
synthesis mainly include citrate lyase, acetyl CoA carboxylase
(ACC) and fatty acid synthase (FAS); the process is as follow:
Citrate is converted by citrate lyase into acetyl-CoA, used for the
synthesis of fatty acids, and oxaloacetate. Synthesis of
malonyl-CoA from acetyl-CoA is catalyzed by ACC. Next, palmitic
acid is synthesized through multiple processes of
condensation-reduction-dehydration-reduction, catalyzed by FAS
(39). Glutamine activates mTORC1
through a special mechanism that requires ADP ribosylation factor 1
and is independent of Rag GTPase to stimulate the lysosomal
localization of mTORC1 and to exert its physiological functions,
such as controlling cell growth, metabolism and autophagy (35). Glutamine participates in complex
signaling pathways through this process and maintains redox
homeostasis mainly by synthesizing glutathione (GSH), an
antioxidant molecule, to resist ROS. When ROS accumulates at high
levels, DNA, proteins and lipids will be degraded (40,41).
GDH1 is another key enzyme involved in glutamine metabolism; it
regulates redox homeostasis through activation of GSH peroxidase 1
(42).
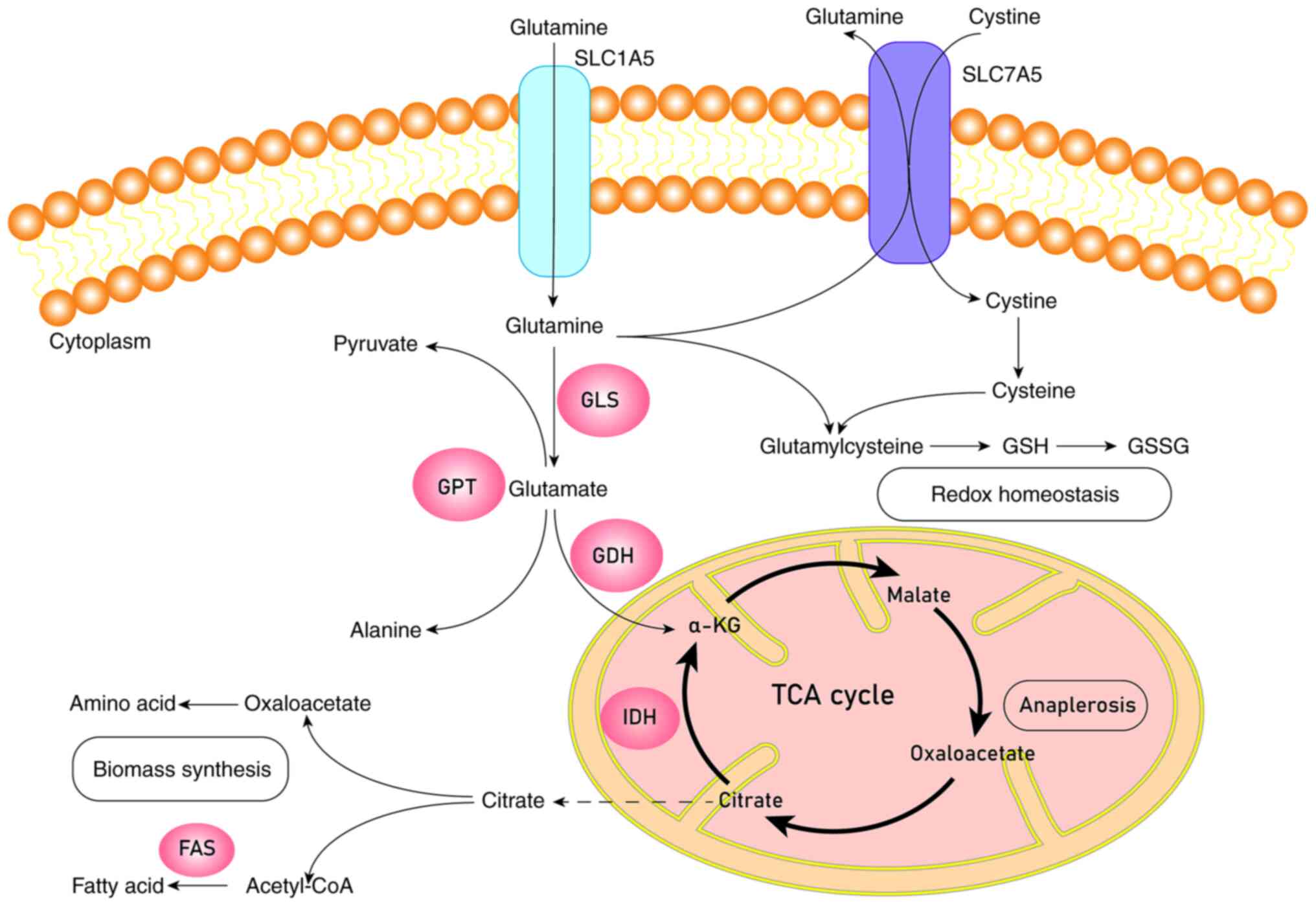 | Figure 1.Schematic diagram of glutamine
metabolism. Glutamine is transported into the cell by SLC1A5 or
SLC7A5 and turns into glutamate catalyzed by transaminases or GLS.
Glutamate will be catalyzed by GDH to generate α-KG. α-KG can enter
into the TCA cycle to complete the process of anaplerosis or be
catalyzed by IDH to produce citrate, which leaves mitochondria for
the biomass synthesis of fatty acids and amino acids. Glutamine and
cystine will synthesize glutathione through a series of catalytic
processes to keep redox homeostasis. GLS, glutaminase; GPT,
glutamic pyruvic transaminase; GDH, glutamate dehydrogenase; IDH,
isocitrate dehydrogenase; FAS, fatty acid synthase; GSH,
glutathione (reduced); GSSH, glutathione (oxidized); α-KG,
α-ketoglutaric acid; TCA cycle, tricarboxylic acid cycle. |
Glutamine metabolism has strong heterogeneity,
reflected in its close relationship with the origin of the tumor
tissue, oncogenes, and the tumor microenvironment (TME) (43). Studies have shown that the oncogene
c-Myc inhibits miR-23a/b and, as a result, the expression of GLS is
increased to promote the glutamine catabolism and to meet the needs
of cancer cell growth and proliferation (44,45).
In KRAS-driven NSCLC, glutamine has less contribution to TCA cycle
than does glucose, meaning that the carbon in TCA cycle mainly
comes from glucose rather than glutamine (26). Interestingly, it is shown that the
mRNA levels of GLS and solute carrier family 1 member 5 (SLC1A5)
increased in KRAS-driven NSCLC, suggesting that these cells are
more dependent on glutamine rather than glucose metabolism
(14). The seemingly contradictory
results may be due to different TMEs and tumor histological
subtypes, which further explains the complexity and heterogeneity
of glutamine metabolism. The complex relationship between glutamine
metabolism, tumor tissue and gene mutation are also reflected in
whether the cancer cells catabolize glutamine to provide energy and
nutrient substrates for cell growth and proliferation or if this
leads to intracellular glutamine accumulation through de
novo glutamine synthesis (31).
For example, in MYC-induced liver cancer, glucose-derived synthesis
of glutamine is suppressed by reducing the level of glutamine
synthetase (GLUL), whereas upregulation of GLS leads to enhanced
catabolism of glutamine. Conversely, in MET-driven liver cancer,
glutamine synthesis is increased. In MYC-induced lung cancer, the
mRNA expression levels of GLUL and GLS are upregulated
simultaneously. However, the effect caused by GLUL overexpression
overrides the effects of GLS upregulation, which leads to
accumulation of glutamine in lung cancer cells (31). There are a number of key processes
in glutamine metabolism. For example, glutamine enters cells
through the SLC1A5 amino acid transporter and is converted into
glutamate, catalyzed by the GLS enzyme. Glutamate is then converted
into α-KG, which enters the process of TCA cycle anaplerosis either
by transaminase or GDH and generates non-essential amino acids,
such as alanine and aspartic acid (46). The enzymes and proteins involved in
these key steps are potential oncotherapy targets.
