Introduction
In cancer cells, aberrant lipid metabolism,
particularly cholesterol, disrupts normal cell signaling and
motility, as cholesterol plays key roles in these processes
(1). The inhibition of the
cholesterol pathway may thus be an efficient strategy which can be
used to limit tumor growth and the metastatic process (2,3). The
do novo synthesis of cholesterol is a complex multi-step
process catalyzed by enzymes (4).
The antitumor efficacy may differ when targeting different nodes
within the cholesterol metabolic pathway due to the nature of
feedback responses elicited, and whether synthesis and transport
processes are simultaneously inhibited (5).
Some researchers have reported that an increased
flux through lanosterol, rather than merely the upregulation of the
cholesterol biosynthesis pathway, leads to more malignant
phenotypes of cancer cells (6). In
the process of the cholesterol anabolic pathway, lanosterol
synthase (LSS) catalyzes the formation of the first cyclized
product, lanosterol, and also the shunt pathway to
24(S),25-epoxycholesterol. Increased 24(S),25-epoxycholesterolby
the partial inhibition of LSS reduces the effects of compensatory
mechanisms to maintain cholesterol by tumor cells due to a higher
affinity for diepoxysqualene than epoxysqualene (7). The inhibition of LSS as a therapeutic
target is potentially advantageous and represents an attractive
target in this regard. RO 48-8071, a specific inhibitor of LSS, has
been shown to inhibit the proliferation of various tumor types
(8,9).
The present study aimed to investigate the effects
of the knockdown of LSS on liver cancer progression and the
possible molecular mechanisms in HepG2 cells.
Materials and methods
Cells and cell culture
The human liver cancer cell line, HepG2, was stored
cultured in a humidified incubator at 37°C with 5% CO2
in DMEM (Gibco; Thermo Fisher Scientific, Inc.) containing 10%
heat-inactivated FBS (cat. no FB15011; Clark Bioscience)
supplemented with penicillin and streptomycin.
Construction of LSS short hairpin
(sh)RNA
In total, three shRNAs were designed targeting human
LSS gene sequences GGACTGCGCTCAACTATGT. The sequences were digested
with restriction endonuclease BamHI at the 5′ end and
HindIII at the 3′ end and inserted into the pRNAT-U6.1/Neo
plasmid. The recombinant plasmids were then transformed into E.
coli DH5α and screened using medium containing ampicillin. The
positive clones were selected with 50 µg/ml ampicillin and expanded
to obtain the recombinant DNA for sequencing. The primers for each
target sequence are presented in Table
I.
 | Table I.Primers used for shRNA
construction. |
Table I.
Primers used for shRNA
construction.
| Primer name | Sequence
(5′à3′) |
|---|
| shLSS | Forward:
GATCCCGGACTGCGCTCAACTATGTTTGATATCCGACATAGTTGAGCGCAGTCCTTTTTTCCAAA |
|
| Reverse:
AGCTTTTGGAAAAAAGGACTGCGCTCAACTATGTCGGATATCAAACATAGTTGAGCGCAGTCCGG |
LSS knockdown by shRNA in HepG2
cells
The confirmed shLSS- pRNAT-U6.1/Neo and
pRNAT-U6.1/Neo vector plasmids were transfected into HepG2 cells
using Lipofectamine 2000 (cat. no. 11668019; Invitrogen; Thermo
Fisher Scientific, Inc.) following the protocol provided by the
manufacturer. A total of 500 ng plasmid DNA diluted in 250 µl
Opti-MEM® medium was mixed together with 5 µl
Lipofectamine® in 50 µl Opti-MEM® and
incubated for 5 min at room temperature. The DNA-lipid complex was
added to HepG2 cells at ~70% confluency, cultured in 24-well
plates. Following a 24-h incubation at 37°C, the cells were
selected with G418 at 500 µl/ml. Viable cells transfected with the
plasmids exhibit green fluorescence under a fluorescence
microscope. Thus, the transfection efficiency was detected using a
fluorescence microscope. The stably transfected HepG2 cell clones
were harvested and LSS expression was detected using western blot
analysis to verify the efficiency of RNA interference by shRNAs
targeting different human LSS gene sequences.
Cell Counting Kit-8 (CCK-8) cell
proliferation assay
Cell proliferation was detected using CCK-8 assay
according to the manufacturer's instructions (cat. no. K1018;
APeXBIO Technology LLC). The stably transfected HepG2 cells, at a
density of 2,000, 3,000 or 6,000 cells in 100 µl 10% FBS/DMEM, were
seeded in 96-well plates and cultured for 48 h. This was followed
by the addition of 10 µl CCK-8 to each well and incubation for 4 h
at 37°C. The absorbance at 450 nm was measured using a microplate
reader (ELx800; BioTek Instruments, Inc.) following gentle mixing
on a shaker.
Cell cycle analysis
Cells were seeded in six-well plates and cultured
routinely to ~80% confluency. The cells were then trypsinized,
washed and fixed in ice-cold 70% ethanol at 4°C for 30 min followed
by treatment with 50 µg/ml propidium iodide (PI, Beijing Biosea
Biotechnology Co., Ltd.) in staining buffer for 30 min at 4°C away
from light. Cell cycle distribution was then analyzed on a BD FACSV
flow cytometer (BD Biosciences). The data obtained from flow
cytometric analyses were examined using ModFit software (v3.1;
Verity Software House, Inc.).
Wound healing assay
Cells were seeded in 24-well plates and cultured to
100% confluency. Scratches were then made in the middle of the
confluent monolayer cells using sterile pipette tips. The debris
was removed by washing with PBS before obtaining images using a
light microscope (Nikon TS2-FL; Nikon Corporation). The cells were
incubated with fresh DMEM with 2% FBS for 48 h and images were
captured (10). The wound area was
measured using Quantity One software (v4.6.6; Bio-Rad Laboratories,
Inc.) and the wound area changes at 48 h were calculated by
measuring the area of the wound at 0 h minus the wound area at 48
h.
Western blot analysis
The cells were harvested, lysed in RIPA lysis buffer
(Sigma-Aldrich; Merck KGaA) on ice and centrifuged at 4°C to yield
total cellular protein. The protein concentration was determined
using BCA assay; Beyotime Institute of Biotechnology).
