Introduction
Intravascular large B-cell lymphoma (IVLBCL) is an
aggressive type of extranodal malignant lymphoma with selective
proliferation of neoplastic lymphoid cells within the vascular
lumina (1). It is divided into
‘Eastern’ and ‘Western’ variants, with the former often being
associated with bone marrow involvement and hemophagocytic
syndrome, and the latter showing central nervous system and skin
involvement (2–4).
Pulmonary involvement of IVLBCL is relatively
frequent at ~60% (5), and the
majority of these cases are secondary lymphomas originating from
systemic diseases. However, initial or predominant pulmonary
presentations are rarely reported (5). With vascular occlusion of the lungs,
patients may show some non-specific clinical presentations, such as
hypoxemia, shortness of breath or fever, which may also occur in a
number of other lung diseases, such as infection, infarction and
diffuse interstitial pneumonia. Thus, primary pulmonary IVLBCL is
highly misdiagnosed in clinical practice, and is often confirmed at
autopsy or by lung biopsy (6). The
present study summarized 4 cases of primary pulmonary IVLBCL and
reviewed the previous literature in this area. The patients of the
present study were diagnosed based on clinical and computed
tomographic (CT) evaluations, as well as the laboratory and
pathological examinations at Nanjing Drum Tower Hospital (Nanjing,
China). These cases presented at this hospital between March 2010
and March 2017.
Case report
Case 1
A 69-year-old female patient was referred to Nanjing
Drum Tower Hospital in March 2010 with a 2-month history of
coughing with white sputum without cause and progressive dyspnea on
exertion. In addition, the patient experienced fever (37.3-39.8°C)
for a duration of 1 week. A physical examination revealed cyanosis
of the lips, edema of the lower limbs, clear consciousness, no skin
rashes, nodules or hemorrhagic spots and no hepatosplenomegaly.
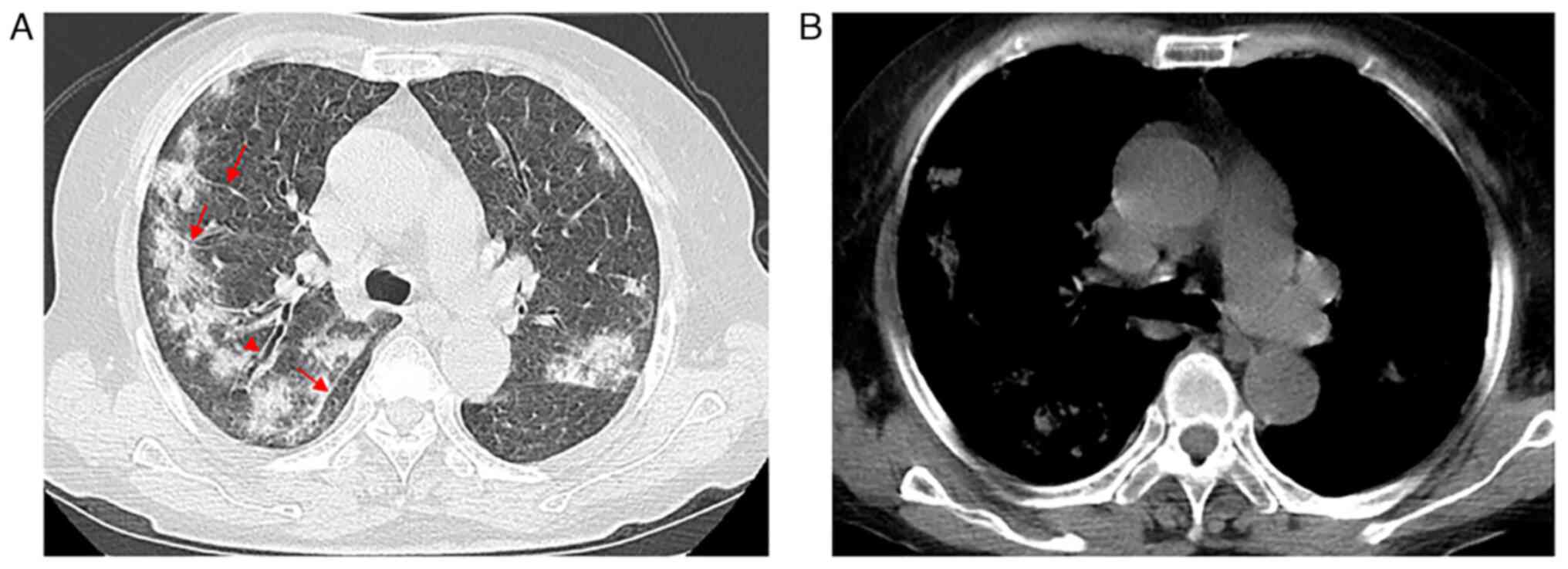
After admission, CT examination was performed. The CT images
demonstrated increased attenuation in bilateral lung parenchyma
with multiple ground-glass opacities (GGOs) and part progression to
consolidation (Fig. 1A), especially
on the superior lobes. Interlobular septal thickening and partial
thickening of bronchovascular bundles as well as ‘tram track-like’
changes were observed (Fig. 1A),
along with no lymph node enlargement within the mediastinum or
hilus (Fig. 1B). During
hospitalization, treatment for infection using imipenem/cilastatin
sodium (0.5 g/8 h) with intermittent use of acetaminophen tablets
(0.5 g) for fever reduction was administered, but no notable effect
was revealed. Finally, pulmonary IVLBCL was diagnosed using
transbronchial lung biopsy (TBLB). Abnormal lymphoid cells
distributed within the blood vessels were observed under a light
microscope, and the diagnosis of pulmonary IVLBCL was further
confirmed through immunohistochemical staining. The patient
rejected chemotherapy treatment and died due to respiratory failure
shortly (20 days) after diagnosis.
