Introduction
AT-rich interactive domain-containing protein 1a
(ARID1a) is a functionally relevant component of the
switch/sucrose non-fermentable (SWI-SNF) chromatin remodeller
complex. Access to various genes is regulated via this complex
(1,2). ARID1a is one of the most
frequently mutated genes in carcinomas and is considered a tumour
suppressor gene (3,4). Approximately 50% of clear cell ovarian
cancers show ARID1a mutations and over 90% of ARID1a
mutations that occur in ovarian cancers are nonsense or frame-shift
mutations that result in loss of protein expression (5,6).
Immunohistochemical analyses visualising ARID1a protein in tissue
are therefore well suited to reveal underlying gene alterations.
Several publications describe the relevance of the ARID1a
mutation in adenocarcinomas of the stomach, which occur
predominantly in the microsatellite unstable (MSI)- and
Epstein-Barr virus (EBV)-associated subtypes (7–10).
However, molecular alteration of ARID1a is likely to
represent a biologically minor epiphenomenon of the already highly
mutated or epigenetically altered tumours in these subgroups.
Little data are available on the significance in oesophageal
adenocarcinoma (EAC). Our group, as well as another, have shown
that ARID1a alterations occur in ~10% of EAC, including MSI
tumours (compare in more detail in ‘Discussion’) (11,12).
When Drage et al describe a clustering of the
medullary phenotype in ARID1a loss EAC, this may merely describe
the underlying MSI phenotype (11).
The clinical and molecular significance of ARID1a loss in the
non-MSI group of EAC is entirely unclear. However, this distinction
is becoming increasingly clinically relevant. There is mounting
evidence that tumours with functional alteration of ARID1a qualify
for various therapies. Discussed is the possibility of an increased
response probability of immune checkpoint inhibitors (ICI)
targeting PD-L1/PD-1 or PARP inhibitors (for further therapy
options, see also ‘Discussion’) (13,14).
However, if ARID1a alteration is an epiphenomenon in MSI
tumours, the predictor of increased treatment response is the
underlying high MSI and not the ARID1a alteration. MSI
tumours also qualify for ICI therapy based on their high PD-L1
expression in the tumour, and the additional determination of
ARID1a is probably of little value. Thus, separation of
ARID1a-altered tumours independent of MSI is reasonable.
The relevant questions are i) how frequently is
ARID1a alteration found in non-MSI-EAC? ii) what is the level of
PD-L1 expression in this group? iii) what morphological, clinical,
and additional molecular characteristics are found in this
subgroup? iv) what is the impact of neoadjuvant therapy regimens
used in EAC?
The present work is the first to describe the
clinical, molecular, and morphological characteristics of
therapy-relevant non-MSI ARID1a loss EAC. To this end, we examined
a very large cohort of 875 patients with EAC and additional data
from the TCGA cohort.
Materials and methods
Patients
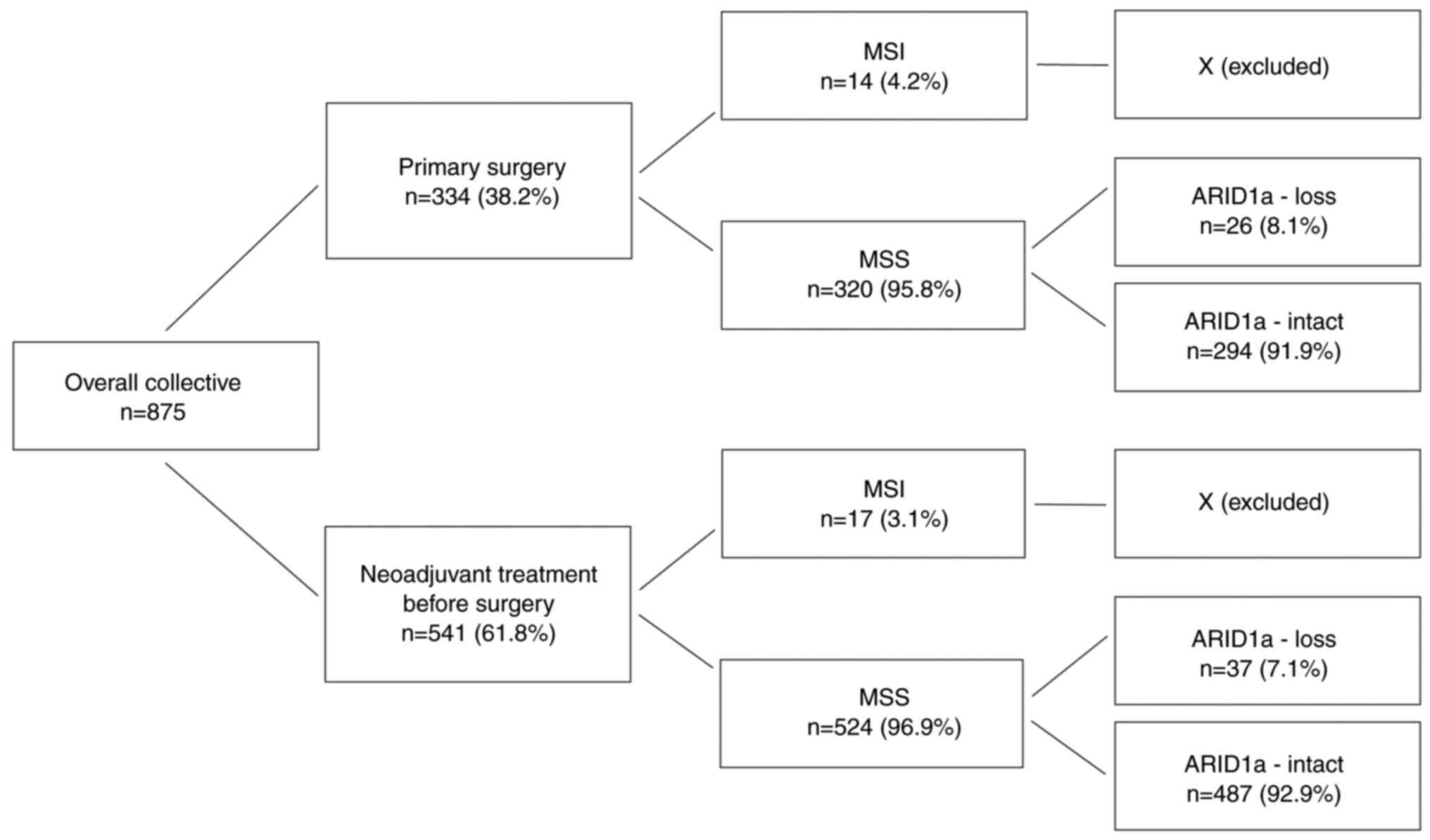
We analysed formalin-fixed, paraffin embedded
material from 875 patients with EAC who underwent primary surgical
resection or resection after neoadjuvant therapy between 1999 and
2018 at the Department of General, Visceral and Cancer Surgery,
University of Cologne, Germany (Table
I, Fig. 1). The majority of
patients were male (88.1%). The average age at surgery was 61.2
years (31–83 years). The standard surgical procedure was
laparotomic or laparoscopic gastrolysis and right transthoracic en
bloc esophagectomy including two-field lymphadenectomy of
mediastinal and abdominal lymph nodes. Reconstruction was performed
by high intrathoracic esophagogastrostomy as described previously
(15).
