Colorectal cancer (CRC) is one of the most common
cancers of the digestive system worldwide and can occur at any age.
According to global Cancer Epidemic Statistics released in 2020 by
the International Agency for Research on Cancer of the World Health
Organization (GLOBOCAN2020) (1), an
estimated 1,931,600 new cases and 935,200 deaths associated with
CRC were reported globally, ranking third and second, respectively,
in terms of morbidity and mortality. The classical
Tumor-Node-Metastasis (TNM) staging system is still an important
reference for clinicians to assess patient prognosis and provide
individualized treatment (2,3);
however, this staging method has limitations, with increasing
evidence of significant differences in the prognosis of patients
with CRC with the same stage and pathological type (4). In 1999, the National Cancer Institute
of the United States proposed the concept of the molecular
classification of tumors, using molecular characteristics through
molecular analysis techniques (5).
At present, the detection of KRAS, BRAF, microsatellite instability
(MSI) and their mutations contributes to the clinical management of
CRC and the selection of personalized drugs (6,7). The
use of novel targeted and cytotoxic drugs has extended the median
overall survival (OS) of patients with advanced CRC to 25–30 months
(8). Despite its widespread use,
the clinical translation of single molecular markers is not always
consistent, which may be related to differences in data processing
and algorithms applied to different patient cohorts, sample
preparation methods and gene expression platforms. With the
progress of molecular sequencing and gene molecular mechanism
research, scientists have used a more organized and universal way
of defining current disease patterns; the characterization of tumor
biological characteristics, namely consensus molecular subtype
(CMS) typing (9), has promoted
cancer classification transition from ‘mutation-centered’ to
‘transcriptome-based’ molecular subtypes. CRC molecular typing
plays an increasingly important role in the era of individualized
precision medicine. Emphasis has been placed on classifying CRC
based on genetic characteristics, tumor microenvironment and, more
recently, immunological characteristics, with each classification
system having its unique importance and clinical significance. The
present review provides an overview of molecular typing and its
clinical significance.
Current evidence suggests that the occurrence and
development of CRC is a multi-step, multi-stage and multi-gene
process. It is widely considered to result from the interaction of
environmental and genetic factors, as well as from the upregulation
of tumor suppressor genes and proto-oncogenes. Based on the genetic
mutations and the cytogenetic background of the genome, molecular
typing based on the CRC genome is affected by the presence of the
following: Chromosomal instability (CIN), microsatellite
instability (MSI), CpG island methylator phenotype (CIMP) and
molecular markers.
CIN has been documented in most sporadic CRCs
(Sp-CRCs) and tumors with APC germline mutations, with an APC
mutation rate of only 1%. Nevertheless, little is known about
whether CIN is an independent predictor of familial CRC. Some
researchers concluded that although the sensitivity of CIN
prediction for familial CRC was acceptable, it was not sufficient
to be an independent predictor (10,17,18).
One study found no significant difference in CIN between familial
CRC cases and non-familial, control CRC cases (P=0.50) (18).
A substantial number of CRCs, known as interval CRCs
(I-CRCs), are diagnosed in the period shortly after a negative
colonoscopy result (i.e., no detectable polyps or CRC) and prior to
the recommended follow-up screening (19). According to the American Cancer
Society, ~5,200 Americans were diagnosed with an I-CRC in 2014, and
nearly 2,000 succumbed to the disease (20). This particular type of CRC may be
associated with genetic defects inducing genome instability, or may
be a specific type of Sp-CRC. In response to this uncertainty,
researchers performed a matching comparison experiment of
I-CRC/Sp-CRC cases and found that CIN occurred in 80–85% of Sp-CRCs
and I-CRCs, and the latter frequently exhibited gains and losses in
chromosomes 8, 11 and 17 (20). One
possible explanation is the inaccurate detection of certain
polyps/tumors or similar clinical features leading to negative
colonoscopies. Another explanation is that I-CRCs represent a
distinct tumor subset with both CIN and MSI phenotypes, and that
these two molecular features may play a synergistic role.
Furthermore, the interval between colonoscopy and screening could
also be an additional explanation.
The CIN phenotype tends to be more of a predictive
tool in clinical practice, and patients with CIN-positive CRC have
shown poor OS and progression-free survival outcomes, regardless of
ethnic background, anatomical location and fluorouracil (5-FU)
chemotherapy efficacy (21).
Watanabe et al (15)
retrospectively reviewed the expression of MSI and CIN in 1,103
patients and concluded that the CIN phenotype could be used as an
independent risk factor for DFS and OS in stage II/III patients,
and CIN-high could be used as a predictor of a poor prognosis. Only
8% of patients with CRC were either in stage I or IV. CIN is a
driver of the metastasis of human cancer cells, which has been
preliminarily verified in breast and lung cancer models (22); however, the progression pattern of
the CIN phenotype in breast cancer and lung adenocarcinoma is not
applicable in CRC. Orsetti et al (23) used array comparative genomic
hybridization to analyze a group of 162 patients with CIN CRC,
consisting of 131 primary cancer cases evenly distributed in stages
I to IV, 31 metastases (28/31 formed a
primary-tumor/matched-metastasis pair) and 14 adenomas. The results
showed that the increased level of genomic instability represented
by CIN was not entirely consistent with the progression from stage
I to IV during the histopathological examination. In addition, with
study of the molecular mechanism of CIN, the genetic variation or
abnormal expression of some molecules that maintain chromosomal
stability may become therapeutic targets and diagnostic markers
(22,24–27).
High levels of CIN are not conducive to the proliferation of tumor
cells. It is widely considered that drugs could induce higher
levels of CIN phenotype, leading to the spontaneous death of tumor
cells. Heat shock protein 90 inhibitors may achieve this effect by
inducing higher aneuploidy and limiting tumor cell growth (28). A phase II trial (29) showed that patients with CRC did not
respond well to docetaxel (Taxotere®), which may be
attributed to the fact that 85% of CRC tumors were of CIN type, and
aneuploidy was less receptive to taxanes than diploid karyotype
(21), associated with increased
taxane resistance caused by abnormal spindle examination points in
CIN (30).
Microsatellites are short nucleotide repeats (1–6
repeat units) that are heritable, unstable and highly polymorphic
in the human genome (31). MSI
refers to the change in the number of microsatellite tandem repeats
within a certain location in certain cells. Importantly, if the DNA
mismatch repair genes (MMR) show germline mutations or LOH, errors
from microsatellite replication will be retained (MMR-Deficient
(dMMR)/MSI) (32). MSI in CRC
includes the majority of hereditary non-polyposis CRC (HNPCC) and
15% of Sp-CRCs (32,33). MSI commonly occurs in two situations
(34): The first is the germline
mutation of MMRs MutL homolog 1 (MLH1), MutS homolog 2 (MSH2),
(MSH6) or Postmeiotic segregation increased 2(PMS2), and the other
is hypermethylation of the MLH1 gene promoter region, which are
predominantly cases of Sp-CRC showing dMMR. According to the
microsatellite expression, MSI can be divided into three types:
Microsatellite high instability (MSI-H), microsatellite low
instability (MSI-L) and microsatellite stability (MSS). The two
main techniques used to determine dMMR/MSI status include
immunohistochemistry (IHC), designed to detect dMMR status, and
molecular testing, which determines MSI status (35). Overwhelming evidence substantiates
that MSI CRC often presents as poorly differentiated carcinoma and
mucinous adenocarcinoma, mostly in the proximal colon with
peritumoral lymphocyte infiltration (31,36–38).
The incidence in younger adults (patient younger
than 50 years), early-onset CRC (EOCRC) is rising alarmingly. EOCRC
is an important reflection of the younger trend of gastrointestinal
tumors. Long-term tumor burden is becoming increasingly severe for
patients with EOCRC. In previous studies, in younger patients,
metastatic tumors represented an increasing proportion of all tumor
stages (39). Compared with general
patients with CRC, patients with EOCRC had a higher frequency of
dMMR/MSI-H and a higher proportion of wild-type (WT) KRAS and BRAF,
as well as a higher BRAF V600E mutation rate (40,41).
Taken together, these results support changing the average-risk
screening age from 50 to 45 years for all patients, with molecular
characterization being an important breakthrough for clinical
intervention. It is well established that patients with MSI-H CRC
have a better prognosis and longer survival time than patients with
either MSI-L or MSS CRC. Guastadisegni et al (42) analyzed the survival status of 12,782
patients with CRC and concluded that patients with MSI-H CRC had
improved OS and DFS times. A meta-analysis involving >7,500
patients showed that MSI-positive tumors were superior to
MSI-negative tumors in terms of MSI and survival assessment,
suggesting that the genomic molecular marker status can be
independently analyzed to assess prognosis (43). Current guidelines recommend
harnessing MSI-H to guide CRC adjuvant therapy and improve the
quality of individualized treatment. In this respect, according to
the National Comprehensive Cancer Network® (NCCN)
guidelines (44,45), patients with stage II MSI-H CRC may
have an improved prognosis but may not benefit from 5-FU-assisted
chemotherapy. Kim et al (46) performed MSI and MMR detection, and
prognosis analysis on 135 patients who received FOLFOX-assisted
chemotherapy (adjuvant oxaliplatin, 5-FU and leucovorin therapy)
after radical resection of CRC. The results showed that DFS and OS
times were not significantly prolonged in patients with
MSI-H/MMR-deficient (MMR-D) CRC compared with patients with
MSI-L/MMR-intact (MMR-I) CRC. It was not investigated whether
patients with MSI-L/MMR-I CRC would benefit more from 5-FU
chemotherapy. Guastadisegni et al (42) hypothesized that patients with MSS
CRC would benefit more from 5-FU chemotherapy than patients with
MSI-H CRC. In a retrospective study of 6,964 patients with stage II
CRC (47), an attempt was made to
determine the relationship between 5-FU-based adjuvant
chemotherapy, primary tumor laterality, MSI status and OS. The
results showed that for MSS-positive tumors, adjuvant chemotherapy
was significantly associated with improved patient 5-year OS rate
[hazard ratio (HR), 0.47; P<0.001], even in the absence of other
risk characteristics. By contrast, there was no significant
association between adjuvant chemotherapy and OS in patients with
MSI-positive CRC (HR, 0.85; P=0.671). It is difficult to judge the
sensitivity of patients to 5-FU based solely on MSI status, and
multiple stable expression markers are needed for a comprehensive
analysis. In recent years, immunotherapy has been increasingly used
to treat MSI CRC (48,49). In 2017, the US Food and Drug
Administration approved pembrolizumab to treat inoperable or
metastatic dMMR/MSI-H solid tumors based on the high response rates
observed in five clinical trials (50–55).
Nivolumab was introduced in dMMR/MSI-H metastatic CRC (mCRC) in the
same year (56). Frameshift
peptides generated by frameshift mutations caused by MSI-H are
highly immunogenic and respond well to programmed death
receptor-1(PD1)/programmed death ligand 1 (PD-L1) inhibitors. In
2015, a study showed that the MSI status of tumors is closely
related to the effect of immunotherapy (57). From later-line monotherapy
(Keynote-164, CheckMate-142) and later-line dual-drug therapy
(CheckMate-142), to first-line monotherapy (Keynote-177) and
first-line dual-drug therapy (CheckMate-142), the role of
immunotherapy in the treatment of dMMR/MSI-H CRC is expanding
(53,56,58,59).
