Introduction
Head and neck squamous cell carcinoma (HNSCC) is a
common malignancy with ~600,000 cases new each year worldwide.
Despite advances in screening, diagnosis and multimodal therapy,
the 5-year survival rate for HNSCC is ~50% and is mainly associated
with locally advanced progression and disease screening (1). Traditional clinicopathological
parameters, such as tumor size, vascular infiltration and tumor
node metastasis stage, do not aid in prediction of individual
outcomes or determining risk stratification (2). Therefore, identification of novel
potential biomarkers is required for predicting the occurrence and
progression of locally advanced HNSCC.
MicroRNAs (miRNAs or miRs) are involved in the
post-transcriptional regulation of gene expression via complete or
partial base pairing with the 3′untranslated region of target genes
(3). Circular RNAs (circRNAs) are
closed, long, non-coding RNAs that are formed via direct reverse
splicing of precursor mRNA that function by regulating RNA
transcription and protein production, as well as sponging miRNAs
(4). Notably, previous studies
revealed that circRNAs participate in cancer pathogenesis by
sponging miRNAs that target mRNA and circRNAs exhibit potential as
predictive biomarkers and therapeutic targets for cancer treatment
(5,6). In addition, a previous study
demonstrated that circRNA/miRNA/mRNA networks are involved in the
development and progression of multiple types of malignancy
(7). Results of a meta-analysis
demonstrated that nine circRNAs may serve as potential prognostic
markers for HNSCC and these are associated with lower overall
survival (OS) and poor clinicopathological outcomes in patients
with HNSCC (8). circRNAs are
involved in the pathogenesis and progression of HNSCC through
multiple mechanisms. For example, circCORO1C promotes laryngeal
squamous cell carcinoma progression by modulating the
let-7c-5p/PBX3 axis (9). Thus,
further investigations into the specific molecular mechanisms
underlying circRNA/miRNA/mRNA regulation are required to aid
discovery of effective therapeutic targets.
With the development of next-generation sequencing
(NGS), research has focused on comprehensive genomic analysis based
on tumor molecular features, such as somatic mutations and copy
number variation. Exploring the interactions between various RNAs
may provide novel insights into the initiation and progression of
HNSCC. Thus, the present study performed NGS on six pairs of HNSCC
cancer and adjacent healthy tissues to detect RNAs with high levels
of differential expression. Potential interactions between
circRNAs/miRNAs/mRNAs were predicted using bioinformatics and
online databases. The present study aimed to provide a novel
theoretical basis for understanding the molecular regulatory role
of circRNAs in HNSCC.
Materials and methods
Patient information and sample
acquisition
In total, six pairs of HNSCC and adjacent healthy
tissue were obtained from patients (4 male and 2 female patients,
aged 40–82 years) admitted to The Binzhou Medical University
Hospital (Binzhou, China) and The West China Hospital of
Stomatology, Sichuan University (Sichuan, China) between December
2020 and January 2021. The adjacent tissues were selected at ~5 mm
from the edge of the lesion tissue, and their size was 3–5 mm.
Notably, patients had not received preoperative chemotherapy or
radiotherapy. All samples were obtained during surgery and
immediately stored in liquid nitrogen and were confirmed to be
HNSCC following histopathological examination. Written informed
consent was obtained from patients and relatives of patients >18
years of age, where required.
Inclusion criteria were as follows: i) Primary
sites: Cheek, tongue, lip, gum, floor of mouth, tongue and tonsil
parapharyngeal; ii) patients diagnosed with SCC by routine
pathology after surgery in the Department of Pathology, Affiliated
Hospital of Binzhou Medical College; iii) patients who underwent
the first operation without radiotherapy and/or chemotherapy before
surgery; iv) patients with preoperative clinical stages of T3 and
T4; and v) patient's with informed consent. Exclusion criteria were
as follows: i) Patients with recurrence and metastasis who were
re-admitted for surgery; ii) patients who had received surgical
treatment in other hospitals and were re-admitted to Affiliated
Hospital of Binzhou Medical College for surgical treatment.
Whole-genome sequencing analysis
Firstly, ribosomal RNA was removed from the total
RNA, and then RNase R enzyme was used to break the RNA into short
fragments of 250–300 bp. The first strand of cDNA was synthesized
using the fragmented RNA as the template and random
oligonucleotides as the primer, and then RNase H was used to
degrade the RNA strand. The second strand of cDNA was synthesized
by dNTPs (dUTP, dATP, dGTP and dCTP) in the system of DNA
polymerase I. The purified double-stranded cDNA was end-repaired,
A-tailed and connected to sequencing joints, and 350–400 bp cDNA
was screened using AMPure XP beads (no. AG21101; Accurate
Biotechnology Co., Ltd.; Hunan Aikerui Bioengineering Co., Ltd.).