 | Table I.Patient's characteristics. |
Table I.
Patient's characteristics.
| Characteristic | Overall
collective | ARID1a loss | ARID1a intact | P-value |
|---|
| Total | 843 | 100% | 63 | 7.5% | 780 | 92.5% |
|
| Sex |
|
|
|
|
|
| 0.070 |
|
Male | 743 | 88.1% | 60 | 8.1% | 683 | 91.9% |
|
|
Female | 100 | 11.9% | 3 | 3.0% | 37 | 97.0% |
|
| Age, years |
|
|
|
|
|
| 0.076 |
|
≤65 | 460 | 54.6% | 28 | 5.7% | 432 | 94.3% |
|
|
>65 | 383 | 45.4% | 35 | 9.1% | 348 | 90.9% |
|
| Neoadjuvant
therapy |
|
|
|
|
|
| 1.000 |
| No | 320 | 37.9% | 26 | 8.1% | 294 | 91.9% |
|
|
Yes | 523 | 62.1% | 37 | 7.1% | 487 | 92.9% |
|
| Tumour stage |
|
|
|
|
|
| 0.819 |
|
pT1 | 154 | 18.4% | 11 | 7.1% | 143 | 92.9% |
|
|
pT2 | 155 | 18.5% | 11 | 7.1% | 144 | 92.9% |
|
|
pT3 | 500 | 59.7% | 40 | 8.0% | 460 | 92.0% |
|
|
pT4 | 29 | 3.5% | 1 | 3.4% | 28 | 96.6% |
|
| Lymph node
metastasis |
|
|
|
|
|
| 0.265 |
|
pN0 | 331 | 39.3% | 31 | 9.4% | 300 | 90.6% |
|
|
pN1 | 267 | 31.7% | 14 | 5.2% | 253 | 94.8% |
|
|
pN2 | 120 | 14.2% | 10 | 8.3% | 110 | 91.7% |
|
|
pN3 | 125 | 14.8% | 8 | 6.4% | 117 | 96.6% |
|
| UICC |
|
|
|
|
|
| 0.736 |
| 1 | 110 | 13.1% | 11 | 10.0% | 99 | 90.0% |
|
| 2 | 101 | 12.1% | 8 | 7.9% | 9 | 92.1% |
|
| 3 | 383 | 45.7% | 26 | 6.8% | 357 | 93.2% |
|
| 4 | 244 | 29.1% | 18 | 7.4% | 226 | 92.6% |
|
Patients with advanced oesophageal cancer (cT3, cNx,
M0) received preoperative chemoradiation (5-FU, cisplatin, 40 Gy as
treated in the area prior the CROSS trial) or chemotherapy alone.
All patients were followed up according to a standardised protocol.
During the first 2 years, patients were followed up clinically in
the hospital every 3 months. Afterwards, annual exams were carried
out. Follow-up examinations included a detailed history, clinical
evaluation, abdominal ultrasound, chest X-ray, and additional
diagnostic procedures as required. Follow-up data were available
for all patients. Patient characteristics are given in Table I. Depending on the effect of
neoadjuvant chemo- or radio-chemotherapy, there is a preponderance
of minor responders in the tissue microarrays (TMAs), defined as
histopathological residual tumour of ≥10% (16).
Immunohistochemistry
All tumours were analysed for protein expression
using appropriate immunohistochemical antibodies: ARID1a (clone EPR
13501, rabbit, EDTA buffer 1:1,000, on automated Leica Bond
stainer) and PD-L1 (clone E1L3N, rabbit, EDTA buffer 1:400, on
automated Leica Bond stainer). Inflammatory cells and fibroblasts
served as internal controls. Only complete loss of ARID1a with
concomitant positive expression of the proteins in peritumoral
tissue was scored. An appropriate in situ technique (EBER, Leica
PB0589, ready-to-use on automated Leica Bond stainer) against EBV
RNA was used, which showed no EBV-positive EAC in our cohort.
TMA as a screening method
Tissue samples of 875 EACs were converted to a TMA
format as previously described (17,18).
In brief, tissue cylinders with a diameter of 1.2 mm each were
punched from selected tumour tissue blocks using a self-constructed
semi-automated precision instrument and embedded in empty recipient
paraffin blocks. 4 µm sections of the resulting TMA blocks were
transferred to an adhesive-coated slide system (Instrumedics Inc.,
Hackensack, NJ) for immunohistochemistry.
Tumour whole slide analysis
All tumours with a loss of ARID1a in the tumour cell
nuclei at the TMA were examined for their ARID1a loss on tumour
whole slides. Possible heterogeneous protein loss within the tumour
or their corresponding lymph node metastasis could thus also be
determined.
On whole tumour slides, the combined positive score
(CPS) was used for the PD-L1 expression in tumour tissue. The CPS
was also applied in all relevant recent studies (e.g., checkmate
649 study; see ‘Discussion’) and considers PD-L1 expression on
tumour cells, as well as on specific inflammatory cells (e.g.,
macrophages). Tumours were classified into four different PD-L1
expression groups (CPS <1 (negative), CPS 1–5, CPS 5–10, and CPS
>10).