The efficacy of nivolumab plus ipilimumab in the treatment of
patients with advanced dMMR/MSI-H CRC is reflected in the 2021 NCCN
guidelines and the 2022 American Society of Clinical Oncology
(ASCO) conference report (60).
Dual immunotherapy can effectively reduce the occurrence of drug
resistance, while B2M or JAK1/2 gene mutations associated with
resistance to traditional immunotherapy do not affect the benefit
of MSI-H CRC to PD-1 antibodies (61); however, immunotherapy will not be
effective for the treatment of MSI-H mucinous adenocarcinoma CRC.
Due to the persistence or potential toxicity of immunotherapy, a
balance between efficacy and toxicity is necessary. It is worth
mentioning that since immune checkpoint inhibitors (ICIs) are not
effective in MMR-proficient (pMMR)/MSS mCRC, MMR IHC and MSI
testing should be performed prior to ICI initiation to minimize the
chance of pMMR/MSS tumors being misdetected as dMMR/MSI (62,63).
Lynch syndrome is an aggressive autosomal dominant genetic disorder
with an ~80% lifetime risk of cancer recurrence caused by germline
mutations in the MMR genes (MLH1, MSH2, MSH6 and PMS2) (64). The NCCN guidelines recommend MMR or
MSI testing for Lynch syndrome in all patients with a history of
CRC (44,45). Some researchers consider that the
relationship between MSI and CIN is not independent, and that both
can be expressed in one patient with CRC, namely a patient with
MSI-positive/CIN-positive CRC, although this is rare (65). The frequency of
MSI-positive/CIN-positive tumors was recorded as 12 (Sp-CRCs) and
14% (I-CRCs), respectively (19).
Furthermore, ~25% of patients presented with
MSI-negative/CIN-negative CRC (66–69). A
study stratified survival by CIN and MSI status, and concluded that
the univariate survival benefit of stage II and III CRC associated
with MSI-positive status was not independent of CIN status during
multivariable analysis (21,67).
Future experiments designed for the three forms of genomic
instability [CIN, MSI and CpG island methylator phenotype (CIMP)]
may clarify the relationship between the three.
Mutations and genomic instability contribute to
inter-tumor heterogeneity. The sub-clonal phenomenon that exists
throughout tumor progression is called intra-tumor heterogeneity
(ITH). It has been reported that ITH can be detected in almost all
cancer types and is associated with tumor prognosis and drug
resistance (70–73). A typical example of ITH is the
molecular differences between primary tumors and metastases, such
as MMR pattern or MSI status. After assessing the MMR status of
mCRC, fewer patients with mCRC showed heterogeneity of MMR status
between the primary and corresponding metastatic sites (11.9 and
18.7%, respectively), among which patients with peritoneal
metastasis tended to exhibit this feature. Furthermore, the
prevalence of heterogeneous MMR phenotypes in primary tumors with
dMMR was significantly higher than that with pMMR (P<0.001)
(74,75). However, it is noteworthy that
various factors, such as the expertise of the pathologists, the
quality of the tumor tissue sampling and staining can contribute to
these discrepancies.
CpG islands are regions rich in cytosine (C) and
guanine (G) dinucleotides in the gene, with CG content >50%,
length >250–550 base pairs, and a CpG value of 0.6 or greater
(76–78). It has been shown that the
pathogenesis of CRC is related to DNA methylation, with
hypomethylation of genes in non-promoter regions and
hypermethylation of genes in promoter regions (79). DNA hypomethylation can lead to
oncogene activation, gene marker deletion and chromosomal
stability. Hypermethylation of promoter sequences interferes with
the normal expression of tumor suppressor genes and DNA repair
genes, which is known as epigenetic silencing (80). This hypermethylated phenotype is
called CIMP. Various classification criteria have been developed to
describe the tumor characteristics of CIMP, each with unique
molecular and oncological characteristics, and no gold standard has
been established. Weisenberger et al (81) divided CRC into CIMP-positive and
CIMP-negative CRC, according to five gene combinations (CACNA1G,
IGF2, NEUROG1, RUNX3 and SOCS1). Similarly, Ogino et al
(82) classified CRC as CIMP-High
(CIMP-H), CIMP-Low (CIMP-L) and CIMP-negative based on eight gene
combinations (RUNX3, CACNA1G, IGF2, MLH1, NEUROG1, CRABP1, SOCS1,
and CDKN2A). This classification has been confirmed to be
genetically associated with TP53, KRAS, BRAF, MSI and specific
histological types (poorly differentiated or mucinous) (83,84);
however, the relationship with CIN remains unclear. An increasing
body of evidence suggests that CIMP-type molecular pathways mostly
occur in the proximal colon, mainly in elderly women (84–86).
Moreover, it has been observed that the frequency of gene
hypermethylation in normal colon mucosa in women and elderly
patients with CRC is higher than that in men and young patients
(86).
Different methods of CIMP identification and
experimental populations may result in different pathological
characteristics. Weisenberger et al (86) showed that the right colon is a
high-risk site for CIMP, and this classification is associated with
advanced age in women, with the hallmark mutation of BRAF (V600E),
loss of hypermethylation of the MLH1 promoter and loss of TP53.
There is ample evidence suggesting that CIMP is closely associated
with prognosis, but it remains unclear whether this association is
positive or negative. Ogino et al (87) showed that CIMP-H was an independent
predictor of colon cancer-specific low mortality. During the
stratified analysis, CIMP-H was associated with significantly
reduced colon cancer-specific mortality regardless of MSI and BRAF
status. However, a more recent meta-analysis (88) involving 15,315 patients with CRC
confirmed that CIMP-H CRC was associated with poorer OS/DFS/PFS/RFS
times than CIMP-L/negative CRC. Furthermore, a survival
disadvantage was observed in terms of OS, especially in stage
III–IV and pMMR tumors. In addition to its prognostic value, the
role of CIMP in the prediction of the chemotherapy response is
another issue requiring resolution. Much controversy surrounds the
efficacy of 5-FU-based chemotherapy against CIMP-positive CRC. Cha
et al (89) showed that CIMP
was associated with adverse outcomes for patients receiving
chemotherapy for mCRC. Jover et al (90) concluded that CIMP-positive patients
did not significantly benefit from 5-FU-based adjuvant chemotherapy
after following up 302 patients with CRC, while CIMP-negative
patients receiving chemotherapy experienced significantly prolonged
DFS times. Iacopetta et al (91) reported contrasting findings that
patients with CIMP-positive CRC can benefit from 5-FU treatment,
mainly related to the association between CIMP positivity and
intracellular folic acid metabolism, and gene silencing caused by
DNA methylation. A recent study claimed that CIMP-positive tumors
are potentially more responsive to the topoisomerase-inhibitor,
irinotecan (92). Although CIMP has
been reported as a potential prognostic biomarker for drug
decision-making, overall research on treating CIMP-positive tumors
with hypomethylating drugs appears to be limited. Indeed, the lack
of a widely accepted CIMP phenotype and the correct stratification
of patients with CRC according to the CIMP status are key issues
for future CRC trials. Since CIMP was shown to be a tissue-specific
phenomenon, DNA methylation information obtained only by array
probes or other low-density techniques is far from sufficient, and
new analysis methods, such as PacBio single-molecule real-time
sequencing or nanopore sequencing are needed (93). Assuming that the CIMP pattern is
stable across tumor sections and that epigenetic drug delivery and
protection against side effects are improved, optimal antitumor
activity is expected for CIMP-positive tumors.
Some patients benefit from the wide application of
molecular targeted therapy, and identifying molecular markers is an
important prerequisite for screening patients with CRC who can
benefit from targeted drugs.
The RAS gene is the most common proto-oncogene in
human tumors. RAS can encode a group of small molecular proteins
homologous to G proteins, called RAS proteins. When a RAS protein
is mutated, it cannot normally complete its signal-mediated
transduction process, and abnormal cell growth, differentiation and
material transport occur, leading to uncontrolled proliferation and
carcinogenesis. Importantly, the KRAS mutation rate in CRC is
30–50% (94,95). Sugimoto et al (96) hypothesized that as a precancerous
lesion of CRC, the progression of laterally spreading
tumor-granular was closely associated with RAS gene mutations, with
RAS mutation rates up to 54.1%. The predictive significance of RAS
mutant (mt) on the anti-EGFR drug response rate and survival time
in patients with mCRC has been confirmed in previous studies
(97–100). According to the NCCN guidelines,
all patients with mCRC should be tested for the genotype of RAS
(KRAS, NRAS) and BRAF mutations in tumor tissue (6,7). After
resection of metastases, a negative association between RAS
mutations and patient survival has previously been reported
(101). Importantly, patients with
any known KRAS (exon 2 or non-exon 2) or NRAS mutation should not
be treated with cetuximab or panitumumab, while other targeted
therapies, such as bevacizumab, can still be used. It has been
established that patients with mCRC carrying WT RAS can benefit
from anti-EGFR therapy with prolonged OS and PFS times (102–104). In an analysis of patients with CRC
of stage III MSS who received FOLFOX chemotherapy plus or minus
cetuximab, KRAS was used as a marker of a poor prognosis (105). The RAS status was also included in
the CMS classification established in 2015 (9). CMS analysis of patients with mCRC KRAS
(exon 2 WT) in a previous FIRE-3 study showed that the CMS3 and
CMS4 subgroups responded significantly better to cetuximab than to
bevacizumab (106).
BRAF is an important transduction factor in the EGFR
signaling pathways of RAS, RAF, MEK, MRK and MAPK, and regulates
various physiological processes of cell growth, differentiation and
apoptosis. The mutation rate of BRAF in CRC is ~10% (107), and BRAF mutations are associated
with the proximal colon and MSI (81,94,108).
BRAF mt is often associated with a poor prognosis (109–111). A study demonstrated that patients
with CRC in BRAF mt stage III have a higher risk of recurrence
(112). By contrast, a study by
Birgisson et al (113)
reported that patients with CRC with both MSI and BRAF (V600E)
mutations had a low recurrence rate, while researchers observed
significantly higher recurrence rates in patients with MSI and KRAS
mutations. Consistently, Seppälä et al (34) showed that in patients with stage
I–II CRC, BRAF (V600E) mutation in conjunction with MSS is
negatively associated with quality of life, and the prognostic
potential of MSI negates the harmful effects of BRAF (V600E) and
presents a positive prognosis. The IHC assay found that MLH1
expression was lost, but BRAF (V600E) was present, which excluded
Lynch syndrome. This finding may be attributed to patients with
MLH1 promoter methylation generally having BRAF mutations, and BRAF
mutations almost always occur at a single site, V600E. BRAF
mutations generally occur in patients with Sp-CRC, but not in
patients with Lynch syndrome. The NCCN guidelines recommend that
patients with mCRC should be tested for BRAF in tumor tissue
(6,7). For patients with mCRC and RAS WT/BRAF
mt, the guidelines do not recommend treatment with anti-EGFR
monotherapy or in combination with chemotherapy agents.