Subsequently, ~30 µg lysate were loaded on each lane of 12%
SDS-PAGE and transferred onto a PVDF membrane. Following a 2-h
blocking in 5% non-fat milk in PBST (0,05% Tween-20 in PBS) at room
temperature, the membrane was incubated with primary antibody (LSS,
cat. no. sc-514507, 1:500; cyclin B1, cat. no. sc-245, 1:500, both
Santa Cruz; cyclin E, cat. no. sc-377100, 1:500, Santa Cruz, CA;
GRP78, cat. no. ab21685, 1:500, Abcam, Cambridge, UK; CLPX 1, cat.
no. PA5-79052, 1:500, Thermo Fisher Scientific, Inc., Waltham, MA;
Bcl-2, cat. no. 9662S, 1:400, Cell Signaling Technology, Danvers,
MA; NF-κB, cat. no. ab32536, 1:500, Abcam, Cambridge, UK; endocan,
cat. no. sc-515304, 1:300, Santa Cruz, CA; pSrc, cat. no. 12432S,
1:500, Cell Signaling Technology, Danvers, MA; Src, cat. no. 2109S,
1:500, Cell Signaling Technology, Danvers, MA; pAKT, cat. no.
sc-514032, 1:500, Santa Cruz, CA; AKT, cat. no. sc-5298, 1:500,
Santa Cruz, CA; pERK, cat. no. sc-81492, 1:500, Santa Cruz, CA;
ERK1/2, cat. no. sc-514302, 1:500, Santa Cruz, CA; β-actin, cat.
no. sc-47778, 1:1,000, Santa Cruz, CA.) at 4°Covernight and
HRP-conjugated secondary antibody (1:10,000, goat anti-rabbit IgG
with cat. no. AP132P and goat anti-mouse IgG with cat. no. AP130,
Sigma Aldrich, Merck KGaA) for 2 h at room temperature. The
SuperSignal™ West Femto Trial kit (Thermo Fisher Scientific, Inc.)
and Chemi Scope (Clinx Science Instrument Co., Ltd.) were used to
visualize the signals and obtain images of the bands. The specific
bands were quantified using Quantity One software (v4.6.6; Bio-Rad
Laboratories, Inc.).
Statistical analysis
SPSS version 15.0 software (SPSS, Inc.) was used to
analyze all the data in the present study. Significant differences
between groups were analyzed using one-way ANOVA followed by
Tukey's post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Knockdown of LSS in HepG2 cells is
successful and effective
The shRNA sequences targeting LSS were constructed
into the pRNAT-U6.1/Neo plasmid and the recombinant plasmids were
sequenced. The sequences of each recombinant plasmids containing
LSS shRNA are presented in Table
II. The confirmed plasmids were transfected into HepG2 cells
and selected using G418 to obtain stable-transfection cell clones.
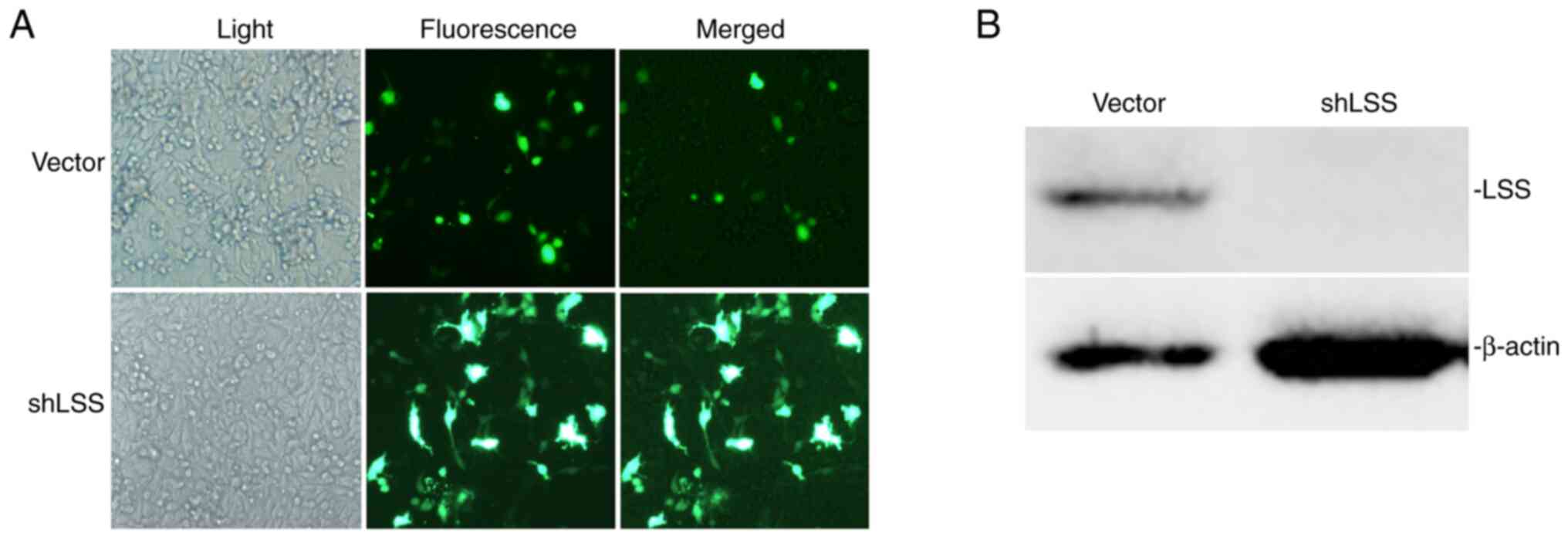
Cells stably transfected with LSS shRNA exhibited an evident
decrease in LSS expression. Together with labeled green fluorescent
protein observed under a fluorescence microscope, it could be
concluded that LSS knockdown cell line was successful and that LSS
expression was effectively suppressed by shRNA targeting LSS
(Fig. 1).
| Table II.Sequences of recombinant plasmids
containing LSS shRNA. |
Table II.