The histomorphological growth patterns were
described (according to WHO 2019): a) tubular and papillary, b)
solid, c) mucinous, d) poorly cohesive (including signet ring cell
tumours), e) others (including rhabdoid-like features as described
before) (19). If a tumour had
multiple growth patterns, the individual patterns were considered
from a proportion of 10% of the total tumour (e.g., tubular and
mucinous).
Mismatch-repair-protein
status/MSI
We have analysed all tumours for their
mismatch-repair-protein status/MSI for a previous publication
[compare (20)]. In brief we
screened for the mismatch-repair-protein-Status using proper
immunohistochemical antibodies for MLH1 (clone: M1 Ventana), MSH2
(G219-1129), PMS2 (EPR3947) and MSH6 (Clone44, Ventana) on Ventana
Benchmark stainers. Microsatellite status was determined using an
in-house PCR protocol with primers for the Bethesda markers,
including the mononucleotide markers BAT25 and BAT26 or the
dinucleotide markers D5S346, D2S123, D17S250, D10S197, D18S58, and
D13S153 and the tetranucleotide marker MYC. The methods used are
also listed in detail in this publication.
TP53 status of the tumours
The TP53 status of the tumours was carried out as
already described in detail (21).
In brief, for the p53-status immunohistochemistry (IHC) was
performed using the primary antibody specific for TP53 (DAKO, clone
DO-7). The intensity of the TP53 staining was scored manually by
two pathologists (A.Q. and H.L.) according to a 3-tier scoring
system. Discrepant results were resolved by consensus review. For a
smaller proportion of tumours, we additionally used next-generation
sequencing for TP53, exons 5–8.
Analysis of the TCGA collective
TCGA data were obtained from the GDC Data Portal
website (22). For mutation
analyses, we used open-access Mutect2 data. The MSI status was
determined by quantifying frameshift mutations in the form of short
insertions and deletions (indels) in mononucleotide repeats (MNRs).
Only indels with a length of one base were considered. MNR were
analysed for indels above a length of three bases. Cases with more
than 100 indels were classified as MSI tumours. In the assessment
of ARID1a and TP53 mutation status, nonsense,
missense, nonstop, and frameshift mutations were considered. If
corresponding mutations were detected, the respective cases were
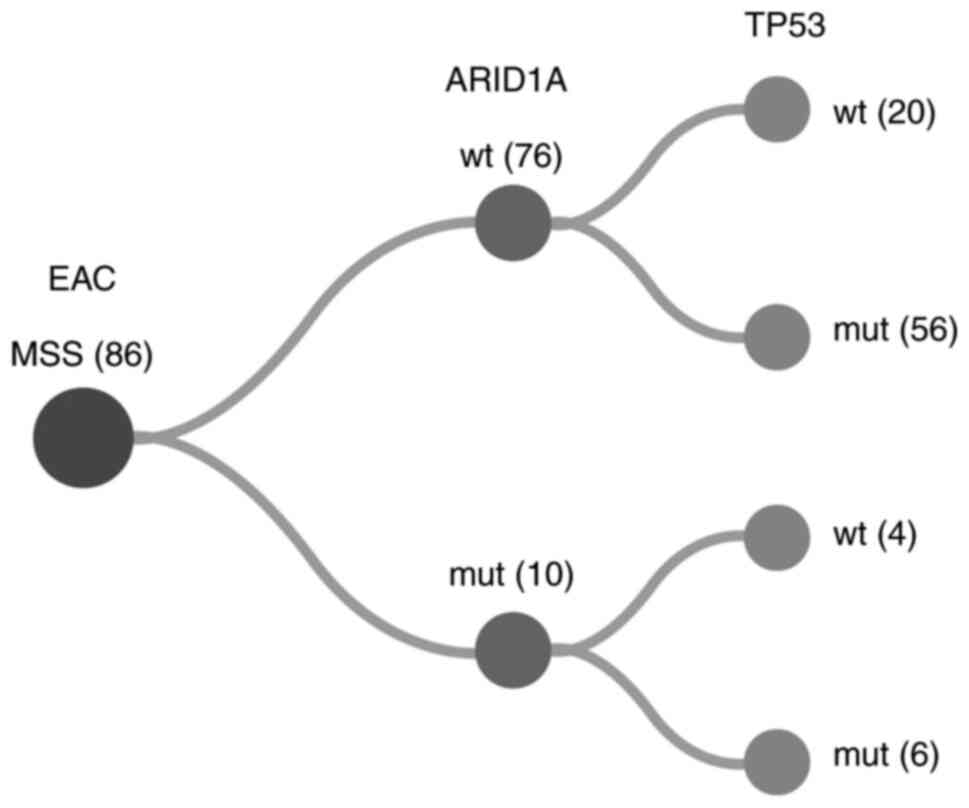
classified as mutated (Fig. 2).
Statistical analysis
Patient data were prospectively collected. Overall
survival was evaluated from the date of surgery until death.
Kaplan-Meier curves were generated and compared using a log-rank
test. Patient data with no events or lost follow up were censored
at the last known date. A two-sided P-value <0.05 was considered
as statistically significant. SPSS package version 25 (IBM, Armonk,
New York) was used for all statistical analyses.
Results
Patient baseline characteristics
Thirty-one patients (n=31; 3.5%) showed MSI and were
excluded from further analysis. Twenty-one MSI tumours (67.7%)
showed concurrent ARID1a loss. In the subgroup of 844
microsatellite-stable (MSS) EACs, we detected loss of ARID1a in 63
cases (7.5%; P<0.001). This distribution was also true in both
subgroups of neoadjuvant and primary surgery patients (Table I, Fig.
3).
It was already known from previous analyses of the
tumour cohort that there was no case of MSH2/MSH6 failure, and
clinically there was no known case of Lynch syndrome (12,20).
Clinicopathological data is depicted in Table I. Patients were predominantly men
(n=744, 88.2%; women n=101, 11.8%). The median age of the
proficient-Mismatch-Repair/Microsatellite-stability
(MMR-p/MSS)-patient cohort at the time of diagnosis was 63.4 years
(range 27.8-87.8 years). In 524 patients (62.0%), a neoadjuvant
treatment (chemo- or radio-chemotherapy) was performed before
surgery.