Significance of glutamine metabolism in
NSCLC
TCA cycle anaplerosis
The main purpose of glucose metabolism is to
generate lactate, which can store a vast amount of carbon,
resulting in a reduction of carbon sources entering the TCA cycle
and a continuous flow of intermediate TCA cycle products into the
cytoplasm for the synthesis of biological macromolecules, such as
lipids, nucleotides and proteins; as such, glucose metabolism leads
to TCA cycle cataplerosis (16,36).
Glutamine provides a source of carbon to maintain the TCA cycle,
which is used to produce sufficient ATP and metabolic derivatives
(12). TCA cycle anaplerosis is the
key to metabolism; through this process, cells can obtain
substrates for the synthesis of various biological macromolecules,
such as oxaloacetate and citrate (16,47).
Biomass synthesis
Cell proliferation requires the synthesis of a large
amount of proteins, nucleic acids and lipids (11). Glutamine generates other
non-essential amino acids through catalysis by transaminases. It is
reported that ~50% of the non-essential amino acids in cells come
from glutamine metabolism (48).
Glutamine-derived α-KG is catalyzed by isocitrate dehydrogenases
(IDHs) with the consumption of NADPH to produce citrate, which
flows into the cytoplasm for lipid or nucleotide synthesis
(49). Glutamine metabolism can
produce a large amount of NADPH, providing reducing substances for
the synthesis of lipids and nucleotides (50). Glutamine also provides a nitrogen
source for nucleotide synthesis (34). The lipids in cells are mainly
derived from glucose, followed by glutamine (36).
Activation of the mTOR signaling
pathway
The mTOR signaling pathway promotes the anabolism of
biological macromolecules, such as lipids, proteins and
nucleotides, by integrating intracellular and extracellular
signals, and inhibits autophagy to sustain cell survival (51). It has been shown that the activation
of mTOR requires bidirectional transport of glutamine (52). Glutamine activates mTORC1
independently of rag GTPase and recruits it into the lysosome
(53). Glutamine is regarded as a
sensitive regulatory signal of mTORC1 complex, promoting the growth
of cancer cells (13).
Metabolic process of glutamine in
NSCLC
Glutamine metabolism in NSCLC involves related
enzymes and transporters. One of the hallmarks of cancer is the
reprogramming of energy metabolism for the regulation of
metabolism-related enzymes and transporters at transcriptional or
post-transcriptional level (54).
For example, SLC15A is the primary transporter of glutamine
entering into the cell. Statistical analysis revealed an
association between SCL15A overexpression with poor prognosis in
patients with NSCLC, making SLC15A a potentially important
prognostic marker (55). SLC7A11,
an antiporter of glutamate and other non-essential amino acids, is
overexpressed in NSCLC; it may enhance the antioxidant stress
ability of cancer cells and increase the consumption of glutamine,
which is also known as glutamine addiction and is one of the
characteristics of NSCLC metabolism (56). In addition, extracellular proteins
are degraded through micropinocytosis to obtain glutamine for cell
growth (48).
In BRAFV600E-driven lung adenocarcinoma,
autophagy sustains mitochondrial glutamine metabolism, consistent
with some ‘autophagy-addicted’ NSCLC cells (24). There is abnormal expression of a
certain type of enzymes directly or indirectly related to glutamine
metabolism in NSCLC cells. For example, GLS is a major enzyme
involved in glutamine metabolism, in which it catalyzes the
conversion of glutamine to glutamate. A number of oncogenes
modulate glutamine metabolism by regulating the expression of GLS.
For example, c-Myc increases the expression of GLS by inhibiting
miR-23a/b to promote intracellular glutamine metabolism (30). Lactate can also stabilize HIF-2α to
activate c-Myc in oxygenated/oxidative cancer cells, which
indirectly enhances the expression of GLS (57). GLS1 is a rate-limiting enzyme of
glutaminolysis in some glutamine-dependent tumor cells. GLS1 has
two splice variants, of which KAC and GAC are the dominant isoforms
in NSCLC (58). The ratio of GAC to
KAC increases significantly in tumor cells, which has been shown to
be due to the decrease of KAC expression. However, whether GAC
expression is increased remains to be determined. Knockdown of GAC
has greater effects on tumor growth inhibition compared to
knockdown of KAC, thus, the targeted therapy of GAC may have
broader prospects (58).
Glutamate is converted into α-KG in two different
pathways catalyzed by either transaminase accompanied by the
production of other non-essential amino acids, or by GDH1
accompanied by generation of the reducing substance NADPH (12). GDH1 is the rate limiting enzyme for
oxidative degradation of glutamate. The decrease of GDH1 activity
limits the flow of carbon into TCA cycle (59). Glutamic-pyruvic transaminase (GPT)
is a type of transaminase that converts glutamate into alanine
through transamination. It was reported that the expression and
distribution of GPT was significantly increased in NSCLC cells that
were resistant to glutamine metabolic inhibitors (60). Other study showed that alanine
metabolism can compensate for the intermediates of the TCA cycle
and the metabolic derivatives during glutamine deprivation to
maintain cell growth and survival (61). Pyruvate carboxylase catalyzes the
conversion of pyruvate into oxaloacetate; it is highly expressed in
early NSCLC cells and it selectively activates GLS. Previous study
also have shown that knockdown of pyruvate carboxylase leads to
inhibition of cell growth and the activity of the TCA cycle owing
to the reduction of intermediates, lipid and nucleotide synthesis,
and the imbalance of GSH which may lead to ROS excess (62). In NSCLC, the abundant expression of
NADPH oxidase 4 induces GLS (glutamine catabolism) and GSH
synthesis at the transcriptional and the post-transcriptional
level; subsequently, GSH synthesis is increased leading to
resistance of tumor cells to oxidative stress (63).
Metabolic heterogeneity of glutamine in
NSCLC
Tumor metabolic heterogeneity
Although the Warburg effect describes the basic
pattern of glucose metabolism in tumor cells, tumor heterogeneity
indicates that metabolism is not a specific metabolic map (54). The association between tumor cell
metabolism and the TME is one of the new characteristics of tumors
(10). Specific tumor metabolism
not only supports the growth and energy of cancer cells, but also
serves a key role in the production of an immunosuppressive TME.
Therefore, tumor metabolism is a way for cancer cells to escape
antitumor immune responses. Glutamine metabolism not only promotes
cell growth, but also creates a TME that benefits from tumor immune
escape (64). Inhibition of
glutamine metabolism can prevent tumor immune escape. Results of
glutamine antagonist experiments revealed the previously unknown
difference in metabolic plasticity between cancer cells and
effector T cells, which can be used as a ‘metabolic checkpoint’ for
tumor immunotherapy (65). Tumor
stromal cells have flexible and adaptive metabolism involving the
synthesis of glutamine using atypical carbon and nitrogen sources
in a nutrient-deficient TME, supporting glutamine-dependent cell
growth (66).
NSCLC metabolic heterogeneity
Glutamine metabolism in NSCLC is heterogeneous;
there is higher glutamine consumption and metabolism in AC compared
to SCC (14). EGFR and KRAS
mutations are more common in AC cells with increased expression of
GLS and SLC1A5 and decreased expression of GLUT (14). On the contrary, TP53 mutations are
more common in SCC cells, suggesting that AC NSCLC cell lines
mainly metabolize glutamine, whereas SCC cell lines mainly
metabolize glucose (14). In
cultured cells, glucose metabolism mainly produces lactate and
glutamine mainly maintains the TCA cycle. However, in KRAS-driven
NSCLC in vivo, production of lactate and progress of the TCA
cycle is facilitated by glucose metabolism, whereas glutamine makes
little contribution to the TCA cycle. The difference between in
vivo and in vitro glucose metabolism implies the effect
of the TME on NSCLC metabolism, even exceeding the gene phenotype
(26). One of the internal factors
that affect the heterogeneity of tumor metabolism includes
provision of oxygen and substrates through different perfusion
levels, exceeding the tumor genotype. In highly perfused NSCLC
cells, lactate can be used as a substrate of respiratory chain,
whereas in lowly perfused areas, lactate is produced and secreted
through glycolysis (67).