The above two markers (RAS and BRAF) have been
investigated in depth in previous studies, and PIK3CA and HER2 have
also attracted the attention of clinicians. Current evidence
suggests that the mutation rate of PIK3CA is 15–20% (114). In 2004, researchers reported a
high frequency of PIK3CA mutations in human cancer cells such as
breast cancer (frequency 7.1-35.5%), colorectal cancer
(16.9-30.6%), ovarian cancer (33%), lung cancer (0.6–20%), among
others, and subsequent studies identified PIK3CA as a risk factor
for numerous types of cancer, including CRC (115). Currently, the American Society for
Clinical Pathology, College of American Pathologists, Association
for Molecular Pathology and ASCO guidelines (116) on the evaluation of molecular
biomarkers of CRC do not recommend routine PIK3CA testing for
treatment outside of clinical trials. In vitro cell and
animal experiments have shown that some classical PIK3CA
inhibitors, such as wortmannin, LY294002 and rapamycin, exhibit
anti-tumor growth; however, the serious toxic side effects and drug
resistance have limited further clinical trials (117,118). In addition, tumor cells treated
with PI3KCA inhibitors tend to stop growing rather than undergo
apoptosis, allowing tumor cells to develop drug resistance in
various ways. It was shown that patients with PIK3CA-mutant CRC had
a poor prognosis (119). An
increasing body of evidence suggests that the benefit of aspirin in
controlling overall CRC mortality may be more significant in
PIK3CA-mutated CRC (114,120). HER2 is a proto-oncogene encoding a
185-kDa plasma membrane-bound tyrosine kinase receptor, a member of
the EGFR gene family. HER2 amplification/upregulated expression can
be detected in 3–5% of patients with RAS WT mCRC, mutually
exclusive to KRAS, NRAS and BRAF mutations, and highly consistent
between primary and metastatic tumors (121). HER2 has long been considered a
marker of a poor prognosis, but multiple previous studies have
shown that HER2 positivity is not significantly associated with
prognosis (122–124). Accordingly, no consensus has been
reached on its predictive effect. In patients with WT RAS,
HER2-positive mCRC, the NCCN guidelines recommend trastuzumab
combined with lapatinib/pertuzumab (6,7). In
addition, HER2 amplification has been reported as one of the causes
of patient resistance to EGFR therapeutics, and HER2 may serve as a
negative predictor of anti-EGFR therapy (125). Therefore, anti-HER2 therapy may be
a more reasonable option for patients with mCRC tested for HER2
upregulated expression before treatment with cetuximab and
panitumumab. More recently, trastuzumab deruxtecan (DS-8201) has
shown promising and long-lasting activity in patients with
refractory, HER2-positive mCRC, including patients previously
treated with HER2-targeted therapy, as preliminarily confirmed in
the phase II trial DESTINY-CRC01 (126).
In most cases, a single marker characterized by
mutations is insufficient to explain the heterogeneity in patients
with CRC. In an attempt to refine the molecular map, combinations
of multiple markers have the opportunity to overcome this
limitation. One study (127)
attempted to combine BRAF, PIK3CA and RAS testing and increased the
proportion of patients benefiting from anti-EGFR treatment from
36.1 to 41.2%. The selection of drugs targeting the altered
multiple gene targets in the MAPK pathway is expected to reduce
drug resistance and improve the response rate. Besides, molecular
classification using these biomarkers can be used to classify
patients. For example, Gil-Raga et al (128) used BRAF (V600E), RAS and MMR
status to divide 105 cases of stage I–III CRC into five molecular
subtypes to identify differences in prognosis. In addition, it is
widely thought that CRC is an umbrella diagnosis encompassing
numerous rare disease subtypes, in a context where the complexity
of molecular markers is increasing, and the combination of
different markers emphasizes the significance of comprehensive
genetic testing (125,129).
The fundamental significance of molecular typing is
to better guide CRC-targeted therapy, prolong DFS, and improve
patient prognosis and quality of life. Discussion of Molecular
typing alone is less comprehensive, and individualized assessment
of disease conditions inevitably takes into account demographic
characteristics, clinicopathological characteristics, molecular
markers, lifestyle and nutritional factors, and chemical agents.
Subsequent classification reports, such as the Jass Classification
of CRC (130), CCS Classification
(131), Ogino Classification
System (132) and Mangi
Classification of CRC (133),
mostly used CIMP, MSI, CIN, RAS and other markers as prototypes to
explore the future direction of neoadjuvant therapy and targeted
therapy in the clinical environment.
Due to the heterogeneity of tumors, patients often
exhibit strong differences in response to classical treatment
regimens, resulting in a weak response, poor tolerance and even
death. Previously established classification methods, such as Jass
Classification, Ogino Classification, Mangi Classification and CCS
Classification, may be appropriate for a certain group of
individuals since they were based on studies with heterogeneous
inclusion and exclusion criteria, tumor type, classification and
mechanism (134). In previous
years, with the development of sequencing technologies, Guinney
et al (9) optimized data
processing, algorithm bias, sample preparation method and
classification basis, standardized the differences in queue
selection, and analyzed genetic data and tumor types from multiple
platforms. Finally, a CMS typing method was proposed. This
classification method is currently recognized as the most robust
classification method, mainly based on tumor biology rather than
clinical results, since it can easily capture the inherent
biomolecular heterogeneity of CRC and is expected to become the
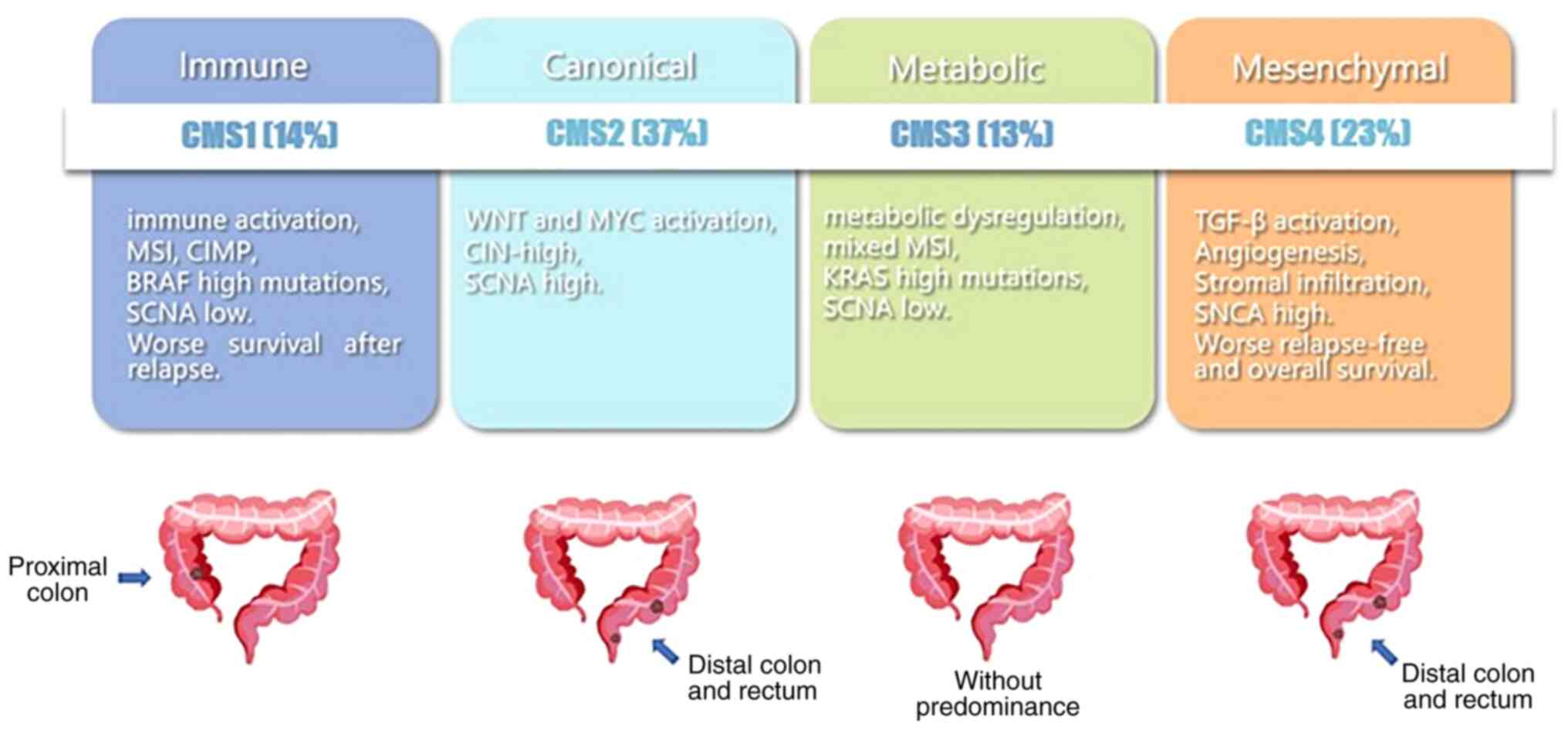
basis of immunotherapy or targeted therapy. There are 5 subtypes
(CMS1, CMS2, CMS3, CMS4 and mixed type) with distinct molecular
properties and clinical characteristics. CMS1 (microsatellite
instability immune, approximately 14% of CRCs) CRC is hypermutated
and associated with both MSI and strong immune activation. CMS2
(canonical, approximately 37% of CRCs) CRC displays epithelial
differentiation and strong upregulation of Wnt and Myc downstream
targets, both of which are classic pathways for the development and
progression of CRC. CMS3 (metabolic, approximately 13% of CRCs) CRC
is associated with epithelial and marked metabolic abnormalities.
The enrichment of multiple metabolic characteristics in epithelial
CRC cells is consistent with the occurrence of KRAS-activated
mutations, which have been described as inducing significant
metabolic adaptations. In CMS4 (mesenchymal, approximately 23% of
CRCs) CRC, transforming growth factor-β (TGF-β) is activated with
enhanced interstitial invasion and angiogenesis. Finally, mixed
type (approximately 13% of CRCs) CRC represents a transitional
phenotype or intratumoral heterogeneity (9). The main hallmarks of CMS are briefly
described in Fig. 1.
CMS includes numerous significant genetic and
biological indicators, and is human-centered to predict survival
and guide drug use. The selection of samples (left half, right half
or rectum), the selection of multiple gene states or indicators,
method of detection, and how to conduct classification analysis are
still issues to be considered in prospective studies. There is a
long way to go before CMS can be truly brought into clinical
practice.
Using proteomics to study CRC, multiple proteins and
their interactions can simultaneously be studied to reveal the
biological mechanisms that occur at the protein level in the
pathological environment of cells. Compared with genomics and
transcriptomics, protein, as the main executor of human life
activities, more directly reflect the biological phenomena. Protein
expression is dynamic and diverse (150,151). Often, changes in abundance or
function are not parallel to gene changes. Given the existence of
multi-level regulation from the mRNA to the protein level, the
transcription level cannot fully reflect protein expression level
(152,153). Importantly, proteomic analysis
enables observation of the biological behavior of cells directly,
which is the basis of establishing proteomic molecular typing.