Case 2
A 68-year-old male with a past medical history of
diabetes for ~20 years presented at Nanjing Drum Tower Hospital in
May 2012 with a 2-month history of coughing with scantly white
sputum, chest tightness, dyspnea on exertion and a recurrent
low-grade fever. The patient experienced weight loss (~3 kg) during
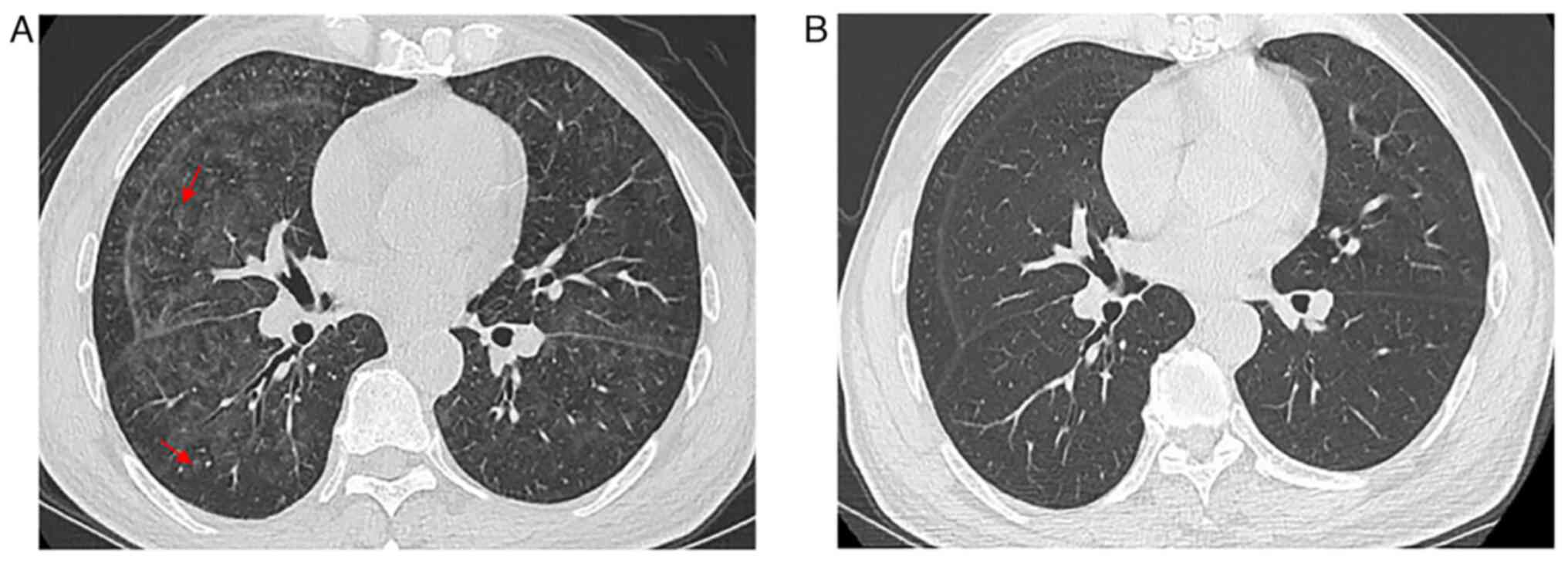
these 2 months. The patient underwent chest CT. Initial CT imaging
evaluation indicated a ground pattern in a mosaic distribution and
small centrilobular nodules (Fig.
2A). The patient received anti-inflammatory treatment. However,
the symptoms did not significantly improve. Bone marrow aspiration
followed by biopsy indicated that the patient had pancytopenia, and
a fluorescent in situ hybridization study detected clonal
rearrangements of Igκ-VJ and Igκ-V/in, without immunoglobulin heavy
locus/BCL2, BCL6 and c-MYC fragmentations or TP53 deletion. TBLB
was performed, which demonstrated only a small number of atypical
cells. Primary pulmonary IVLBCL was eventually confirmed using open
lung biopsy. An incision was made in the fourth intercostal space
along the anterior axillary line of the left side of the chest, and
lung tissues were respectively excised from the lingual and dorsal
segments of the left upper lobe and the dorsal segment of the left
lower lobe. The patient started receiving rituximab,
cyclophosphamide, doxorubicin, vincristine and prednisolone
(R-CHOP) treatment but experienced an allergic reaction after being
injected with rituximab (600 mg). Rituximab was removed from the
treatment plan, and after improvement with anti-allergy treatment,
the patient received 6 cycles of CHOP [1.2 g cyclophosphamide,
intravenously (iv) day l; 90 mg doxorubicin, iv day l; 4 mg
vincristine, iv day l; 50 mg prednisolone, orally twice per day,
days 1–5] treatment, with each cycle lasting for 3 weeks. At
completion of the sixth cycle, the disease was in complete
remission. As visualized using a CT scan, the bilateral lungs were
clear (Fig. 2B).