Loss of ARID1a in MSS-EAC
Loss of ARID1a was detectable in 63 patients (7.5%)
(Fig. 1). In cross table analysis
for the entire patient cohort, a correlation between ARID1a loss
and clinical parameter could not be revealed (Table I). Subgroup analyses were performed
for patients after neoadjuvant therapy and patients after primary
surgery without preoperative therapy. Loss of ARID1a was not
associated with any of the analysed clinical parameter, neither in
the neoadjuvant group nor in the primary surgery group.
Molecular characteristics
The co-occurrence of ARID1a loss and TP53 mutations
is a rare event. Within the group after neoadjuvant treatment,
AIRD1a loss TP53 wild-type tumours were observed in 15
patients (14.2%), whereas ARID1a loss in TP53-mutated
tumours were seen in five patients (5.3%; P<0.057). In patients
without neoadjuvant treatment, a similar distribution was observed.
ARID1a loss in TP53 wild-type tumours was seen in 13
patients (19.7%) and in TP53-mutated tumours in three
patients (4.0%; P=0.003).
AIRD1a loss and prognosis
Loss of ARID1a is not associated with a shortened
overall survival (OS) (P=0.568). Median OS for the entire patient
cohort is 30.9 months (95% confidence interval (95%CI) 27.2-34.7
months) in patients with intact ARID1a and 23.7 months (95%CI
10.8-36.6 months) in patients with ARID1a loss tumours. A survival
difference is also not detectable when stratifying patients
according to neoadjuvant therapy or primary surgery and ARID1a loss
(P=0.237 and P=0.505, respectively).
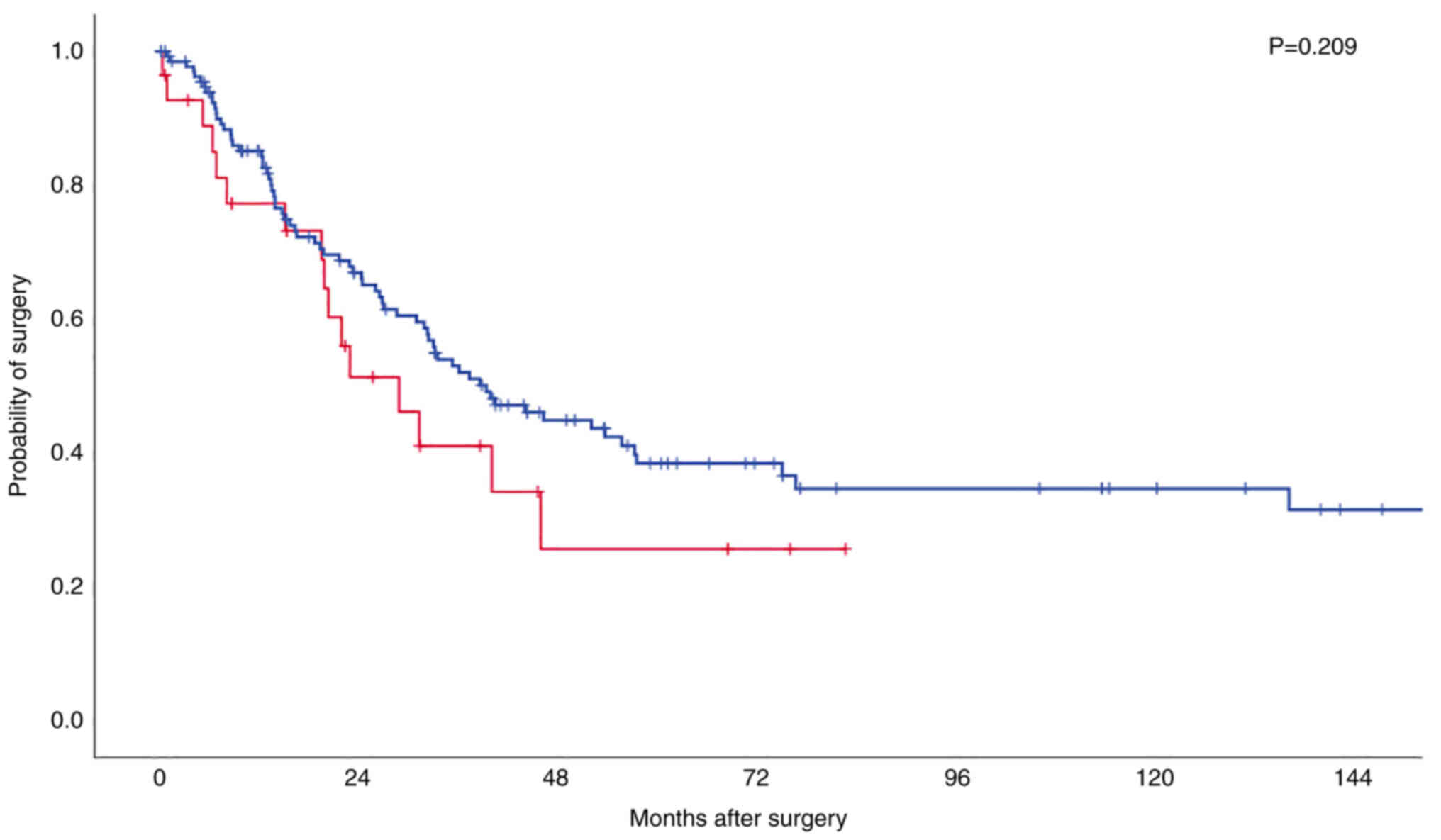
Neither in TP53 wild-type tumours nor in
TP53-mutated tumours did ARID1a loss show significant impact
on the OS, though a trend towards shortened OS in the group of
TP53 wild-type tumours could be observed in the Kaplan-Meier
survival analysis (P=0.209; Fig. 4,
Kaplan-Meier-Curve).
Morphological subtypes of ARID1a loss
MSS-EAC
Within the subcohort of MSS-EACs with ARID1a loss,
the following distribution regarding morphological patterns was
observed: 51.6% (32/63) tubular/papillary, 11.3% (7/63) solid
growth, 11.3% (7/63) poorly cohesive, and mixed 25.8% (16/63).