The second strand of cDNA containing U was degraded by USER enzyme,
and the library was obtained by PCR amplification (ABI2720; Applied
Biosystems; USA). The extraction method used was a TRNzol Universal
Reagent+small column (cat. no. DP424+RK177-A). The kit was a NEB
Next® Multiplex Small RNA Library Prep Set for
Illumina® (cat. no. NEB E7300L). The sequencing
instrument platform was an Illumina Novaseq 6000 (Illumina, Inc.)
and the sequencing strategy was SE50. The sequencing of smallRNA in
the current project was conducted from the 5′-3′ direction
(forward, 5′-GTTCAGAGTTCTACAGTCCGACGATC-3′ and reverse,
5′-AGATCGGAAGAGCACACGTCT-3′). The reagents used for PCR were all
contained in the NEB Next® Multiplex Small RNA Library
Prep Set for Illumina. A chain-specific library has many
advantages, such as the same amount of data can obtain more
effective information; more accurate gene quantification,
localization and annotation information can be obtained. An Agilent
2100 bioanalyzer (Agilent Technologies, Inc.) was used to detect
the insert size of the library, and the insert size was about
250–300 bp, which was in line with the expectation. The effective
concentration of the library was >2 nM, which ensured the
quality of the sample library. NGS was used to detect
differentially expressed circRNAs, miRNAs and mRNAs in HNSCC and
adjacent healthy tissues. Sample detection, library construction
and testing and computer sequencing were completed by Beijing
Novogene Technology Co., Ltd. Briefly, a strand-specific library
was constructed by removing ribosomal RNA. Following library
inspection, Illumina PE150 sequencing was performed in accordance
with effective centralization and data output requirements. RNA
sequencing (RNAseq) data were compared and analyzed using Tophat2
(version 2.0.8) (10) software.
edgeR (version 4.2.2) (11) was
used for analysis of differentially expressed RNAs; differentially
expressed circRNAs, miRNAs and mRNAs were screened based on
P<0.05 and |log2FC|>1. Hierarchical clustering was performed
based on all differentially expressed transcripts to construct
expression profiles. The expression values of all samples were
clustered and visualized in heat maps using hierarchical
clustering. The mainstream hierarchical clustering method was used
to convert log10 (FPKM+1) values and cluster them.
Gene Ontology (GO) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) analysis
To explore the biological pathways of potential
target genes, GOseq (http://www.geneontology.org/) (12) and KEGG Orthology Based Annotation
System (version 2.0) (13) software
were used to perform GO and KEGG pathway enrichment analysis of
target genes. GO is a comprehensive database describing gene
function, which is divided into molecular function (MF), biological
process (BP) and cellular component (CC). The results are presented
as bar graphs and bubble plots.
circRNA/miRNA/mRNA network
construction
miRNAs associated with the development and prognosis
of HNSCC were screened using OncomiR (oncomir.org). MiRanda (v3.3a)
(14) software was used for
predicting the top five differentially expressed circRNA-targeted
miRNAs. MiRanda and RNAhybrid (v2.1) (15) were used for target gene prediction
of key miRNAs. Venn analysis was further performed through the
R-VennDiagram package (ggplot2, version3.3.6; VennDiagram, version
1.7.3).
Visualization of miRNA intersection
results
mRNAs associated with HNSCC prognosis were
downloaded from Gene Expression Profiling Interactive Analysis
(GEPIA; gepia.cancer-pku.cn/) for analysis. circRNA/miRNA and
miRNA/mRNA pairs were merged and Cytoscape (version 3.5.1)
(https://cytoscape.org/) was used to visualize the
topological network of circRNA/miRNA/mRNA.
Targeted binding strength
analysis
mRNA and miRNA sequences were downloaded from
National Center for Biotechnology Information (NCBI;
ncbi.nlm.nih.gov/) and miRbase (mirbase.org/index.shtml),
respectively. NCBI Blast (blast.ncbi.nlm.nih.gov/Blast.cgi) was
used to compare sequences of hsa_ circ_0035431 and hsa_circ_0035432
to screen out key circRNAs with potential roles in HNSCC. The
binding sequence and strength of miRNA/mRNA was analyzed using R22
(version 2; cm.jefferson.edu/rna22/).
Comprehensive analysis of online
databases and The Cancer Genome Atlas (TCGA)
GEPIA (gepia.cancer-pku.cn/) and UALCAN
(ualcan.path.uab.edu/) were used to compare transcription levels of
FUT6 and CGNL1 in HNSCC tissue and healthy samples, with further
analysis by subtypes and stages. Patients with complete expression
data of FUT6 and CGNL1 were screened using TCGA
(portal.gdc.cancer.gov/) database. Pair expression (ggplot2,
version 3.3.6; stats, version 4.2.1; car, version 3.1–0), logistic
regression (astats, version 4.2.1) and prognosis analysis
(survival, version 3.3.1; survminer, ggplot2, version 3.3.6) of
FUT6 and CGNL1 in healthy and tumor tissue was performed using R
software (version 4.2.1; R Core Team) and associated R packages to
evaluate association with clinical variables and patient
survival.
Cell culture
Oral squamous Hok and oral squamous cell carcinoma
SCC25 and HSC2 cell lines were purchased from the American Type
Culture Collection and stored in liquid nitrogen (−196°C) in the
Medical Research Center, Binzhou Medical University Hospital. All
cell lines were cultured in DMEM (VivaCell Biotechnology GmbH)
supplemented with 10% fetal bovine serum (HyClone; Cytiva) and 1%
penicillin/streptomycin (100 mg/ml; VivaCell Biotechnology GmbH) at
37°C in 5% CO2.
