Introduction
The incidence of renal cell carcinoma (RCC) is
increasing (1,2). Approximately one in five patients with
RCC miss surgery at the time of diagnosis, and almost one in three
patients with localized RCC will have recurrence after resection,
these patients have a 5-year survival rate of <10% and are not
sensitive to radiation or chemotherapy (3). Tyrosine kinase inhibitors (TKIs) are
the main treatment choice for advanced renal cancer. Sorafenib is
the first multi-target TKI approved for the treatment of renal
cancer with dual antitumor activity. Sorafenib has direct antitumor
activities by inhibiting RAF/MEK/ERK signaling, and it also acts on
VEGFR, platelet-derived growth factor (PDGFR) and other targets to
inhibit tumor angiogenesis (4).
However, some patients with RCC do not respond sufficiently to
sorafenib treatment, and most patients develop resistance and
disease progression over time, even if sorafenib is initially
effective (5). Therefore, there is
an urgent need to elucidate the underlying mechanisms of sorafenib
sensitivity regulation and to explore effective strategies to
improve clinical diagnosis and treatment of renal cancer.
Cyclooxygenase-2 (COX-2) has low expression in
normal cells, but high expression in inflammation and tumor cells
(6,7). COX-2 leads to increased synthesis and
secretion of prostaglandin E2 (PGE2), which in turn activates cell
growth and inhibits apoptosis (8).
COX-2 is associated with the occurrence and progression of various
cancer types (9), including
squamous cell carcinoma, cholangiocarcinoma, endometrial carcinoma
and hepatocellular carcinoma. COX-2 regulates the transcription of
EGFR through transcriptional activator protein-1 (10). It also plays a role in the process
of drug resistance (11,12). Celecoxib, a COX-2 specific
inhibitor, is one of the most widely used and promising drugs for
cancer therapy (13). However,
little research has been conducted on its use in the treatment of
renal cancer. The objective of the present study was to investigate
whether the combination of celecoxib may improve the efficacy of
sorafenib in the treatment of RCC.
A stress granule (SG) is a highly dynamic
membrane-free structure in cells that contains mRNA encoding
stress-adaptive proteins and a variety of RNA-binding proteins
(RBPs), including Ras GTPase-activating protein-binding protein 1
(G3BP1), human antigen R (HuR), T-cell-restricted intracellular
antigen-1 (TIA-1) and Tristetraprolin (TTP) (14). SGs can redistribute intracellular
resources under stress conditions, stabilize mRNA and regulate
expression of genes that promote cell survival (14,15).
In tumor cells, SGs can promote cell resistance to stress and
promote tumor survival and progression (16,17).
Several SG components have been found to be overexpressed in
tumors, and their expression levels can be used to predict clinical
outcomes (18,19). In addition, previous studies have
shown that SGs may confer chemotherapy resistance to tumor cells
(20,21). It has been reported that COX-2 mRNA
can be captured by SGs (22). SGs
represent a novel target for developing therapies to suppress COX-2
protein expression. It will also be important to study the role of
SGs on the expression of other genes involved in tumor
pathogenesis. In the present study, the efficacy of sorafenib in
combination with the COX-2 inhibitor, celecoxib, on renal cancer
cells was investigated and the mechanism of COX-2 upregulation
induced by sorafenib was explored.
Materials and methods
Cell culture
The 786-O and ACHN renal cancer cell lines were
obtained from The Cell Bank of Type Culture Collection of The
Chinese Academy of Sciences. 786-O cells were cultured in RPMI 1640
(Wisent Biotechnology) and ACHN cells were cultured in DMEM (Wisent
Biotechnology) under standard procedures. All complete media were
supplemented with 10% (v/v) FBS (Gibco; Thermo Fisher Scientific,
Inc.), 100 U/ml penicillin and 100 µg/ml streptomycin (Wisent
Biotechnology) at 37°C with 5% CO2.
RNA interference and lentivirus
Small interfering RNAs (siRNAs) of COX-2 and G3BP1
were purchased from Generay Biotech Co., Ltd. The specific siRNA
were as follows: COX-2 siRNA-1 sense, 5′-GAGCAGUUGUUCCAGACAATT-3′;
COX-2 siRNA-1 antisense, 5′-UUGUCUGGAACAACUGCUCTT-3′; COX-2 siRNA-2
sense, 5′-GAUUGAAGAUUAUGUGCAATT-3′; COX-2 siRNA-2 antisense,
5′-UUGCACAUAAUCUUCAAUCTT-3′; G3BP1 siRNA-1 sense,
5′-GGAGGAGUCUGAAGAAGAATT-3′; G3BP1 siRNA-1 antisense,
5′-UUCUUCUUCAGACUCCUCCTT-3′; G3BP1 siRNA-2 sense,
5′-GCCUGAGCCAGUAUUAGAATT-3′; G3BP1 siRNA-2 antisense,
5′-UUCUAAUACUGGCUCAGGCTT-3′; Control siRNA sense,
5′-UUCUCCGAACGUGUCACGUTT-3′; Control siRNA antisense,
5′-ACGUGACACGUUCGGAGAATT-3′. ACHN cells were transiently
transfected with the siRNAs of target genes and negative control
siRNA using INTERFERin (Polyplus-transfection SA). The cells were
inoculated in a six-well plate, and when the density was about
30–50%. Diluted 2.2 pmoles of siRNA duplexes into 200 µl of medium
without serum. Added 8 µl of INTERFERin to the 200 µl of siRNA
duplexes and immediately homogenized by vortexing for 10 sec.
Incubated for 10 min at room temperature. Added 2 ml of fresh
pre-warmed complete medium and 200 µl of transfection mix per well
and incubated the plate at 37°C. Changed to fresh medium after 12 h
and continued to culture for 24 or 48 h. G3BP1 short hairpin RNA
(shRNA) and control shRNA lentivirus were purchased from Shanghai
GeneChem, Co., Ltd. The sequences of the shRNA was as follows:
G3BP1 shRNA, CCTGATGATTCTGGAACTT; Control shRNA,
TTCTCCGAACGTGTCACGT. The plasmid information is
hU6-MCS-Ubiquitin-firefly_Luciferase-IRES-puromycin. ACHN cells
(MOI: 15) were inoculated in 12-well plate and transduced with
lentivirus (Virus titer: G3BP1 shRNA, 4.5E+08T U/ml; Control shRNA,
3.0E+08T U/ml.) after the density reached 20%. Added 3 µl of G3BP1
shRNA lentivirus or 4.5 µl of control shRNA lentivirus, 20 µl of
cotransfection reagent HitransG P (GeneChem, Co., Ltd.) and 500 µl
of fresh medium per well and incubated the plate at 37°C. Changed
to complete culture medium after 10 h. After 48 h of transfection,
2 µg/ml puromycin (Thermo Fisher Scientific, Inc.) was used to
select for positively transfected cells. After 7 days of selection
with puromycin, the surviving cells were used to determine the
knockdown efficiency.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from ACHN and 786-O cells
using TRIzol (Vazyme Biotech Co., Ltd.). Reverse transcriptions
were performed using HiScript RT SuperMix (Vazyme Biotech Co.,
Ltd.). The procedure was as follows: 50°C for 15 min; 85°C for 5
sec; 4°C for 10 sec. qPCR was performed using ChamQ Universal SYBR
qPCR Master Mix (Vazyme Biotech Co., Ltd.), on an ABI QuantStudio 6
Flex Real-Time PCR System (Thermo Fisher Scientific, Inc.). The PCR
was as follows: 95°C for 30 sec and 45 cycles of 95°C for 10 sec;
60°C for 30 sec. The fold change of the mRNA levels was calculated
using the 2−ΔΔCq method. The specific primers were as
follows: COX-2 forward, 5′-CTATCACTGGCATCCCCTTCT-3′; COX-2 reverse,
5′-CTTTCTGTACTGCGGGTGGAA-3′; G3BP1 forward,
5′-AGAGGTGAGGTCCGTCTGAA-3′; G3BP1 reverse,
5′-TTATCTCGTCGGTCGCCTTC-3′; actin forward,
5′-CATGTACGTTGCTATCCAGGC-3′; actin reverse,
5′-CTCCTTAATGTCACGCACGAT-3′.