Tumours with mixed pattern harboured at least 10% of two different
growth patterns. No predominant mucinous pattern was seen.
In this cohort MSI-like features (medullary
phenotype, increased tumour-infiltrating lymphocytes, peritumoral
lymphoid follicle formation) were seen in 9.7% (6/63) cases (see
discussion).
Heterogeneity of ARID1a loss in
MSS-EAC
In most cases of MSS-EACs with ARID1a loss, the
tumours showed homogenous loss of ARID1a expression (58/63, 92%). A
heterogeneous loss of ARID1a expression was seen in only five cases
(5/63, 8%).
PD-L1 expression CPS
In the overall cohort, ~40% of ARID1a loss MSS
tumours showed no PD-L1 expression (CPS <1). This number is not
relevantly affected by neoadjuvant therapy (for details, see
Table II). The six tumours with
MSI-like phenotype were PD-L1 positive. They showed a CPS from a
minimum of 5 up to 100.
| Table II.Expression of PD-L1. |
Table II.
Expression of PD-L1.
| PD-L1 combined
positive score | Total cohort
(n=50) | Primary surgery
(n=21) | Neoadjuvant
treatment (n=29) |
|---|
| 0 | 20 (40%) | 9 (43%) | 11 (38%) |
| 1-5 | 16 (32%) | 7 (33%) | 9 (31%) |
| 5-10 | 10 (20%) | 2 (10%) | 8 (28%) |
| >10 | 4 (8%) | 3 (14%) | 1 (3%) |
Analysis of the TCGA collective
For the analysis of TCGA cases, subtyping and
mutation data of the GDC Data Portal were available for 184 EACs.
These were subclassified into 96 squamous cell and 88
adenocarcinomas. In agreement with the results of previous papers,
two adenocarcinomas and one squamous cell carcinoma exhibited MSI
(see ‘Methods’) (23). Analysing
the mutation status, among the MSS adenocarcinomas, we detected
TP53 mutations in 72% and ARID1a mutations in 12% of
the cases. A simultaneous occurrence of ARID1a and
TP53 mutations was observed in 7% of all tumours (Fig. 2). In the subgroup of 10
ARID1a-mutated EACs, six cases also harboured mutations in
TP53.
Discussion
Since ARID1a loss tumours in the GI tract occur
frequently in the context of MSI (and additionally in gastric
carcinoma associated with EBV), many consider prognostic or
morphologic aspects are overlaid by the characteristics of MSI or
EBV. In our cohort of 875 EAC, only 31 tumours were proven to be
dMMR/MSI (3.5%). It was already known from previous analyses of the
tumour cohort that there was no case of MSH2/MSH6 failure and
clinically there was no known case of Lynch syndrome (20), so MLH1 was only analysed for the
detection of defective mismatch-repair protein status (20). This is in line with the literature
reporting MSI in EAC of 1–5%. There is no EBV-associated EAC
consistent with previous publications (24,25).
We detected loss of ARID1a expression in ~10% of
EAC, in accordance with the previous work of Drage et al
(11). This finding is also in line
with further studies and data by the TCGA describing ARID1a
alteration in 10–13% (26–28).
Drage et al only considered primary operated
EAC. This also explains the long period of time considered in this
paper, ranging from 1989 to 2011. Today, the majority of EAC are
treated with neoadjuvant therapy. Knowledge of the frequency and
characteristics of neoadjuvantly treated EAC with concurrent ARID1a
loss is discussed here for the first time.
The extent of 7.5% MSS-EAC with ARID1a loss is
unaffected by neoadjuvant therapy. This suggests that in an
operable patient population, ARID1a loss tumours are not strikingly
more chemo-sensitive, as we would otherwise find them in a
significantly lower volume after neoadjuvant treatment has
occurred.
According to the TCGA data, all ARID1a
alterations are either deep deletions or truncating mutations. This
fact and the high percentage of ARID1a deficient tumours in the
upper GI tract (10–17%) can be taken as a good indication that the
loss of ARID1a is important for tumour biology. Furthermore, it
also explains well that immunohistochemistry is indeed able to
reliably represent an underlying genomic alteration of the
ARID1a gene via the lack of protein detection in tumour cell
nuclei. Since the extent of mutation and protein loss is comparable
in different collectives, other possibilities, such as epigenetic
downregulation of ARID1a at least do not seem to play a
major role. As histopathologists, we strive to define morphological
characteristics in the same molecular subgroups. This works well,
for example, in MSI tumours that show clustered tumours with
so-called medullary features or highly inflamed tumours in which
lymphocytes show close spatial adjacency to tumour cells. The
latter is also found in Epstein-Barr virus-associated
carcinomas-appropriately referred to in WHO as carcinomas with
lymphoid stroma. One paper claimed to find characteristics of the
medullary phenotype also clustered in ARID1a-deficient carcinomas
of the upper GI tract. We cannot actuate this in our collective.
Since the previous publication [Drage et al (11)] did not distinguish between MSI and
MSS-ARID1a deficient tumours, we evaluate the accumulation of
medullary features in their collective as an expression of tumour
microsatellite instability only. Thus, the following statement is
relevant: ARID1a-deficient tumours are not predictable by
morphological criteria. If this subgroup is indeed therapeutically
relevant in the future, immunohistochemical or molecular testing
must be performed to detect ARID1a alteration. According to our
data, histomorphology is not able to perform a reliable
preselection.
We did not find rhabdoid-like features as considered
in a study of gastrointestinal tract carcinomas with SWI/SNF loss
(19).
In agreement with Drage et al we see no
prognostic relevance of ARID1a loss in EAC, even when considering
the overall collective. This applies to primary operated and
neoadjuvant pre-treated tumours (11).
The notion that an ARID1a mutation occurs
mutation-exclusively and does not occur concomitantly with a
TP53 mutation has been described mainly in carcinomas of the
internal genitalia. This is not true for adenocarcinoma of the
oesophagus. (29,30). While in endometrioid endometrial
carcinomas ARID1a loss and mutations in TP53 are almost
mutually exclusive, this is not the case in EAC (31). In our collective, as well as in the
TCGA-cohort we analysed, mutations in TP53 are found to be
simultaneously manifest. Interestingly, in the subgroup of TP53
wild-type EAC we find a tendency towards an unfavourable prognosis
(but even there without statistical significance, P=0.209).