Sequences of recombinant plasmids
containing LSS shRNA.
| Target
sequences | DNA sequencing of
cloned fragments (5′à3′) |
|---|
| h-LSS-shRNA
(GGACTGCGCTCAACTATGT) |
AAAATTCTTGGGTAGTTTGCAGTTTTAAATTATGTTTTAAAATGGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTGGGTTTATATATCTTGTGGAAAGGACGCGGGATCCCGGACTGCGCTCAACTATGTTTGATATCCGACATAGTTGAGCGCAGTCCTTTTTTCCAAAAGCTTAAGTTTAAACCGCTGATCAGCCTCGACTGTGCCTTCTAAATAGTAATCAATTACGGGGTCATTAGTTCATAGCCCATATATGGAGTTCCGCGTTACATAACTTACGGTAAATGGC |
LSS knockdown inhibits the
proliferation and migration but induces the apoptosis of HepG2
cells
Fundamentally, the disruption of cell growth and
proliferation are basic characteristics of malignant tumors. CCK-8
cell proliferation assay revealed a cytostatic effect of LSS
knockdown on HepG2 cells. The viability of the HepG2 cells was
significantly reduced following LSS knockdown when compared with
the vector control-transfected cells, with initial seeding
densities at 2,000, 3,000 or 6,000 cells per well (Fig. 2C).
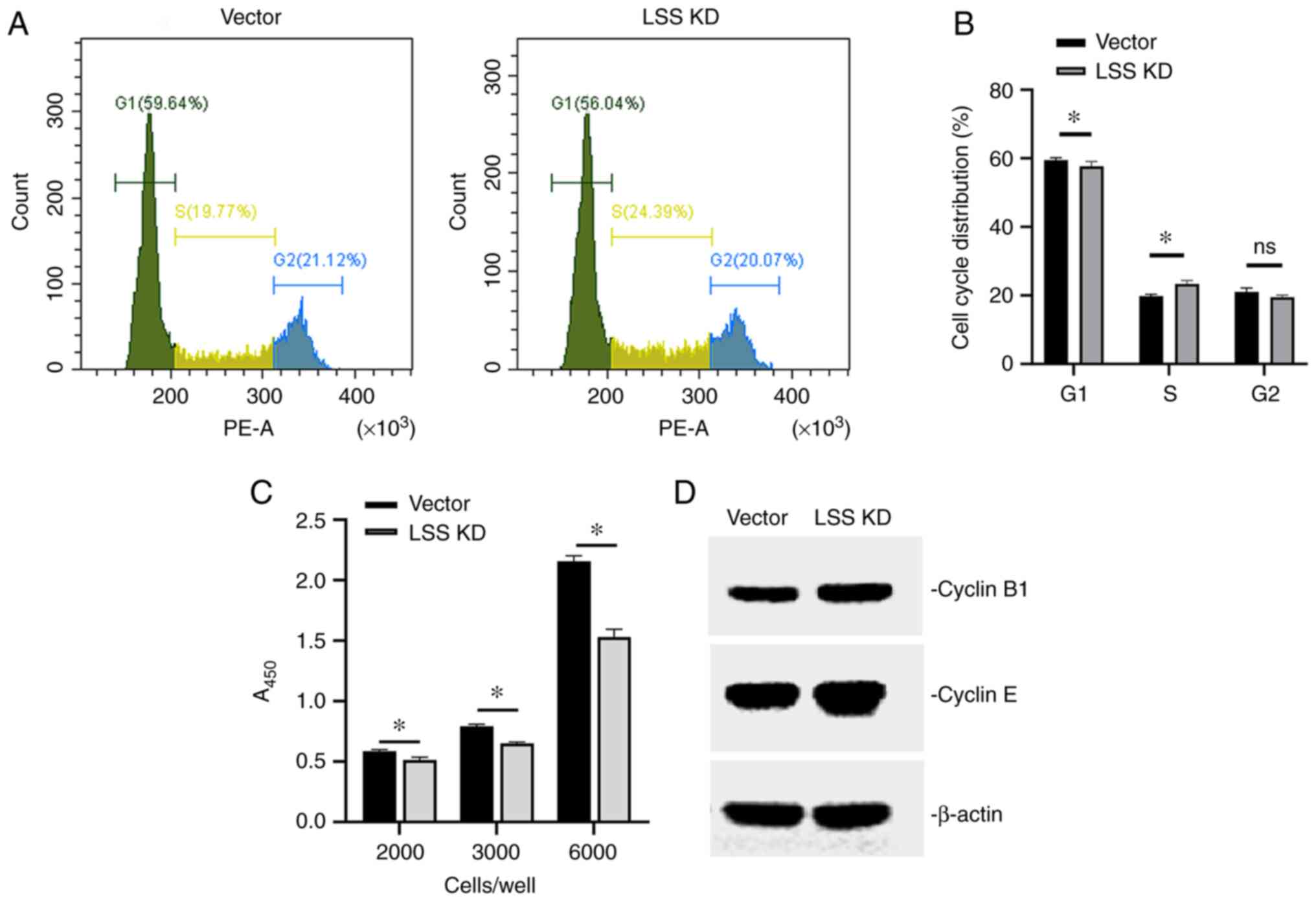 | Figure 2.Effects of LSS knockdown on cell
cycle, cell viability and cell cyclin expression. (A)
Representative cell cycle distribution of transfected HepG2 cells
using flow cytometric analyses, and (B) quantification of the
results. *P<0.05, LSS knockdown vs. vector control. (C) Cell
Counting Kit-8 assay was used to examine the effects of LSS
knockdown on HepG2 cell viability, with initial seeding densities
at 2,000, 3,000 or 6,000 cells per well. *P<0.05. (D) Western
blot analysis of cyclin levels in transfected HepG2 cells. Vector,
HepG2 cells with vector control; LSS KD, HepG2 cells subjected to
LSS knockdown using shRNA; LSS, lanosterol synthase; shRNA, short
hairpin RNA. |
Cell cycle analysis using flow cytometry revealed
that the proportion of cells in the G1 phase was significantly
lower than that of the vector control, whereas the proportion of
cells in the S phase was higher. The cell proportion in the G2/M
phase was slightly decreased in the cells following LSS knockdown,
although no significant difference was detected between groups
(Fig. 2A and B). This indicated
that cells subjected to LSS knockdown were blocked in the S phase.
Although there was genomic DNA replication, the cells could not
enter the division phase and thus, an inhibitory effect on cell
proliferation was observed.