Antibodies
Antibodies against G3BP1 (WB, 1:1,000; IF, 1:300;
cat. no. 13057-2-AP) and HuR (IF, 1:300; cat. no. 66549-1-Ig) were
from Proteintech Group, Inc. The antibody against COX-2 (WB,
1:1,000; cat. no. 12282S) was from Cell Signaling Technology, Inc.
Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Alexa Fluor 488
secondary antibody (IF, 1:800; cat. no. A32731) and Goat anti-Mouse
IgG (H+L) Cross-Adsorbed Alexa Fluor 594 Secondary Antibody (IF,
1:800; cat. no. A11005) were from Thermo Fisher Scientific, Inc.
Anti-rabbit IgG, HRP-linked Antibody (WB, 1:2,000; cat. no. 7074S)
and Anti-mouse IgG, HRP-linked Antibody (WB, 1:2,000; cat. no.
7076S) were from Cell Signaling Technology, Inc.
Western blotting
Total cell extracts were lysed on ice using a lysis
buffer (containing proteinase and phosphatase inhibitors; Beyotime
Biotech Co., Ltd.). Protein concentrations were measured using the
BCA Kit (Vazyme Biotech Co., Ltd.). Samples were mixed with
SDS-PAGE Sample Loading Buffer (5X, Beyotime Biotech Co., Ltd.) and
denatured at 95°C for 5 min, then resolved on 10% gels using
SDS-PAGE in running buffer (Sangon Biotech Co., Ltd. Tris, 3 g;
Glycine, 14.4 g; SDS, 1 g; Fix the volume to 1 liter with DD
water.). Proteins were transferred to polyvinylidene difluoride
(PVDF) membranes (Bio-Rad Laboratories, Inc.). After blocking with
5% skimmed milk at room temperature for 1 h, the PVDF membranes
were incubated with primary antibodies at 4°C overnight, washed
with PBST and then incubated with secondary antibodies at room
temperature for 1 h. After washing with PBST 3 times, signals were
detected using an enhanced chemiluminescence system (Vazyme Biotech
Co., Ltd.) and acquired by ChemiScope 3300 Mini Imaging System
(Clinx Science Instruments Co., Ltd.).
Immunofluorescence (IF)
ACHN and 786-O cells were fixed with 4%
paraformaldehyde for 15 min at room temperature, then washed with
PBS 3 times and permeabilized with 0.3% Triton X-100 for 20 min at
room temperature. Samples were blocked with 3% BSA (Sangon Biotech
Co., Ltd.) for 30 min at room temperature and incubated with
specific primary antibodies overnight at 4°C in 3% BSA. After
washing, the fluorescently labelled secondary antibodies were
incubated for 1 h at room temperature. After further washing,
nuclei were stained with DAPI at room temperature for 10 min.
Slides were observed and imaged using the EVOS FL Auto 2.0 Imaging
System (Thermo Fisher Scientific, Inc.).
MTT assay
Cell viability was measured using an MTT assay. ACHN
cells in logarithmic phase were cultured in 96-well plates
(4×103 cells per well). Cells were exposed to sorafenib
and celecoxib (Selleck Chemicals) at the indicated concentration
for 48 h. After discarding the cell culture media, cells were
washed and then incubated in 0.5 mg/ml MTT (Sangon Biotech Co.,
Ltd.) for 2 h at 37°C. MTT crystals were dissolved in DMSO (Sangon
Biotech Co., Ltd.). The absorbance per well was proportional to the
cell viability. The absorption value was measured at a wavelength
of 490 nm using the infinite M200Pro system (Tecan Group,
Ltd.).
Cell apoptosis
Cell apoptosis was detected using the annexin
V-Alexa Fluor 647/propidium iodide double staining method. The
adherent ACHN cells were digested and collected after drug
treatment. The cells were washed and resuspended with 1X binding
buffer (Yeasen Biotech Co., Ltd.). Annexin V-Alexa Fluor 647 and
propidium iodide staining solution (Yeasen Biotech Co., Ltd.) were
added. A total of 10,000 cells per sample were analyzed by flow
cytometry (NovoCyte, 2060R, ACEA Bioscience, Inc.; Agilent)
following the reaction at room temperature and avoiding light. The
data were analyzed using FlowJo v10 (FlowJo LLC).
RNA fluorescence in situ hybridization
(FISH)
COX-2 mRNAs were detected using FISH mixed
probe (RIBO Biotech Co., Ltd.) and an RNAScope kit (RIBO Biotech
Co., Ltd.). The ACHN cells were aliquoted into 8-well chamber
slides and treated with 20 µM sorafenib for 1.5 h when the cell
density reached 60%. Except for sorafenib, the treatment of the
control group was the same as that for the experimental group. Then
cells were fixed with 4% paraformaldehyde for 20 min at 4°C. After
permeation with 0.3% Triton X-100 for 15 min at room temperature
and pre-hybridization with prehybridized buffer (RIBO Biotech Co.,
Ltd.) for 30 min at 37°C, the COX-2 mRNA mixed probes were
added and incubated at 45°C overnight. Then, slides were washed
with washing buffer (RIBO Biotech Co., Ltd.) at 45°C and incubated
with G3BP1 antibody at 4°C overnight, and then the goat anti-Rabbit
IgG (H+L) Highly Cross-Adsorbed Alexa Fluor 488 secondary antibody
and goat anti-Mouse IgG (H+L) Cross-Adsorbed Alexa Fluor 594
secondary antibody at room temperature for 1 h. Nuclei were stained
with DAPI at room temperature for 10 min. The slides were observed
and imaged using the EVOS FL Auto 2.0 Imaging System.
Actinomycin D (Act D) chase
experiment
ACHN cells were treated with 10 µM sorafenib for 2 h
to upregulate the mRNA levels of COX-2 and then washed with
PBS twice before 5 µg/ml Act D (Sigma-Aldrich; Merck KGaA) was
added. After pre-incubation with Act D for 30 min, cells were
additionally treated for the indicated times (2 and 4 h) with
vehicle or with sorafenib. At the indicated time, the cells were
harvested and total RNA was extracted. RT-qPCR was used to detect
the COX-2 mRNA levels.
Xenograft tumor models
Male nude mice (4–6 weeks old, weighing 20.1±1.8 g)
were purchased from GemPharmatech. The animal experiments were
approved by The Ethical Committee of Nanjing Drum Tower Hospital
(Medical School of Nanjing University, Nanjing, China; project ID,
2021-640-01) and were conducted in accordance with The National
Institute of Health Guide for the Care and Use of Laboratory
Animals and The Institutional Animal Care and Use Committee of
Nanjing Drum Tower Hospital. Mice were raised in pathogen-free
animal facilities at 20–24°C, 50% relative humidity and 12 h of
light and dark cycle. Mice had free access to water and food, and
the health and behavior of the animals were monitored every day. In
the in vivo experiment of drug combination (n=6), ACHN cells
(3×106 cells in 100 µl PBS) were inoculated
subcutaneously in the right flank of each mouse. One week after
tumor implantation, the tumor volume of mice reached >50
mm3, mice were randomized and divided into four groups
with similar starting mean tumor volumes: Control, Sorafenib (30
mg/kg), Celecoxib (50 mg/kg), sorafenib (30 mg/kg) plus celecoxib
(50 mg/kg). The medicine was given by intragastric administration
three times a week for 22 days. In the in vivo experiment of
administration after knocking down G3BP1 (n=7), the mice were
divided into four groups: ACHNCtrlKD with control
treatment, ACHNCtrlKD with sorafenib (30 mg/kg)
treatment, ACHNG3BP1KD with control treatment,
ACHNG3BP1KD with sorafenib (30 mg/kg) treatment. The
lentivirus knockdown stable cell line ACHNG3BP1KD and
control cell line ACHNCtrlKD were amplified and
4×106 cells were suspended in 100 µl PBS and inoculated
subcutaneously in the right flank of each mouse. One week after
tumor implantation, the tumor volume of mice reached >50
mm3, at which time the medicine was given to the mice.