Therapeutic targeting of glutamine
metabolism in NSCLC
In addition to the classical metabolic substrate,
glucose, glutamine is another important nutrient, making cancer
cells glutamine-dependent by synthesizing biological
macromolecules, supplementing TCA cycle substrates and maintaining
redox balance. Simultaneously, it is also one of the hallmarks of
NSCLC. Therefore, targeting the transport and metabolism of
glutamine will become a promising therapeutic strategy for the
treatment of NSCLC (34).
Glutamine targeted traditional
therapy
The first step of glutamine influx or efflux is
through the various glutamine transporters (68). SLC1A5 is the main amino acid
transporter of glutamine influx. Overexpression of SLC1A5 was
associated with poor prognosis of patients with NSCLC (69). Blocking its function can achieve an
antitumor effect by suppressing the consumption of glutamine,
inducing cell apoptosis through inherent pathways and preventing
cell growth through oxidative stress, serving as an effective
therapeutic target (55).
L-γ-glutamyl-p-nitroanilide (GPNA), an inhibitor of SLC1A5, has
shown potential antitumoral effects through the inhibition of
glutamine influx and by acting as a competitive inhibitor of SLC7A5
[also known as human L-type amino acid transporter 1 (LAT1)],
inhibiting the influx of essential amino acids (70). This inhibition may lead to changes
in intracellular amino acid composition and mTORC1 activity
(69,70). SLC7A5 is responsible for
transporting glutamine out of the cells and transporting essential
amino acids into the cells. A previous study has shown that, as a
bidirectional transporter of glutamine, SLC1A5 cooperates with
SLC7A5 and serves an important role in amino acid transportation,
mTORC1 activation and regulation of cell growth (52). An inhibitor of SLC7A5,
2-aminobicyclo-(2,2,1)-heptane-2-carboxylic acid (BCH), induces
cancer cell apoptosis by preventing the activation of mTORC1 which
is glutamine-dependent, therefore achieving its antitumor effect
(71). SLC7A11 is a transporter of
cysteine/glutamine, which is overexpressed in NSCLC. It synthesizes
GSH by transporting cysteine to resist oxidative stress, and its
overexpression induces metabolic dependency on glucose and
glutamine synchronously, which is also a process of glutamine
addiction in NSCLC (56,72). An SLC7A11 inhibitor, sulfasalazine,
has shown its potential effect through the inhibition of GSH
synthesis and the disturbance of the redox balance in KRAS-mutant
lung AC (73).
Key enzymes of the glutamine metabolism process are
promising therapeutic targets that have been widely studied, and
some mature drugs have been used in clinical treatment (48). GLS is one of the most crucial
enzymes that convert glutamine into glutamate to provide substrates
for the TCA cycle. GLS and its isoforms have shown that it serves
an irreplaceable role in glutamine metabolism, making it a
potential therapeutic target (74).
6-Diazo-5-oxo-L-norleucine (DON), is a first-generation drug of
glutamine analogues that can widely inhibit the enzymes used by
glutamine, including GLS, transaminase and GLUL, and has
therapeutic effects on patients with cancer. However, its clinical
application is limited owing to oral toxicity, gastrointestinal
toxicity, leukopenia and thrombocytopenia (75). More importantly, since glutamine is
an important neurotransmitter, DON has a series of inevitable
CNS-associated side effects (76).
DON-derived drugs not only maintain their efficient therapeutic
effect, but reduce toxicity and side effects, which are being
investigated further (77). For
example, the compound JHU083 not only inhibits tumor growth, but
also enhances the effect of immunotherapy by improving the function
and efficiency of immune cell responses, which is mainly exerted by
CD8+ T cells (65).
Bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide
(BPTES) is another GLS inhibitor that selectively inhibits GLS but
does not inhibit KGA; however, the structure of BPTES, being
different from that of either glutamine or glutamate, will not
cause toxicity owing to interaction with other transporters or
receptors (78,79). CB-839 (telaglenastat) is a potential
and selective GLS inhibitor that shows a positive result in the
treatment of triple-negative breast cancer and other
glutamine-dependent cancers, such as NSCLC (80).
Combined therapeutic strategy
Surgery, chemotherapy, and radiotherapy are still
the first-line treatment approaches for NSCLC; emerging
immunotherapy and molecular-targeted therapy approaches also have
hopeful prospects (5). However, all
types of therapeutic strategies have their limitations and
selectivity. Therefore, the combination of targeting glutamine
metabolism with traditional chemotherapeutic drugs, such as
cisplatin, paclitaxel, cyclophosphamide, gemcitabine and pemetrexed
or the EGFR TKIs, including erlotinib and gefitinib, may be
beneficial (19,80,81).
Radiotherapy has great potential for the treatment of NSCLC, but
not all patients can benefit from it. Recently, a study has shown
that the GLS inhibitor CB-839 can improve the sensitivity of
patients with NSCLC to radiotherapy by reducing the secretion of
the intracellular antioxidant molecule GSH (82). Moreover, CB-839 can also be combined
with some molecular-targeted therapeutic drugs to improve their
antitumor effect. For example, the combination of CB-839 and
selumetinib was shown to improve antitumoral effects in KRAS-driven
NSCLC (83). The outcome of
combined treatment is reflected by the redox stress caused by the
decrease of mitochondrial membrane potential and the increase of
the ROS levels, as well as the energy stress induced by the
inhibition of glycolysis and glutamine metabolism. Meanwhile,
combined treatment can inhibit the phosphorylation process of AKT,
inducing autophagy, and eventually leading to cancer cell death
(83). The combination of CB-839
and the EGFR inhibitor erlotinib, can lead to energy stress and
metabolic risk through autophagy and activation of AMPK. In
addition, the lower expression of MYC and HIF-1α, and their
downstream targets GLUT1 and SLC1A5, indicate that the combination
of these two drugs may block the metabolic process of two basic
nutrients, glucose and glutamine, inducing cell death (84). MLN0128 (sapanisertib) is an
antitumor drug that inhibits mTOR and glycolysis. However,
GSK-3α/β, a central regulator in lung SCC cells, can promote the
expression of GLS and glutamine metabolism by upregulating c-MYC
and c-JUN to make cancer cells adapt to chronic mTOR and glycolysis
inhibition (85). Therefore, CB-839
is used to block glutamine metabolism, avoid drug resistance and
achieve unique antitumor effect due to simultaneous inhibition of
glycolysis and glutamine metabolism (85).
It is well known that ATP production depends on
cytosol NADP; NSCLC cells obtain ATP from cytosol NADH through the
malate-aspartate shuttle system using glutamate, which is the
catalysate of GLS1. GLS1 inhibitors, such as BPTES, reduce ATP
production through glutamate deficiency, and finally inhibit cancer
cell growth (86). Combined with
the thymidylate synthase inhibitor 5-FU, GLS1 inhibitors have
synthetic lethal effects (87).
EGFR TKIs are targeted therapeutic drugs for patients with NSCLC
with EGFR mutations; however, secondary drug resistance is the main
reason for the limited antitumor effect (88). The expression of SLC1A5 increased
after treatment with the EGFR TKI almonertinib (89), which may be related to secondary
resistance since glutamine can activate the downstream signal of
EGFR, such as mTOR. SLC1A5 inhibitors, such as GPNA, can enhance
the antitumor effect of almonertinib independently of the presence
of EGFR mutations, and the combination of these two drugs may be
the reason for inducing apoptosis by inhibiting autophagy (89). The GAC inhibitor 968 can reverse the
secondary resistance of erlotinib by preventing glutamine
metabolism; their combination not only restricts glutamine and
glycolytic metabolism, but also maximizes the antitumor effect and
safety, which provides a direction for the treatment of patients
with NSCLC resistant to EGFR TKIs (81).