A heterogeneous distribution of ARID1a-deficient and
ARID1a-proficient tumour clones in the same tumour is the exception
in EAC. Homogeneity also applies to their lymph node metastases.
The homogeneous occurrence of ARID1a loss clones within the tumour
and its metastases is particularly significant for effective
therapeutic intervention. The more homogeneous a therapeutically
relevant change occurs in the tumour, the more likely it can be
assumed that the therapy will be effective.
For example, Her2/neu is more often not
homogeneously expressed in EAC, in contrast to breast carcinoma.
The lack of homogeneity is likely one reason for the only moderate
benefit of trastuzumab in OS of just under 3 months in EAC and a
major reason for the failure of the GATSBY study (32,33).
MSI carcinomas of the colon, stomach, and oesophagus
would be effectively treated with ICIs directed against PD-1 or
PD-L1 in most cases. From a therapeutic perspective, concurrent
ARID1a failure in the MSI subgroup is probably irrelevant. Thus, at
~7.5% ARID1a loss, MSS-EAC represent a relevant tumour subgroup. In
ARID1a-altered tumours of different entities, different agents have
been described as (potentially) effective (EZH2 inhibition, HDAC6
inhibition, PARP inhibition, PIK3CA pathway inhibitor, and
PD-1/PD-L1 inhibitors). In malignant extrarenal rhabdoid tumours,
which typically show a failure of the SMARCB1 (INI-1) subunit of
the SWI/SNF complex, phase 2 clinical trials are ongoing to
investigate the efficacy of inhibition of EZH2 methyltransferase as
a catalytic subunit of the Polycomb complex (34). Bitler et al then also
describe the synthetic lethality of EZH2 methyltransferase
inhibition in ARID1a mutant tumours (35). Shen et al have been able to
demonstrate the efficacy of PARP inhibition (e.g., Olaparib) in
ARID1a-deficient tumours in vitro and in vivo
(36).
There is evidence that ARID1a is involved in the
repair of DNA double-strand breaks (similar to BRCA1 and BRCA2).
The enzyme PARP works in the same way. Loss of function of ARID1a
with concomitant therapeutic blockade of PARP could be lethal to
the tumour cell (as has been successfully used therapeutically in
BRCA-deficient ovarian cancers). Specific HDAC6 inhibitory small
molecules are in clinical trials in haematologic tumours (37).
In clear cell ovarian cancer with ARID1a loss, cell
culture experiments and mouse models have also demonstrated the
efficacy of this class of compounds. Furthermore, cell culture
analyses have shown that loss of ARID1a protein renders tumour
cells highly sensitive to inhibition for PI3K and AKT inhibitors
(38,39).
Some work has also discussed the relevance of PD-L1
expression in the context of ARID1A deficiency and the
effectiveness of the corresponding checkpoint inhibitors (14).
According to our results, ARID1a-altered EAC are not
disproportionately frequent or particularly marked PD-L1 positive
tumours. Approximately 60% of tumours in our collective are PD-L1
positive (CPS >1), 32% show a CPS of 1–5, 20% of 5–10, and 8% of
>10. Thus, slightly fewer PD-L1 positive tumours are found
compared to the Checkmate 649 study, which looked at a molecularly
unselected collective of gastric carcinomas and gastroesophageal
transition carcinomas. The Checkmate 649 study also measured PD-L1
using the CPS-Score and showed the efficacy of nivolumab in PD-L1
positive upper GI-tract tumours (40). Whether ARID1a-deficient EAC could
nevertheless particularly benefit from PD-1/PD-L1 blockade therapy
will have to be shown by future studies or retrospective subgroup
analyses of already completed studies.
We have investigated the significance of different
altered SWI/SNF proteins, including ARID1a, on over 600 EACs in a
previous two-year-old study (12).
In that study, we found that ARID1a can fail in MSI tumours but
also independently of MSI in oesophageal cancer. This publication
was the basis of the present work. Similarly to Drage et al
(11) we had not clearly
distinguished between characteristics of MSS and MSI carcinomas in
the previous publication, an inaccuracy that we resolve with this
work. Here we focus exclusively on alteration of ARID1a and MSS-EAC
in the current manuscript.
A limitation of our current study is that we
surveyed ARID1a status only at the protein level. It may be that
some mutations induce a functionless ARID1a protein that is still
recognised by the antibody used. The proportion of ARID1a-deficient
tumours would then be somewhat higher than we have described. This
may be supported by studies reporting ARID1a mutation status
at 13% (rather than 10%).
However, this once again highlights the importance
of this subgroup in EAC. Another limitation is the retrospective
nature and single-centre analysis. However, we think that the
number of the EACs considered (N=875) may provide relevant
information despite these limitations. We have deliberately
refrained from digitally supported image analyses, as image
analyses are not helpful in these cases from our point of view.
Today, the PD-L1 TPS score can already be determined well by image
analysis; this does not work comparably well for the CPS score.
However, histopathologists are able to determine both growth
patterns and the CPS score in the tumour with high concordance.
This was also done in all studies (e.g., PD-L1 determination in the
Checkmate 649 study). For our work, we believe it made sense to
provide morphologists with comparative analytics.
In conclusion, according to analysis of a very large
tumour collective (N=875), at least 10% of EAC are
ARID1a-deficient, the majority of which are MSS (7.5%, MSS/MMR-p).
A specific morphologic phenotype is absent (e.g., there is no
clustering of tumours with mucinous or rhabdoid differentiation or
so-called medullary features). However, there is strong evidence
that this tumour subgroup is particularly sensitive to some agents
(such as PARP or anti-PD-L1 checkpoint inhibitors). Since
personalised therapeutics are largely lacking in EAC, clinical
trials investigating the efficacy of these therapeutics,
specifically in this subgroup, are useful (biomarker-based
trials).
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets generated during and/or analysed during
the current study are available from the corresponding author on
reasonable request.