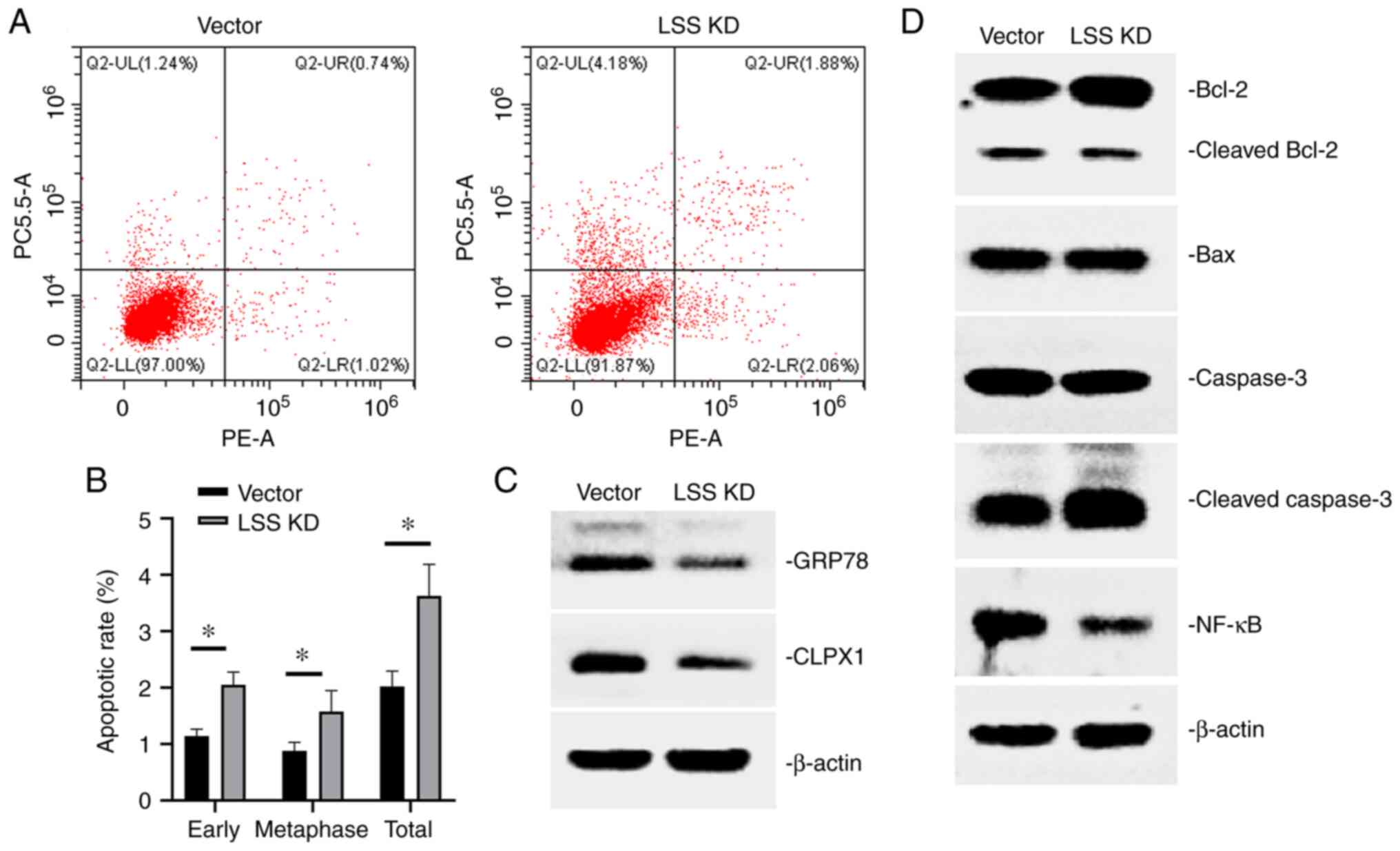
Moreover, flow cytometry was performed to analyze
the extent of apoptosis. As shown in Fig. 3A and B, there were higher
proportions of apoptotic cells in the cells subjected to LSS
knockdown than in the cells transfected with the control vector,
whether in the early stages or in metaphase. The migratory
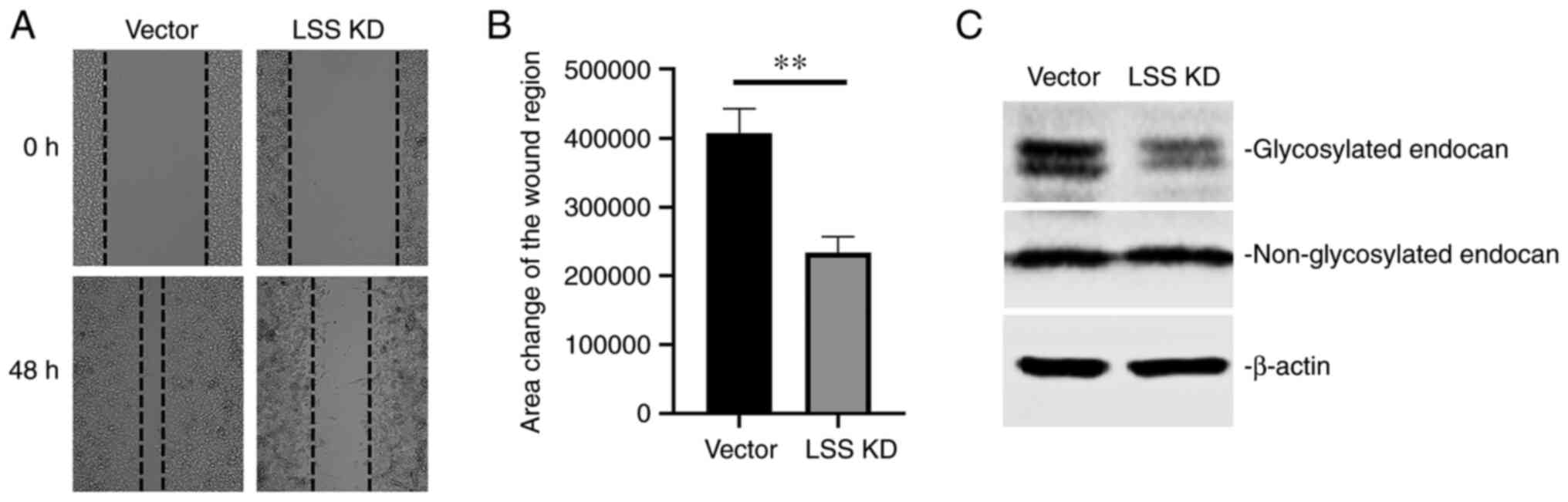
potential is another characteristic of cancer cells. Wound healing
assay demonstrated a reduced migratory ability of HepG2 cells
following stable transfection with LSS shRNA (Fig. 4A and B).