In addition to targeting intracellular metabolic
processes, the metabolism of cells in the TME has attracted the
interest of researchers because the relationship between cell
metabolism and TME is regarded as one of the emerging hallmarks of
cancer (10). Cancer-associated
fibroblasts (CAFs) are the most common cells in the TME; they
exhibit metabolic flexibility and adaptability reflected by the
synthesis of glutamine using atypical carbon and nitrogen sources
in a nutrient-deprived TME to maintain the growth of glutamine
dependent cells (90). The
combination therapy between inhibiting glutamine synthesis in CAFs
cells by targeting GLUL and blocking glutamine metabolism in tumor
cells is a hopeful prospect (66).
Glutamine metabolism supports the rapid growth of cancer cells and
creates a TME conducive to tumor immune escape (66). The different phenotype induced by
glutamine antagonists between cancer cells and T cells provide an
opportunity for emerging immunotherapies (91). The function of glutamine antagonists
in cancer cells is to induce intracellular hypoxia, acidosis and
nutrient deprivation in cancer cells, whereas in effector T cells,
glutamine antagonists promote intracellular antioxidant metabolism
and induce transformation of T cells into long-lived, high-activity
and high-memory subtypes; this is shown by the abnormal
overexpression of activation markers, memory markers,
anti-apoptotic proteins and transcription factors (65). Therefore, glutamine antagonists,
such as DON and JHU038, may not only improve immunotherapy but also
enhance inherent antitumor responses. Based on the data
aforementioned, combined therapy is a promising strategy (92).
Targeting the sweet spot
Apart from the traditional targeted drugs that aim
at inhibiting glutamine transporters and GLS, an increasing number
of promising targets directly or indirectly involved in glutamine
metabolic pathway have been studied, with the aims of achieving an
antitumor effect by blocking the glutamine-dependent metabolism in
tumor cells (61,93). Although using GLS as a target for
immunotherapy is promising, there are still some limitations. GLS
depends on glutamine metabolism and is a rate-limiting enzyme of
some cancer cells, including hepatocellular carcinoma cells,
colorectal cancer cells and NSCLC (86). The role of GLS has been widely
studied and, until recently, selective inhibitors targeting GLS2
have been shown to induce autophagy and apoptosis by inhibiting the
mTORC1 signaling pathway, making GLS2 a potential therapeutic
target (92). Overexpression of
GLS2 may contribute to drug resistance following treatment of
breast cancer cells with the GLS inhibitor CB-839 (94). GPT2 is a key enzyme that catalyzes
the reversible transformation of alanine and pyruvate. It was
reported that the expression and distribution of GPT2 was
significantly increased in patients with NSCLC who were resistant
to glutamine metabolic inhibitors (61). A study also showed that, under
glutamine-deficient conditions, alanine metabolism is involved in
the production of TCA cycle intermediates and metabolic derivatives
to maintain cell growth and survival. The GPT2 inhibitor
L-cycloserine can effectively improve the sensitivity of NSCLC to
GLS inhibitors (61).
Glutamine-dependency in tumor cells is partly
determined by the process of TCA cycle anaplerosis. In
MYC-transformed cells, the carbon provided by glutamine enters the
TCA cycle mainly through transamination, which exposes the
vulnerable points during metabolism and provides an opportunity for
targeting the inhibition of transaminase (95). The therapeutic effect of the
transaminase inhibitor aminooxyacetate further enhances the
confidence of researchers, and the transaminase inhibitor may have
promising development prospects (96). TCA cycle replenishment is a vital
process in intracellular metabolism because it allows cells to
obtain substrates from the TCA cycle for the synthesis of
macromolecules. TCA replenishment involves the use of glutamine or
oxaloacetate, which is the product of pyruvate under the catalysis
of pyruvate carboxylase (PC) (16);
thus, targeting the suppression of PC activity may also have a
similar effect. Interestingly, researchers have shown that PC is
highly expressed in early NSCLC cells, and knockdown of PC leads to
reduced cell growth and proliferation and lower TCA cycle activity
due to the reduction of compensatory intermediates, resulting in
the reduction of lipid and nucleotide synthesis and the imbalance
of GSH, which may lead to the excessive production of ROS (62). The function of glutamine in
maintaining intracellular redox balance is mainly through GSH
synthesis to combat ROS produced in cells. GDH1 is an essential
enzyme that assists glutamine in maintaining intracellular redox
balance by catalyzing the reversible oxidative deamination process
of L-glutamine to α-KG. GDH1 regulates redox balance by controlling
intracellular levels of α-KG and glutaminolysis (42). Targeting GDH1 disrupts intracellular
redox homeostasis and suppresses the growth and proliferation of
tumor cells. In NSCLC, the abundant expression of NOX4 induces the
increase of GLS and GSH synthetase (GSS) both at mRNA and protein
levels; NOX4 is the key enzyme catalyzing GSH synthesis, resulting
in increased GSH synthesis, which is the basis for cancer cells to
resist oxidative stress and gain oxidative resistance (63). Therefore, it is a promising therapy
acting by accelerating the efflux of glutamine and selectively
depriving GSH (97), and NOX4 may
also be a novel therapeutic target.
Patient stratification and predictive
markers
Tumor metabolism is not a specific metabolic map,
which means that targeted metabolic therapy is not fixed (54). Therefore, the treatment of patients
with NSCLC should be selective and specific to maximize the
treatment effect, making patient stratification particularly
important (98). Stratification
mainly includes tumor histological subtypes and specific driver
gene mutations. Owing to the metabolic heterogeneity of glutamine,
we hypothesize that glutamine inhibitors may be more effective for
the treatment of patients with either EGFR or KRAS mutations.
Among the Asian population, the proportion of EGFR
mutations in lung AC ranges between 45 and 75% (99). For patients with EGFR mutations,
EGFR-TKIs are the first-line treatment of choice. However,
secondary drug resistance is an obstacle for achieving a
satisfactory therapeutic effect, mainly due to the presence of EGFR
T790M mutation, which represents a marker of secondary drug
resistance and the amplification of MET gene expression (81). The combination of traditional
EGFR-TKIs and glutamine inhibitors may provide a good solution to
this problem. The issue of drug resistance could be prevented by
combining gefitinib, a therapeutic drug for patients with NSCLC
with EGFR mutations, and BCH, a SLC7A5 inhibitor that reduces the
phosphorylation level of mTOR and its downstream molecules
(93). Moreover, SLC7A5 inhibitors
could also be used for the treatment of patients with NSCLC without
EGFR mutations (93). The
transcription factor p53 is a tumor suppressor, and mutations are
often identified in lung SCC (14).
GLS2 is a unique target gene of p53; it regulates glutamine
metabolism, energy production and intracellular ROS levels
(100). In NSCLC, p53 inhibits
tumor growth by promoting pyroptosis (101). Moreover, p53 mutations are related
to resistance of NSCLCs to EGFR TKIs (102). Hence, the status of p53 can be
considered as a target molecule to improve the therapeutic effect
of EGFR TKIs against NSCLC (102).
KRAS-driven cancer accounts for ~35% of lung AC
cases (103). In NSCLC cells,
simultaneous presence of mutations in KRAS, LKB1 and KEAP1 is
possible and these are sensitive to GLS inhibitors due to glutamine
dependency, making them promising targets for therapy in the future
(29). Mutations in other genes,
such as IDH, are rare in NSCLC, but these are often associated with
smoking history and KRAS mutations (104). The prognosis of NSCLC is generally
poor, but numerous studies have revealed that the expression levels
of some molecules can be used as markers to predict the prognosis
and therapeutic effect. For example, SLC1A5 is an independent
marker of poor prognosis, particularly in patients with NSCLC AC
who are undergoing surgery (105).
The expression of SLC1A5 is significantly higher in non-AC cases
and depends on a number of factors, including sex (particularly
males) and presence of advanced tumors (105). Another transporter of glutamine is
SLC7A11; its high expression is associated with KEAP1 mutations and
abnormal expression of BAP1. These biological markers help identify
tumors with high expression of SLC7A11, which may subsequently
benefit patients with the combination of radiotherapy or
chemotherapy and SLC7A11 inhibition (72). In KRAS-driven NSCLC, the expression
of glutamine-dependent genes, especially glutamic-oxaloacetate
transaminase 1 (GOT1) and malic enzyme 1 (ME1), is increased to
maintain redox balance. A study has shown that ME1 and GOT1 are
predictive markers of sensitivity to radiotherapy (106). Therefore, KRAS-driven cancer cells
are sensitive to radiotherapy during glutamine deprivation
(106). In addition to the markers
mentioned above, the level of GPT2 can be used to identify those
patients with NSCLC who will benefit the most from the treatment
with GLS inhibitors (61). The
purpose of patient stratification and biological predictive markers
is to achieve individualized treatment as well as to obtain
therapeutic effects and prognosis.