The medicine was given by intragastric administration three times a
week for 16 days. Considering animal welfare, action should be
taken to reduce the pain of animals. The mice were euthanized with
intraperitoneal injection of 60 mg/kg pentobarbital followed by
rapid cervical dislocation. Mice were judged to be dead if they
were not breathing and did not exhibit nerve reflexes. Tumor volume
was calculated using the formula: V=½ × L × W2, where L
is the maximum diameter and W is the minimum diameter of the
tumor.
Statistical analysis
All data were presented as mean ± SD. Two group
comparisons were performed by unpaired Student's t-test.
Statistical analyses involving multiple group comparisons were
performed using one-way ANOVA followed by Dunnett's or Tukey's post
hoc test. All data analyses were performed with GraphPad Prism 7
(Dotmatics). P<0.05 was considered to indicate a statistically
significant difference.
Results
COX-2 expression levels determine
sensitivity of renal cancer cells to sorafenib
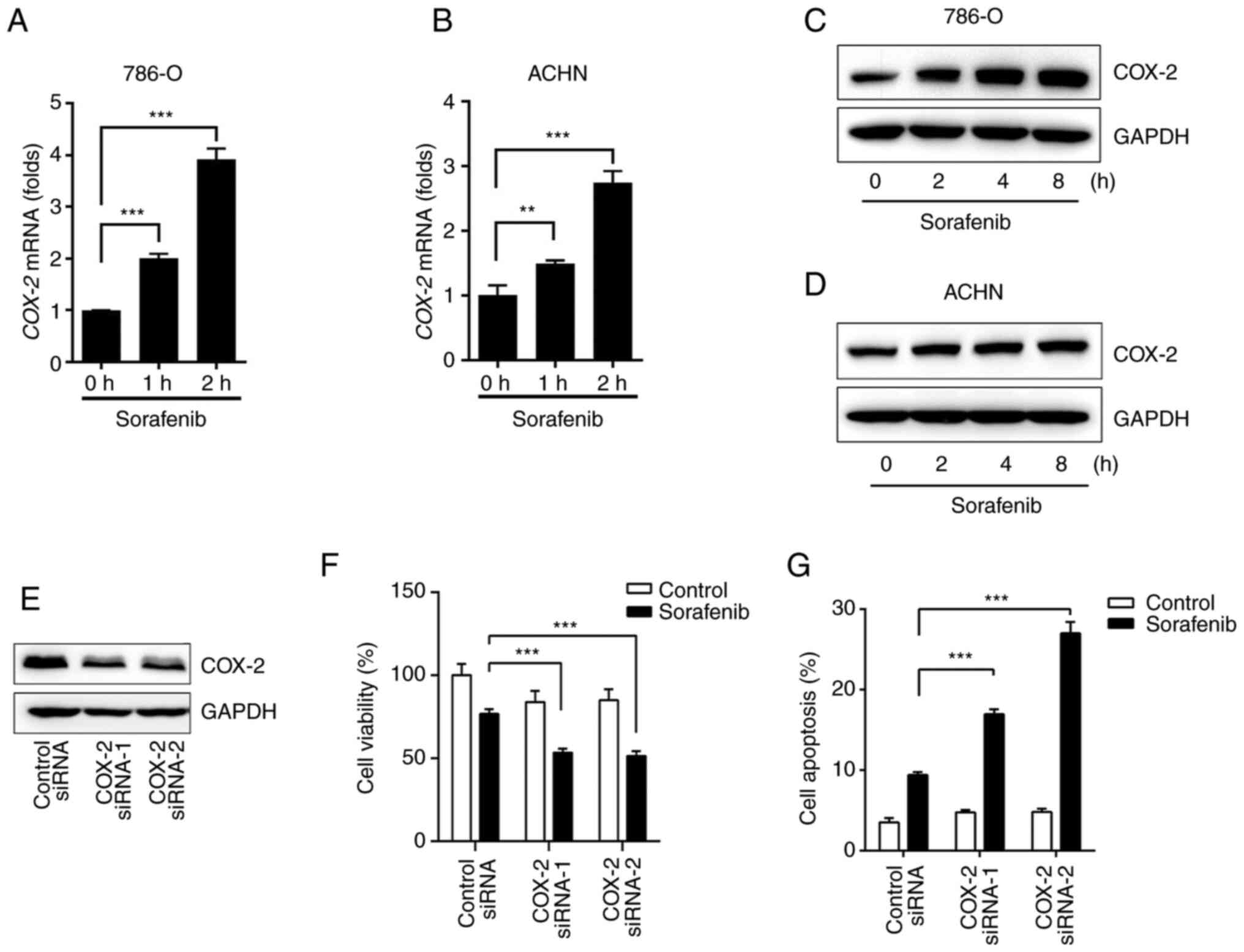
By examining COX-2 levels before and after sorafenib
treatment, it was found that sorafenib rapidly increased mRNA and
protein levels of COX-2 in ACHN and 786-O cells (Fig. 1A-D). COX-2 plays a crucial role in
the progression of renal cancer (23) and a previous study has suggested
that COX-2 decreases the sensitivity of sorafenib in hepatocellular
carcinoma (24). In the present
study, it was found that the cytotoxicity of sorafenib was affected
by the presence of COX-2. After the silencing of COX-2 (Fig. 1E), the specificity effect of COX-2
was indicated by the significant attenuation of ACHN cell survival
after sorafenib treatment (Fig.
1F). By using flow cytometry analysis, sorafenib-induced
apoptosis was also notably increased in ACHN cells after the
silencing of COX-2 expression (Figs.
1G and S1A).
Celecoxib enhances the cytotoxicity of
sorafenib against RCC
Increasing evidence has implied that COX-2
inhibitors have potent antitumor effects (9). A case report has also described 2
desmoid tumor patients with multiple recurrences after a
combination medical and surgery, who had a major objective response
to a combination therapy of celecoxib and sorafenib (25). These findings highlight the
possibility that celecoxib may enhance the response to sorafenib in
renal cancer. To assess the effect of sorafenib and celecoxib on
the viability of human renal cancer cells, dose-dependent
inhibition of cell activity with sorafenib and celecoxib was
conducted. Cell viability was significantly inhibited after 48 h of
sorafenib treatment when the dose exceeded 8 µM (Fig. 2A). Celecoxib had no significant
effect on cell viability within 50 µM (Fig. 2B). Thus, 10 µM sorafenib and 30 µM
celecoxib were selected for combination therapy using dosage in
vitro. In the MTT assay, the combination of sorafenib and
celecoxib displayed significantly increased cytotoxicity compared
to sorafenib alone (Fig. 2C). This
synergy also occurred when celecoxib was applied in combination
with sorafenib during cell apoptosis experiments (Figs. 2D and S1B).