A seventh cycle of chemotherapy began with CHOP
(same drug dosage as aforementioned) within 3 months of the
completion of the sixth cycle as the patient was admitted to
hospital with a fever, cough and chest discomfort. At 2 weeks after
discharge after the seventh cycle, an additional three cycles of
CHOP (same drug dosage as aforementioned) were provided at 1-week
intervals. An additional three cycles of ifosfamide (8 g, iv day
2), carboplatin (500 mg, iv day 2) and etoposide (0.16 g, iv days
1–3) were administered only 1 month after the latest completion of
chemotherapy for the recurrence of cough, sputum and fever. The
patient's clinical symptoms improved markedly after the treatment.
However, the patient was hospitalized again 1 year later due to
fever and infection secondary to myelosuppression. Eventually, the
treatment failed, and the patient passed away.
Case 3
A 65-year-old male patient was admitted to Nanjing
Drum Tower Hospital in March 2017 with a 3-month history of
progressive dyspnea and cough. The patient had no prior history of
lung disease, chest pain, intermittent fever or night sweating. No
focal findings were noted on physical examination, especially of
the skin and central nervous system, and lymph nodes were not
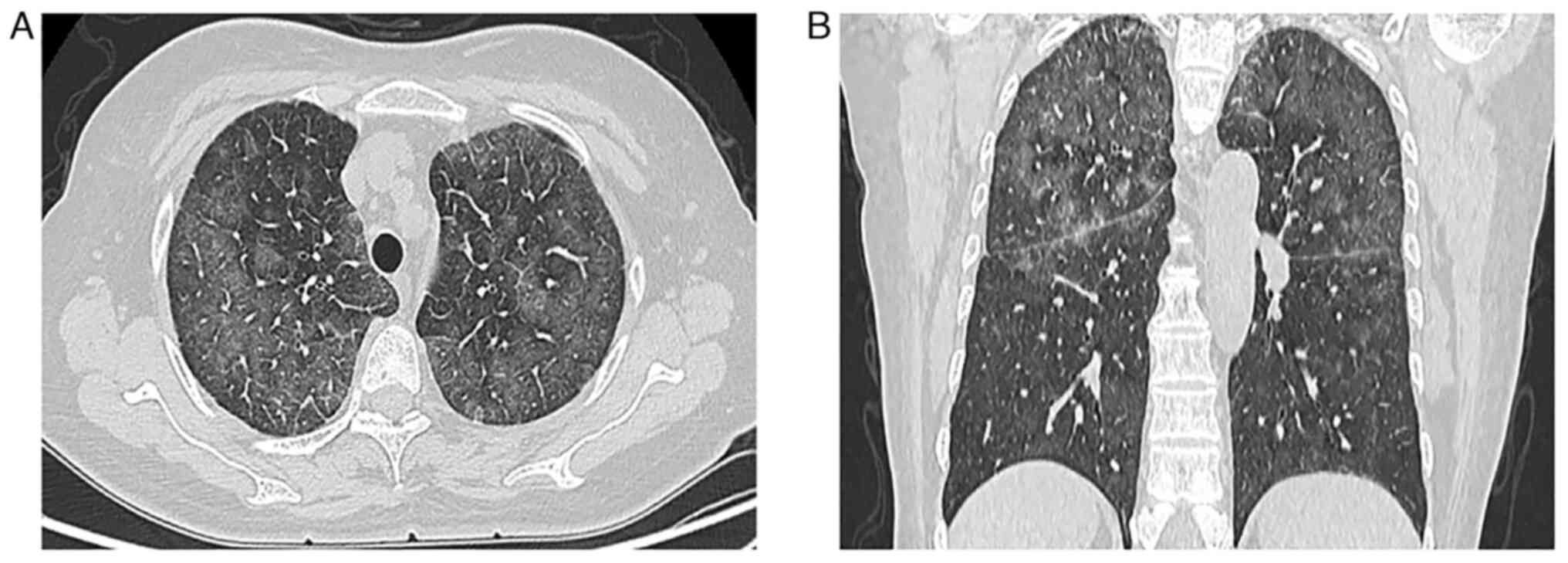
palpable. A CT examination revealed diffuse ground-glass
attenuation and thickened interlobular septa in the upper lungs
(Fig. 3A and B). No abnormal soft
tissue mass was observed in the mediastinum, bilateral hilus and
axilla. Whole-body positron emission tomography-computed tomography
(PET-CT) scan revealed only mild 18F-fluorodeoxyglucose
(FDG) uptake in both lungs. The patient was mistakenly diagnosed
with pneumonia and received wide-spectrum antibiotics treatment (2
g cefazoxime sodium, twice per day) for half a month. The patient's
symptoms continued, and the treatment had no effect. The patient
was discharged from hospital after diagnosis of IVLBCL using TBLB
and did not return for follow-up treatment and examination.