RNA extraction and reverse
transcription-quantitative (RT-q)PCR
Total RNA was extracted from all tissue and cell
samples using AG RNA ex Pro Reagent (Accurate Biotechnology Co.,
Ltd.; Hunan Aikerui Bioengineering Co., Ltd.) Total RNA was
reverse-transcribed into cDNA using an Evo M-MLV RT kit with gDNA
Clean for qPCR II (Accurate Biotechnology Co., Ltd.). The reverse
transcription reaction conditions were as follows: i) 42°C for 2
min; store at 4°C; ii) 37°C for 15 min; 85°C for 5 sec; store at
4°C. hsa-miR-940 was reverse-transcribed using miRNA 1st Strand
cDNA Synthesis kit (Accurate Biotechnology Co., Ltd.). The reverse
transcription reaction condition was 37°C for 60 min, 85°C for 5
min and storage at 4°C. qPCR was performed using a SYBR®
Green Premix Pro Taq HS qPCR kit (Accurate Biotechnology Co.,
Ltd.). The following thermocycling conditions were used: 40 cycles
of 95°C for 30 sec, 95°C for 5 sec and 60°C for 30 sec. RT-qPCR
data were collected using the CFX96 Real-Time PCR Detection System
(Bio-Rad Laboratories, Inc.) and expression levels were quantified
using the 2-ΔΔCq method (16). β-actin and U6 were used as internal
controls for mRNA and miRNA, respectively. All primers were