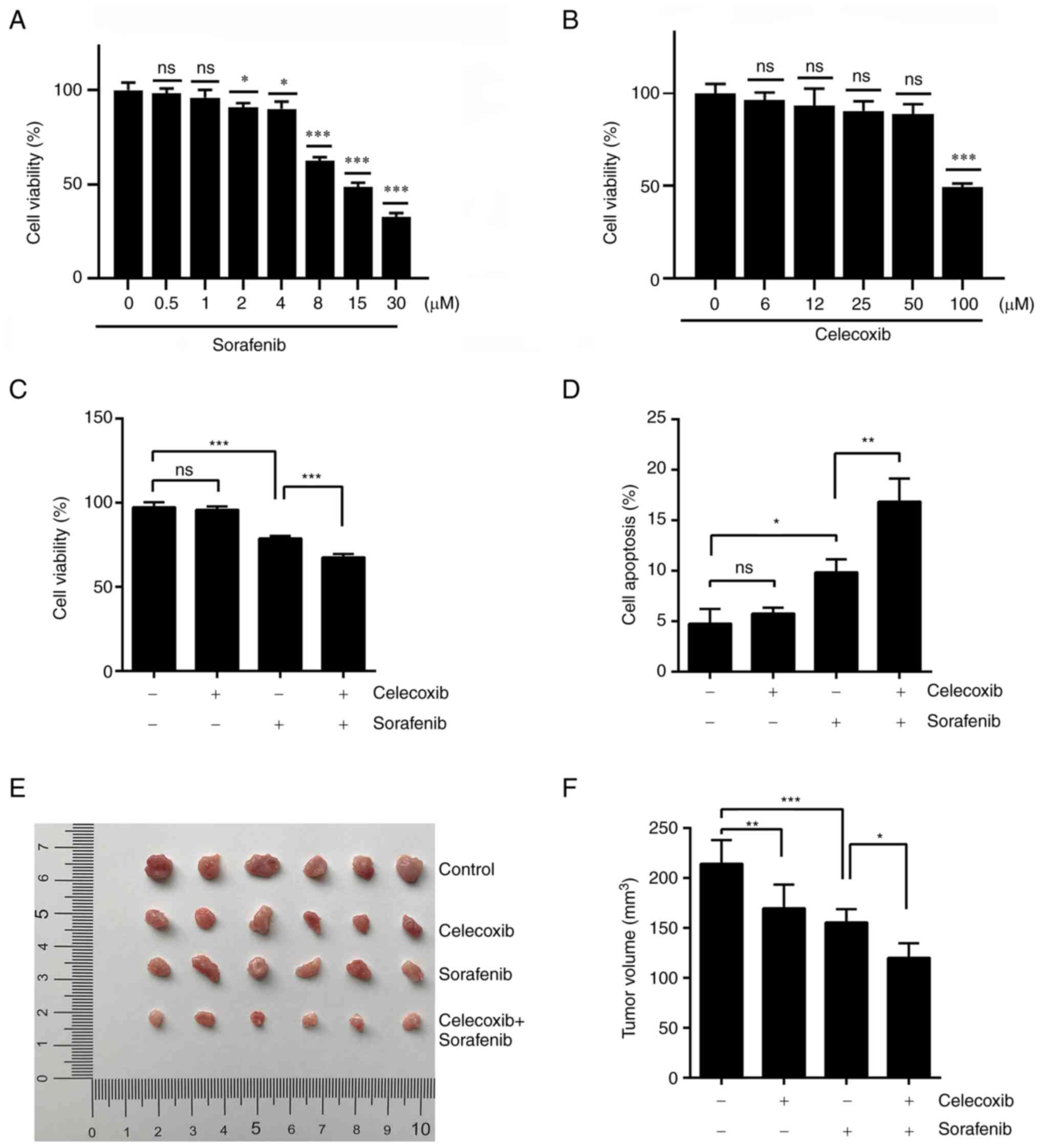 | Figure 2.Celecoxib in combination with
sorafenib for the treatment of renal cell carcinoma. (A) ACHN cells
were treated with sorafenib for 48 h at 0, 0.5, 1, 2, 4, 8, 15 and
30 µM. (B) ACHN cells were treated with celecoxib for 48 h at 0, 6,
12, 25, 50 and 100 µM. Cell viability was examined by MTT assay and
the viability of untreated cells was arbitrarily set at 100%. (C)
ACHN cells were treated with 30 µM celecoxib, 10 µM sorafenib or 30
µM celecoxib + 10 µM sorafenib for 48 h. Cell viability was
examined by MTT assay. (D) Cell apoptosis was detected by flow
cytometry. (E) Nude mice were randomized and divided into four
groups with similar starting mean tumor volumes. The groups were:
Control, 30 mg/kg sorafenib, 50 mg/kg celecoxib and 30 mg/kg
sorafenib + 50 mg/kg celecoxib. Tumors were resected and measured
after 22 days of drug treatment. (F) Statistical Analysis of tumor
Volume in (E). Data represent mean ± SD. *P<0.05, **P<0.01,
***P<0.001. ns, not significant. |
To confirm the synergistic effect of the combination
therapy in vivo, nude mice were inoculated with ACHN cells
to produce tumor-bearing models. Consistent with the previous in
vitro results, combined celecoxib and sorafenib treatments
significantly slowed the tumor growth of ACHN xenografts compared
with sorafenib alone (Fig. 2E). An
analysis of the tumors removed after experiments demonstrated that
combined celecoxib and sorafenib treatments resulted in a
significant decrease in tumor volume compared with sorafenib alone
(Fig. 2F). Overall, these results
demonstrated that the combination of celecoxib and sorafenib lead
to a significant synergistic effect on tumor growth inhibition.
Sorafenib induces the formation of
SGs
The mechanism by which sorafenib upregulates COX-2
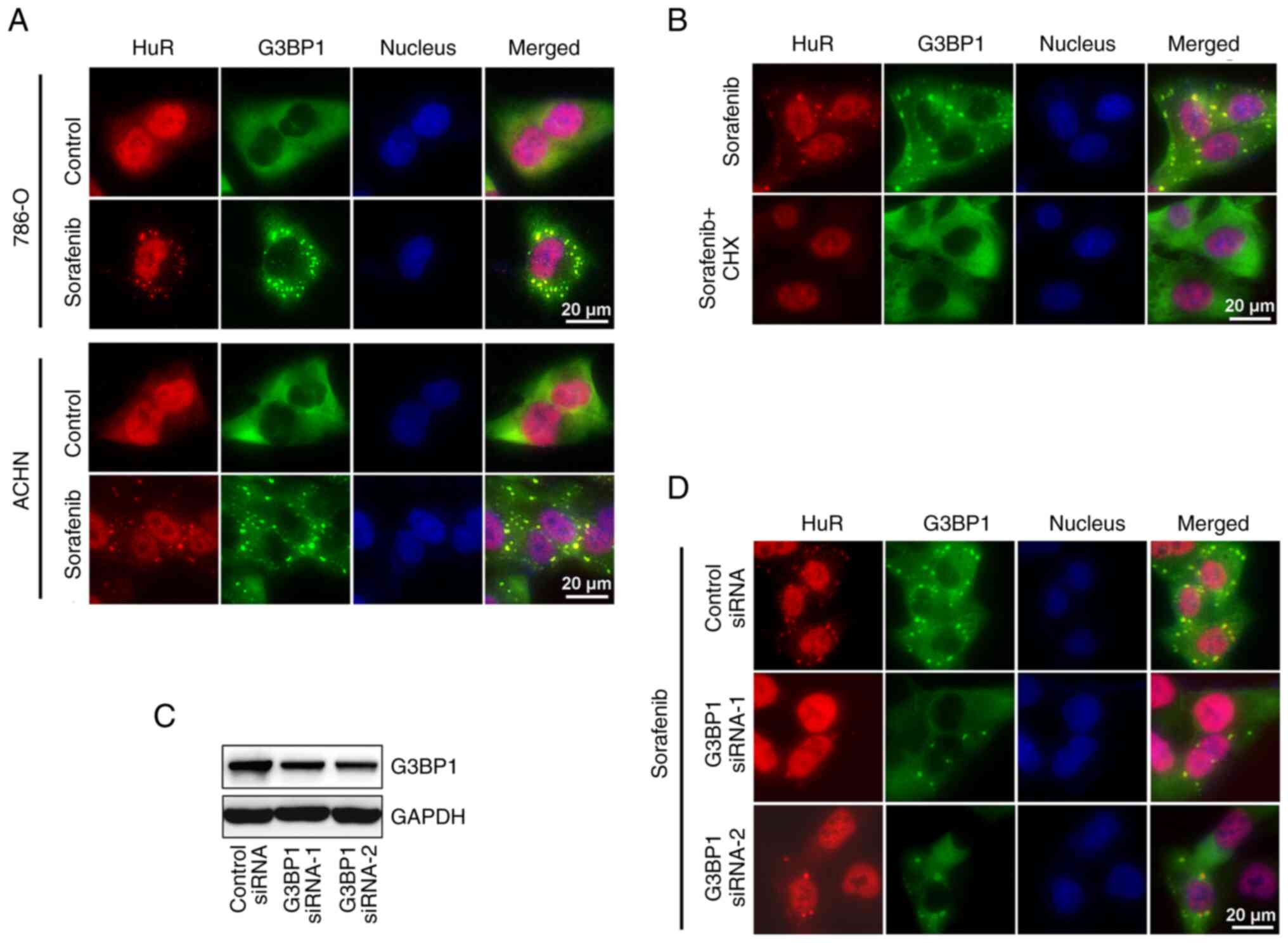
expression was further explored. It was observed that sorafenib can
induce HuR translocation to the cytoplasm and the formation of
numerous small foci in the cytoplasm. Using IF analysis, it was
observed that these foci were co-localized with SG markers, HuR and
G3BP1 (Fig. 3A). The formation of
these small cytoplasmic foci was inhibited in cells treated with
the SG inhibitor, cycloheximide (Fig.