Conclusions
As the most abundant non-essential amino acid in the
circulation, glutamine serves an indispensable role in the
metabolism of some tumors. It has an important role in supporting
cell growth and proliferation, activating signal transduction
pathways, and maintaining redox balance. Since the publication of
the Warburg effect, which proposed concept of aerobic glycolysis
and glutamine addiction, there has been an increase in the number
of studies on tumor metabolism. NSCLC cells are dependent on
glutamine through the metabolic reprogramming caused by the
mutation of oncogenes or tumor suppressor genes, which makes them
unable to grow normally under glutamine deprivation. The metabolic
reprogramming of tumor cells can promote cell growth and
proliferation, as well as lead to the malignant progress of tumor
and the emergence of drug resistance. Recently, targeting the key
enzymes and transporters of glutamine metabolism with specific
inhibitors showed pharmacological progress and has led to clinical
trials, which revealed surprising results regarding treatment
(summarized in Table I). However,
the metabolic vulnerability of non-cancer cells and the metabolic
plasticity of cancer cells in the TME should be taken into account.
The metabolic reprogramming and flexibility of cancer cells may
limit drug efficacy, and the impact on immune cells may limit the
attack on cancer cells. Therefore, a better understanding of cancer
cells and immune cell metabolism is required. Considering the
glutamine dependence of NSCLC cells, CB-839, BPTES and DON are also
increasingly used in treatment, but with mixed results. These drugs
may be beneficial to some patients due to the important role of
glutamine in cell growth. However, treatment is limited by the
heterogeneity of NSCLC metabolism and not all patients may benefit.
Therefore, we propose a therapy scheme including patient
stratification and screening of special biological predictive
markers. Patient stratification includes the classification of
tissue and gene mutation subtypes, aiming to achieve individualized
medical treatment and maximize the benefits for each patient.
Biological predictive markers can be used to identify the most
suitable treatment approach and make predictions regarding
prognosis. Combined therapy strategies are a promising way to
improve the effectiveness of treatment and may potentially be the
leading treatment approach in the future. The combination of
glutamine inhibitors with emerging immunotherapy or
targeted-therapy drugs may not only improve their antitumor effect,
but also may reduce drug resistance and increase sensitivity. In
addition, new targets which are directly or indirectly associated
with glutamine metabolism require further exploration.
 | Table I.Summary of preclinical tools and
clinical therapeutic drugs targeting different process of glutamine
metabolism. |
Table I.
Summary of preclinical tools and
clinical therapeutic drugs targeting different process of glutamine
metabolism.
| First author/s,
year | Class | Drug | Mechanism | Stage | Target | (Refs.) |
|---|
| Magill et
al, 1957 | Glutamine
analogue | DON | Widely inhibit the
glutamine metabolic enzyme | Limited due to
toxicity | Enzyme used by
glutamine | (75) |
| Leone et al,
2019 |
| JHU083 | Pro-drug of
DON | Preclinical
compound tool |
| (65) |
| Robinson et
al, 2007 | GLS inhibitors | BPTES | Inhibition of
GLS | Preclinical
tool | GLS | (78) |
| Gross et al,
2014 |
| CB-839 |
| Phase I and II |
| (80) |
| Caiola et
al, 2020 | Transaminase
inhibitors | L-cycloserine | Inhibition of TCA
cycle | Preclinical
tool | GPT2 | (61) |
| Moreadith and
Lehninger, 1984 |
| AOA | anaplerosis | Preclinical
tool | Aminobutyrate
aminotransferase | (96) |
| Hassanein et
al, 2013 | Glutamine
transporters inhibitors | GPNA | Inhibition of
glutamine transport | Preclinical
tool | SLC1A5 (also known
as ASCT2) | (69) |
| Wise and Thompson,
2010 |
| BCH | Inhibition of
essential amino acids | Preclinical
tool | SLC7A5 (also known
as LAT1) | (71) |
| Hu et al,
2020 |
| Sulfasalazine | Inhibition of
cysteine-glutamine transport | Phase II for breast
cancer | SLC7A11 (also known
as xCT) | (73) |
This review described the identification of
glutamine as a promising target for treatment by emphasizing its
role in NSCLC cell metabolism. In addition to the traditional drugs
targeting glutamine metabolic enzymes and transporters, new sweet
spots, along with the concept of combination and individualized
therapy, are also described. Inhibition of glutamine as a targeted
therapy for NSCLC is promising, but still faces new challenges.
Cancer and normal cells share many pathways, making selective
inhibition particularly important. Future studies should not only
consider glutamine metabolism in normal cells, but also target
glutamine addiction in NSCLC cells, which will maximize the
therapeutic effect and reduce side effects. Moreover, the
heterogeneity of NSCLC metabolism and the difference in individual
gene expression lead to the absence of a metabolic map suitable for
everyone. 18F-flurodeoxyglucose provides useful information by
tracking glucose metabolism in vivo based on the increase of
glucose uptake and glycolysis by cancer cells (107). L-[5-11C]-glutamine may
be used as an indicator for in vivoglutamine-dependent tumor
metabolism (108). Therefore, the
progress of glutamine-targeted therapy also depends on imaging
technology.
Although the heterogeneity of cancer metabolism
limits the application of metabolism-targeted drugs to some extent,
glutamine-targeted therapy still has potential. Glutamine
supplement treatment may be beneficial to prevent mucositis in
patients receiving radiotherapy and chemotherapy. For example,
glutamine can be used to prevent chemotherapeutic or radioactive
esophagitis in patients with esophageal cancer (109). At present, metabolic tracking
technologies, such as spectroscopy and PET/CT, have rapidly been
developed. With the development of metabonomic research, the
heterogeneity of cancer cell metabolism will be further explored.
At the same time, the application of genetic testing and biomarker
testing will help us choose the patients who would benefit the most
from treatment.
Acknowledgements
Not applicable.
Funding
This work was supported by the grants from The National Natural
Science Foundation of China (grant no. 32170793, 82160133, 31960147
and 31760329), Jiangxi Provincial Natural Science Foundation (grant
no. 20212BAB206086, 20212ACB216005, 20224ACB216013 and
20224BAB206007).
Availability of data and materials
Not applicable.
Authors' contributions
LZ, QZha and QZhu wrote the manuscript; YZ and YL
revised the manuscript and XH revised and confirmed the final
version of the manuscript. All authors read and approved the final
version of the manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global Cancer Statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schabath MB and Cote ML: Cancer progress
and priorities: Lung cancer. Cancer Epidemiol Biomarkers Prev.
28:1563–1579. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ganti AK, Klein AB, Cotarla I, Seal B and
Chou E: Update of incidence, prevalence, survival, and initial
treatment in patients with non-small cell lung cancer in the US.
JAMA Oncol. 7:1824–1832. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou Y, Du Q, Zhao Q, Zhang M, Qin X,
Jiang Y and Luan Y: A heme-regulatable chemodynamic nanodrug
harnessing transcription factor Bach1 against lung cancer
metastasis. J Colloid Interface Sci. 610:698–708. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Duma N, Santana-Davila R and Molina JR:
Non-Small cell lung cancer: Epidemiology, screening, diagnosis, and
treatment. Mayo Clin Proc. 94:1623–1640. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Goldstraw P, Chansky K, Crowley J,
Rami-Porta R, Asamura H, Eberhardt WE, Nicholson AG, Groome P,
Mitchell A, Bolejack V, et al: The IASLC Lung Cancer Staging
Project: Proposals for Revision of the TNM Stage Groupings in the
Forthcoming (Eighth) Edition of the TNM Classification for Lung
Cancer. J Thorac Oncol. 11:39–51. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Otto AM: Warburg effect(s)-a biographical
sketch of Otto Warburg and his impacts on tumor metabolism. Cancer
Metab. 4:52016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Boroughs LK and DeBerardinis RJ: Metabolic
pathways promoting cancer cell survival and growth. Nat Cell Biol.