designed by Accurate Biotechnology Co., Ltd. (Table I).
 | Table I.Sequences of primers for reverse
transcription-quantitative PCR. |
Table I.
Sequences of primers for reverse
transcription-quantitative PCR.
| Gene | Primer sequence
(5′-3′) |
|---|
| FUT6 | F:
GACGATCCCACTGTGTACCCTA |
|
| R:
TGTTAAAAGGCCACGTCCACAG |
| CGNL1 | F:
GCAGATGGAGGACAAGGTGTCT |
|
| R:
CTCGCAGCTCTCTCCTGAAGT |
| hsa-miR-940 | F:
AGGGCCCCCGCTCCCCAA |
| circRNA_ | F:
GGAAATAACCAACTGGAACAGTGA |
| 0035431 | R:
TAGGAGCCTGCCTTGGAGTTC |
| β-actin | F:
TGGCACCCAGCACAATGAA |
|
| R:
CTAAGTCATAGTCCGCCTAGAAGCA |
| U6 | F:
CTCGCTTCGGCAGCACA |
|
| R:
AACGCTTCACGAATTTGCGT |
Statistical analysis
RT-qPCR data were analyzed by Graph-pad Prism 8.0
software (Graphpad Inc.; Dotmatics). Three independent replicate
experiments were performed, and data are presented as the mean ±
SD. The tissue samples were statistically analyzed by paired
Student's t-test. One-way ANOVA with Dunnett's post hoc test was
used for comparison of cell samples. P<0.05 was considered to
indicate a statistically significant difference.
Results
Screening of differentially expressed
RNA
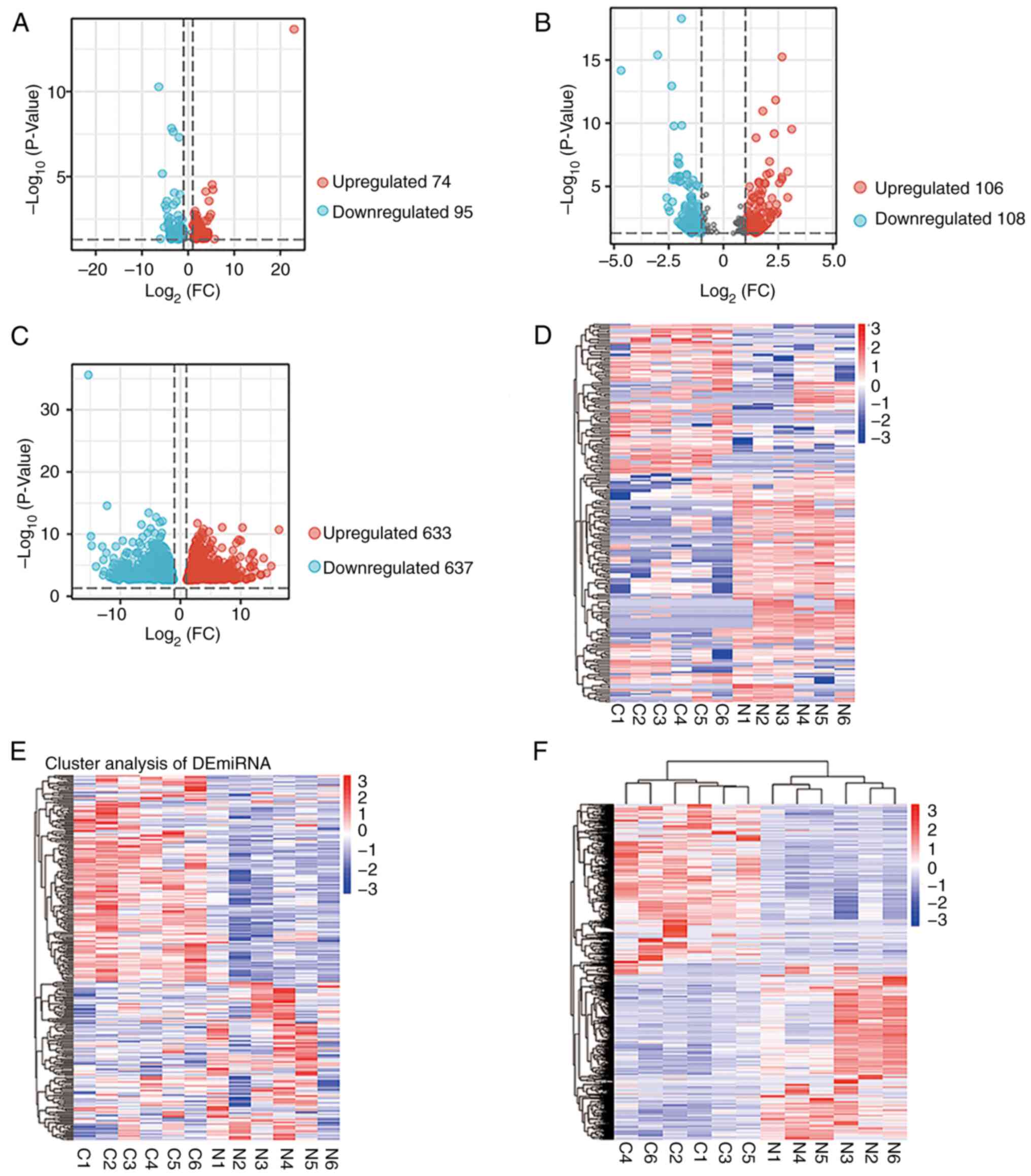
Whole-genome transcriptional sequencing identified