Notably, Zhang et al (154)
performed consistent clustering of proteomics data of 95 CRC tissue
specimens, and divided CRC into five categories, A-E, describing
the correlation between CRC and genome. Categories A, D and E were
closely related to CIN, while B and C were closely related to MSI.
More recently, Li et al (155) analyzed the clinical tissues of 146
patients with CRC, including 70 patients with mCRC, and divided CRC
into three subtypes (CC1, CC2 and CC3) at the proteomic level. The
three subtypes were associated with different clinical prognoses
and molecular characteristics. For example, patients with CC3 CRC
have a worse prognosis than CC1/2 patients, similar to mCRC. In
addition, the results were consistent with those of CMS. Further
phosphoproteomic profiling identified cases with mCRC. In addition,
multi-omics collaboration and appropriate drug sensitivity tests
are expected to identify an accurate target for drug selection for
specific tumor types. The discovery of unique molecular markers and
effective drug targets is still expected to integrate proteome and
phosphorylated proteome analysis, and systematic validation in
large tumor cohorts, and needs to be compared with established
diagnostic methods to select the best or optimize the current
diagnostic markers, which is the limit of their clinical
translation (155,156). In addition, proteomics research
has limitations such as complex preparation, inconvenient storage,
difficult data analysis and poor repeatability, making it difficult
for this method to become the mainstream method of diagnosis or
medication guidance.
The significance of CRC molecular typing lies in
predicting survival, customizing precision treatment strategies and
predicting drug resistance. Although exciting progress has been
made in the molecular typing of CRC, the clinical transformation of
molecular typing, especially CMS typing, still faces great
challenges and opportunities. At present, there is no unified
system for the molecular typing of CRC, and there are crossovers
among various subtypes. Therefore, it is essential to form a
unified molecular typing by integrating the advantages of numerous
molecular typing methods and summarizing the similarities and
differences of various subgroups of CRC. Moreover, there is a lack
of clinical detection methods, and the current mainstream molecular
typing method is not applicable during clinical practice due to its
lack of convenience and economical costs. Moreover, the process of
CMS diagnosis by clinicians and pathologists is cumbersome, with a
dearth of specific biomarkers to determine CMS, which fails to
achieve the effect of strong practicability, easy availability and
high accuracy of prediction shown by HER2, estrogen receptor and
progesterone receptor in breast cancer management (157). Screening for fairly robust
biomarkers is a multi-stage process involving biomarker discovery,
model development, laboratory validation and prospective study
validation. Besides, the current classifiers and gene maps should
be optimized and simplified to find the most advantageous
predictive genes in the list of characteristic genes. For example,
based on the CCS typing of CRC, five markers (CDX2, FRMD6, HTR2B,
ZEB1 and KER) were selected from a total of 146 characteristic
genes in the CCS classifier, and were subjected to IHC staining and
further classified in combination with MSI status (158). The accuracy of molecular
diagnostic tests largely depends on the quality of
paraffin-embedded tissues and the identification of tumor-rich
regions, which pose an additional task for pathologists (159,160).
Single omics data can only provide a limited insight
into the intrinsic molecular characteristics of CRC, whereas
integration of multiple omics offers a more comprehensive approach
to understanding tumor heterogeneity, uncovering the intricate
regulatory pathways of the disease and ultimately improving the
accuracy of CRC classification (157,161–165). The Cancer Genome Atlas has
multi-omics data from >10,000 patient samples across 33 types of
tumors, providing a comprehensive data set to explore the origin
and progression of tumors from multiple angles (166,167). This presents both opportunities
and challenges for the integration of multi-omics analysis. A
comprehensive analysis of muscle-infiltrating bladder cancer showed
that multi-omics classification was more effective than mRNA-based
clustering analysis for guiding clinical decision-making (157,168). Banias et al (159) described three molecular subtypes
of CRC (epithelial, mesenchymal and mixed) based on the IHC markers
E-cadherin, β-catenin, maspin and vimentin. A novel relationship
between intracellular maspin expression and MSI, tumor budding and
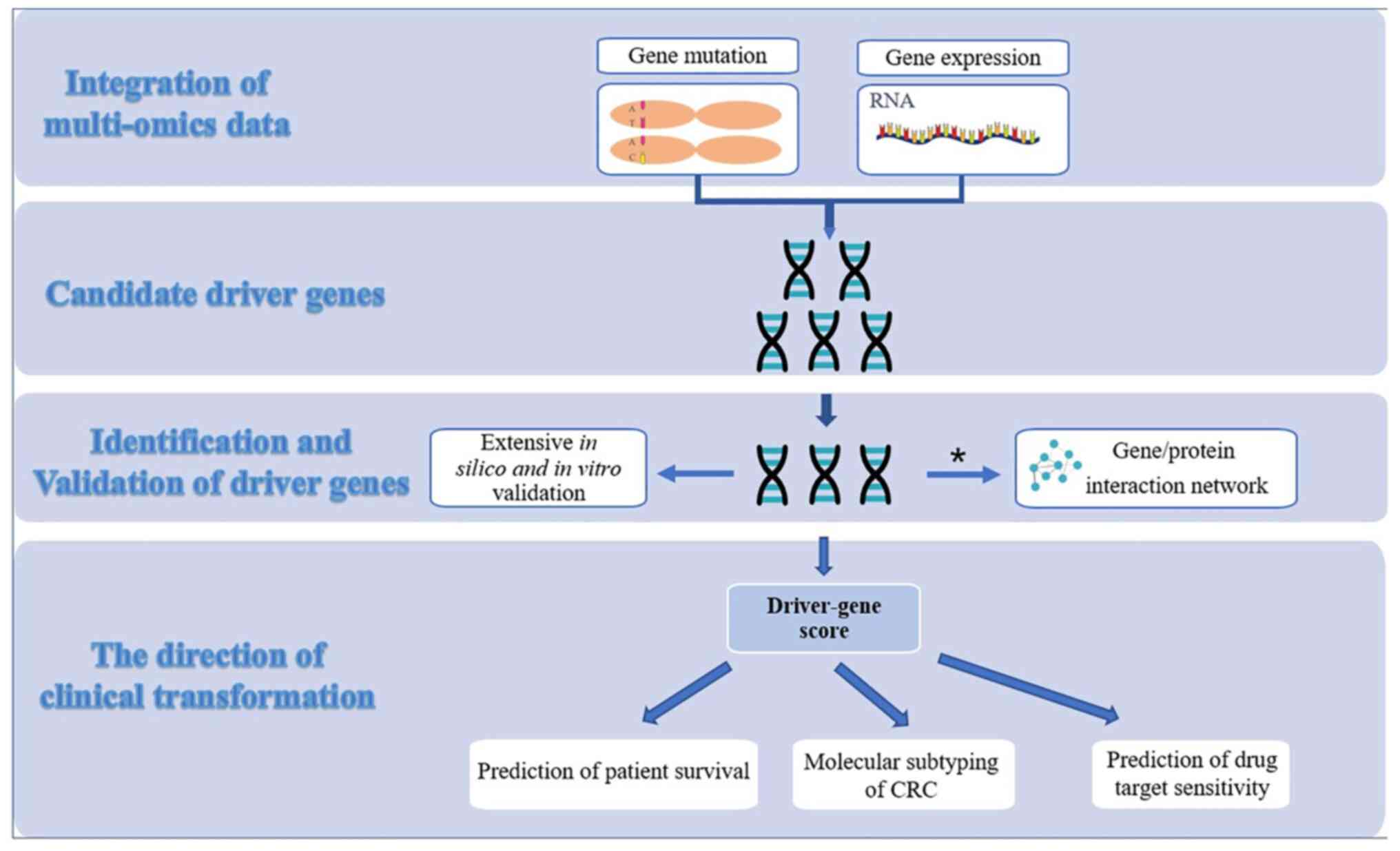
prognosis has far-reaching clinical significance. Integrative
approaches through network-based analysis have been previously
developed to predict the functional impact of driver gene mutations
(169–171) (Fig.
2). Successful transformation of routine clinical practice in
molecular typing still requires multidisciplinary and multi-omics
collaboration between basic cancer research, bioinformatics and
clinical research.
Not applicable.
This study was funded by the National Natural Science Foundation
of China (grant nos. 62276084 and 82103030).
Not applicable.
CLW, HZ and YXL reviewed the literature and collated
the relevant data. HZ and YXL made substantial contributions to
conception and design of the present study and participated in the
collection and interpretation of relevant data. CLW and HQH
designed the article structure. CLW and GYW wrote the original
draft. HQH, YLW and GYW reviewed and edited the manuscript. All
authors read and approved the final manuscript. Data authentication
is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Quirke P, Williams GT, Ectors N, Ensari A,
Piard F and Nagtegaal I: The future of the TNM staging system in
colorectal cancer: Time for a debate? Lancet Oncol. 8:651–657.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Amin MB, Edge SB, Greene FL, Byrd DR,
Brookland RK, Washington MK, Gershenwald JE, Compton CC, Hess KR,
Sullivan DC, et al: AJCC Cancer Staging Manual. 8th edition.
Springer; New York, NY: 2017, View Article : Google Scholar
|
|
4
|
Bae JM, Kim JH and Kang GH: Molecular
subtypes of colorectal cancer and their clinicopathologic features,
with an emphasis on the serrated neoplasia pathway. Arch Pathol Lab
Med. 140:406–412. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
National Cancer Institute (NCI), .
Director's Challenge: Toward a molecular classification of tumors
[J/0L]. https://grants.nih.gov/grants/guide/rfa-files/RFA-CA-98-027.html1999.
|
|
6
|
Benson AB, Venook AP, Al-Hawary MM, Arain
MA, Chen YJ, Ciombor KK, Cohen S, Cooper HS, Deming D, Farkas L, et
al: Colon cancer, version 2.2021, NCCN clinical practice guidelines
in oncology. J Natl Compr Canc Netw. 19:329–359. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Benson AB, Venook AP, Al-Hawary MM, Arain
MA, Chen YJ, Ciombor KK, Cohen S, Cooper HS, Deming D,
Garrido-Laguna I, et al: NCCN guidelines insights: rectal cancer,
version 6.2020. J Natl Compr Canc Netw. 18:806–815. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cohen R, Pudlarz T, Delattre JF, Colle R
and André T: Molecular targets for the treatment of metastatic
colorectal cancer. Cancers (Basel). 12:23502020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Guinney J, Dienstmann R, Wang X, de
Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda
G, Angelino P, et al: The consensus molecular subtypes of
colorectal cancer. Nat Med. 21:1350–1356. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lengauer C, Kinzler KW and Vogelstein B:
Genetic instability in colorectal cancers. Nature. 386:623–627.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Miao T, Wang Z, Sang N, Xiong R and Cao S:
Clinical significance of flow cytometric deoxyribonucleic acid
measurements of deparaffinized specimens in bladder tumors. Eur
Urol. 21:98–102. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mitelman F, Johansson B, Mandahl N and
Mertens F: Clinical significance of cytogenetic findings in solid
tumors. Cancer Genet Cytogenet. 95:1–8. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zeng WJ, Liu GY, Xu J, Zhou XD, Zhang YE
and Zhang N: Pathological characteristics, PCNA labeling index and
DNA index in prognostic evaluation of patients with moderately
differentiated hepatocellular carcinoma. World J Gastroenterol.