3B), and they were therefore finally characterized as SGs.
SGs are assembled by liquid-liquid phase separation,
which results from unevenly distributed interactions across the
core protein-RNA network. G3BP1 is the central protein of this
network (26). In the present
study, ACHN cells were transfected with specific siRNA to knockdown
G3BP1 expression (Fig. 3C). As
expected, the formation of SGs was inhibited in cells with a
decreased expression of G3BP1 (Fig.
3D).
SGs sequester and stabilize COX-2 mRNA
in renal cancer cells
SGs contain translationally stalled mRNAs and RBPs,
such as HuR, which bind to mRNAs and modulate their stability
(22). Treatment of renal cancer
cells with sorafenib resulted in a rapid upregulation of
COX-2 mRNA (Fig. 1A and B).
A corresponding increase in the COX-2 protein level was also
detected only 2 h after sorafenib treatment (Fig. 1C and D). A previous study has
reported that COX-2 mRNA can be sequestered by SGs (22). To determine whether the increase in
COX-2 in sorafenib-treated renal cancer cells was due to the
stabilization of COX-2 mRNA by SGs, sorafenib-treated cells
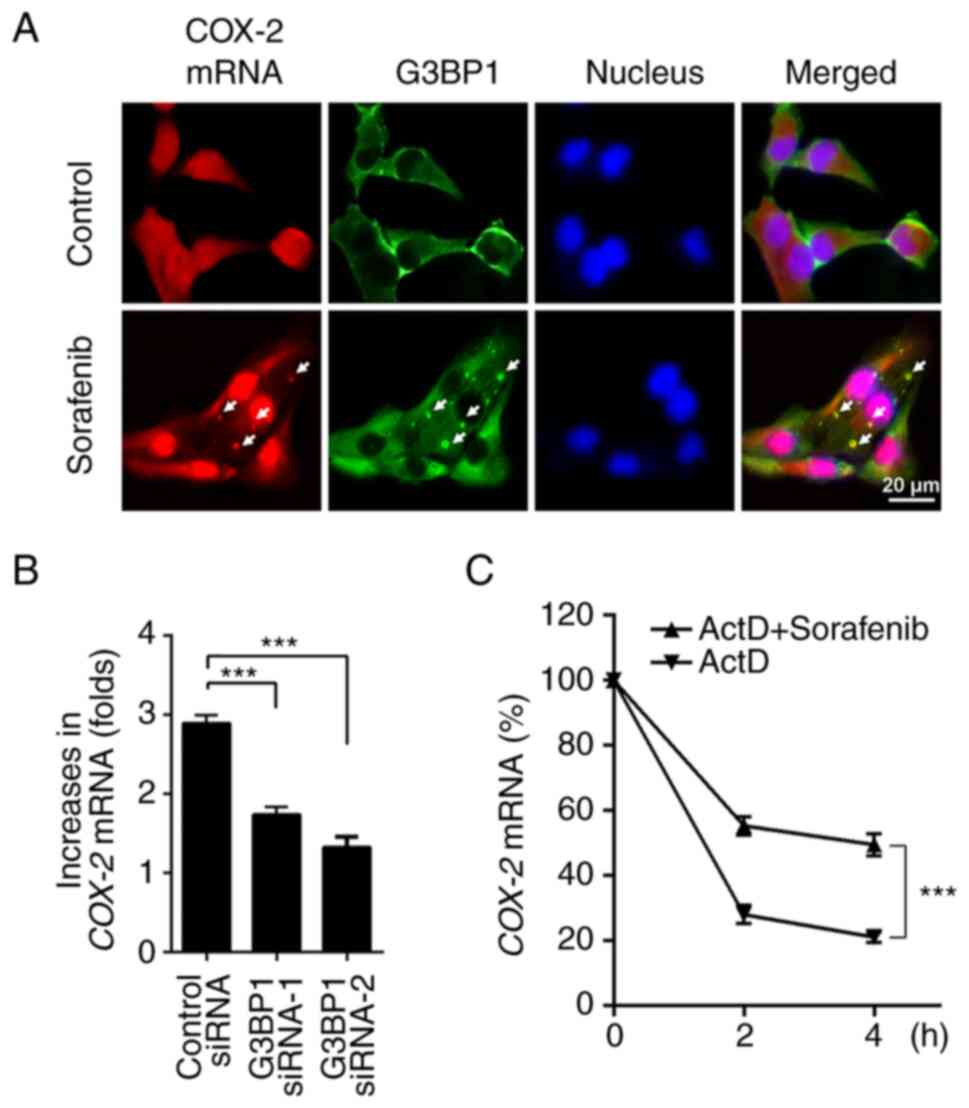
were examined by FISH of COX-2 mRNA and IF of G3BP1 protein.
Subsequently, granular distribution of COX-2 mRNA was
notably co-localized with G3BP1 protein (Fig. 4A).
Of note, sorafenib-enhanced COX-2 mRNA levels
were significantly suppressed after interfering with SG formation
by knocking down G3BP1 (Fig. 4B).
This result demonstrated that SGs were required for the
upregulation of COX-2 induced by sorafenib. The effect of SGs on
the stability of COX-2 mRNA was analyzed using 5 ug/ml Act
D. ACHN cells were treated with sorafenib for 2 h to allow the
upregulation of COX-2 mRNA. After pre-incubation with Act D,
cells were additionally treated with vehicle or sorafenib, and then
the COX-2 mRNA levels were determined at the indicated
times. The half-life of COX-2 mRNA in renal cancer cells
increased after sorafenib treatment and the decay of COX-2
mRNA was prevented by sorafenib therapy (Fig. 4C). These findings support the
suggestion that sorafenib upregulates COX-2 expression as its mRNA
is sequestrated and stabilized by SGs.
SGs protect cells from
sorafenib-induced cell death
To examine whether SG formation protects cancer
cells, silencing of G3BP1 expression in ACHN cells was performed to
disrupt SG formation (Fig. 3D).
Silencing G3BP1 markedly promoted sorafenib-induced cell death
(Fig. 5A). In addition,
sorafenib-induced cell apoptosis increased after the expression of
G3BP1 decreased (Figs. 5B and
S1C).