6,750 circRNAs, 265 miRNAs and 19,816 mRNAs. In total, 169
circRNAs, 214 miRNAs and 1,270 mRNAs were differentially expressed.
Volcano plots were used to illustrate differential expression
(Fig. 1A-C). The expression
patterns of circRNAs, miRNAs and mRNAs were distinguished using
hierarchical clustering analysis. circRNA, miRNA and mRNA
expression patterns in HNSCC differed from those in adjacent
healthy tissues (Fig. 1D-F).
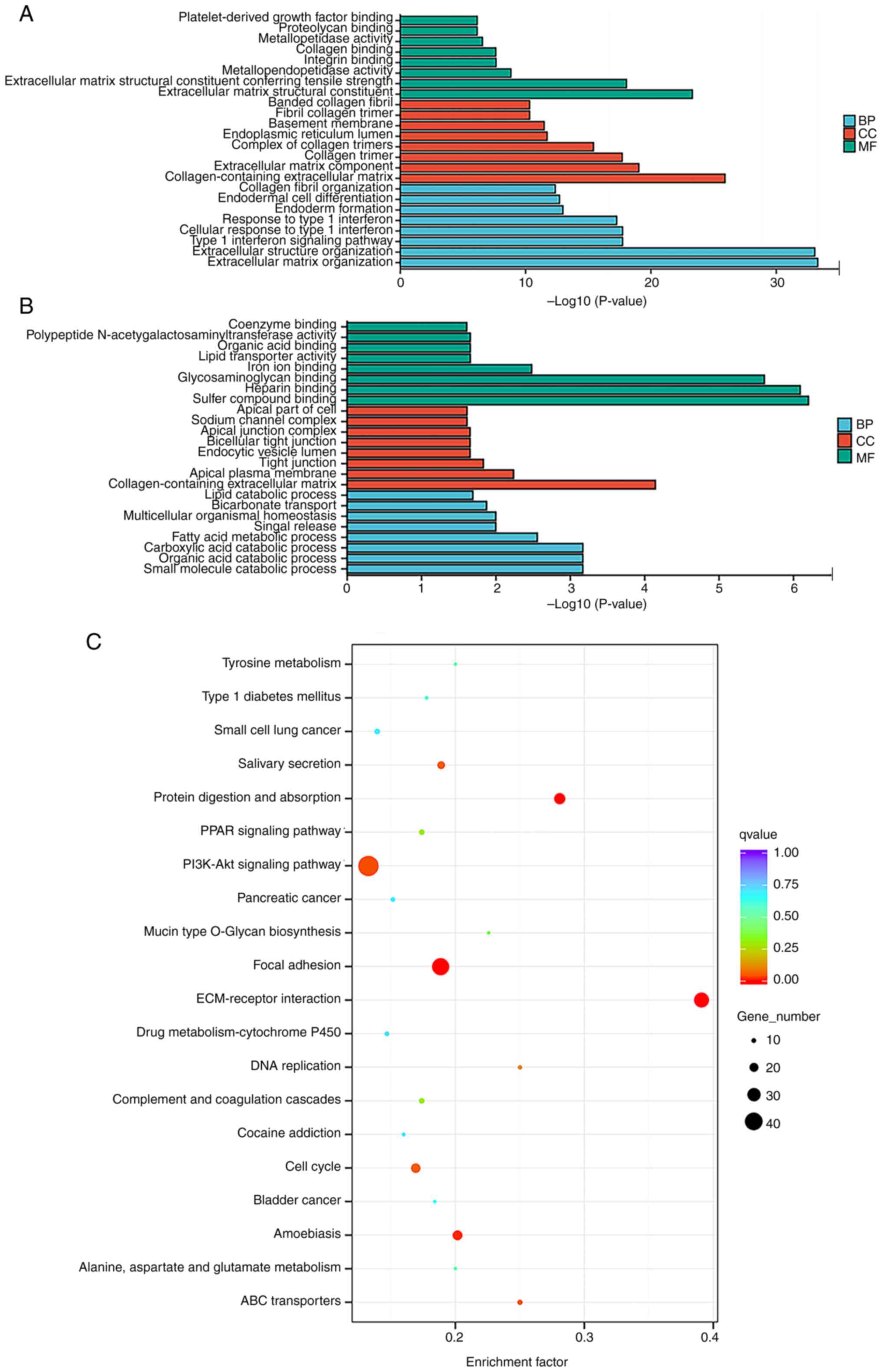
GO and KEGG analysis
Functions of the majority of circRNAs are yet to be
annotated and functional prediction of circRNAs is largely based on
the annotation of the protein codes they interact with. As such, GO
and KEGG analysis of dysregulated mRNAs may predict the function of
circRNAs (17). GO analysis in the
present study demonstrated that DEGs were primarily enriched in BP
terms such as ‘extracellular structure organization’ (GO:0043062),
‘extracellular matrix organization’ (GO:0030198) and ‘type 1
interferon signaling pathway’ (GO: 0060337). The most abundant CC
terms included ‘collagen-containing extracellular matrix’
(GO:0062023), ‘collagen trimer’ (GO:0005581) and ‘extracellular