8:1040–1044. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lengauer C, Kinzler KW and Vogelstein B:
Genetic instabilities in human cancers. Nature. 396:643–649. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Watanabe T, Kobunai T, Yamamoto Y, Matsuda
K, Ishihara S, Nozawa K, Yamada H, Hayama T, Inoue E, Tamura J, et
al: Chromosomal instability (CIN) phenotype, CIN high or CIN low,
predicts survival for colorectal cancer. J Clin Oncol.
30:2256–2264. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li JA, Liu BC, Song Y and Chen X: Cyclin
A2 regulates symmetrical mitotic spindle formation and centrosome
amplification in human colon cancer cells. Am J Transl Res.
10:2669–2676. 2018.PubMed/NCBI
|
|
17
|
Grady WM: Genomic instability and colon
cancer. Cancer Metastasis Rev. 23:11–27. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sunde L, Bisgaard ML, Soll-Johanning H,
Jacobsen NO, Bolund L, Skouv J and Lynge E: Familial colorectal
cancer, can it be identified by microsatellite instability and
chromosomal instability? -A case-control study. Cancer Biomark.
5:197–205. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cisyk AL, Nugent Z, Wightman RH, Singh H
and McManus KJ: Characterizing microsatellite instability and
chromosome instability in interval colorectal cancers. Neoplasia.
20:943–950. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cisyk AL, Penner-Goeke S, Lichtensztejn Z,
Nugent Z, Wightman RH, Singh H and McManus KJ: Characterizing the
prevalence of chromosome instability in interval colorectal cancer.
Neoplasia. 17:306–316. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Walther A, Houlston R and Tomlinson I:
Association between chromosomal instability and prognosis in
colorectal cancer: A meta-analysis. Gut. 57:941–950. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bakhoum SF, Ngo B, Laughney AM, Cavallo
JA, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, et
al: Chromosomal instability drives metastasis through a cytosolic
DNA response. Nature. 553:467–472. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Orsetti B, Selves J, Bascoul-Mollevi C,
Lasorsa L, Gordien K, Bibeau F, Massemin B, Paraf F, Soubeyran I,
Hostein I, et al: Impact of chromosomal instability on colorectal
cancer progression and outcome. BMC Cancer. 14:1212014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Leber B, Maier B, Fuchs F, Chi J, Riffel
P, Anderhub S, Wagner L, Ho AD, Salisbury JL, Boutros M and Krämer
A: Proteins required for centrosome clustering in cancer cells. Sci
Transl Med. 2:33ra382010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Levine MS, Bakker B, Boeckx B, Moyett J,
Lu J, Vitre B, Spierings DC, Lansdorp PM, Cleveland DW, Lambrechts
D, et al: Centrosome amplification is sufficient to promote
spontaneous tumorigenesis in mammals. Dev Cell. 40:313–322.e5.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Raab MS, Breitkreutz I, Anderhub S,
Ronnest MH, Leber B, Larsen TO, Weiz L, Konotop G, Hayden PJ, Podar
K, et al: GF-15, a novel inhibitor of centrosomal clustering,
suppresses tumor cell growth in vitro and in vivo. Cancer Res.
72:5374–5385. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Warner SL, Bearss DJ, Han H and Von Hoff
DD: Targeting Aurora-2 kinase in cancer. Mol Cancer Ther.
2:589–595. 2003.PubMed/NCBI
|
|
28
|
Chen G, Bradford WD, Seidel CW and Li R:
Hsp90 stress potentiates rapid cellular adaptation through
induction of aneuploidy. Nature. 482:246–250. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pazdur R, Lassere Y, Soh LT, Ajani JA,
Bready B, Soo E, Sugarman S, Patt Y, Abbruzzese JL and Levin B:
Phase II trial of docetaxel (Taxotere) in metastatic colorectal
carcinoma. Ann Oncol. 5:468–470. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Swanton C, Tomlinson I and Downward J:
Chromosomal instability, colorectal cancer and taxane resistance.
Cell Cycle. 5:818–823. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Thibodeau SN, Bren G and Schaid D:
Microsatellite instability in cancer of the proximal colon.
Science. 260:816–819. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Aaltonen LA, Peltomäki P, Mecklin JP,
Järvinen H, Jass JR, Green JS, Lynch HT, Watson P, Tallqvist G,
Juhola M, et al: Replication errors in benign and malignant tumors
from hereditary nonpolyposis colorectal cancer patients. Cancer
Res. 54:1645–1648. 1994.PubMed/NCBI
|
|
33
|
Mori Y, Selaru FM, Sato F, Yin J, Simms
LA, Xu Y, Olaru A, Deacu E, Wang S, Taylor JM, et al: The impact of
microsatellite instability on the molecular phenotype of colorectal
tumors. Cancer Res. 63:4577–4582. 2003.PubMed/NCBI
|
|
34
|
Seppälä TT, Böhm JP, Friman M, Lahtinen L,
Väyrynen VM, Liipo TK, Ristimäki AP, Kairaluoma MV, Kellokumpu IH,
Kuopio TH and Mecklin JP: Combination of microsatellite instability
and BRAF mutation status for subtyping colorectal cancer. Br J
Cancer. 112:1966–1975. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Evrard C, Messina S, Sefrioui D, Frouin É,
Auriault ML, Chautard R, Zaanan A, Jaffrelot M, De La Fouchardière
C, Aparicio T, et al: Heterogeneity of mismatch repair status and
microsatellite instability between primary tumour and metastasis
and its implications for immunotherapy in colorectal cancers. Int J
Mol Sci. 23:44272022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kazama Y, Watanabe T, Kanazawa T, Tanaka
J, Tanaka T and Nagawa H: Microsatellite instability in poorly
differentiated adenocarcinomas of the colon and rectum:
Relationship to clinicopathological features. J Clin Pathol.
60:701–704. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gryfe R, Kim H, Hsieh ET, Aronson MD,
Holowaty EJ, Bull SB, Redston M and Gallinger S: Tumor
microsatellite instability and clinical outcome in young patients
with colorectal cancer. N Engl J Med. 342:69–77. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kim H, Jen J, Vogelstein B and Hamilton
SR: Clinical and pathological characteristics of sporadic
colorectal carcinomas with DNA replication errors in microsatellite
sequences. Am J Pathol. 145:148–156. 1994.PubMed/NCBI
|
|
39
|
Montminy EM, Zhou M, Maniscalco L, Heda R,
Kim MK, Patel SG, Wu XC, Itzkowitz SH and Karlitz JJ: Shifts in the
proportion of distant stage early-onset colorectal adenocarcinoma
in the United States. Cancer Epidemiol Biomarkers Prev. 31:334–341.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bailey CE, Hu CY, You YN, Bednarski BK,
Rodriguez-Bigas MA, Skibber JM, Cantor SB and Chang GJ: Increasing
disparities in the age-related incidences of colon and rectal
cancers in the United States, 1975-2010. JAMA Surg. 150:17–22.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jin Z, Dixon JG, Fiskum JM, Parekh HD,
Sinicrope FA, Yothers G, Allegra CJ, Wolmark N, Haller D, Schmoll
HJ, et al: Clinicopathological and molecular characteristics of
early-onset stage III colon adenocarcinoma: An analysis of the
ACCENT database. J Natl Cancer Inst. 113:1693–1704. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Guastadisegni C, Colafranceschi M, Ottini
L and Dogliotti E: Microsatellite instability as a marker of
prognosis and response to therapy: A meta-analysis of colorectal
cancer survival data. Eur J Cancer. 46:2788–2798. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Popat S, Hubner R and Houlston RS:
Systematic review of microsatellite instability and colorectal
cancer prognosis. J Clin Oncol. 23:609–618. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
NCCN Clinical Practice Guideline in
Oncology, . Version 3.2021. Available at. Colon. Cancer.NCCN.org
|
|
45
|
NCCN Clinical Practice Guideline in
Oncology, . Version 1.2021. Available at. Rectal. Cancer.NCCN.org
|
|
46
|
Kim ST, Lee J, Park SH, Park JO, Lim HY,
Kang WK, Kim JY, Kim YH, Chang DK, Rhee PL, et al: Clinical impact
of microsatellite instability in colon cancer following adjuvant
FOLFOX therapy. Cancer Chemother Pharmacol. 66:659–667. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Koenig JL, Toesca DAS, Harris JP, Tsai CJ,
Haraldsdottir S, Lin AY, Pollom EL and Chang DT: Microsatellite
instability and adjuvant chemotherapy in stage II colon cancer. Am
J Clin Oncol. 42:573–580. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
De'Angelis GL, Bottarelli L, Azzoni C,
De'Angelis N, Leandro G, Di Mario F, Gaiani F and Negri F:
Microsatellite instability in colorectal cancer. Acta Biomed.
89:97–101. 2018.
|
|
49
|
Taieb J, Svrcek M, Cohen R, Basile D,
Tougeron D and Phelip JM: Deficient mismatch repair/microsatellite
unstable colorectal cancer: Diagnosis, prognosis and treatment. Eur
J Cancer. 175:136–157. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Coupez D, Hulo P, Touchefeu Y, Bossard C
and Bennouna J: Pembrolizumab for the treatment of colorectal
cancer. Expert Opin Biol Ther. 20:219–226. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chung HC, Ros W, Delord JP, Perets R,
Italiano A, Shapira-Frommer R, Manzuk L, Piha-Paul SA, Xu L,
Zeigenfuss S, et al: Efficacy and safety of pembrolizumab in
previously treated advanced cervical cancer: Results from the phase
II KEYNOTE-158 study. J Clin Oncol. 37:1470–1478. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Le DT, Uram JN, Wang H, Bartlett BR,
Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et
al: PD-1 blockade in tumors with mismatch-repair deficiency. N Engl
J Med. 372:2509–2520. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Le DT, Yoshino T, Jäger D, Andre T,
Bendell JC, Wang R, Kang SP, Koshiji M and Diaz LA: KEYNOTE-164:
Phase II study of pembrolizumab (MK-3475) for patients with
previously treated, microsatellite instability-high advanced
colorectal carcinoma. J Clin Oncol. 34 (Suppl 4):TPS7872016.
View Article : Google Scholar
|
|
54
|
Muro K, Chung HC, Shankaran V, Geva R,
Catenacci D, Gupta S, Eder JP, Golan T, Le DT, Burtness B, et al:
Pembrolizumab for patients with PD-L1-positive advanced gastric
cancer (KEYNOTE-012): A multicentre, open-label, phase 1b trial.