Multiple proteins associated with
proliferation, cell cycle regulation, apoptosis and migration are
altered by LSS knockdown in HepG2 cells
The aforementioned results demonstrated that HepG2
cells subjected to LSS knockdown exhibited a decreased
proliferative activity and migratory potential, as well as an
increased apoptotic rate with cell cycle arrest at S phase. These
findings suggested that LSS may be closely related to liver cancer
and may play a role in the malignant biological behavior of liver
cancer cells such as a high proliferative and invasive ability. LSS
loss of function exerted inhibitory effects on the development and
progression of liver cancer. The expression of proteins associated
with proliferation, apoptosis and migration was examined by western
blot analysis to reveal the possible molecules involved in the
inhibitory effects of LSS knockdown on HepG2 cells.
The analysis of cell cycle distribution revealed
that the cells subjected to LSS knockdown were blocked in the S
phase (Fig. 2A and B), which may
suggest that although there was genomic DNA replication, the cells
could not enter the division phase and thus, an inhibitory effect
on proliferation was observed. As HepG2 cells subjected to LSS
knockdown were arrested in the S phase, as detected using flow
cytometry, the expression of cyclin B1 and cyclin E was then
detected. Cyclin B1 is known to be highly expressed in cells in the
S phase and cyclin E has been proven to regulate the G2/M phase
transition. An increased expression of cyclin B1 and cyclin E was
found in the HepG2 cells following LSS knockdown (Fig. 2D), suggesting that cyclin B1 and
cyclin E may participate in the S phase cell cycle arrest following
LSS knockdown.
The mitochondrial apoptotic pathway is one of the
most critical pathways mediating cell apoptosis. In HepG2 cells
subjected to LSS knockdown, a higher apoptotic ratio was observed.
Molecular mechanistic analysis revealed an activated mitochondrial
apoptotic pathway, as evidenced by an altered Bcl-2, Bax, caspase
and NF-κB expression (Fig. 3D). The
expression of GRP78 and CLPX1, endoplasmic reticulum and
mitochondrial unfolded protein reaction-related proteins, was
decreased in cells subjected to LSS knockdown, mediating cell
apoptosis (Fig. 3C).
Furthermore, the endocan level was investigated.
This has been proven to play differential roles in various
malignancies. Western blot analysis revealed two specific bands
corresponding to the glycosylated endocan at 50 kDa and
non-glycosylated endocan at 20 kDa, respectively. In cells
subjected to LSS knockdown, there was a decreased glycosylated
endocan level when compared with the vector control-transfected
cells, with no change of non-glycosylated endocan (Fig. 4C). This suggested LSS loss of
function inhibited HepG2 cell migration by decreasing the ratio of
glycosylated and non-glycosylated endocan.
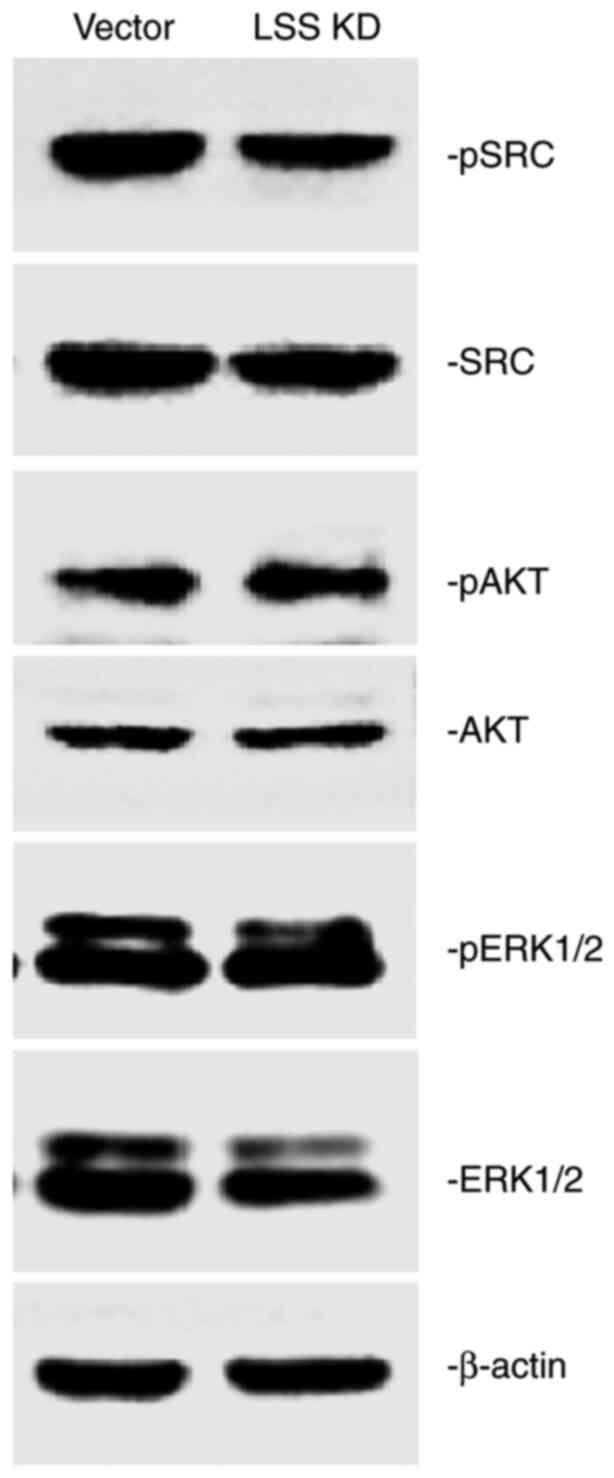
Src/MAPK is one of the possible
signaling pathway by which LSS KD plays its roles in HepG2
cells
The aforementioned results demonstrated that several
proteins are involved in the inhibitory effects on HepG2 liver
cancer cells mediated by LSS knockdown. The exploration of the
related mechanisms of signal transduction in HepG2 cells (Fig. 5) demonstrated that pSrc was
downregulated by LSS knockdown, while no difference was observed in
the level of total Src compared with the vector control-transfected
cells. No marked differences were detected in p-AKT and p-ERK
levels between the cells transfected with the vector control or
those subjected to LSS knockdown. However, there was a decreased
ERK level in the HepG2 cells with LSS knockdown when compared with
the vector control-transfected cells. These results suggest a
possible signaling mechanism, which involves the deactivation of
the Src/MAPK pathway by LSS knockdown in HepG2 cells.