17:351–359. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pavlova NN and Thompson CB: The emerging
hallmarks of cancer metabolism. Cell Metab. 23:27–47. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hensley CT, Wasti AT and DeBerardinis RJ:
Glutamine and cancer: Cell biology, physiology, and clinical
opportunities. J Clin Invest. 123:3678–3684. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mohamed A, Deng X, Khuri FR and Owonikoko
TK: Altered glutamine metabolism and therapeutic opportunities for
lung cancer. Clin Lung Cancer. 15:7–15. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Meijer TWH, Looijen-Salamon MG, Lok J, van
den Heuvel M, Tops B, Kaanders JHAM, Span PN and Bussink J: Glucose
and glutamine metabolism in relation to mutational status in NSCLC
histological subtypes. Thorac Cancer. 10:2289–2299. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kroemer G and Pouyssegur J: Tumor cell
metabolism: Cancer's Achilles' heel. Cancer Cell. 13:472–482. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
DeBerardinis RJ, Lum JJ, Hatzivassiliou G
and Thompson CB: The biology of cancer: Metabolic reprogramming
fuels cell growth and proliferation. Cell Metab. 7:11–20. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
DeBerardinis RJ and Chandel NS:
Fundamentals of cancer metabolism. Sci Adv. 2:e16002002016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yoshida GJ: Metabolic reprogramming: The
emerging concept and associated therapeutic strategies. J Exp Clin
Cancer Res. 34:1112015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cruz-Bermudez A, Laza-Briviesca R,
Vicente-Blanco RJ, García-Grande A, Coronado MJ, Laine-Menéndez S,
Palacios-Zambrano S, Moreno-Villa MR, Ruiz-Valdepeñas AM, Lendinez
C, et al: Cisplatin resistance involves a metabolic reprogramming
through ROS and PGC-1α in NSCLC which can be overcome by OXPHOS
inhibition. Free Radic Biol Med. 135:167–181. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Solanki HS, Babu N, Jain AP, Bhat MY,
Puttamallesh VN, Advani J, Raja R, Mangalaparthi KK, Kumar MM,
Prasad TSK, et al: Cigarette smoke induces mitochondrial metabolic
reprogramming in lung cells. Mitochondrion. 40:58–70. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pavlova NN, Zhu J and Thompson CB: The
hallmarks of cancer metabolism: Still emerging. Cell Metab.
34:355–377. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Judd J, Abdel Karim N, Khan H, Naqash AR,
Baca Y, Xiu J, VanderWalde AM, Mamdani H, Raez LE, Nagasaka M, et
al: Characterization of KRAS mutation subtypes in non-small cell
lung cancer. Mol Cancer Ther. 20:2577–2584. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kawada K, Toda K and Sakai Y: Targeting
metabolic reprogramming in KRAS-driven cancers. Int J Clin Oncol.
22:651–659. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Strohecker AM, Guo JY, Karsli-Uzunbas G,
Price SM, Chen GJ, Mathew R, McMahon M and White E: Autophagy
sustains mitochondrial glutamine metabolism and growth of
BrafV600E-driven lung tumors. Cancer Discov. 3:1272–1285. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Commisso C, Davidson SM, Soydaner-Azeloglu
RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin
JA, Thompson CB, et al: Macropinocytosis of protein is an amino
acid supply route in Ras-transformed cells. Nature. 497:633–637.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Davidson SM, Papagiannakopoulos T,
Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK,
O'Brien JP, Pierce KA, et al: Environment impacts the metabolic
dependencies of ras-driven non-small cell lung cancer. Cell Metab.
23:517–528. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dowling CM, Zhang H, Chonghaile TN and
Wong KK: Shining a light on metabolic vulnerabilities in non-small
cell lung cancer. Biochim Biophys Acta Rev Cancer. 1875:1884622021.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Faubert B, Vincent EE, Griss T, Samborska
B, Izreig S, Svensson RU, Mamer OA, Avizonis D, Shackelford DB,
Shaw RJ and Jones RG: Loss of the tumor suppressor LKB1 promotes
metabolic reprogramming of cancer cells via HIF-1α. Proc Natl Acad
Sci USA. 111:2554–2559. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Galan-Cobo A, Sitthideatphaiboon P, Qu X,
Poteete A, Pisegna MA, Tong P, Chen PH, Boroughs LK, Rodriguez MLM,
Zhang W, et al: LKB1 and KEAP1/NRF2 pathways cooperatively promote
metabolic reprogramming with enhanced glutamine dependence in
KRAS-Mutant lung adenocarcinoma. Cancer Res. 79:3251–3267. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gao P, Tchernyshyov I, Chang TC, Lee YS,
Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT and
Dang CV: c-Myc suppression of miR-23a/b enhances mitochondrial
glutaminase expression and glutamine metabolism. Nature.
458:762–765. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yuneva MO, Fan TW, Allen TD, Higashi RM,
Ferraris DV, Tsukamoto T, Matés JM, Alonso FJ, Wang C, Seo Y, et
al: The metabolic profile of tumors depends on both the responsible
genetic lesion and tissue type. Cell Metab. 15:157–170. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hua Q, Wang D, Zhao L, Hong Z, Ni K, Shi
Y, Liu Z and Mi B: AL355338 acts as an oncogenic lncRNA by
interacting with protein ENO1 to regulate EGFR/AKT pathway in
NSCLC. Cancer Cell Int. 21:5252021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hua Q, Mi B, Xu F, Wen J, Zhao L, Liu J
and Huang G: Hypoxia-induced lncRNA-AC020978 promotes proliferation
and glycolytic metabolism of non-small cell lung cancer by
regulating PKM2/HIF-1α axis. Theranostics. 10:4762–4778. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vanhove K, Derveaux E, Graulus GJ,
Mesotten L, Thomeer M, Noben JP, Guedens W and Adriaensens P:
Glutamine addiction and therapeutic strategies in lung cancer. Int
J Mol Sci. 20:2522019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Choi YK and Park KG: Targeting glutamine
metabolism for cancer treatment. Biomol Ther (Seoul). 26:19–28.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
DeBerardinis RJ, Mancuso A, Daikhin E,
Nissim I, Yudkoff M, Wehrli S and Thompson CB: Beyond aerobic
glycolysis: Transformed cells can engage in glutamine metabolism
that exceeds the requirement for protein and nucleotide synthesis.
Proc Natl Acad Sci USA. 104:19345–19350. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xiao D, Zeng L, Yao K, Kong X, Wu G and
Yin Y: The glutamine-alpha-ketoglutarate (AKG) metabolism and its
nutritional implications. Amino Acids. 48:2067–2080. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Metallo CM, Gameiro PA, Bell EL, Mattaini
KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L,
et al: Reductive glutamine metabolism by IDH1 mediates lipogenesis
under hypoxia. Nature. 481:380–384. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kuhajda FP: Fatty acid synthase and
cancer: New application of an old pathway. Cancer Res.
66:5977–5980. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gorrini C, Harris IS and Mak TW:
Modulation of oxidative stress as an anticancer strategy. Nat Rev
Drug Discov. 12:931–947. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Harris IS, Treloar AE, Inoue S, Sasaki M,
Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA,
et al: Glutathione and thioredoxin antioxidant pathways synergize
to drive cancer initiation and progression. Cancer Cell.
27:211–222. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jin L, Li D, Alesi GN, Fan J, Kang HB, Lu
Z, Boggon TJ, Jin P, Yi H, Wright ER, et al: Glutamate
dehydrogenase 1 signals through antioxidant glutathione peroxidase
1 to regulate redox homeostasis and tumor growth. Cancer Cell.