Case 4
A 60-year-old female patient presented at Nanjing
Drum Tower Hospital in August 2015 with a 1-month history of
progressive dyspnea and a 20-day history of intermittent high
fever, with a maximum temperature of ~39°C. The patient had no
history of lung disease and was a non-smoker. No cyanosis,
clubbing, neurological abnormalities, nor cutaneous lesions were
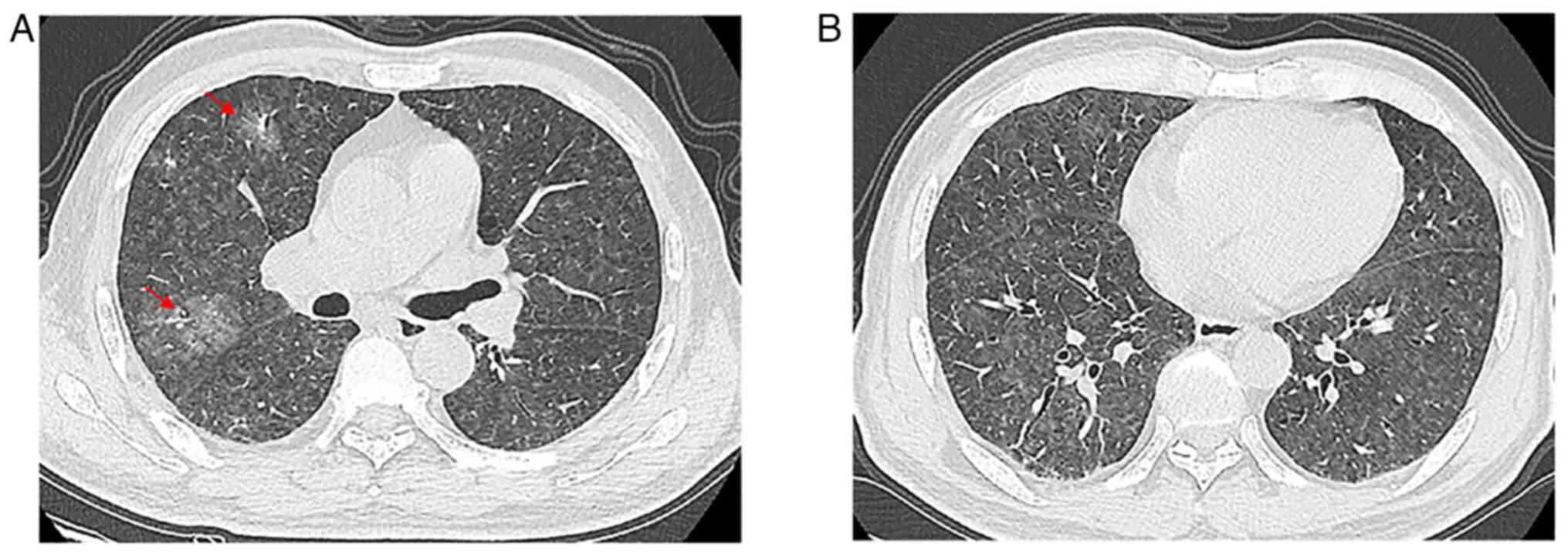
observed on physical examination. A high revolution chest CT (HRCT)
examination was performed, and the images revealed pulmonary
nodules with part-solid diffused GGOs in the lungs without pleural
involvement (Fig. 4A and B).
Additional examinations of whole-body CT examination yielded normal
findings. The patient was diagnosed with interstitial pneumonia and
the infection was treated with moxifloxacin hydrochloride (0.4
g/day) and sodium chloride injection solution for 7 days, and
intermittent use of indomethacin (0.05 g) for antipyretic
treatment, but the symptoms did not notably improve. Since there
were no specific clinical or imaging findings and anti-inflammatory
therapy failed, the patient underwent TBLB examination (of the
superior lobe of the right lung). Light microscopy revealed a
diffuse intravascular proliferation of atypical B cells and,
combined with immunohistochemistry, IVLBCL was diagnosed. After a
short period of glucocorticoids (40 mg methylprednisolone sodium
succinate per day) treatment, the dyspnea initially improved, and
the treatment appeared to be working. Unfortunately, the patient
eventually discharged and gave up treatment due to a severe and
uncontrollable lung infection.
Laboratory results
Laboratory data demonstrated that the patients had
anemia (n=4, 100%), thrombocytopenia (n=4, 100%), pancytopenia
(n=3, 75%) and hypoxemia (n=4, 100%). Laboratory findings were
notable for marked high serum lactate dehydrogenase (LDH) levels,
elevated erythrocyte sedimentation rates (ESRs) and increased
C-reactive protein (CRP) levels (Table
I).
 | Table I.Overview of the 4 cases of primary
pulmonary intravascular large B-cell lymphoma. |
Table I.
Overview of the 4 cases of primary
pulmonary intravascular large B-cell lymphoma.
| Case no. | Age, years | Sex | Hb, g/l | Plt,
×109/l | SaO2,
% | LDH, IU/l | CRP, mg/l | ESR, mm/h | Treatment and
outcome |
|---|
| 1 | 69 | Female | 87 | 107 | 91 | 2,250 | 88 | 97 | None; deceased
shortly (20 days) after diagnosis |
| 2 | 68 | Male | 102 | 96 | 93 | 1,449 | 13 | 106 | R-CHOP, CHOP, ICE;
Deceased 17 months after diagnosis |
| 3 | 65 | Male | 110 | 111 | 92 | 886 | 27 | 92 | N/A (outside
consultation) |
| 4 | 60 | Female | 90 | 102 | 94 | 1,542 | 80 | 80 | N/A (outside
consultation) |
Histopathological results
All patients underwent TBLB, and partial tissue
biopsies of different segments of the lung lobes were performed.
Only 1 patient underwent open lung biopsy. The pathological
analysis showed mild expansion of the capillary lumen within
alveolar and peribronchial interstitial tissue by a proliferation
of large tumor cells. The intravascular tumor cells had multiple
(1–3) nucleoli, little cytoplasm, high nucleus
to cytoplasm ratios and irregular nuclear contours. Mitoses,
including atypical forms, were observed.