Lancet Oncol. 17:717–726. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
O'Neil BH, Wallmark JM, Lorente D, Elez E,
Raimbourg J, Gomez-Roca C, Ejadi S, Piha-Paul SA, Stein MN, Abdul
Razak AR, et al: Safety and antitumor activity of the anti-PD-1
antibody pembrolizumab in patients with advanced colorectal
carcinoma. PLoS One. 12:e01898482017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Overman MJ, McDermott R, Leach JL, Lonardi
S, Lenz HJ, Morse MA, Desai J, Hill A, Axelson M, Moss RA, et al:
Nivolumab in patients with metastatic DNA mismatch repair-deficient
or microsatellite instability-high colorectal cancer (CheckMate
142): An open-label, multicentre, phase 2 study. Lancet Oncol.
18:1182–1191. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Le DT, Durham JN, Smith KN, Wang H,
Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et
al: Mismatch repair deficiency predicts response of solid tumors to
PD-1 blockade. Science. 357:409–413. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Diaz LA Jr, Shiu KK, Kim TW, Jensen BV,
Jensen LH, Punt C, Smith D, Garcia-Carbonero R, Benavides M, Gibbs
P, et al: Pembrolizumab versus chemotherapy for microsatellite
instability-high or mismatch repair-deficient metastatic colorectal
cancer (KEYNOTE-177): Final analysis of a randomised, open-label,
phase 3 study. Lancet Oncol. 23:659–670. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lenz HJ, Van Cutsem E, Luisa Limon M, Wong
KYM, Hendlisz A, Aglietta M, Garcia-Alfonso P, Neyns B, Luppi G,
Cardin DB, et al: First-line nivolumab plus low-dose ipilimumab for
microsatellite instability-high/mismatch repair-deficient
metastatic colorectal cancer: The phase II CheckMate 142 study. J
Clin Oncol. 40:161–170. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Josef LH, Parikh AR, Spigel DR, Cohn AL,
Yoshino T, Kochenderfer MD, Elez E, Shao SH, Deming DA, Holdridge
RC, et al: Nivolumab (NIVO) + 5-fluorouracil/leucovorin/oxaliplatin
(mFOLFOX6)/bevacizumab (BEV) versus mFOLFOX6/BEV for first-line
(1L) treatment of metastatic colorectal cancer (mCRC): Phase 2
results from CheckMate 9X8. J Clin Oncol. 40 (Suppl 4):S82022.
View Article : Google Scholar
|
|
61
|
Zhang C, Li D, Xiao B, Zhou C, Jiang W,
Tang J, Li Y, Zhang R, Han K, Hou Z, et al: B2M and JAK1/2-mutated
MSI-H colorectal carcinomas can benefit from Anti-PD-1 therapy. J
Immunother. 45:187–193. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Cohen R, Hain E, Buhard O, Guilloux A,
Bardier A, Kaci R, Bertheau P, Renaud F, Bibeau F, Fléjou JF, et
al: Association of primary resistance to immune checkpoint
inhibitors in metastatic colorectal cancer with misdiagnosis of
microsatellite instability or mismatch repair deficiency status.
JAMA Oncol. 5:551–555. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Luchini C, Bibeau F, Ligtenberg MJL, Singh
N, Nottegar A, Bosse T, Miller R, Riaz N, Douillard JY, Andre F and
Scarpa A: ESMO recommendations on microsatellite instability
testing for immunotherapy in cancer, and its relationship with
PD-1/PD-L1 expression and tumour mutational burden: A systematic
review-based approach. Ann Oncol. 30:1232–1243. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Hajirawala L and Barton JS: Diagnosis and
management of lynch syndrome. Dis Colon Rectum. 62:403–405. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Mouradov D, Domingo E, Gibbs P, Jorissen
RN, Li S, Soo PY, Lipton L, Desai J, Danielsen HE, Oukrif D, et al:
Survival in stage II/III colorectal cancer is independently
predicted by chromosomal and microsatellite instability, but not by
specific driver mutations. Am J Gastroenterol. 108:1785–1793. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Goel A, Arnold CN, Niedzwiecki D, Chang
DK, Ricciardiello L, Carethers JM, Dowell JM, Wasserman L, Compton
C, Mayer RJ, et al: Characterization of sporadic colon cancer by
patterns of genomic instability. Cancer Res. 63:1608–1614.
2003.PubMed/NCBI
|
|
67
|
Sinicrope FA, Rego RL, Halling KC, Foster
N, Sargent DJ, La Plant B, French AJ, Laurie JA, Goldberg RM,
Thibodeau SN and Witzig TE: Prognostic impact of microsatellite
instability and DNA ploidy in human colon carcinoma patients.
Gastroenterology. 131:729–737. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Diep CB, Thorstensen L, Meling GI,
Skovlund E, Rognum TO and Lothe RA: Genetic tumor markers with
prognostic impact in Dukes' stages B and C colorectal cancer
patients. J Clin Oncol. 21:820–829. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Rowan A, Halford S, Gaasenbeek M, Kemp Z,
Sieber O, Volikos E, Douglas E, Fiegler H, Carter N, Talbot I, et
al: Refining molecular analysis in the pathways of colorectal
carcinogenesis. Clin Gastroenterol Hepatol. 3:1115–1123. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Sobral D, Martins M, Kaplan S, Golkaram M,
Salmans M, Khan N, Vijayaraghavan R, Casimiro S, Fernandes A,
Borralho P, et al: Genetic and microenvironmental intra-tumor
heterogeneity impacts colorectal cancer evolution and metastatic
development. Commun Biol. 5:9372022. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Almendro V, Cheng YK, Randles A, Itzkovitz
S, Marusyk A, Ametller E, Gonzalez-Farre X, Muñoz M, Russnes HG,
Helland A, et al: Inference of tumor evolution during chemotherapy
by computational modeling and in situ analysis of genetic and
phenotypic cellular diversity. Cell Rep. 6:514–527. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Morris LG, Riaz N, Desrichard A,
Şenbabaoğlu Y, Hakimi AA, Makarov V, Reis-Filho JS and Chan TA:
Pan-cancer analysis of intratumor heterogeneity as a prognostic
determinant of survival. Oncotarget. 7:10051–10063. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Stanta G and Bonin S: Overview on clinical
relevance of intra-tumor heterogeneity. Front Med (Lausanne).
5:852018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
He WZ, Hu WM, Wang F, Rong YM, Yang L, Xie
QK, Yang YZ, Jiang C, Qiu HJ, Lu JB, et al: Comparison of mismatch
repair status between primary and matched metastatic sites in
patients with colorectal cancer. J Natl Compr Canc Netw.
17:1174–1183. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Huang Q, Yu T, Li L, Zhang Q, Zhang S, Li
B, Li X, Xiao W and Liu G: Intraindividual tumor heterogeneity of
mismatch repair status in metastatic colorectal cancer. Appl
Immunohistochem Mol Morphol. 31:84–93. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Costello JF, Frühwald MC, Smiraglia DJ,
Rush LJ, Robertson GP, Gao X, Wright FA, Feramisco JD, Peltomäki P,
Lang JC, et al: Aberrant CpG-island methylation has non-random and
tumour-type-specific patterns. Nat Genet. 24:132–138. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Gardiner-Garden M and Frommer M: CpG
islands in vertebrate genomes. J Mol Biol. 196:261–282. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Weber M, Hellmann I, Stadler MB, Ramos L,
Pääbo S, Rebhan M and Schübeler D: Distribution, silencing
potential and evolutionary impact of promoter DNA methylation in
the human genome. Nat Genet. 39:457–466. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Hinoue T, Weisenberger DJ, Lange CP, Shen
H, Byun HM, Van Den Berg D, Malik S, Pan F, Noushmehr H, van Dijk
CM, et al: Genome-scale analysis of aberrant DNA methylation in
colorectal cancer. Genome Res. 22:271–282. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Herman JG and Baylin SB: Gene silencing in
cancer in association with promoter hypermethylation. N Engl J Med.
349:2042–2054. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Weisenberger DJ, Siegmund KD, Campan M,
Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D,
Buchanan D, et al: CpG island methylator phenotype underlies
sporadic microsatellite instability and is tightly associated with
BRAF mutation in colorectal cancer. Nat Genet. 38:787–793. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Ogino S, Kawasaki T, Kirkner GJ, Kraft P,
Loda M and Fuchs CS: Evaluation of markers for CpG island
methylator phenotype (CIMP) in colorectal cancer by a large
population-based sample. J Mol Diagn. 9:305–314. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Issa JP: CpG island methylator phenotype
in cancer. Nat Rev Cancer. 4:988–993. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Samowitz WS: The CpG island methylator
phenotype in colorectal cancer. J Mol Diagn. 9:281–283. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Bae JM, Kim MJ, Kim JH, Koh JM, Cho NY,
Kim TY and Kang GH: Differential clinicopathological features in
microsatellite instability-positive colorectal cancers depending on
CIMP status. Virchows Arch. 459:55–63. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Weisenberger DJ, Levine AJ, Long TI,
Buchanan DD, Walters R, Clendenning M, Rosty C, Joshi AD, Stern MC,
LeMarchand L, et al: Association of the colorectal CpG island
methylator phenotype with molecular features, risk factors, and
family history. Cancer Epidemiol Biomarkers Prev. 24:512–519. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Ogino S, Nosho K, Kirkner GJ, Kawasaki T,
Meyerhardt JA, Loda M, Giovannucci EL and Fuchs CS: CpG island
methylator phenotype, microsatellite instability, BRAF mutation and
clinical outcome in colon cancer. Gut. 58:90–96. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Wang J, Deng Z, Lang X, Jiang J, Xie K, Lu
S, Hu Q, Huo Y, Xiong X, Zhu N and Zhang W: Meta-analysis of the
prognostic and predictive role of the CpG island methylator
phenotype in colorectal cancer. Dis Markers. 2022:42548622022.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Cha Y, Kim KJ, Han SW, Rhee YY, Bae JM,
Wen X, Cho NY, Lee DW, Lee KH, Kim TY, et al: Adverse prognostic
impact of the CpG island methylator phenotype in metastatic
colorectal cancer. Br J Cancer. 115:164–171. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Jover R, Nguyen TP, Pérez-Carbonell L,
Zapater P, Payá A, Alenda C, Rojas E, Cubiella J, Balaguer F,
Morillas JD, et al: 5-Fluorouracil adjuvant chemotherapy does not
increase survival in patients with CpG island methylator phenotype
colorectal cancer. Gastroenterology. 140:1174–1181. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Iacopetta B, Kawakami K and Watanabe T:
Predicting clinical outcome of 5-fluorouracil-based chemotherapy
for colon cancer patients: Is the CpG island methylator phenotype
the 5-fluorouracil-responsive subgroup? Int J Clin Oncol.
13:498–503. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Jahan Z, Benthani FA, Currey N, Parker HW,
Dahlstrom JE, Caldon CE and Kohonen-Corish MRJ: MCC gene silencing
is a CpG island methylator phenotype-associated factor that
predisposes colon cancer cells to irinotecan and olaparib. Cancers
(Basel). 14:28592022. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Pawel K and Maria Małgorzata S: CpG island
methylator phenotype-a hope for the future or a road to nowhere?
Int J Mol Sci. 23:8302022. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Cancer Genome Atlas Network, .