matrix component’ (GO:0044420). The most enriched MFs included
‘extracellular matrix structural constituent’ (GO:0005201),
‘extracellular matrix structural constituent conferring tensile
strength’ (GO:0030020) and ‘glycosaminoglycan binding’
(GO:0005539). The eight most significantly enriched GO terms for
up- and downregulated mRNAs in BP, CC and MF categories are
displayed in Fig. 2.
KEGG pathway analysis identified 263 pathways
associated with dysregulated mRNAs, which were primarily enriched
in the ‘PI3K-Akt signaling pathway’, ‘focal adhesion’ and
‘ECM-receptor interaction pathways’, demonstrating the key role of
these factors in HNSCC. The top 20 most enriched pathways for
protein-coding gene dysregulation are listed in Fig. 2.
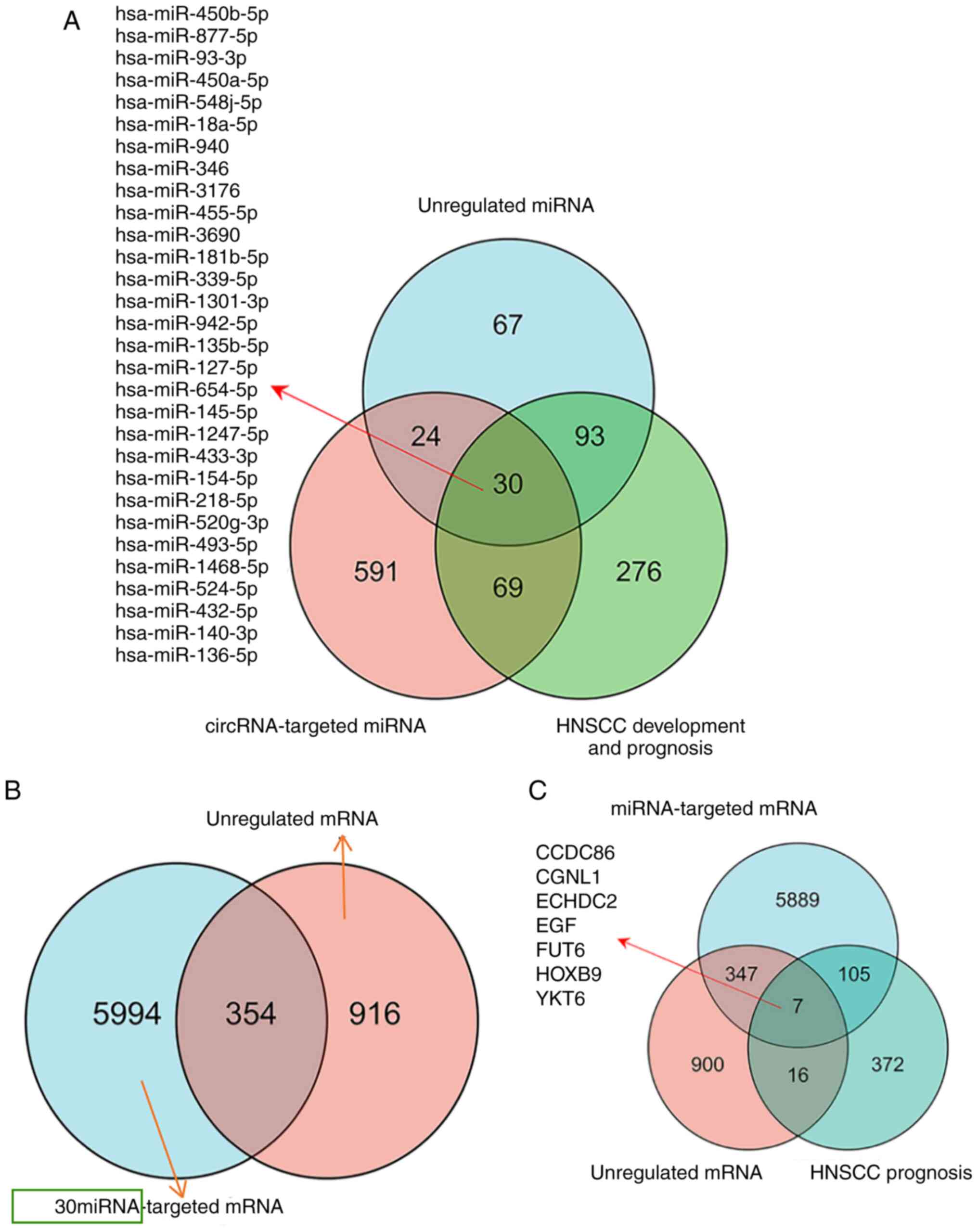
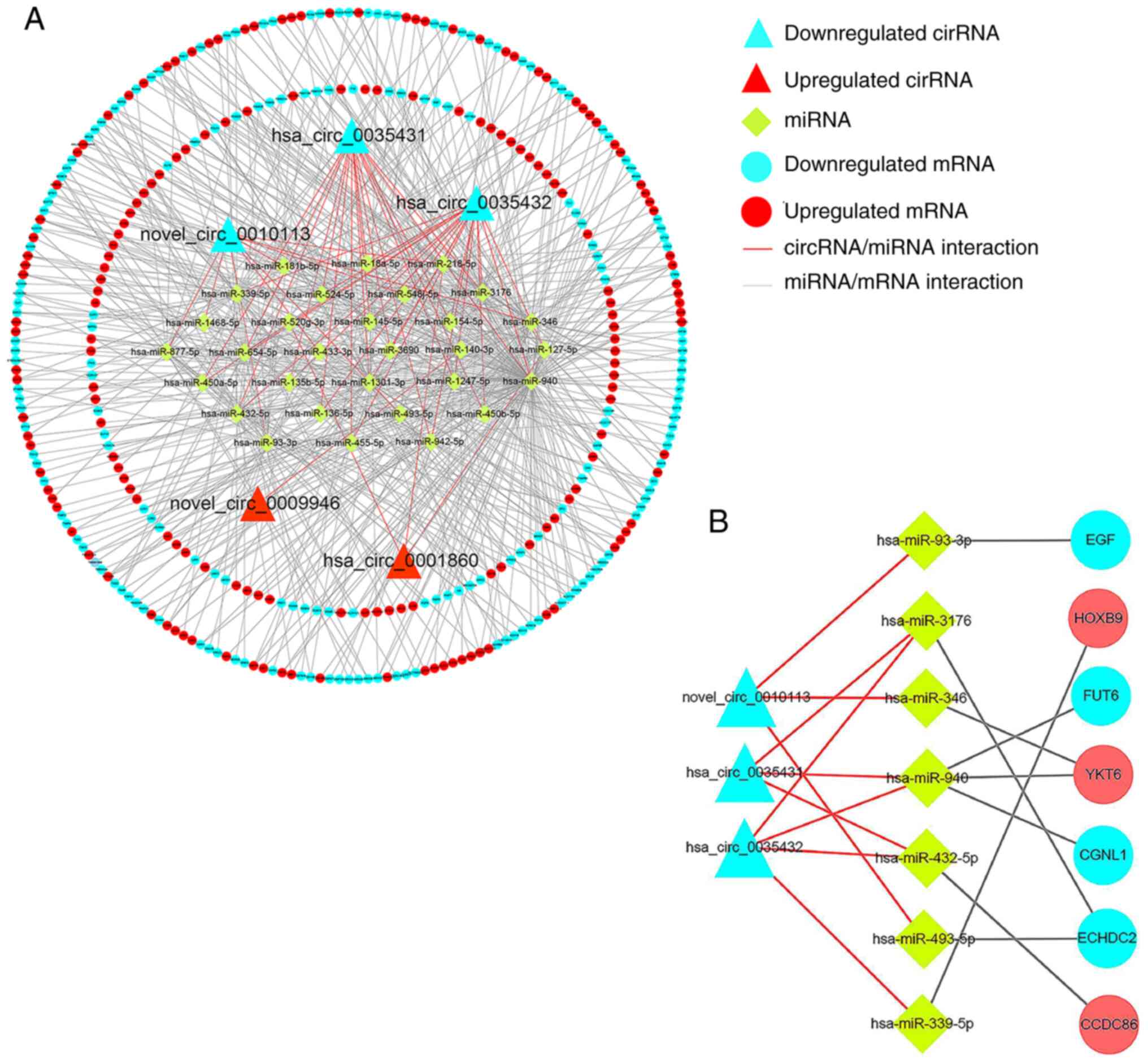
Construction of the circRNA/miRNA/mRNA
network
The target genes of the top five differentially
expressed circRNAs were identified using miRanda (Table II) and the results demonstrated 714
target-binding miRNAs. Oncomir database predicted 468 miRNAs
associated with the progression and prognosis of HNSCC.
Differentially expressed miRNAs were overlapped with results of the
databases to obtain 30 key miRNAs (Fig.