Immunohistochemical results
The tissues were incubated in 10% neutral formalin
fixative for overnight fixation at room temperature for 12 h. After
slicing the sample to a thickness of 4 µm and performing
deparaffinization and hydration pretreatments, 3% hydrogen peroxide
was added to cover the part to be stained with peroxidase blocking
agent, and the sample was incubated at room temperature for 30 min.
Mouse primary antibody was added to the sample and incubated
overnight at 4°C. Rabbit secondary antibody, with a horseradish
peroxidase conjugate, was added to the sample and incubated at room
temperature for 30 min. DAB (Ultraview Universal DAB Kit; Roche
Tissue Diagnostics) was used for 10 sec of chromogenic staining,
followed by staining with hematoxylin for 4–8 min at room
temperature. Finally, the location and intensity of the markers
were observed under an optical microscope. The primary antibodies
selected include: CD20 (cat. no. ZM-0039), CD79a (cat. no.
ZA-0293), CD31 (cat. no. ZM-0044), CD3 (cat. no. ZM-0417), CD10
(cat. no. ZM-0283), CD5 (cat. no. ZM-0280), Bcl-6 (cat. no.
ZM-0011), and Mum-1 (cat. no. ZM-0401), all purchased from OriGene
Technologies, Inc.
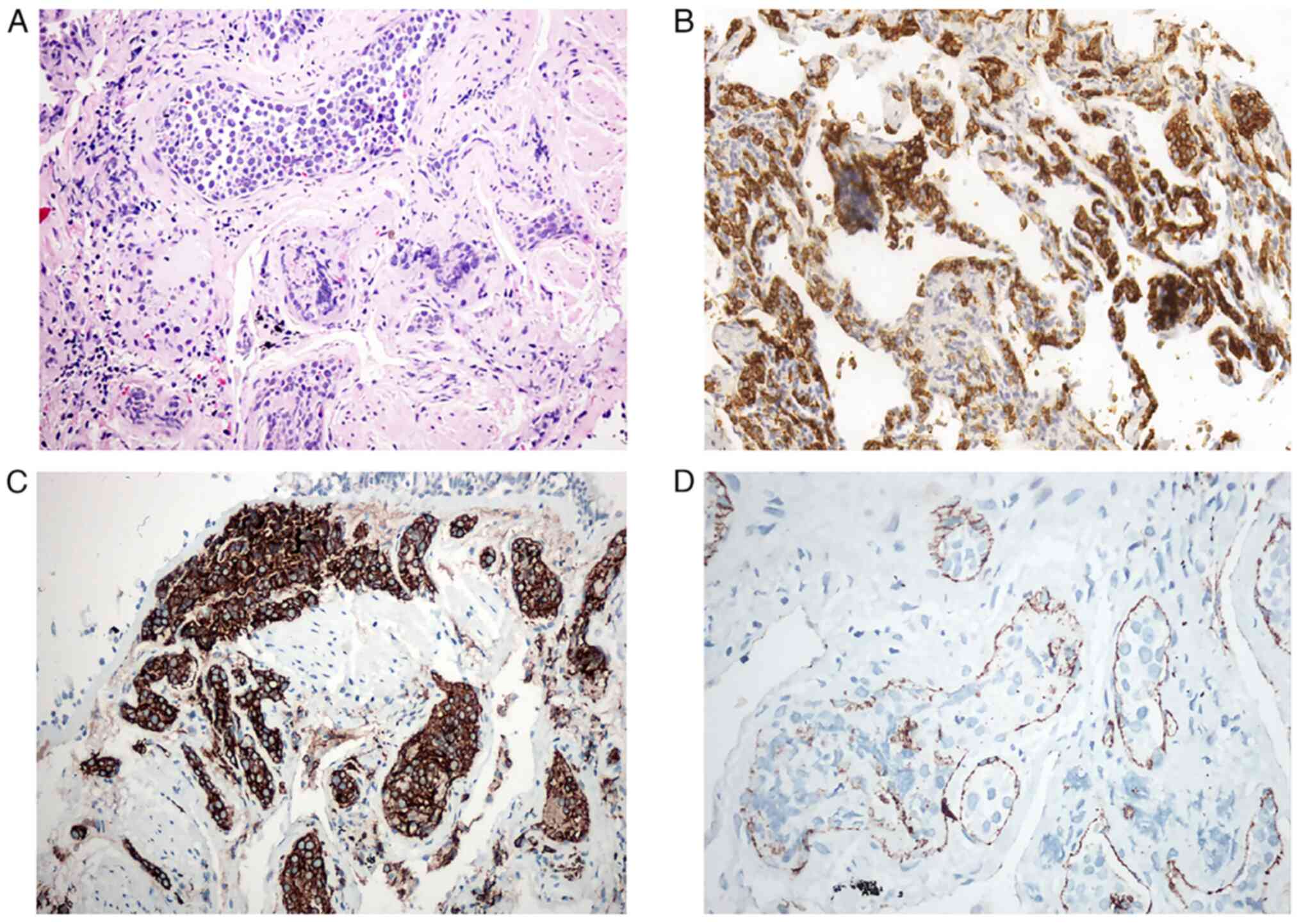
H&E and immunohistochemistry staining revealed
tumor cells that were positive for CD20 (n=4, 100%) and CD79a (n=3,
75%), which identified all four tumors as a lymphoma of B cell
lineage (Fig. 5A-C). In addition,
the tumor cells of all 4 cases were negative for the T cell
markers, CD3 and cytokeratin (n=4, 100%). CD31 was positive in the
small vessel endothelium of case 2 (Fig. 5D). Only 1 case was considered to
have a germinal center B cell (GCB) phenotype, as the cells were
positive for CD10, BCL6 and multiple myeloma oncogene 1 (Mum-1)
(Table II).
| Table II.Immunohistochemical analysis for the 4
cases of primary pulmonary intravascular large B-cell lymphoma. |
Table II.