Authors' contributions
JR, JB, TZ, RB and AQ made substantial contributions
to conception and design. JR and AQ were responsible for the
authenticity of all raw data. JR, AQ and BU were responsible for
analysis and interpretation of data. AQ, JR, RB and TZ wrote the
main manuscript. FG, CJB, SS and WS were responsible for the data
collection, and reviewed the text. All authors have been involved
in drafting the manuscript or revising it critically for important
intellectual content. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Approval was obtained from the University of Cologne
Ethics Committee (approval nos. 20-1583 and 10-242). We confirm
that informed consent was obtained from all subjects and/or their
legal guardians.
Patient consent for publication
Patients gave their written consent to usage of
their tumor specimens.
Competing interests
All authors declare that they had no competing
interests.
References
|
1
|
Wang W, Xue Y, Zhou S, Kuo A, Cairns BR
and Crabtree GR: Diversity and specialization of mammalian SWI/SNF
complexes. Genes Dev. 10:2117–2130. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wilson BG and Roberts CW: SWI/SNF
nucleosome remodellers and cancer. Nat Rev Cancer. 11:481–492.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wu JN and Roberts CW: ARID1A mutations in
cancer: Another epigenetic tumour suppressor? Cancer Discovery.
3:35–43. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lawrence MS, Stojanov P, Polak P, Kryukov
GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH,
Roberts SA, et al: Mutational heterogeneity in cancer and the
search for new cancer-associated genes. Nature. 499:214–218. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jones S, Wang TL, Shih Ie M, Mao TL,
Nakayama K, Roden R, Glas R, Slamon D, Diaz LA Jr, Vogelstein B, et
al: Frequent mutations of chromatin remodeling gene ARID1A in
ovarian clear cell carcinoma. Science. 330:228–231. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Guan B, Gao M, Wu CH, Wang TL and Shih Ie
M: Functional analysis of in-frame indel ARID1A mutations reveals
new regulatory mechanisms of its tumour suppressor functions.
Neoplasia. 14:986–993. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang K, Kan J, Yuen ST, Shi ST, Chu KM,
Law S, Chan TL, Kan Z, Chan AS, Tsui WY, et al: Exome sequencing
identifies frequent mutation of ARID1A in molecular subtypes of
gastric cancer. Nat Genet. 43:1219–1223. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zang ZJ, Cutcutache I, Poon SL, Zhang SL,
McPherson JR, Tao J, Rajasegaran V, Heng HL, Deng N, Gan A, et al:
Exome sequencing of gastric adenocarcinoma identifies recurrent
somatic mutations in cell adhesion and chromatin remodeling genes.
Nat Genet. 44:570–574. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang SC, Ng KF, Chang IY, Chang CJ, Chao
YC, Chang SC, Chen MC, Yeh TS and Chen TC: The clinicopathological
significance of SWI/SNF alterations in gastric cancer is associated
with the molecular subtypes. PLoS One. 16:e02453562021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Abe H, Maeda D, Hino R, Otake Y, Isogai M,
Ushiku AS, Matsusaka K, Kunita A, Ushiku T, Uozaki H, et al: ARID1A
expression loss in gastric cancer: Pathway-dependent roles with and
without Epstein-Barr virus infection and microsatellite
instability. Virchows Arch. 461:367–377. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Drage MG, Tippayawong M, Agoston AT, Zheng
Y, Bueno R, Hornick JL, Odze RD and Srivastava A: Morphological
features and prognostic significance of ARID1A-deficient esophageal
adenocarcinomas. Arch Pathol Lab Med. 141:970–977. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schallenberg S, Bork J, Essakly A, Alakus
H, Buettner R, Hillmer AM, Bruns C, Schroeder W, Zander T, Loeser
H, et al: Loss of the SWI/SNF-ATPase subunit members SMARCF1
(ARID1A), SMARCA2 (BRM), SMARCA4 (BRG1) and SMARCB1 (INI1) in
oesophageal adenocarcinoma. BMC Cancer. 20:122020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Park Y, Chui MH, Suryo Rahmanto Y, Yu ZC,
Shamanna RA, Bellani MA, Gaillard S, Ayhan A, Viswanathan A,
Seidman MM, et al: Loss of ARID1A in Tumor cells renders selective
vulnerability to combined ionizing radiation and PARP inhibitor
therapy. Clin Cancer Res. 25:5584–5594. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge
Z, Nagel ZD, Zou J, Wang C, Kapoor P, et al: ARID1A deficiency
promotes mutability and potentiates therapeutic antitumor immunity
unleashed by immune checkpoint blockade. Nat Med. 24:556–562. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hölscher AH, Schneider PM, Gutschow C and
Schröder W: Laparoscopic ischemic conditioning of the stomach for
esophageal replacement. Ann Surg. 245:241–246. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schneider PM, Metzger R, Schaefer H,
Baumgarten F, Vallbohmer D, Brabender J, Wolfgarten E,
Bollschweiler E, Baldus SE, Dienes HP, et al: Response evaluation
by endoscopy, rebiopsy, and endoscopic ultrasound does not
accurately predict histopathologic regression after neoadjuvant
chemoradiation for esophageal cancer. Ann Surg. 248:902–908. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Simon R, Mirlacher M and Sauter G: Tissue
microarrays. Biotechniques. 36:98–105. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Helbig D, Ihle MA, Pütz K, Tantcheva-Poor
I, Mauch C, Büttner R and Quaas A: Oncogene and therapeutic target
analyses in atypical fibroxanthomas and pleomorphic dermal
sarcomas. Oncotarget. 7:21763–21774. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Agaimy A, Daum O, Märkl B, Lichtmannegger
I, Michal M and Hartmann A: SWI/SNF Complex-deficient
Undifferentiated/Rhabdoid carcinomas of the gastrointestinal tract:
A Series of 13 cases highlighting mutually exclusive loss of
SMARCA4 and SMARCA2 and Frequent Co-inactivation of SMARCB1 and
SMARCA2. Am J Surg Pathol. 40:544–553. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Quaas A, Rehkaemper J, Rueschoff J, Pamuk
A, Zander T, Hillmer A, Siemanowski J, Wittig J, Buettner R, Plum
P, et al: Occurrence of high microsatellite-instability/mismatch
repair deficiency in nearly 2,000 human adenocarcinomas of the
gastrointestinal tract, pancreas, and bile ducts: A study from a
Large German comprehensive cancer Center. Front Oncol.