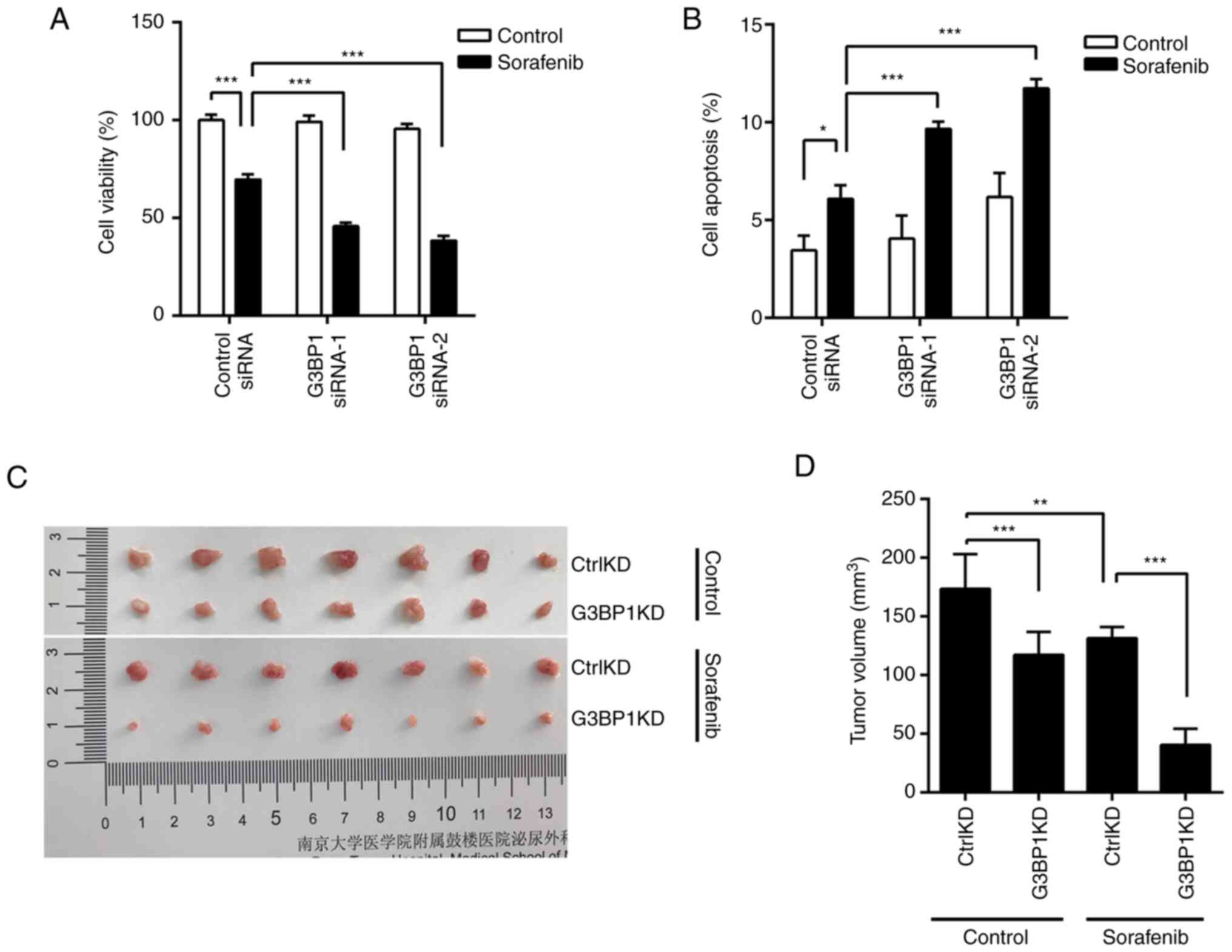 | Figure 5.SGs protect renal cancer cells from
sorafenib-induced cell death. (A) Control or G3BP1-silenced ACHN
cells were treated with 10 µM sorafenib for 48 h and cell viability
was examined by MTT assay. (B) After treatment, cell apoptosis was
detected by flow cytometry. (C) Nude mice were randomized and
divided into four groups with similar starting mean tumor volumes:
ACHNCtrlKD with control treatment, ACHNCtrlKD
with 30 mg/kg sorafenib treatment, ACHNG3BP1KD with
control treatment, ACHNG3BP1KD with 30 mg/kg sorafenib
treatment. Tumors were resected and measured after 16 days of drug
treatment. (D) Statistical Analysis of tumor Volume in (C). Data
represent mean ± SD. *P<0.05, **P<0.01, ***P<0.001. ns,
not significant. Ctrl, control; KD, knockdown; G3BP1, Ras
GTPase-activating protein-binding protein 1; siRNA, small
interfering RNA. |
To confirm the protective effect of SGs in
vivo, G3BP1 expression was stably knocked down in ACHN cells to
obtain ACHNG3BP1KD cells alongside control
ACHNCtrlKD cells, and then their response to sorafenib
was tested. The cells were inoculated into nude mice to produce
tumor-bearing models. Knockdown of G3BP1 significantly suppressed
the tumor growth of ACHN xenografts (Fig. 5C). In addition, sorafenib treatment
of ACHNG3BP1KD cells resulted in a marked reduction in
tumor volume compared with the treatment of ACHNCtrlKD
cells (Fig. 5D). Overall, these
data suggest that sorafenib-induced SGs have a protective effect on
renal cancer cells.
Discussion
COX-2 has been reported to modulate the sensitivity
of sorafenib to liver cancer (24).
In the present study, it was demonstrated that sorafenib rapidly
upregulated COX-2 expression, which decreased the sensitivity of
renal cancer cells to sorafenib. COX-2 inhibitors have potent
antitumor effects (27). Therefore,
the COX-2 inhibitor, celecoxib, in combination with sorafenib in
the treatment of RCC was explored. In both cell and animal
experiments, it was demonstrated that the combination therapy was
significantly better than sorafenib alone in the treatment of RCC.
Moreover, it was demonstrated that sorafenib could induce the
formation of SGs in renal cancer cells, and the upregulation of
COX-2 expression was dependent on the formation of SGs. SGs are
membrane-free structures in cells that selectively protect and
stabilize pro-survival mRNAs, and it has been reported that
COX-2 mRNA can be captured by SGs (22). In the present study, it was
demonstrated that COX-2 mRNA colocalized with
sorafenib-induced SGs in the cytoplasm and the half-life of
COX-2 mRNA was significantly increased after sorafenib
treatment. Disruption of SG formation by knocking down G3BP1
expression significantly promoted the cytotoxicity of sorafenib in
renal cancer. These results indicate that sorafenib can selectively
stabilize mRNAs by inducing SG formation, subsequently increasing
the level of COX-2 and the viability of renal cancer cells.
Therefore, a regulatory mechanism for the effect of sorafenib on
COX-2 levels has been uncovered and new light has been shed on
renal cancer therapy.
COX-2 commonly exerts a role in promoting cancer
(28–30). For example, COX-2 is a well-known
promoter of proliferation, angiogenesis, apoptosis inhibition and
immune suppression in melanoma (31). COX-2 expression in breast cancer is
associated with the increase of blood vessels and the elevated
expression of the angiogenesis marker, VEGF (32,33).
COX-2 also plays an essential role in the progression of renal
cancer (23,34). In the present study, it was found
that sorafenib rapidly and significantly increased COX-2 expression
in RCC cells. COX-2 has also previously been found to be important
in tumor immunosuppression and is associated with lower
infiltration of immune cells in melanoma tissues and a shorter
survival time (35,36). The COX-2/mPGES1/PGE2 pathway can
regulate the expression of PD-L1 in tumor-associated macrophages
and myeloid-derived suppressor cells (30). Immunotherapy offers new hope for
patients with cancer. However, the mechanisms of immune regulation
in the renal tumor microenvironment and their interaction with
molecularly targeted therapeutic agents require further
investigation.
COX-2 also exerts a crucial role in the promotion of
cancer cell resistance to chemotherapy (9). In the present study, it was
demonstrated that the upregulated expression of COX-2 promoted the
survival of renal cancer cells and decreased the cytotoxicity of
sorafenib. These results highlight the high potential of the COX-2
inhibitor, celecoxib, in the treatment of renal cancer,
particularly for enhancing the response to sorafenib. In mice
bearing human RCC xenografts, COX-2 inhibition can extend the
therapeutic effect of the VEGFR inhibitor, sunitinib (37). Celecoxib combined with sorafenib
significantly inhibited proliferation and induced apoptosis of
hepatocellular carcinoma cells (38). A combined treatment of sorafenib and
celecoxib had a more notable tumor suppression effect than the
single drug group in mice with lung cancer (39). In the present study, it was
demonstrated that the combined treatment with celecoxib
significantly enhanced the cytotoxicity of sorafenib on renal
carcinoma cells. COX-2 inhibitors are inexpensive, have tolerable
side effects and can sensitize cancer cells to chemotherapy
(40). A single-center randomized
controlled clinical phase III trial recommended celecoxib for the
mitigation of sorafenib-related skin toxicity in patients with
hepatocellular carcinoma (41).