Immunohistochemical analysis for the 4
cases of primary pulmonary intravascular large B-cell lymphoma.
|
| Antibodies |
|---|
|
|
|
|---|
| Case no. | CD20 | CD3 | CD10 | CD79a | Mum-1 | Ki-67, %
positive | BCL6 | CK |
|---|
| 1 |
Positive | Negative | Negative |
Positive | Negative | 40 | Negative | Negative |
| 2 |
Positive | Negative | Negative |
Positive |
Positive | 70 | Negative | Negative |
| 3 |
Positive | Negative |
Positive |
Positive |
Positive | 50 |
Positive | Negative |
| 4 |
Positive | Negative | Negative | Negative |
Positive | 20 | Negative | Negative |
Discussion
In 1959, Pfleger and Tappeiner first reported a type
of malignant lymphoma, known as ‘malignant angioendotheliomatosis’
(1,2,7).
Malignant angioendotheliomatosis is an uncommon subcategory of
extranodal large B-cell lymphoma derived from blood vessels and
features the presence of large tumor cells (4,8,9). A
study recommended differentiating the classification of ‘classical
IVLBCL’ and ‘IVLBCL associated with hemophagocytic syndrome’ due to
the crossover of clinical manifestations, without being limited to
regional differences in Europe and Asia (4). ‘Classical IVLBCL’ is characterized by
cutaneous and/or neurological involvement. The features of ‘IVLBCL
associated with hemophagocytic syndrome’ are hemophagocytic
syndrome, bone marrow involvement, fever, hepatosplenomegaly,
and/or thrombocytopenia (4).
Pulmonary IVLBCL without evidence of extrapulmonary
involvement is uncommonly reported and is difficult to diagnose
antemortem (10,11). It predominantly occurs in the 6th
decade of life and is likely to get worse if not treated with an
adequate remedy. The clinical manifestations of pulmonary IVLBCL
are non-specific, and share numerous clinical features with
pulmonary tumor embolism, which are often experienced in both
diseases, including dyspnea, fever, cough, night sweats, tachypnea
and hypoxemia (3,5,6,12–14).
The immortal proliferation cells (abnormally proliferating B
lymphocytes) play a central role in the development of pulmonary
tumor embolism, and the presence of pulmonary hypertension and cor
pulmonale are almost irreversibly associated with blood clots that
occur in the lung (12). Pulmonary
function testing typically measures reduced diffusion capacity for
carbon monoxide compared with normal baselines (13). In addition, serum LDH, ESR and CRP
levels are often elevated by different degrees in IVLBCL (1,4,11). All
these results may indicate that a patient is suffering from a type
of lymph-proliferative disease.
Compared with other pulmonary lymphoma, IVLBCL
generally does not involve lymphadenopathy or a localizing solid
mass, as the lymphomatous cells mainly involve the pulmonary
arteries and capillary beds (14).
Results of CT assessments are diverse and can be inconspicuous or
show GGO and interstitial infiltration (11,12,14).
In the cases of the present study, patchy areas of GGO existed on
patient presentation and this accentuated the bilateral lung
attenuation resulting from pulmonary vascular obstruction. In case
1, the chest HRCT revealed bilateral disease that was
pneumonia-like, which progressed partly to consolidation. Due to
local interlobular septal thickening along with the thickening of
bronchovascular bundles, new GGO indicated lymphatic and
hematological spread. The other 3 cases showed bilateral GGOs,
micronodules and thickened interlobular septa in the lungs without
pleural involvement, which suggested that the disease may spread
along lymphatic structures. In case 2, the pulmonary shadows
completely disappeared after a short-term chemotherapy schedule,
which supported the diagnosis of IVLBCL. The etiology of GGO
(heterogeneous and partially consolidated) in all four patients
remains unclear. Malignant cells may invade the alveolar space,
resulting in consolidation (increased density) on CT imaging.
Increased diffuse density in bilateral lungs with GGOs and
thickening of interstitial septum needs to be distinguished from
non-neoplastic lesions, such as interstitial diseases and
mechanical pneumonia, which may also present as progressive dyspnea
with cough and other manifestations of obstructive ventilation.
TBLB and bronchoalveolar lavage fluid can be used to aid diagnosis.
Undoubtably, IVLBCL needs to be differentiated from venous
thromboembolism and other intravascular malignancies of the lung,
including lymphomatoid granulomatosis, angiocentric lymphoma and
pulmonary involvement by acute and chronic lymphocytic leukemias
(3). At this point of the patient
examination, the clinical manifestations and CT characteristics may
be non-specific, and immunohistochemistry will provide great help
for the correct diagnosis.
PET-CT is used in the early diagnosis of isolated
pulmonary IVLBCL (11,15) as it can differentiate IVLBCL from
other lung diseases, such as idiopathic pulmonary fibrosis and
pneumonia. PET-CT scans typically show a high metabolic activity of
tumor cells in IVLBCL, which is similar to that observed for
diffuse large B-cell lymphoma (DLBCL), as IVLBCL falls under the
category of DLBCL (16). At
present, PET has a high ability to differentiate lymph nodes,
especially invasive lymph node lesions, and almost all the lymph
nodes and extranodal organs can be found through a single
examination. PET has been widely used in the diagnosis, staging and
restaging, efficacy evaluation and prognosis prediction of
lymphoma. However, only 1 case in the present study presented
pulmonary mild 18F-FDG uptake on PET, which was
atypical.