Comprehensive molecular characterization of human colon and rectal
cancer. Nature. 487:330–337. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Andreyev HJ, Norman AR, Cunningham D,
Oates J, Dix BR, Iacopetta BJ, Young J, Walsh T, Ward R, Hawkins N,
et al: Kirsten ras mutations in patients with colorectal cancer:
The ‘RASCAL II’ study. Br J Cancer. 85:692–696. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Sugimoto T, Ohta M, Ikenoue T, Yamada A,
Tada M, Fujishiro M, Ogura K, Yamaji Y, Okamoto M, Kanai F, et al:
Macroscopic morphologic subtypes of laterally spreading colorectal
tumors showing distinct molecular alterations. Int J Cancer.
127:1562–1569. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Amado RG, Wolf M, Peeters M, Van Cutsem E,
Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, et
al: Wild-type KRAS is required for panitumumab efficacy in patients
with metastatic colorectal cancer. J Clin Oncol. 26:1626–1634.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Bokemeyer C, Bondarenko I, Makhson A,
Hartmann JT, Aparicio J, de Braud F, Donea S, Ludwig H, Schuch G,
Stroh C, et al: Fluorouracil, leucovorin, and oxaliplatin with and
without cetuximab in the first-line treatment of metastatic
colorectal cancer. J Clin Oncol. 27:663–671. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Douillard JY, Oliner KS, Siena S,
Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham
D, Jassem J, et al: Panitumumab-FOLFOX4 treatment and RAS mutations
in colorectal cancer. N Engl J Med. 369:1023–1034. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Lièvre A, Bachet JB, Boige V, Cayre A, Le
Corre D, Buc E, Ychou M, Bouché O, Landi B, Louvet C, et al: KRAS
mutations as an independent prognostic factor in patients with
advanced colorectal cancer treated with cetuximab. J Clin Oncol.
26:374–379. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Osumi H, Shinozaki E, Suenaga M, Matsusaka
S, Konishi T, Akiyoshi T, Fujimoto Y, Nagayama S, Fukunaga Y, Ueno
M, et al: RAS mutation is a prognostic biomarker in colorectal
cancer patients with metastasectomy. Int J Cancer. 139:803–811.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Stintzing S, Modest DP, Rossius L, Lerch
MM, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U,
Al-Batran SE, Heintges T, et al: FOLFIRI plus cetuximab versus
FOLFIRI plus bevacizumab for metastatic colorectal cancer (FIRE-3):
A post-hoc analysis of tumour dynamics in the final RAS wild-type
subgroup of this randomised open-label phase 3 trial. Lancet Oncol.
17:1426–1434. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Van Cutsem E, Lenz HJ, Köhne CH, Heinemann
V, Tejpar S, Melezinek I, Beier F, Stroh C, Rougier P, van Krieken
JH and Ciardiello F: Fluorouracil, leucovorin, and irinotecan plus
cetuximab treatment and RAS mutations in colorectal cancer. J Clin
Oncol. 33:692–700. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Venook AP, Niedzwiecki D, Lenz HJ,
Innocenti F, Fruth B, Meyerhardt JA, Schrag D, Greene C, O'Neil BH,
Atkins JN, et al: Effect of first-line chemotherapy combined with
cetuximab or bevacizumab on overall survival in patients with KRAS
wild-type advanced or metastatic colorectal cancer: A randomized
clinical trial. JAMA. 317:2392–2401. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Taieb J, Le Malicot K, Penault-Llorca FM,
Bouche O, Shi Q, Thibodeau SN, Tabernero J, Mini E, Goldberg RM,
Folprecht G, et al: Prognostic value of BRAF V600E and KRAS exon 2
mutations in microsatellite stable (MSS), stage III colon cancers
(CC) from patients (pts) treated with adjuvant FOLFOX+/-cetuximab:
A pooled analysis of 3934 pts from the PETACC8 and N0147 trials. J
Clin Oncol. 33 (Suppl 15):S35072015. View Article : Google Scholar
|
|
106
|
Stintzing S, Wirapati P, Lenz HJ,
Neureiter D, Fischer von Weikersthal L, Decker T, Kiani A, Kaiser
F, Al-Batran S, Heintges T, et al: Consensus molecular subgroups
(CMS) of colorectal cancer (CRC) and first-line efficacy of FOLFIRI
plus cetuximab or bevacizumab in the FIRE3 (AIO KRK-0306) trial.
Ann Oncol. 30:1796–1803. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Tejpar S, Bertagnolli M, Bosman F, Lenz
HJ, Garraway L, Waldman F, Warren R, Bild A, Collins-Brennan D,
Hahn H, et al: Prognostic and predictive biomarkers in resected
colon cancer: Current status and future perspectives for
integrating genomics into biomarker discovery. Oncologist.
15:390–404. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Yamauchi M, Morikawa T, Kuchiba A, Imamura
Y, Qian ZR, Nishihara R, Liao X, Waldron L, Hoshida Y, Huttenhower
C, et al: Assessment of colorectal cancer molecular features along
bowel subsites challenges the conception of distinct dichotomy of
proximal versus distal colorectum. Gut. 61:847–854. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Chen KH, Lin YL, Liau JY, Tsai JH, Tseng
LH, Lin LI, Liang JT, Lin BR, Hung JS, Chang YL, et al: BRAF
mutation may have different prognostic implications in early- and
late-stage colorectal cancer. Med Oncol. 33:392016. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Van Cutsem E, Köhne CH, Láng I, Folprecht
G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D,
Tejpar S, et al: Cetuximab plus irinotecan, fluorouracil, and
leucovorin as first-line treatment for metastatic colorectal
cancer: Updated analysis of overall survival according to tumor
KRAS and BRAF mutation status. J Clin Oncol. 29:2011–2019. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Venderbosch S, Nagtegaal ID, Maughan TS,
Smith CG, Cheadle JP, Fisher D, Kaplan R, Quirke P, Seymour MT,
Richman SD, et al: Mismatch repair status and BRAF mutation status
in metastatic colorectal cancer patients: A pooled analysis of the
CAIRO, CAIRO2, COIN, and FOCUS studies. Clin Cancer Res.
20:5322–5330. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Sinicrope FA, Shi Q, Allegra CJ, Smyrk TC,
Thibodeau SN, Goldberg RM, Meyers JP, Pogue-Geile KL, Yothers G,
Sargent DJ and Alberts SR: Association of DNA mismatch repair and
mutations in BRAF and KRAS with survival after recurrence in stage
III colon cancers: A secondary analysis of 2 randomized clinical
trials. JAMA Oncol. 3:472–480. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Birgisson H, Edlund K, Wallin U, Påhlman
L, Kultima HG, Mayrhofer M, Micke P, Isaksson A, Botling J,
Glimelius B and Sundström M: Microsatellite instability and
mutations in BRAF and KRAS are significant predictors of
disseminated disease in colon cancer. BMC Cancer. 15:1252015.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Liao X, Lochhead P, Nishihara R, Morikawa
T, Kuchiba A, Yamauchi M, Imamura Y, Qian ZR, Baba Y, Shima K, et
al: Aspirin use, tumor PIK3CA mutation, and colorectal-cancer
survival. N Engl J Med. 367:1596–1606. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Thorpe LM, Yuzugullu H and Zhao JJ: PI3K
in cancer: Divergent roles of isoforms, modes of activation and
therapeutic targeting. Nat Rev Cancer. 15:7–24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Sepulveda AR, Hamilton SR, Allegra CJ,
Grody W, Cushman-Vokoun AM, Funkhouser WK, Kopetz SE, Lieu C,
Lindor NM, Minsky BD, et al: Molecular biomarkers for the
evaluation of colorectal cancer: Guideline from the american
society for clinical pathology, college of American pathologists,
association for molecular pathology, and the American society of
clinical oncology. J Clin Oncol. 35:1453–1486. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Akinleye A, Avvaru P, Furqan M, Song Y and
Liu D: Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer
therapeutics. J Hematol Oncol. 6:882013. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Carew JS, Kelly KR and Nawrocki ST:
Mechanisms of mTOR inhibitor resistance in cancer therapy. Target
Oncol. 6:17–27. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Liao X, Morikawa T, Lochhead P, Imamura Y,
Kuchiba A, Yamauchi M, Nosho K, Qian ZR, Nishihara R, Meyerhardt
JA, et al: Prognostic role of PIK3CA mutation in colorectal cancer:
Cohort study and literature review. Clin Cancer Res. 18:2257–2268.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Tougeron D, Sha D, Manthravadi S and
Sinicrope FA: Aspirin and colorectal cancer: Back to the future.
Clin Cancer Res. 20:1087–1094. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Valtorta E, Martino C, Sartore-Bianchi A,
Penaullt-Llorca F, Viale G, Risio M, Rugge M, Grigioni W,
Bencardino K, Lonardi S, et al: Assessment of a HER2 scoring system
for colorectal cancer: Results from a validation study. Mod Pathol.
28:1481–1491. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Tu J, Yu Y, Liu W and Chen S: Significance
of human epidermal growth factor receptor 2 expression in
colorectal cancer. Exp Ther Med. 9:17–24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Kavanagh DO, Chambers G, O'Grady L, Barry
KM, Waldron RP, Bennani F, Eustace PW and Tobbia I: Is
overexpression of HER-2 a predictor of prognosis in colorectal
cancer? BMC Cancer. 9:12009. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Seo AN, Kwak Y, Kim DW, Kang SB, Choe G,
Kim WH and Lee HS: HER2 status in colorectal cancer: Its clinical
significance and the relationship between HER2 gene amplification
and expression. PLoS One. 9:e985282014. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Sveen A, Kopetz S and Lothe RA:
Biomarker-guided therapy for colorectal cancer: Strength in
complexity. Nat Rev Clin Oncol. 17:11–32. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Siena S, Di Bartolomeo M, Raghav K,
Masuishi T, Loupakis F, Kawakami H, Yamaguchi K, Nishina T, Fakih
M, Elez E, et al: Trastuzumab deruxtecan (DS-8201) in patients with
HER2-expressing metastatic colorectal cancer (DESTINY-CRC01): a
multicentre, open-label, phase 2 trial. Lancet Oncol. 22:779–789.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
De Roock W, Claes B, Bernasconi D, De
Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V,
Papamichael D, Laurent-Puig P, et al: Effects of KRAS, BRAF, NRAS,
and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy
in chemotherapy-refractory metastatic colorectal cancer: A
retrospective consortium analysis. Lancet Oncol. 11:753–762. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Gil-Raga M, Jantus-Lewintre E, Gallach S,
Giner-Bosch V, Frangi-Caregnato A, Safont-Aguilera MJ,
Garde-Noguera J, Zorraquino-Pina E, Garcia-Martinez M and
Camps-Herrero C: Molecular subtypes in early colorectal cancer
associated with clinical features and patient prognosis. Clin
Transl Oncol. 20:1422–1429. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Ros J, Saoudi N, Salvà F, Baraibar I,
Alonso G, Tabernero J and Elez E: Ongoing and evolving clinical
trials enhancing future colorectal cancer treatment strategies.