Longer PFS was achieved in the celecoxib + sorafenib combination
group, although overall survival time was not prolonged in clinical
practice. Clinical trials combining celecoxib and sorafenib to
delay the progression of renal cancer should therefore be
considered.
Sorafenib can act on both wild-type and V599E mutant
RAF (42). The level of VEGFR/PDGFR
is still inhibited in RCC after sorafenib treatment (43). However, Some compensatory signaling
pathways such as PI3K-AKT-mTOR can promote renal cell survival and
cancer progression (44). To the
best of our knowledge, there is still no marker that predicts renal
cancer response to drug therapy. Sorafenib treatment creates a
pressure environment and the surviving cancer cells are more
aggressive (45). This adaptation
requires cancer cells to survive through their own stress response
mechanisms. The formation of SGs is a mechanism for minimizing
stress damage and increasing cell survival. SGs participate in
inflammatory and apoptotic signaling and promote cell survival
(46,47). Sorafenib also promotes SG production
in various cancer cells, including HeLa, MCF-7, PC3 and LnCaP cells
(48). In the present study, it was
found that sorafenib could induce the assembly of SGs in renal
cancer cells. Moreover, these SGs protected and promoted the
survival of renal cancer cells by selectively stabilizing
COX-2 mRNA and promoting its expression. SG proteins are
involved in processes of translation and mRNA stability. Targeting
SGs may be a novel method of treating disease (49). The specific mechanism of sensitivity
of renal cancer cells to sorafenib, which is regulated by SGs,
needs further exploration as targeting SGs may be a potential
therapeutic strategy for renal cancer therapy.
However, There are inevitably some limitations to
the present study. In the in vitro study, normal renal
cancer cells were used to conduct cell sensitivity experiments
after drug treatments. If the normal renal cancer cells were
replaced with cell lines resistant to sorafenib, it may be closer
to the physiological conditions of drug resistance. In the in
vivo study, it was demonstrated that celecoxib combined with
sorafenib can inhibit the growth of subcutaneous tumors in nude
mice. However, there was a lack of exploration of the molecular
mechanism behind this drug combination. In the molecular mechanism
study, it was demonstrated that SGs can stabilize COX-2 mRNA.
However, it is worth noting that SGs may also play different roles
by regulating the mRNA stability of other molecules, which requires
a more in-depth study.
In summary, sorafenib is an identified drug that can
upregulate COX-2 levels in renal cancer cells. COX-2 promotes the
survival of renal cancer cells. The use of celecoxib, a COX-2
inhibitor, in combination with sorafenib significantly enhanced the
sensitivity of renal cancer cells to sorafenib and improved
efficacy. Clinical trials of celecoxib in combination with
sorafenib for the treatment of renal cancer should be considered.
In the present study, a novel regulatory mechanism for COX-2
expression was uncovered. Sorafenib-induced assembly of SGs in
renal cancer cells is a critical event that promotes COX-2
expression and acts as a stress mechanism against treatment.
Therefore, the assembly of SGs and their role in regulating the
expression of different oncogenes is crucial for additional
exploration. The findings of the present study may provide novel
targets for the treatment of renal cancer.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by The National Natural Science
Foundation of China (grant no. 81972387) and The Nanjing Health
Distinguished Youth Fund (grant no. JQX20002).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HQD, GLW, WMC, WQ, WC and HQG confirm the
authenticity of all the raw data. HQG and WC participated in the
conception and design of the study. HQD and GLW performed all the
experiments with help from WMC and WQ. WC and HQG wrote the
manuscript, performed the statistical analyses and evaluated the
results. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by The Ethical Committee of
Nanjing Drum Tower Hospital (Medical School of Nanjing University,
Nanjing, China; project ID, 2021–640-01), and all experiments
involving mice were conducted in accordance with The National
Institute of Health Guide for the Care and Use of Laboratory
Animals and The Institutional Animal Care and Use Committee of
Nanjing Drum Tower Hospital.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Capitanio U, Bensalah K, Bex A, Boorjian
SA, Bray F, Coleman J, Gore JL, Sun M, Wood C and Russo P:
Epidemiology of renal cell carcinoma. Eur Urol. 75:74–84. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Capitanio U and Montorsi F: Renal cancer.
Lancet. 387:894–906. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu L, Cao Y, Chen C, Zhang X, McNabola A,
Wilkie D, Wilhelm S, Lynch M and Carter C: Sorafenib blocks the
RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor
cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer
Res. 66:11851–11858. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ljungberg B, Albiges L, Abu-Ghanem Y,
Bensalah K, Dabestani S, Fernández-Pello S, Giles RH, Hofmann F,
Hora M, Kuczyk MA, et al: European association of urology
guidelines on renal cell carcinoma: The 2019 update. Eur Urol.
75:799–810. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sano Y, Kogashiwa Y, Araki R, Enoki Y,
Ikeda T, Yoda T, Nakahira M and Sugasawa M: Correlation of
inflammatory markers, survival, and COX2 expression in oral cancer
and implications for prognosis. Otolaryngol Head Neck Surg.
158:667–676. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gurram B, Zhang S, Li M, Li H, Xie Y, Cui
H, Du J, Fan J, Wang J and Peng X: Celecoxib conjugated fluorescent
probe for identification and discrimination of cyclooxygenase-2
enzyme in cancer cells. Anal Chem. 90:5187–5193. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu WK, Sung JJ, Lee CW, Yu J and Cho CH:
Cyclooxygenase-2 in tumorigenesis of gastrointestinal cancers: An
update on the molecular mechanisms. Cancer Lett. 295:7–16. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hashemi Goradel N, Najafi M, Salehi E,
Farhood B and Mortezaee K: Cyclooxygenase-2 in cancer: A review. J
Cell Physiol. 234:5683–5699. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li H, Zhu F, Boardman LA, Wang L, Oi N,
Liu K, Li X, Fu Y, Limburg PJ, Bode AM and Dong Z: Aspirin prevents
colorectal cancer by normalizing EGFR expression. EBioMedicine.
2:447–455. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xiao J, Wang F, Lu H, Xu S, Zou L, Tian Q,
Fu Y, Lin X, Liu L, Yuan P, et al: Targeting the COX2/MET/TOPK
signaling axis induces apoptosis in gefitinib-resistant NSCLC
cells. Cell Death Dis. 10:7772019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tong D, Liu Q, Wang LA, Xie Q, Pang J,
Huang Y, Wang L, Liu G, Zhang D, Lan W and Jiang J: The roles of
the COX2/PGE2/EP axis in therapeutic resistance. Cancer Metastasis
Rev. 37:355–368. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Khafaga AF, Shamma RN, Abdeen A, Barakat
AM, Noreldin AE, Elzoghby AO and Sallam MA: Celecoxib repurposing
in cancer therapy: Molecular mechanisms and nanomedicine-based
delivery technologies. Nanomedicine (Lond). 16:1691–1712. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bley N, Lederer M, Pfalz B, Reinke C,
Fuchs T, Glaß M, Möller B and Hüttelmaier S: Stress granules are
dispensable for mRNA stabilization during cellular stress. Nucleic
Acids Res. 43:e262015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Protter DSW and Parker R: Principles and
properties of stress granules. Trends Cell Biol. 26:668–679. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Grabocka E and Bar-Sagi D: Mutant KRAS
enhances tumor cell fitness by upregulating stress granules. Cell.
167:1803–1813.e12. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thedieck K, Holzwarth B, Prentzell MT,
Boehlke C, Kläsener K, Ruf S, Sonntag AG, Maerz L, Grellscheid SN,
Kremmer E, et al: Inhibition of mTORC1 by astrin and stress
granules prevents apoptosis in cancer cells. Cell. 154:859–874.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Somasekharan SP, El-Naggar A, Leprivier G,
Cheng H, Hajee S, Grunewald TG, Zhang F, Ng T, Delattre O,
Evdokimova V, et al: YB-1 regulates stress granule formation and
tumor progression by translationally activating G3BP1. J Cell Biol.