Histological findings, such as pulmonary veins,
peribronchial arterioles and accumulation of malignant lymphocytes,
are key for the definitive diagnosis of primary pulmonary IVLBCL
(3,13). Moreover, small vessels filled with
large B cells are also confirmed using H&E staining. B cell
markers (CD19, CD20, CD79a and PAX5) are usually expressed while T
cell markers (CD3 and CD4) are typically negative on
immunohistochemical examination (3,9). CD31
mainly marks endothelial tissue and can be used to identify benign
and malignant vasogenic tumors. Meanwhile, CD31 is not expressed in
non-vasogenic tumors, so it has high specificity and sensitivity
for studying vasogenic tumors (17). Pathologically, IVLBCL and DLBCL have
similar cytology and sometimes the same immunophenotypes.
Nonetheless, combined with CT findings, DLBCL often presents as a
localized solid mass and is easy to identify. In the present study,
1 patient was positive for CD31, indicating endovascular origin of
the tumor. CD10 or BCL6 are hallmarks for the determination for GCB
and non-GCB (3,10). Based on the Hans algorithm, cases of
CD10+ VLBCL are categorized as GCB-type (1,10,18),
thus, the large B cells in case 3 were
CD10+/Bcl-6+/Mum-1+, which is classified as a
GCB-type in immunophenotypical analysis. Moreover, GCB phenotypes
with patterns such as
CD10+/BCL6+/Mum-1+,
CD10+/BCL6−/Mum-1+,
CD10+/BCL-6−/Mum-1−,
CD10+/BCL6+/Mum-1− and
CD10−/BCL-6+/Mum-1−, are
considered to indicate an improved prognosis compared with that of
a non-GCB phenotype (18,19). In total, ~20% of cases of IVLBCL
have been classified as GCB-type (1), which corresponds with the results of
the present study.
In the case of malignant tumor, a high Ki-67
proliferation index (PI) in neoplastic cells is closely associated
with poor prognosis. At present, the majority of researchers agree
that a 20% cut-off PI for Ki-67 in lymphoma is clinically
meaningful (20). The Ki-67 PI was
≥20% (20–70%) in the present study. In addition, in the present
study, the patient of case 2 underwent genetic testing, which
demonstrated no specific chromosomal alterations and no clonal
rearrangement of the variable region of the immunoglobulin heavy
chain gene, but a genetic recombination of Igκ-VJ and Igκ-V/in was
detected, indicating abnormal lymphocyte proliferation (9). Structural abnormalities of chromosomes
1 (1p), 6 and 18 (trisomy 18) have also been previously reported
(3).
With an improved clinical awareness and the
development of immunohistochemical technology, it is possible to
confirm the diagnosis of this rare disease during the lifetime of
the patient. The present study performed a diagnosis of 4 living
patient cases of primary pulmonary IVLBCL using lung tissue
biopsies (3 cases by TBLB and 1 case by open lung biopsy). IVLBCL
is a diffuse lung disease and, even though TBLB is already highly
diagnostic, due to the limited availability of lung tissue samples
from minimally invasive procedures leading to a failed accurate
diagnosis, a surgical lung biopsy may ultimately be performed.
Furthermore, we hypothesize that it will be possible for IVLBCL to
be cured in the future. Treatment applied in combination with
chemotherapy for intermediate and high-grade lymphomas has achieved
success in the field of long-term disease-free survival (3,10). In
addition, combined systemic chemotherapy, similar to that for
diffuse large B-cell lymphoma, may help treat IVLBCL with long-term
disease-free survival as a clinical outcome (13).
In conclusion, the present study demonstrated that
primary pulmonary IVLBCL should be considered in differential
diagnoses, especially when GGOs are associated with small nodules
and thickened interlobular septa in the upper lung. Other general
clinical manifestations include fever, night sweats and weight
loss, high elevations in serum LDH, ESR and CRP in laboratory
examination and no response to anti-inflammatory treatments. A
histopathological examination of lung biopsy samples is needed to
confirm the definitive diagnosis and therapeutic procedures. The
present study also demonstrated the potential diagnostic
effectiveness of TBLB in early diagnosis and the relative
non-invasiveness of the technique, compared with open chest
surgery. Taken together, a standard regimen for lymphoma treatment
may result in an overall improved clinical response.
Acknowledgements
Not applicable.
Funding
This project was supported by a key project grant by The Medical
Science and Technology Development Foundation (grant no.
ZKX21023).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
requests.
Authors' contributions
MZ and YC contributed to manuscript writing and
editing, and data collection; HF analyzed and interpreted the
patient data; JS performed the pathological examinations; BZ
contributed significantly to the concept and design of the study;
XM contributed to conceptualization and supervision and was
responsible for revision of the manuscript for important
intellectual content. All authors have read and approved the final
version of the manuscript. MZ, YC and XM confirm the authenticity
of all the raw data.
Ethics approval and consent to
participate
Since this study was a retrospective study, we
applied to exempt patients from informed consent, and it was
supervised and approved by the Ethics Committee of Nanjing Drum
Tower Hospital (Nanjing, China; approval no. 2022-009-01).
Patient consent for publication
Since this study was a retrospective study, we
applied to exempt patients from informed consent for publication,
and it was approved by the Ethics Committee of Nanjing Drum Tower
Hospital (Nanjing, China; approval no. 2022-009-01).