Expert Opin Investig Drugs. 31:235–247. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Jass JR: Classification of colorectal
cancer based on correlation of clinical, morphological and
molecular features. Histopathology. 50:113–130. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
De Sousa E, Melo F, Wang X, Jansen M,
Fessler E, Trinh A, de Rooij LP, de Jong JH, de Boer OJ, van
Leersum R, Bijlsma MF, et al: Poor-prognosis colon cancer is
defined by a molecularly distinct subtype and develops from
serrated precursor lesions. Nat Med. 19:614–618. 2013. View Article : Google Scholar
|
|
132
|
Ogino S and Goel A: Molecular
classification and correlates in colorectal cancer. J Mol Diagn.
10:13–27. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Mangi FH, Shaikh TA, Soria D, Waryah AM,
Ujjan ID, Qureshi JN and Syed BM: Novel molecular classification of
colorectal cancer and correlation with survival. Saudi J Biol Sci.
29:3929–3936. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Singh MP, Rai S, Pandey A, Singh NK and
Srivastava S: Molecular subtypes of colorectal cancer: An emerging
therapeutic opportunity for personalized medicine. Genes Dis.
8:133–145. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Ten Hoorn S, de Back TR, Sommeijer DW and
Vermeulen L: Clinical value of consensus molecular subtypes in
colorectal cancer: A systematic review and meta-analysis. J Natl
Cancer Inst. 114:503–516. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Valenzuela G, Canepa J, Simonetti C, Solo
de Zaldivar L, Marcelain K and González-Montero J: Consensus
molecular subtypes of colorectal cancer in clinical practice: A
translational approach. World J Clin Oncol. 12:1000–1008. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Allen WL, Dunne PD, McDade S, Scanlon E,
Loughrey M, Coleman H, McCann C, McLaughlin K, Nemeth Z, Syed N, et
al: Transcriptional subtyping and CD8 immunohistochemistry
identifies poor prognosis stage II/III colorectal cancer patients
who benefit from adjuvant chemotherapy. JCO Precis Oncol.
2018.PO.17.00241. 2018. View Article : Google Scholar
|
|
138
|
Li Y, Yao Q, Zhang L, Mo S, Cai S, Huang D
and Peng J: Immunohistochemistry-based consensus molecular subtypes
as a prognostic and predictive biomarker for adjuvant chemotherapy
in patients with stage II colorectal cancer. Oncologist.
25:e1968–e1979. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Roepman P, Schlicker A, Tabernero J,
Majewski I, Tian S, Moreno V, Snel MH, Chresta CM, Rosenberg R,
Nitsche U, et al: Colorectal cancer intrinsic subtypes predict
chemotherapy benefit, deficient mismatch repair and
epithelial-to-mesenchymal transition. Int J Cancer. 134:552–562.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Dunne PD, O'Reilly PG, Coleman HG, Gray
RT, Longley DB, Johnston PG, Salto-Tellez M, Lawler M and McArt DG:
Stratified analysis reveals chemokine-like factor (CKLF) as a
potential prognostic marker in the MSI-immune consensus molecular
subtype CMS1 of colorectal cancer. Oncotarget. 7:36632–36644. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Borelli B, Fontana E, Giordano M,
Antoniotti C, Lonardi S, Bergamo F, Pietrantonio F, Morano F,
Tamburini E, Boccaccino A, et al: Prognostic and predictive impact
of consensus molecular subtypes and CRCAssigner classifications in
metastatic colorectal cancer: A translational analysis of the
TRIBE2 study. ESMO Open. 6:1000732021. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Del Rio M, Mollevi C, Bibeau F, Vie N,
Selves J, Emile JF, Roger P, Gongora C, Robert J, Tubiana-Mathieu
N, et al: Molecular subtypes of metastatic colorectal cancer are
associated with patient response to irinotecan-based therapies. Eur
J Cancer. 76:68–75. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Okita A, Takahashi S, Ouchi K, Inoue M,
Watanabe M, Endo M, Honda H, Yamada Y and Ishioka C: Consensus
molecular subtypes classification of colorectal cancer as a
predictive factor for chemotherapeutic efficacy against metastatic
colorectal cancer. Oncotarget. 9:18698–18711. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Yuki S, Gamoh M, Denda T, Takashima A,
Takahashi S, Nakamura M, Ohori H, Yamaguchi T, Kobayashi Y, Baba H,
et al: Analysis of consensus molecular subtypes (CMS)
classification in the TRICOLORE trial: A randomized phase III trial
of S-1 and irinotecan (IRI) plus bevacizumab (Bmab) versus mFOLFOX6
or CapeOX plus Bmab as first-line treatment for metastatic
colorectal cancer (mCRC). J Clin Oncol. 38 (Suppl 4):S1692020.
View Article : Google Scholar
|
|
145
|
Mooi JK, Wirapati P, Asher R, Lee CK,
Savas P, Price TJ, Townsend A, Hardingham J, Buchanan D, Williams
D, et al: The prognostic impact of consensus molecular subtypes
(CMS) and its predictive effects for bevacizumab benefit in
metastatic colorectal cancer: Molecular analysis of the AGITG MAX
clinical trial. Ann Oncol. 29:2240–2246. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Lenz HJ, Ou FS, Venook AP, Hochster HS,
Niedzwiecki D, Goldberg RM, Mayer RJ, Bertagnolli MM, Blanke CD,
Zemla T, et al: Impact of consensus molecular subtype on survival
in patients with metastatic colorectal cancer: Results from
CALGB/SWOG 80405 (alliance). J Clin Oncol. 37:1876–1885. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Aderka D, Stintzing S and Heinemann V:
Explaining the unexplainable: Discrepancies in results from the
CALGB/SWOG 80405 and FIRE-3 studies. Lancet Oncol. 20:e274–e283.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Lan Y, Zhang D, Xu C, Hance KW, Marelli B,
Qi J, Yu H, Qin G, Sircar A, Hernández VM, et al: Enhanced
preclinical antitumor activity of M7824, a bifunctional fusion
protein simultaneously targeting PD-L1 and TGF-β. Sci Transl Med.
10:eaan54882018. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Mehrvarz Sarshekeh A, Lam M, Zorrilla IR,
Holliday EB, Das P, Kee BK, Overman MJ, Parseghian CM, Shen JPYC,
Tam A, et al: Consensus molecular subtype (CMS) as a novel integral
biomarker in colorectal cancer: A phase II trial of bintrafusp alfa
in CMS4 metastatic CRC. J Clin Oncol. 38 (15 Suppl):S40842020.
View Article : Google Scholar
|
|
150
|
Eisenberg D, Marcotte EM, Xenarios I and
Yeates TO: Protein function in the post-genomic era. Nature.
405:823–826. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
von Mering C, Krause R, Snel B, Cornell M,
Oliver SG, Fields S and Bork P: Comparative assessment of
large-scale data sets of protein-protein interactions. Nature.
417:399–403. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Barbieri I and Kouzarides T: Role of RNA
modifications in cancer. Nat Rev Cancer. 20:303–322. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Eisenberg E: Proteome diversification by
RNA editing. Methods Mol Biol. 2181:229–251. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi
Z, Chambers MC, Zimmerman LJ, Shaddox KF, Kim S, et al:
Proteogenomic characterization of human colon and rectal cancer.
Nature. 513:382–387. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Li C, Sun YD, Yu GY, Cui JR, Lou Z, Zhang
H, Huang Y, Bai CG, Deng LL, Liu P, et al: Integrated omics of
metastatic colorectal cancer. Cancer Cell. 38:734–747.e9. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Ciardiello F, Ciardiello D, Martini G,
Napolitano S, Tabernero J and Cervantes A: Clinical management of
metastatic colorectal cancer in the era of precision medicine. CA
Cancer J Clin. 72:372–401. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Wang W, Kandimalla R, Huang H, Zhu L, Li
Y, Gao F, Goel A and Wang X: Molecular subtyping of colorectal
cancer: Recent progress, new challenges and emerging opportunities.
Semin Cancer Biol. 55:37–52. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Trinh A, Trumpi K, De Sousa EMF, Wang X,
de Jong JH, Fessler E, Kuppen PJ, Reimers MS, Swets M, Koopman M,
et al: Practical and robust identification of molecular subtypes in
colorectal cancer by immunohistochemistry. Clin Cancer Res.
23:387–398. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Banias L, Jung I, Chiciudean R and Gurzu
S: From dukes-MAC staging system to molecular classification:
Evolving concepts in colorectal cancer. Int J Mol Sci. 23:94552022.
View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Chen G, Yang Z, Eshleman JR, Netto GJ and
Lin MT: Molecular diagnostics for precision medicine in colorectal
cancer: Current status and future perspective. Biomed Res Int.
2016:98506902016. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Drier Y and Domany E: Do two
machine-learning based prognostic signatures for breast cancer
capture the same biological processes? PLoS One. 6:e177952011.
View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Ein-Dor L, Kela I, Getz G, Givol D and
Domany E: Outcome signature genes in breast cancer: Is there a
unique set? Bioinformatics. 21:171–178. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Haury AC, Gestraud P and Vert JP: The
influence of feature selection methods on accuracy, stability and
interpretability of molecular signatures. PLoS One. 6:e282102011.
View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Huang S, Chaudhary K and Garmire LX: More
is better: Recent progress in multi-omics data integration methods.
Front Genet. 8:842017. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Vucic EA, Thu KL, Robison K, Rybaczyk LA,
Chari R, Alvarez CE and Lam WL: Translating cancer ‘omics’ to
improved outcomes. Genome Res. 22:188–195. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Akbani R, Ng PK, Werner HM, Shahmoradgoli
M, Zhang F, Ju Z, Liu W, Yang JY, Yoshihara K, Li J, et al: A
pan-cancer proteomic perspective on the cancer genome atlas. Nat
Commun. 5:38872014. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Cancer Genome Atlas Research Network, .
Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA,
Ellrott K, Shmulevich I, Sander C and Stuart JM: The cancer genome
atlas pan-cancer analysis project. Nat Genet. 45:1113–1120. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Robertson AG, Kim J, Al-Ahmadie H,
Bellmunt J, Guo G, Cherniack AD, Hinoue T, Laird PW, Hoadley KA,
Akbani R, et al: Comprehensive molecular characterization of
muscle-invasive bladder cancer. Cell. 171:540–556.e25. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Suo C, Hrydziuszko O, Lee D, Pramana S,
Saputra D, Joshi H, Calza S and Pawitan Y: Integration of somatic
mutation, expression and functional data reveals potential driver
genes predictive of breast cancer survival. Bioinformatics.
31:2607–2613. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Bertrand D, Chng KR, Sherbaf FG, Kiesel A,
Chia BK, Sia YY, Huang SK, Hoon DS, Liu ET, Hillmer A and Nagarajan
N: Patient-specific driver gene prediction and risk assessment
through integrated network analysis of cancer omics profiles.
Nucleic Acids Res. 43:e442015. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Pavel AB, Sonkin D and Reddy A:
Integrative modeling of multi-omics data to identify cancer drivers
and infer patient-specific gene activity. BMC Syst Biol. 10:162016.
View Article : Google Scholar : PubMed/NCBI
|