208:913–929. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gao X, Jiang L, Gong Y, Chen X, Ying M,
Zhu H, He Q, Yang B and Cao J: Stress granule: A promising target
for cancer treatment. Br J Pharmacol. 176:4421–4433. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao J, Fu X, Chen H, Min L, Sun J, Yin J,
Guo J, Li H, Tang Z, Ruan Y, et al: G3BP1 interacts with YWHAZ to
regulate chemoresistance and predict adjuvant chemotherapy benefit
in gastric cancer. Br J Cancer. 124:425–436. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shi Q, Zhu Y, Ma J, Chang K, Ding D, Bai
Y, Gao K, Zhang P, Mo R, Feng K, et al: Prostate cancer-associated
SPOP mutations enhance cancer cell survival and docetaxel
resistance by upregulating Caprin1-dependent stress granule
assembly. Mol Cancer. 18:1702019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ansari MY and Haqqi TM: Interleukin-1β
induced stress granules sequester COX-2 mRNA and regulates its
stability and translation in human OA chondrocytes. Sci Rep.
6:276112016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kaminska K, Szczylik C, Lian F and
Czarnecka AM: The role of prostaglandin E2 in renal cell cancer
development: Future implications for prognosis and therapy. Future
Oncol. 10:2177–2187. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dong XF, Liu TQ, Zhi XT, Zou J, Zhong JT,
Li T, Mo XL, Zhou W, Guo WW, Liu X, et al: COX-2/PGE2 axis
regulates HIF2α activity to promote hepatocellular carcinoma
hypoxic response and reduce the sensitivity of sorafenib treatment.
Clin Cancer Res. 24:3204–3216. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Benech N, Walter T and Saurin JC: Desmoid
tumors and celecoxib with sorafenib. N Engl J Med. 376:2595–2597.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang P, Mathieu C, Kolaitis RM, Zhang P,
Messing J, Yurtsever U, Yang Z, Wu J, Li Y, Pan Q, et al: G3BP1 is
a tunable switch that triggers phase separation to assemble stress
granules. Cell. 181:325–345.e28. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tołoczko-Iwaniuk N, Dziemiańczyk-Pakieła
D, Nowaszewska BK, Celińska-Janowicz K and Miltyk W: Celecoxib in
cancer therapy and prevention-review. Curr Drug Targets.
20:302–315. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun Y, Dai H, Chen S, Zhang Y, Wu T, Cao
X, Zhao G, Xu A, Wang J and Wu L: Disruption of chromosomal
architecture of cox2 locus sensitizes lung cancer cells to
radiotherapy. Mol Ther. 26:2456–2465. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tong D, Liu Q, Liu G, Xu J, Lan W, Jiang
Y, Xiao H, Zhang D and Jiang J: Metformin inhibits
castration-induced EMT in prostate cancer by repressing
COX2/PGE2/STAT3 axis. Cancer Lett. 389:23–32. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Prima V, Kaliberova LN, Kaliberov S,
Curiel DT and Kusmartsev S: COX2/mPGES1/PGE2 pathway regulates
PD-L1 expression in tumor-associated macrophages and
myeloid-derived suppressor cells. Proc Natl Acad Sci USA.
114:1117–1122. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tudor DV, Bâldea I, Lupu M, Kacso T,
Kutasi E, Hopârtean A, Stretea R and Gabriela Filip A: COX-2 as a
potential biomarker and therapeutic target in melanoma. Cancer Biol
Med. 17:20–31. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lala PK, Nandi P and Majumder M: Roles of
prostaglandins in tumor-associated lymphangiogenesis with special
reference to breast cancer. Cancer Metastasis Rev. 37:369–384.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Davies G, Salter J, Hills M, Martin LA,
Sacks N and Dowsett M: Correlation between cyclooxygenase-2
expression and angiogenesis in human breast cancer. Clin Cancer
Res. 9:2651–2656. 2003.PubMed/NCBI
|
|
34
|
Li Z, Zhang Y, Kim WJ and Daaka Y: PGE2
promotes renal carcinoma cell invasion through activated RalA.
Oncogene. 32:1408–1415. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim SH, Roszik J, Cho SN, Ogata D, Milton
DR, Peng W, Menter DG, Ekmekcioglu S and Grimm EA: The COX2
effector microsomal PGE2 synthase 1 is a regulator of
immunosuppression in cutaneous melanoma. Clin Cancer Res.
25:1650–1663. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mao Y, Poschke I, Wennerberg E, Pico de
Coaña Y, Egyhazi Brage S, Schultz I, Hansson J, Masucci G,
Lundqvist A and Kiessling R: Melanoma-educated CD14+ cells acquire
a myeloid-derived suppressor cell phenotype through COX-2-dependent
mechanisms. Cancer Res. 73:3877–3887. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang X, Zhang L, O'Neill A, Bahamon B,
Alsop DC, Mier JW, Goldberg SN, Signoretti S, Atkins MB, Wood CG
and Bhatt RS: Cox-2 inhibition enhances the activity of sunitinib
in human renal cell carcinoma xenografts. Br J Cancer. 108:319–326.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cervello M, Bachvarov D, Lampiasi N,
Cusimano A, Azzolina A, McCubrey JA and Montalto G: Novel
combination of sorafenib and celecoxib provides synergistic
anti-proliferative and pro-apoptotic effects in human liver cancer
cells. PLoS One. 8:e655692013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang H, Li Z and Wang K: Combining
sorafenib with celecoxib synergistically inhibits tumor growth of
non-small cell lung cancer cells in vitro and in
vivo. Oncol Rep. 31:1954–1960. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shi L, Xu L, Wu C, Xue B, Jin X, Yang J
and Zhu X: Celecoxib-induced self-assembly of smart
albumin-doxorubicin conjugate for enhanced cancer therapy. ACS Appl
Mater Interfaces. 10:8555–8565. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen JC, Wang JC, Pan YX, Yi MJ, Chen JB,
Wang XH, Fu YZ, Zhang YJ, Xu L, Chen MS, et al: Preventive effect
of celecoxib in sorafenib-related hand-foot syndrome in
hepatocellular carcinoma patients, a single-center, open-label,
randomized, controlled clinical phase III trial. Am J Cancer Res.
10:1467–1476. 2020.PubMed/NCBI
|
|
42
|
Wilhelm SM, Adnane L, Newell P, Villanueva
A, Llovet JM and Lynch M: Preclinical overview of sorafenib, a
multikinase inhibitor that targets both Raf and VEGF and PDGF
receptor tyrosine kinase signaling. Mol Cancer Ther. 7:3129–3140.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bergers G and Hanahan D: Modes of
resistance to anti-angiogenic therapy. Nat Rev Cancer. 8:592–603.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Rini BI and Atkins MB: Resistance to
targeted therapy in renal-cell carcinoma. Lancet Oncol.
10:992–1000. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fulda S, Gorman AM, Hori O and Samali A:
Cellular stress responses: Cell survival and cell death. Int J Cell
Biol. 2010:2140742010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wippich F, Bodenmiller B, Trajkovska MG,
Wanka S, Aebersold R and Pelkmans L: Dual specificity kinase DYRK3
couples stress granule condensation/dissolution to mTORC1
signaling. Cell. 152:791–805. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Arimoto K, Fukuda H, Imajoh-Ohmi S, Saito
H and Takekawa M: Formation of stress granules inhibits apoptosis
by suppressing stress-responsive MAPK pathways. Nat Cell Biol.
10:1324–1332. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Adjibade P, St-Sauveur VG, Quevillon
Huberdeau M, Fournier MJ, Savard A, Coudert L, Khandjian EW and
Mazroui R: Sorafenib, a multikinase inhibitor, induces formation of
stress granules in hepatocarcinoma cells. Oncotarget.
6:43927–43943. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wang F, Li J, Fan S, Jin Z and Huang C:
Targeting stress granules: A novel therapeutic strategy for human
diseases. Pharmacol Res. 161:1051432020. View Article : Google Scholar : PubMed/NCBI
|