3A). The target gene prediction of 30 miRNAs using miRanda and
RNA hybridization were overlapped with DEGs to obtain 334 mRNAs
(Fig. 3B), constructing a network
including five circRNAs, 30 miRNAs and 334 mRNAs (Fig. 4A). In total, 500 mRNAs associated
with HNSCC prognosis were predicted using GEPIA, overlapping with
the aforementioned 334 overlapped mRNAs, yielding seven key mRNAs
(Fig. 3C). Thus, a network of three
circRNAs, seven miRNAs and seven mRNAs was constructed (Fig. 4B). In total, eight regulatory axes
were determined based on intermolecular regulatory interactions
(Table III).
| Table II.Top five differentially expressed
circRNAs. |
Table II.
Top five differentially expressed
circRNAs.
| circRNA | Regulation | log2FC | P.adj | Chromosome | Gene type | Source gene |
|---|
|
hsa_circ_0035432 | Down | −6.0217 | 0.045209 | 15 | Exonic | CGNL1 |
|
hsa_circ_0035431 | Down | −5.5869 |
6.65×10−6 | 15 | Exonic | CGNL1 |
|
novel_circ_0010113 | Down | −6.3637 |
5.20×10−11 | 6 | Exonic | RIMS1 |
|
hsa_circ_0001860 |
Up | 5.7202 | 0.046254 | 9 | Exonic | ZCCHC7 |
|
novel_circ_0009946 |
Up | 22.9550 |
2.15×10−14 | 6 | Intergenic | NA |
| Table III.circRNA/miR/mRNA regulatory axes. |
Table III.
circRNA/miR/mRNA regulatory axes.
| circRNA | miR | mRNA |
|---|
|
hsa_circ_0035431 | hsa-miR-3176 | ECHDC2 |
|
hsa_circ_0035432 | hsa-miR-3176 | ECHDC2 |
|
hsa_circ_0035431 | hsa-miR-940 | FUT6 |
|
hsa_circ_0035432 | hsa-miR-940 | FUT6 |
|
hsa_circ_0035431 | hsa-miR-940 | CGNL1 |
|
hsa_circ_0035432 | hsa-miR-940 | CGNL1 |
|
novel_circ_0010113 | hsa-miR-93-3p | EGF |
miRNA/mRNA target binding sequence and
intensity analysis
miRNA and mRNA binding regions were determined and
intensity analysis was performed using R22 (Table IV). Compared with that in ECHDC2
and EGF, miRNA and mRNA intensity analysis demonstrated increased
binding of hsa-miR-940 to FUT6 and CGNL1, leading to the
identification of four regulatory axes (Table V). hsa_circ_0035431 and
hsa_circ_0035432 sequences were compared using NCBI and
demonstrated a complete overlap of 1,818 base pairs, with the
sequence of hsa_circ_0035431 being more closely aligned. Therefore,
hsa_circ_0035431/hsa-miR-940/FUT6 and
hsa_circ_0035431/hsa-miR-940/CGNL1 were selected for further
analysis.
| Table IV.miR and mRNA binding regions and
intensity. |
Table IV.
miR and mRNA binding regions and
intensity.
| RNA | miR/mRNA binding
site | Folding energy,
kcal/mol | P-value |
|---|
| FUT6 | 5′-3′:
TGGGA-CCTGTGCCCAGCCTA | −18.50 |
3.51×10−1 |
| hsa-miR-940 | 3′-5′:
CCCCTCGCCCCCGGGACGGAA |
|
|
| FUT6 | 5′-3′:
CTCCAGTGGTGAAGACCCAGCCTG | −15.30 |
2.41×10−2a |
| hsa-miR-940 | 3′-5′:
CCCCTCGCC-C-CC-GGGACGGAA |
|
|
| CGNL1 | 5′-3′:
CAGGAGCAGAAGCAGTTGTCTG | −15.80 |
3.81×10−2a |
| hsa-miR-940 | 3′-5′:
CCCCTCGCCCCCG-GGACGGAA |
|
|
| CGNL1 | 5′-3′:
CTGCAGCTATGGCCTTGTTTG | −16.80 |
1.16×10−2a |
| hsa-miR-940 | 3′-5′:
CCCCTCGCCCCCGGGACGGAA |
|
|
| CGNL1 | 5′-3′:
TCAGAGTGGTCAGCCCAGTTTA | −17.30 |
2.73×10−1 |
| hsa-miR-940 | 3′-5′:
CCCCTCGCC-CCCGGGACGGAA |
|
|
| CGNL1 | 5′-3′: GGGGTGATTCTCACCTCTGCCTG | −19.44 |
2.76×10−2a |
| hsa-miR-940 | 3′-5′: CCCCTC--GCCCCCGGGACGGAA |
|
|
| CGNL1 | 5′-3′:
ATGGTTTGGTCGCTTTGGTTG | −12.60 |
5.27×10−2 |
| hsa-miR-940 | 3′-5′:
CCCCTCGCCCCCGGGACGGAA |
|
|
| EGF | 5′-3′:
ATGGAACTCTGCTCAGC-CAGCAGA | −20.30 |
1.18×10−1 |
| hsa-miR-93-3p | 3′-5′:
GCCCTT--CACGA-TCGAGTCGTCA |
|
|
| EGF | 5′-3′:
TGAGGA-TGGCCAG-GCAGCAGA | −16.20 |
9.61×10−2 |
| hsa-miR-93-3p | 3′-5′:
GC-CCTTCACGATCGAGTCGTCA |
|
|
| ECHDC2 | 5′-3′: AGTGAAGGGCGTGTTC-TGTGCAG | −15.50 |
1.73×10−1 |
| hsa-miR-493-5p | 3′-5′: TTACTTTCGG--ATGGTACATGTT |
|
|
| ECHDC2 | 5′-3′:
CGGG-AGGACC-GGCAAGT | −13.80 |
1.73×10−1 |
| hsa-miR-3176 | 3′-5′:
GGCCATCAGGGTCCGGTCA |
|
|
| Table V.circRNA-miR-mRNA regulatory axes. |
Table V.