27:257–270. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cluntun AA, Lukey MJ, Cerione RA and
Locasale JW: Glutamine metabolism in cancer: Understanding the
heterogeneity. Trends Cancer. 3:169–180. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu W, Le A, Hancock C, Lane AN, Dang CV,
Fan TW and Phang JM: Reprogramming of proline and glutamine
metabolism contributes to the proliferative and metabolic responses
regulated by oncogenic transcription factor c-MYC. Proc Natl Acad
Sci USA. 109:8983–8988. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kerr EM, Gaude E, Turrell FK, Frezza C and
Martins CP: Mutant Kras copy number defines metabolic reprogramming
and therapeutic susceptibilities. Nature. 531:110–113. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Jin L, Alesi GN and Kang S: Glutaminolysis
as a target for cancer therapy. Oncogene. 35:3619–3625. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
DeBerardinis RJ and Cheng T: Q's next: The
diverse functions of glutamine in metabolism, cell biology and
cancer. Oncogene. 29:313–324. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Altman BJ, Stine ZE and Dang CV: From
Krebs to clinic: Glutamine metabolism to cancer therapy. Nat Rev
Cancer. 16:619–634. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yoo HC, Yu YC, Sung Y and Han JM:
Glutamine reliance in cell metabolism. Exp Mol Med. 52:1496–1516.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Alberghina L and Gaglio D: Redox control
of glutamine utilization in cancer. Cell Death Dis. 5:e15612014.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Laplante M and Sabatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Nicklin P, Bergman P, Zhang B,
Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson
C, et al: Bidirectional transport of amino acids regulates mTOR and
autophagy. Cell. 136:521–534. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Jewell JL, Kim YC, Russell RC, Yu FX, Park
HW, Plouffe SW, Tagliabracci VS and Guan KL: Metabolism.
Differential regulation of mTORC1 by leucine and glutamine.
Science. 347:194–198. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Strickaert A, Saiselet M, Dom G, De Deken
X, Dumont JE, Feron O, Sonveaux P and Maenhaut C: Cancer
heterogeneity is not compatible with one unique cancer cell
metabolic map. Oncogene. 36:2637–2642. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hassanein M, Qian J, Hoeksema MD, Wang J,
Jacobovitz M, Ji X, Harris FT, Harris BK, Boyd KL, Chen H, et al:
Targeting SLC1a5-mediated glutamine dependence in non-small cell
lung cancer. Int J Cancer. 137:1587–1597. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Santarpia M, Aguilar A, Chaib I, Cardona
AF, Fancelli S, Laguia F, Bracht JWP, Cao P, Molina-Vila MA,
Karachaliou N and Rosell R: Non-Small-cell lung cancer signaling
pathways, metabolism, and PD-1/PD-L1 Antibodies. Cancers (Basel).
12:14752020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Perez-Escuredo J, Dadhich RK, Dhup S,
Cacace A, Van Hée VF, De Saedeleer CJ, Sboarina M, Rodriguez F,
Fontenille MJ, Brisson L, et al: Lactate promotes glutamine uptake
and metabolism in oxidative cancer cells. Cell Cycle. 15:72–83.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
van den Heuvel AP, Jing J, Wooster RF and
Bachman KE: Analysis of glutamine dependency in non-small cell lung
cancer: GLS1 splice variant GAC is essential for cancer cell
growth. Cancer Biol Ther. 13:1185–1194. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yang C, Sudderth J, Dang T, Bachoo RM,
McDonald JG and DeBerardinis RJ: Glioblastoma cells require
glutamate dehydrogenase to survive impairments of glucose
metabolism or Akt signaling. Cancer Res. 69:7986–7993. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Chen SL, Xue N, Wu MT, Chen H, He X, Li
JP, Liu WL and Dai SQ: Influence of preoperative serum aspartate
aminotransferase (AST) level on the prognosis of patients with
non-small cell lung cancer. Int J Mol Sci. 17:14742016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Caiola E, Colombo M, Sestito G, Lupi M,
Marabese M, Pastorelli R, Broggini M and Brunelli L: Glutaminase
inhibition on NSCLC depends on extracellular alanine exploitation.
Cells. 9:17662020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sellers K, Fox MP, Bousamra M II, Slone
SP, Higashi RM, Miller DM, Wang Y, Yan J, Yuneva MO, Deshpande R,
et al: Pyruvate carboxylase is critical for non-small-cell lung
cancer proliferation. J Clin Invest. 125:687–698. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zeng C, Wu Q, Wang J, Yao B, Ma L, Yang Z,
Li J and Liu B: NOX4 supports glycolysis and promotes glutamine
metabolism in non-small cell lung cancer cells. Free Radic Biol
Med. 101:236–248. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Oh MH, Sun IH, Zhao L, Leone RD, Sun IM,
Xu W, Collins SL, Tam AJ, Blosser RL, Patel CH, et al: Targeting
glutamine metabolism enhances tumor-specific immunity by modulating
suppressive myeloid cells. J Clin Invest. 130:3865–3884. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Leone RD, Zhao L, Englert JM, Sun IM, Oh
MH, Sun IH, Arwood ML, Bettencourt IA, Patel CH, Wen J, et al:
Glutamine blockade induces divergent metabolic programs to overcome
tumor immune evasion. Science. 366:1013–1021. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Yang L, Achreja A, Yeung TL, Mangala LS,
Jiang D, Han C, Baddour J, Marini JC, Ni J, Nakahara R, et al:
Targeting stromal glutamine synthetase in tumors disrupts tumor
microenvironment-regulated cancer cell growth. Cell Metab.
24:685–700. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hensley CT, Faubert B, Yuan Q, Lev-Cohain
N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, et al:
Metabolic heterogeneity in human lung tumors. Cell. 164:681–694.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Bhutia YD and Ganapathy V: Glutamine
transporters in mammalian cells and their functions in physiology
and cancer. Biochim Biophys Acta. 1863:2531–2539. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hassanein M, Hoeksema MD, Shiota M, Qian
J, Harris BK, Chen H, Clark JE, Alborn WE, Eisenberg R and Massion
PP: SLC1A5 mediates glutamine transport required for lung cancer
cell growth and survival. Clin Cancer Res. 19:560–570. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Chiu M, Sabino C, Taurino G, Bianchi MG,
Andreoli R, Giuliani N and Bussolati O: GPNA inhibits the
sodium-independent transport system L for neutral amino acids.
Amino Acids. 49:1365–1372. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Wise DR and Thompson CB: Glutamine
addiction: A new therapeutic target in cancer. Trends Biochem Sci.
35:427–433. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Koppula P, Zhuang L and Gan B: Cystine
transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient
dependency, and cancer therapy. Protein Cell. 12:599–620. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Hu K, Li K, Lv J, Feng J, Chen J, Wu H,
Cheng F, Jiang W, Wang J, Pei H, et al: Suppression of the
SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant
lung adenocarcinoma. J Clin Invest. 130:1752–1766. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Katt WP and Cerione RA: Glutaminase
regulation in cancer cells: A druggable chain of events. Drug
Discov Today. 19:450–457. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Magill GB, Myers WP, Reilly HC, Putnam RC,
Magill JW, Sykes MP, Escher GC, Karnofsky DA and Burchenal JH:
Pharmacological and initial therapeutic observations on
6-diazo-5-oxo-1-norleucine (DON) in human neoplastic disease.
Cancer. 10:1138–1150. 1957. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Dang CV, Le A and Gao P: MYC-induced
cancer cell energy metabolism and therapeutic opportunities. Clin
Cancer Res. 15:6479–6483. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Lemberg KM, Vornov JJ, Rais R and Slusher
BS: We're Not ‘DON’ Yet: Optimal dosing and prodrug delivery of
6-Diazo-5-oxo-L-norleucine. Mol Cancer Ther. 17:1824–1832. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Robinson MM, McBryant SJ, Tsukamoto T,
Rojas C, Ferraris DV, Hamilton SK, Hansen JC and Curthoys NP: Novel
mechanism of inhibition of rat kidney-type glutaminase by
bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide
(BPTES). Biochem J. 406:407–414. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Shukla K, Ferraris DV, Thomas AG, Stathis
M, Duvall B, Delahanty G, Alt J, Rais R, Rojas C, Gao P, et al:
Design, synthesis, and pharmacological evaluation of
bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3
(BPTES) analogs as glutaminase inhibitors. J Med Chem.