11:5694752021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Quaas A, Heydt C, Gebauer F, Alakus H,
Loeser H, Buettner R, Hillmer A, Bruns C, Merkelbach-Bruse S,
Zander T, et al: Genomic characterization of TP53-Wild-type
esophageal carcinoma. Transl Oncol. 12:154–161. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Grossman RL, Heath AP, Ferretti V, Varmus
HE, Lowy DR, Kibbe WA and Staudt LM: Toward a shared vision for
cancer genomic data. N Engl J Med. 375:1109–1112. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bonneville R, Krook MA, Kautto EA, Miya J,
Wing MR, Chen HZ, Reeser JW, Yu L and Roychowdhury S: Landscape of
microsatellite instability across 39 cancer types. JCO Precis
Oncol. 2017: PO.17.00073. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hewitt LC, Inam IZ, Saito Y, Yoshikawa T,
Quaas A, Hoelscher A, Bollschweiler E, Fazzi GE, Melotte V, Langley
RE, et al: Epstein-Barr virus and mismatch repair deficiency status
differ between oesophageal and gastric cancer: A large multi-centre
study. Eur J Cancer. 94:104–114. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Falkenback D, Johansson J, Halvarsson B
and Nilbert M: Defective mismatch-repair as a minor tumorigenic
pathway in Barrett esophagus-associated adenocarcinoma. Cancer
Genet Cytogenet. 157:82–86. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Frankell AM, Jammula S, Li X, Contino G,
Killcoyne S, Abbas S, Perner J, Bower L, Devonshire G, Ococks E, et
al: The landscape of selection in 551 esophageal adenocarcinomas
defines genomic biomarkers for the clinic. Nat Genet. 51:506–516.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dulak AM, Stojanov P, Peng S, Lawrence MS,
Fox C, Stewart C, Bandla S, Imamura Y, Schumacher SE, Shefler E, et
al: Exome and whole-genome sequencing of esophageal adenocarcinoma
identifies recurrent driver events and mutational complexity. Nat
Genet. 45:478–486. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chong IY, Cunningham D, Barber LJ,
Campbell J, Chen L, Kozarewa I, Fenwick K, Assiotis I, Guettler S,
Garcia-Murillas I, et al: The genomic landscape of oesophagogastric
junctional adenocarcinoma. J Pathol. 231:301–310. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Allo G, Bernardini MQ, Wu RC, Shih Ie M,
Kalloger S, Pollett A, Gilks CB and Clarke BA: ARID1A loss
correlates with mismatch repair deficiency and intact p53
expression in high-grade endometrial carcinomas. Modern Pathol.
27:255–261. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guan B, Wang TL and Shih Ie M: ARID1A, a
factor that promotes formation of SWI/SNF-mediated chromatin
remodeling, is a tumour suppressor in gynecologic cancers. Cancer
Res. 71:6718–6727. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bosse T, ter Haar NT, Seeber LM, v Diest
PJ, Hes FJ, Vasen HF, Nout RA, Creutzberg CL, Morreau H and Smit
VT: Loss of ARID1A expression and its relationship with PI3K-Akt
pathway alterations, TP53 and microsatellite instability in
endometrial cancer. Mod Pathol. 26:1525–1535. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Grabsch H, Sivakumar S, Gray S, Gabbert HE
and Müller W: HER2 expression in gastric cancer: Rare,
heterogeneous and of no prognostic value-conclusions from 924 cases
of two independent series. Cell Oncol. 32:57–65. 2010.PubMed/NCBI
|
|
33
|
Thuss-Patience PC, Shah MA, Ohtsu A, Van
Cutsem E, Ajani JA, Castro H, Mansoor W, Chung HC, Bodoky G,
Shitara K, et al: Trastuzumab emtansine versus taxane use for
previously treated HER2-positive locally advanced or metastatic
gastric or gastro-oesophageal junction adenocarcinoma (GATSBY): An
international randomised, open-label, adaptive, phase 2/3 study.
Lancet Oncol. 18:640–653. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Knutson SK, Warholic NM, Wigle TJ, Klaus
CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP,
Copeland RA, et al: Durable tumour regression in genetically
altered malignant rhabdoid tumors by inhibition of
methyltransferase EZH2. Proc Natl Acad Sci USA. 110:7922–7927.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bitler BG, Aird KM, Garipov A, Li H,
Amatangelo M, Kossenkov AV, Schultz DC, Liu Q, Shih Ie M,
Conejo-Garcia JR, et al: Synthetic lethality by targeting EZH2
methyltransferase activity in ARID1A-mutated cancers. Nat Med.
21:231–238. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shen J, Peng Y, Wei L, Zhang W, Yang L,
Lan L, Kapoor P, Ju Z, Mo Q, Shih Ie M, et al: ARID1A deficiency
impairs the DNA damage checkpoint and sensitizes cells to PARP
inhibitors. Cancer Discovery. 5:752–767. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Santo L, Hideshima T, Kung AL, Tseng JC,
Tamang D, Yang M, Jarpe M, van Duzer JH, Mazitschek R, Ogier WC, et
al: Preclinical activity, pharmacodynamic, and pharmacokinetic
properties of a selective HDAC6 inhibitor, ACY-1215, in combination
with bortezomib in multiple myeloma. Blood. 119:2579–2589. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Samartzis EP, Gutsche K, Dedes KJ, Fink D,
Stucki M and Imesch P: Loss of ARID1A expression sensitizes cancer
cells to PI3K- and AKT-inhibition. Oncotarget. 5:5295–5303. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
St Pierre R and Kadoch C: Mammalian
SWI/SNF complexes in cancer: Emerging therapeutic opportunities.
Curr Opin Genet Dev. 42:56–67. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Janjigian YY, Shitara K, Moehler M,
Garrido M, Salman P, Shen L, Wyrwicz L, Yamaguchi K, Skoczylas T,
Campos Bragagnoli A, et al: First-line nivolumab plus chemotherapy
versus chemotherapy alone for advanced gastric, gastro-oesophageal
junction, and oesophageal adenocarcinoma (CheckMate 649): A
randomised, open-label, phase 3 trial. Lancet. 398:27–40. 2021.
View Article : Google Scholar : PubMed/NCBI
|