Discussion
As a rapidly growing tissue, the tumor requires
large quantities of cholesterol for its membrane components, and
for the maintenance of cell morphology and functions. The
derivation of cholesterol in tumor cells may thus be an efficient
strategy with which to limit tumor growth and the metastatic
process. Enzymes involved in cholesterol synthesis efflux have been
found to be increasingly required for cancer cell proliferation, in
addition to their role in maintaining the homeostasis of lipid
metabolism (11).
Although studies have suggested that inhibiting the
cholesterol pathway can be used as a target for tumor therapy, the
antitumor efficacy may differ when targeting different nodes within
the cholesterol metabolic pathway due to the nature of feedback
responses elicited, and whether synthesis and transport processes
are simultaneously inhibited. Some researchers have noted that not
the upregulation of the cholesterol biosynthesis pathway, but
rather an increased flux through lanosterol translates into a more
malignant phenotype of cancer cells (6); this suggests that lanosterol may be a
pro-survival factor for cancer cells. Thus, the present study
focused on biosynthetic intermediate lanosterol for hepatic
carcinoma progression.
A high LSS activity or expression has been reported
to be positively associated with tumor metastasis and to be
predictors of a poor prognosis of patients with cancer (12). As a therapeutic target, the
inhibition of LSS is potentially advantageous as function of LSS is
not limited to cholesterol synthesis, but also catalyzes an
alternate flux through the shunt pathway to
24(S),25-epoxycholesterol. When the conversion of epoxysqualene to
lanosterol is inhibited, part of the accumulated epoxysqualene can
be converted into diepoxysqualene and 24(S),25-epoxycholesterolis
ultimately catalyzed by LSS (13).
Oxysterols act as ligands of certain receptors such as LXRα and
LXRβ, or regulate several cellular signaling pathways acting on
cellular receptors to regulate the transcription of target genes
which are involved in the modulation of TGF-β1, Hedgehog, Wnt or
MAPK signaling pathways to regulate cell proliferation and
apoptosis (14). Increased 24, 25
epoxycholesterol, due to the combined effect of LSS partial
inhibition, and due to a higher affinity for diepoxysqualene than
epoxysqualene, activates cholesterol export mediated by LXR, while
concurrently reducing post-oxidosqualene cholesterol synthesis
(7,15). This ‘double hit’ cholesterol
depletion mechanism may further reduce the effect of compensatory
mechanisms to maintain cholesterol by tumor cells.
It has been reported that the specific inhibitor of
LSS, RO 48-8071, decreased cell growth in various tumor types
including pancreatic ductal adenocarcinoma, glioblastoma, breast
cancer and colon carcinoma, and these inhibitors may thus be
potential anticancer drugs (8,16–19).
The present study investigated the functional importance of LSS in
regulating the proliferation, apoptosis and migration of human
liver cancer cells in vitro, although with limitations that
the effects of RO48-8071 on HepG2 cells have not been tested yet
and the lack of in vivo studies. Inhibition of LSS activity
by shRNA led to a decreased proliferation and led to cell cycle
arrest. Cell cycle dysregulation may be due to the altered
expression of cyclins and may thus result in tumorigenesis and
cancer development. In the present study, it was found that cyclin
B1 and cyclin E expression levels were upregulated in HepG2 cells
following LSS knockdown. It is known that cyclin E promotes the
transition from the G1 phase to the S phase, and that cyclin B1
undergoes dynamic changes throughout the cell cycle; a marked
increase in the levels of these markers is observed when the cells
enter the S phase (20–22). In the present study, following LSS
loss of function in HepG2 cells, the expression of cyclin B1 and
cyclin E increased, regulating the cell cycle in S phase, and
leading to S phase arrest and the inhibition of cell
proliferation.
Moreover, the knockdown of LSS expression markedly
altered the cell apoptotic ratio. Mitochondria are dependent on
accumulation of cholesterol in mitochondrial membranes and also
mitochondrial cholesterol metabolism into oxysterols. The function
of cholesterol in mitochondria is not limited to synthesis of
steroid hormones in steroidogenic tissues or bile acids in the
liver. In mitochondria, cholesterol is metabolized into oxysterols
which regulate multiple pathways such as ERK, Hedgehog, Wnt and
TGF-β1 signaling pathways to regulate cell proliferation and
apoptosis, not limited on synthesis of steroid hormones in
steroidogenic tissues or bile acids in the liver. Thus,
mitochondria are dependent on cholesterol components although lower
cholesterol level in mitochondrial membrane compared with other
bilayers (23). The activated
mitochondrial apoptotic pathway, which is characterized by an
altered Bcl-2, Bax, caspase and NF-κB expression, was observed in
HepG2 cells following LSS knockdown; this may be responsible for
the higher apoptotic ratio in the cells compared with the vector
control-transfected cells. Bcl-2 is known as an
apoptosis-antagonizing protein (24). The present study found that in HepG2
cells subjected to LSS knockdown, Bcl-2 expression was markedly
higher than that in the control cells; thus, more apoptotic cells
were detected in the HepG2 cells subjected to LSS knockdown.
Further analysis revealed a smaller molecule binding to Bcl-2
antibody, which is shown as cleaved Bcl-2. It has been reported
that Bcl-2/Bax and caspase 3 are interrelated and are mutually
restricted in the process of apoptosis transmission (25). Bcl-2 and Bax can not only act as the
upstream regulatory mechanism of caspase-3 and participate in the
regulation of caspase-3 activity but can also act as the direct
substrate of caspase-3, downstream of caspase-3, to form a positive
apoptotic feedback pathway. Bcl-2 protein can be cleaved by
caspase-3 into fragments with Bax; this pro-apoptotic activity
accelerates the process of apoptosis (26). The results of the present study are
inconsistent with this mechanism, which involves a higher level of
total Bcl-2, and a lower level of cleaved Bcl-2, with no change in
the Bax level. However, a higher level of cleaved caspase-3 and
lower intracytoplasmic NF-κB were both detected in cells in which
LSS was knocked down. NF-κB, known as a transcription factor, is
translocated into the nucleus to activate multiple target genes
involved in cell survival and apoptosis (27). This suggested that the mitochondrial
apoptotic pathway participated in the promotion of apoptosis
following LSS knockdown; however, the inconsistent changes in the
level of Bcl-2/Bax warrant further investigation. Moreover, lower
levels of GRP78 (28) and CLPX1
(29) were found in the cells
subjected to LSS knockdown when compared with the vector
control-transfected cells; this suggested that the endoplasmic
reticulum and mitochondrial stress response were also involved in
the apoptotic effects induced by LSS knockdown, and not merely the
classic mitochondrial apoptotic pathway.