55:10551–10563. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Gross MI, Demo SD, Dennison JB, Chen L,
Chernov-Rogan T, Goyal B, Janes JR, Laidig GJ, Lewis ER, Li J, et
al: Antitumor activity of the glutaminase inhibitor CB-839 in
triple-negative breast cancer. Mol Cancer Ther. 13:890–901. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Xie C, Jin J, Bao X, Zhan WH, Han TY, Gan
M, Zhang C and Wang J: Inhibition of mitochondrial glutaminase
activity reverses acquired erlotinib resistance in non-small cell
lung cancer. Oncotarget. 7:610–621. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Boysen G, Jamshidi-Parsian A, Davis MA,
Siegel ER, Simecka CM, Kore RA, Dings RPM and Griffin RJ:
Glutaminase inhibitor CB-839 increases radiation sensitivity of
lung tumor cells and human lung tumor xenografts in mice. Int J
Radiat Biol. 95:436–442. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Xia M, Li X, Diao Y, Du B and Li Y:
Targeted inhibition of glutamine metabolism enhances the antitumor
effect of selumetinib in KRAS-mutant NSCLC. Transl Oncol.
14:1009202021. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Momcilovic M, Bailey ST, Lee JT, Fishbein
MC, Magyar C, Braas D, Graeber T, Jackson NJ, Czernin J, Emberley
E, et al: Targeted Inhibition of EGFR and glutaminase induces
metabolic crisis in EGFR mutant lung cancer. Cell Rep. 18:601–610.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Momcilovic M, Bailey ST, Lee JT, Fishbein
MC, Braas D, Go J, Graeber TG, Parlati F, Demo S, Li R, et al: The
GSK3 signaling axis regulates adaptive glutamine metabolism in lung
squamous cell carcinoma. Cancer Cell. 33:905–921. –e5. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Yu W, Yang X, Zhang Q, Sun L, Yuan S and
Xin Y: Targeting GLS1 to cancer therapy through glutamine
metabolism. Clin Transl Oncol. 23:2253–2268. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Lee JS, Kang JH, Lee SH, Hong D, Son J,
Hong KM, Song J and Kim SY: Dual targeting of glutaminase 1 and
thymidylate synthase elicits death synergistically in NSCLC. Cell
Death Dis. 7:e25112016. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Yoneda K, Imanishi N, Ichiki Y and Tanaka
F: Treatment of non-small cell lung cancer with EGFR-mutations. J
UOEH. 41:153–163. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Liu Y, Ge X, Pang J, Zhang Y, Zhang H, Wu
H, Fan F and Liu H: Restricting glutamine uptake enhances NSCLC
Sensitivity to Third-Generation EGFR-TKI Almonertinib. Front
Pharmacol. 12:6713282021. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Sazeides C and Le A: Metabolic
relationship between cancer-associated fibroblasts and cancer
cells. Adv Exp Med Biol. 1063:149–165. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Cerezo M and Rocchi S: Cancer cell
metabolic reprogramming: A keystone for the response to
immunotherapy. Cell Death Dis. 11:9642020. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Lee YZ, Yang CW, Chang HY, Hsu HY, Chen
IS, Chang HS, Lee CH, Lee JC, Kumar CR, Qiu YQ, et al: Discovery of
selective inhibitors of Glutaminase-2, which inhibit mTORC1,
activate autophagy and inhibit proliferation in cancer cells.
Oncotarget. 5:6087–6101. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Imai H, Kaira K, Oriuchi N, Shimizu K,
Tominaga H, Yanagitani N, Sunaga N, Ishizuka T, Nagamori S,
Promchan K, et al: Inhibition of L-type amino acid transporter 1
has antitumor activity in non-small cell lung cancer. Anticancer
Res. 30:4819–4828. 2010.PubMed/NCBI
|
|
94
|
Lukey MJ, Cluntun AA, Katt WP, Lin MJ,
Druso JE, Ramachandran S, Erickson JW, Le HH, Wang ZE, Blank B, et
al: Liver-Type Glutaminase GLS2 is a druggable metabolic node in
luminal-subtype breast cancer. Cell Rep. 29:76–88. –e7. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Wise DR, DeBerardinis RJ, Mancuso A, Sayed
N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon
SB and Thompson CB: Myc regulates a transcriptional program that
stimulates mitochondrial glutaminolysis and leads to glutamine
addiction. Proc Natl Acad Sci USA. 105:18782–18787. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Moreadith RW and Lehninger AL: The
pathways of glutamate and glutamine oxidation by tumor cell
mitochondria. Role of mitochondrial NAD(P)+-dependent malic enzyme.
J Biol Chem. 259:6215–6221. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Estrela JM, Ortega A and Obrador E:
Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci.
43:143–181. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Yang WH, Qiu Y, Stamatatos O, Janowitz T
and Lukey MJ: Enhancing the efficacy of glutamine metabolism
inhibitors in cancer therapy. Trends Cancer. 7:790–804. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Clay TD, Russell PA, Do H, Sundararajan V,
Conron M, Wright GM, Dobrovic A, Moore MM and McLachlan SA:
Associations between the IASLC/ATS/ERS lung adenocarcinoma
classification and EGFR and KRAS mutations. Pathology. 48:17–24.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Suzuki S, Tanaka T, Poyurovsky MV, Nagano
H, Mayama T, Ohkubo S, Lokshin M, Hosokawa H, Nakayama T, Suzuki Y,
et al: Phosphate-activated glutaminase (GLS2), a p53-inducible
regulator of glutamine metabolism and reactive oxygen species. Proc
Natl Acad Sci USA. 107:7461–7466. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Zhang T, Li Y, Zhu R, Song P, Wei Y, Liang
T and Xu G: Transcription Factor p53 suppresses tumor growth by
prompting pyroptosis in non-small-cell lung cancer. Oxid Med Cell
Longev. 2019:87468952019. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Jung S, Kim DH, Choi YJ, Kim SY, Park H,
Lee H, Choi CM, Sung YH, Lee JC and Rho JK: Contribution of p53 in
sensitivity to EGFR tyrosine kinase inhibitors in non-small cell
lung cancer. Sci Rep. 11:196672021. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Rekhtman N, Ang DC, Riely GJ, Ladanyi M
and Moreira AL: KRAS mutations are associated with solid growth
pattern and tumor-infiltrating leukocytes in lung adenocarcinoma.
Mod Pathol. 26:1307–1319. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Toth LN, de Abreu FB and Tafe LJ:
Non-small cell lung cancers with isocitrate dehydrogenase 1 or 2
(IDH1/2) mutations. Hum Pathol. 78:138–143. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Shimizu K, Kaira K, Tomizawa Y, Sunaga N,
Kawashima O, Oriuchi N, Tominaga H, Nagamori S, Kanai Y, Yamada M,
et al: ASC amino-acid transporter 2 (ASCT2) as a novel prognostic
marker in non-small cell lung cancer. Br J Cancer. 110:2030–2039.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Chakrabarti G: Mutant KRAS associated
malic enzyme 1 expression is a predictive marker for radiation
therapy response in non-small cell lung cancer. Radiat Oncol.
10:1452015. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Almuhaideb A, Papathanasiou N and Bomanji
J: 18F-FDG PET/CT imaging in oncology. Ann Saudi Med. 31:3–13.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Qu W, Oya S, Lieberman BP, Ploessl K, Wang
L, Wise DR, Divgi CR, Chodosh LA, Thompson CB and Kung HF:
Preparation and characterization of L-[5-11C]-glutamine for
metabolic imaging of tumors. J Nucl Med. 53:98–105. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Anderson PM and Lalla RV: Glutamine for
amelioration of radiation and chemotherapy associated mucositis
during cancer therapy. Nutrients. 12:16752020. View Article : Google Scholar : PubMed/NCBI
|