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shimada K, Kinoshita T, Naoe T and
Nakamura S: Presentation and management of intravascular large
B-cell lymphoma. Lancet Oncol. 10:895–902. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pfleger L and Tappeiner J: On the
recognition of systematized endotheliomatosis of the cutaneous
blood vessels (reticuloendotheliosis? Hautarzt. 10:359–363.
1959.(In German). PubMed/NCBI
|
|
3
|
Sanguedolce F, Zanelli M, Zizzo M, Bisagni
A, Soriano A, Cocco G, Palicelli A, Santandrea G, Caprera C, Corsi
M, et al: Primary pulmonary B-Cell lymphoma: A review and update.
Cancers (Basel). 13:4152021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brunet V, Marouan S, Routy JP, Hashem MA,
Bernier V, Simard R, Petrella T, Lamarre L, Théorêt G, Carrier C,
et al: Retrospective study of intravascular large B-cell lymphoma
cases diagnosed in Quebec: A retrospective study of 29 case
reports. Medicine (Baltimore). 96:e59852017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Satoh T, Arai E, Kayano H, Sakaguchi H,
Takahashi N, Tsukasaki K and Yasuda M: Pulmonary intravascular
large B-cell lymphoma accompanying synchronous primary pulmonary
adenocarcinoma and benign interstitial lesions. J Clin Exp Hematop.
59:140–144. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Masood S, Vijayan K and Wheeler YY:
Primary lung intravascular large B-cell lymphoma clinically
mimicking sarcoidosis: A rare case report and review of literature.
Respir Med Case Rep. 29:1009892019.PubMed/NCBI
|
|
7
|
Geer M, Roberts E, Shango M, Till BG,
Smith SD, Abbas H, Hill BT, Kaplan J, Barr PM, Caimi P, et al:
Multicentre retrospective study of intravascular large B-cell
lymphoma treated at academic institutions within the United States.
Br J Haematol. 186:255–262. 2019.PubMed/NCBI
|
|
8
|
Swerdlow SH, Campo E, Pileri SA, Harris
NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz
AD and Jaffe ES: The 2016 revision of the World Health Organization
classification of lymphoid neoplasms. Blood. 127:2375–2390. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Orwat DE and Batalis NI: Intravascular
large B-cell lymphoma. Arch Pathol Lab Med. 136:333–338. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ponzoni M, Campo E and Nakamura S:
Intravascular large B-cell lymphoma: A chameleon with multiple
faces and many masks. Blood. 132:1561–1567. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu F, Wang Z, Xing X, Yu M and Shi B: The
value of 18F-FDG PET/CT in diagnostic procedure of
intravascular large B-cell lymphoma presenting fever of unknown
origin and pulmonary hypertension as an initial manifestation. Clin
Nucl Med. 41:506–507. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang H, Huang Y, Xu CW and Lin L: Clinical
analysis of tumor and non-tumor patients complicated with pulmonary
embolism. Int J Clin Exp Med. 8:18729–18736. 2015.PubMed/NCBI
|
|
13
|
Matea F, Alowami S, Bonert M, Sur M,
Shargall Y and Naqvi AH: Pulmonary intravascular B-cell lymphoma
with angiotropism/angioinvasion mimicking interstitial lung
disease: A clinical dilemma and potential diagnostic challenge.
Case Rep Hematol. 2018:38213922018.PubMed/NCBI
|
|
14
|
Nguyen TT, Sekiguchi H, Yi ES and Ryu JH:
Occult diffuse neoplasm in the lungs: Intravascular large B-cell
lymphoma. Am J Med. 134:926–929. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Spencer J, Dusing R, Yap W, Hill J and
Walter C: Intravascular large B-cell lymphoma presenting with
diffusely increased pulmonary fluorodeoxyglucose uptake without
corresponding CT abnormality. Radiol Case Rep. 14:260–264. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hong JY, Kim HJ, Ko YH, Choi JY, Jung CW,
Kim SJ and Kim WS: Clinical features and treatment outcomes of
intravascular large B-cell lymphoma: A single-center experience in
Korea. Acta Haematol. 131:18–27. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sullivan HC, Edgar MA, Cohen C, Kovach CK,
HooKim K and Reid MD: The utility of ERG, CD31 and CD34 in the
cytological diagnosis of angiosarcoma: An analysis of 25 cases. J
Clin Pathol. 68:44–50. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Robetorye RS, Ramsower CA, Rosenthal AC,
Yip TK, Wendel Spiczka AJ, Glinsmann-Gibson BJ and Rimsza LM:
Incorporation of digital gene expression profiling for
cell-of-origin determination (Lymph2Cx testing) into the routine
work-up of diffuse large B-cell lymphoma. J Hematop. 2:3–10. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Scott DW, Mottok A, Ennishi D, Wright GW,
Farinha P, Ben-Neriah S, Kridel R, Barry GS, Hother C, Abrisqueta
P, et al: Prognostic significance of diffuse large B-cell lymphoma
cell of origin determined by digital gene expression in
formalin-fixed paraffin-embedded tissue biopsies. J Clin Oncol.
33:2848–2856. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
He X, Chen Z, Fu T, Jin X, Yu T, Liang Y,
Zhao X and Huang L: Ki-67 is a valuable prognostic predictor of
lymphoma but its utility varies in lymphoma subtypes: Evidence from
a systematic meta-analysis. BMC Cancer. 14:1532014. View Article : Google Scholar : PubMed/NCBI
|