Endocan is a type of protein which has been
demonstrated to play opposite roles in different types of tumors
(30–32). The roles of promoting or inhibiting
tumor development by endocan may be dependent on the ratio between
glycosylated and non-glycosylated endocan; glycosylated endocan,
but not non-glycosylated form, plays roles as a stimulus in tumor
progression, by promoting the activity of HGF/SF, VEGF and EGFR and
inducing tumor growth and angiogenesis (33–36). A
decreased glycosylated endocan level, but not non-glycosylated
endocan, in HepG2 cells subjected to LSS knockdown revealed that
the glycosylation of endocan was also regulated by LSS loss of
function to inhibit HepG2 cell migration and malignant
behaviors.
As a consequence of LSS knockdown in the HepG2 liver
cancer cell line, the ability of migration and the rate of cell
proliferation were decreased, while the percentage of apoptotic
cells was significantly increased. This phenomenon may be related
to LSS loss of function, at least in part.
Moreover, multiple cellular signaling pathways are
involved in the suppressive effects of inhibitors of the
cholesterol synthesis pathway, as well as in the occurrence and
progression of liver cancer, such as Src, AKT and MAPKs (37,38).
The present study also examined the effects of LSS knockdown by
shRNA on tumor progression. A decreased level of p-Src level and a
lower level of total ERK were observed following LSS knockdown;
however, this was not observed for pERK. In addition, no change was
observed in the levels of p-AKT or AKT in the cells subjected to
LSS knockdown. This suggested that the activities of Src and the
ERK signaling pathways were inhibited by LSS knockdown to regulate
the expression of numerous genes, thus reducing the malignant
potential of HepG2 cells.
In conclusion, the findings of the present study
suggest that LSS loss of function decreases the expression of genes
associated with HepG2 proliferation, apoptosis and migration via
the deactivation of the Src/MAPK signaling pathway. These findings
establish the molecular basis of LSS loss of function in the
inhibition of tumorigenesis and development, and provide a
rationale for targeting LSS and other enzymes catalyzing
cholesterol biosynthesis, as a promising approach for cancer
treatment.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the University
Science Research Project of Anhui Province (grant nos. KJ2020A0143
and KJ2017A195) and the Natural Science Foundation of Anhui
Province (grant nos. 1708085MH212 and 2108085MH266).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SuZ and ShZ designed the study. XS, JZ and HL
performed the experiments. ShZ analyzed data and revised the
manuscript. SuZ drafted the manuscript. ML, LL, ZY, WH, HB, JXu,
JXi, ZX, AM, ZG, YB, QZ and YW collected, analyzed and interpreted
data. All authors have read and approved the final manuscript. SuZ
and ShZ confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Martinez-Outschoorn UE, Sotgia F and
Lisanti MP: Caveolae and signalling in cancer. Nat Rev Cancer.
15:225–237. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Huang B, Song BL and Xu C: Cholesterol
metabolism in cancer: Mechanisms and therapeutic opportunities. Nat
Metabol. 2:132–141. 2020. View Article : Google Scholar
|
|
3
|
Chimento A, Casaburi I, Avena P, Trotta F,
De Luca A, Rago V, Pezzi V and Sirianni R: Cholesterol and its
metabolites in tumor growth: Therapeutic potential of statins in
cancer treatment. Front Endocrinol (Lausanne). 9:8072019.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chang TY, Chang CC, Ohgami N and Yamauchi
Y: Cholesterol sensing, trafficking, and esterification. Annu Rev
Cell Dev Biol. 22:129–157. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kuzu OF, Noory MA and Robertson GP: The
role of cholesterol in Cancer. Cancer Res. 76:2063–2070. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stäubert C, Krakowsky R, Bhuiyan H, Witek
B, Lindahl A, Broom O and Nordström A: Increased lanosterol
turnover: A metabolic burden for daunorubicin-resistant leukemia
cells. Med Oncol. 33:62016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Repa JJ and Mangelsdorf DJ: The liver X
receptor gene team: Potential new players in atherosclerosis. Nat
Med. 8:1243–1248. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ding Z, Gu Y, Huang D, Zhou H, Zhu T, Luo
X, Zhang S, Zhang S and Qian Y: Cholesterol biosynthesis inhibitor
RO 48-8071 inhibits pancreatic ductal adenocarcinoma cell viability
by deactivating the JNK and ERK/MAPK signaling pathway. Mol Med
Rep. 24:8282021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liang Y, Mafuvadze B, Aebi JD and Hyder
SM: Cholesterol biosynthesis inhibitor RO 48-8071 suppresses growth
of hormone-dependent and castration-resistant prostate cancer
cells. Onco Targets Ther. 9:3223–3232. 2016.PubMed/NCBI
|
|
10
|
Zhou XN, Li GM, Xu YC, Zhao TJ and Wu JX:
Knockdown of decoy Receptor 3 impairs growth and invasiveness of
hepatocellular carcinoma cell line of hepG2. Chin Med J (Engl).
129:2623–2629. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cruz PM, Mo H, McConathy WJ, Sabnis N and
Lacko AG: The role of cholesterol metabolism and cholesterol
transport in carcinogenesis: A review of scientific findings,
relevant to future cancer therapeutics. Front Pharmacol. 4:1192013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Phillips RE, Yang Y, Smith RC, Thompson
BM, Yamasaki T, Soto-Feliciano YM, Funato K, Liang Y,
Garcia-Bermudez J, Wang X, et al: Target identification reveals
lanosterolsynthase as a vulnerability in glioma. Proc Natl Acad Sci
USA. 116:7957–7962. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rowe AH, Argmann CA, Edwards JY, Sawyez
CG, Morand OH, Hegele RA and Huff MW: Enhanced synthesis of the
oxysterol 24(S), 25-epoxycholesterol in macrophages by inhibitors
of 2,3-oxidosqualene: Lanosterol cyclase: A novel mechanism for the
attenuation of foam cell formation. Circ Res. 93:717–725. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
de Freitas FA, Levy D, Zarrouk A, Lizard G
and Bydlowski SP: Impact of oxysterols on cell death,
proliferation, and differentiation induction: Current status.
Cells. 10:23012021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Eisele B, Budzinsky R, Muller P, Maier R
and Mark M: Effects of a novel 2,3-oxidosqualene cyclase inhibitor
on cholesterol biosynthesis and lipid metabolism in vivo. J Lipid
Res. 38:564–575. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Staedler D, Chapuis-Bernasconi C, Dehmlow
H, Fischer H, Juillerat-Jeanneret L and Aebi JD: Cytotoxic effects
of combination of oxidosqualene cyclase inhibitors with
atorvastatin in human cancer cells. J Med Chem. 55:4990–5002. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maione F, Oliaro-Bosso S, Meda C, Di
Nicolantonio F, Bussolino F, Balliano G, Viola F and Giraudo E: The
cholesterol biosynthesis enzyme oxidosqualene cyclase is a new
target to impair tumour angiogenesis and metastasis dissemination.
Sci Rep. 5:90542015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mafuvadze B, Liang Y and Hyder SM:
Cholesterol synthesis inhibitor RO 48-8071 suppresses
transcriptional activity of human estrogen and androgen receptor.
Oncol Rep. 32:1727–1733. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liang Y, Besch-Williford C, Aebi JD,
Mafuvadze B, Cook MT, Zou X and Hyder SM: Cholesterol biosynthesis
inhibitors as potent novel anti-cancer agents: Suppression of
hormone-dependent breast cancer by the oxidosqualene cyclase
inhibitor RO 48-8071. Breast Cancer Res Treat. 146:51–62. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sherr CJ: G1 phase progression: Cycling on
cue. Cell. 79:551–555. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Massagué J: G1 cell-cycle control and
cancer. Nature. 432:298–306. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Blagosklonny MV and Pardee AB: The
restriction point of the cell cycle. Cell Cycle. 1:103–110. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Garcia-Ruiz C, Conde de la Rosa L, Ribas V
and Fernandez-Checa JC: Mitochondrial cholesterol and cancer. Semin
Cancer Biol. 73:76–85. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Siddiqui WA, Ahad A and Ahsan H: The
mystery of BCL2 family: Bcl-2 proteins and apoptosis: An update.
Arch Toxicol. 89:289–317. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Desagher S and Martinou JC: Mitochondria
as the central control point of apoptosis. Trends Cell Biol.
10:369–377. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kirsch DG, Doseff A, Chau BN, Lim DS, de
Souza-Pinto NC, Hansford R, Kastan MB, Lazebnik YA and Hardwick JM:
Caspase-3-dependent cleavage of Bcl-2 promotes release of
cytochrome C. J Biol Chem. 274:21155–21161. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wan FY and Lenardo MJ: The nuclear
signaling of NF-kappaB: Current knowledge, new insights, and future
perspectives. Cell Res. 20:24–33. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Samanta S, Yang S, Debnath B, Xue D, Kuang
Y, Ramkumar K, Lee AS, Ljungman M and Neamati N: The
Hydroxyquinoline analogue YUM70 Inhibits GRP78 to induce ER
stress-mediated apoptosis in pancreatic cancer. Cancer Res.
81:1883–1895. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang Y, Zhou L, Safran H, Borsuk R, Lulla
R, Tapinos N, Seyhan AA and El-Deiry WS: EZH2i EPZ-6438 and HDACi
vorinostat synergize with ONC201/TIC10 to activate integrated
stress response, DR5, reduce H3K27 methylation, ClpX and promote
apoptosis of multiple tumor types including DIPG. Neoplasia.
23:792–810. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sumei Z, Shaolong C, Xiang W, Yinliang Q,
Qing Z and Yuan W: Endocan reduces the malign grade of gastric
cancer cells by regulating associated proteins expression. Tumour
Biol. 37:14915–14921. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xu Z, Zhang S, Zhou Q, Wang Y and Xia R:
Endocan, a potential prognostic and diagnostic biomarker of acute
leukemia. Mol Cell Biochem. 395:117–123. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Youssef AA, Issa HA, Omar MZ, Behiry EG,
Elfallah AA, Hasaneen A, Darwish M and Ibrahim DB: Serum human
endothelial cell-specific molecule-1 (endocan) and vascular
endothelial growth factor in cirrhotic HCV patients with liver
cancer as predictors of mortality. Clin Exp Gastroenterol.
11:431–438. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Béchard D, Gentina T, Delehedde M,
Scherpereel A, Lyon M, Aumercier M, Vazeux R, Richet C, Degand P,
Jude B, et al: Endocan is a novel chondroitin sulfate/dermatan
sulfate proteoglycan that promotes hepatocyte growth factor/scatter
factor mitogenic activity. J Biol Chem. 276:48341–48349. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Depontieu F, Grigoriu BD, Scherpereel A,
Adam E, Delehedde M, Gosset P and Lassalle P: Loss of Endocan
tumorigenic properties after alternative splicing of exon 2. BMC
Cancer. 8:142008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang YC, Pan KF, Lee WJ, Chang JH, Tan P,
Gu CC, Chang WM, Yang SF, Hsiao M, Hua KT and Chien MH: Circulating
proteoglycan endocan mediates EGFR-driven progression of non-small
cell lung cancer. Cancer Res. 80:3292–3304. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shin JW, Huggenberger R and Detmar M:
Transcriptional profiling of VEGF-A and VEGF-C target genes in
lymphatic endothelium reveals endothelial-specific molecule-1 as a
novel mediator of lymphangiogenesis. Blood. 112:2318–2326. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ricoult SJ, Yecies JL, Ben-Sahra I and
Manning BD: Oncogenic PI3K and K-Ras stimulate de novo lipid
synthesis through mTORC1 and SREBP. Oncogene. 35:1250–1260. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Parra-Mercado GK, Fuentes-Gonzalez AM,
Hernandez-Aranda J, Diaz-Coranguez M, Dautzenberg FM, Catt KJ,
Hauger RL and Olivares-Reyes JA: CRF1 receptor signaling via the
ERK1/2-MAP and Akt kinase cascades: Roles of Src, EGF receptor, and
PI3-kinase mechanisms. Front Endocrinol (Lausanne). 10:8692019.
View Article : Google Scholar : PubMed/NCBI
|