circRNA-miR-mRNA regulatory axes.
| circRNA | miR | mRNA |
|---|
|
hsa_circ_0035431 | hsa-miR-940 | FUT6 |
|
hsa_circ_0035432 | hsa-miR-940 | FUT6 |
|
hsa_circ_0035431 | hsa-miR-940 | CGNL1 |
|
hsa_circ_0035432 | hsa-miR-940 | CGNL1 |
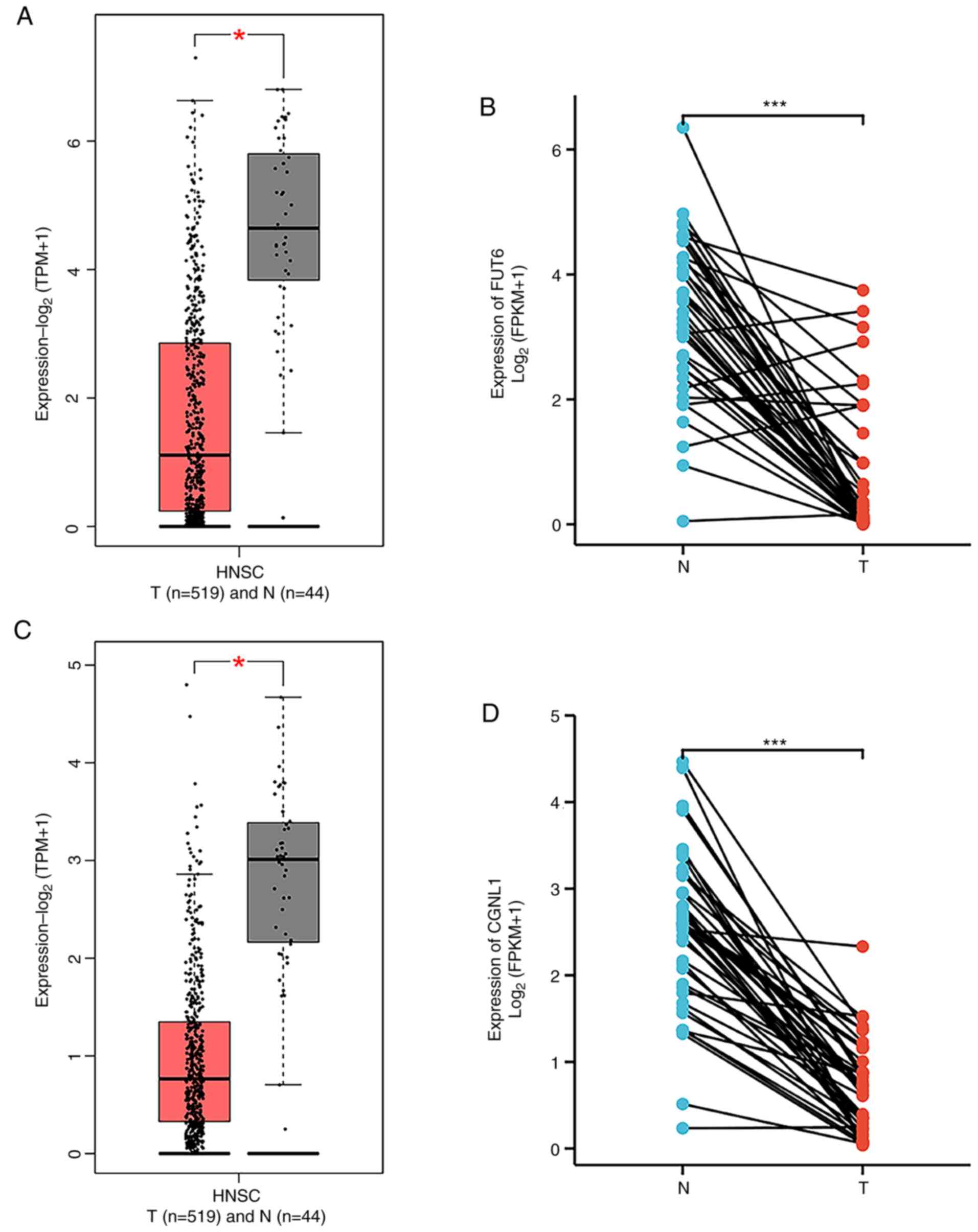
Expression of FUT6/CGNL1 in HNSCC
Transcription levels of FUT6 and CGNL1 in HNSCC were
investigated using the GEPIA2 online database. FUT6 and CGNL1 mRNA
expression levels were significantly decreased in HNSCC compared
with healthy tissue (Fig. 5A and
C). Paired sample analysis using data obtained from TCGA
confirmed the aforementioned findings (Fig. 5B and D).
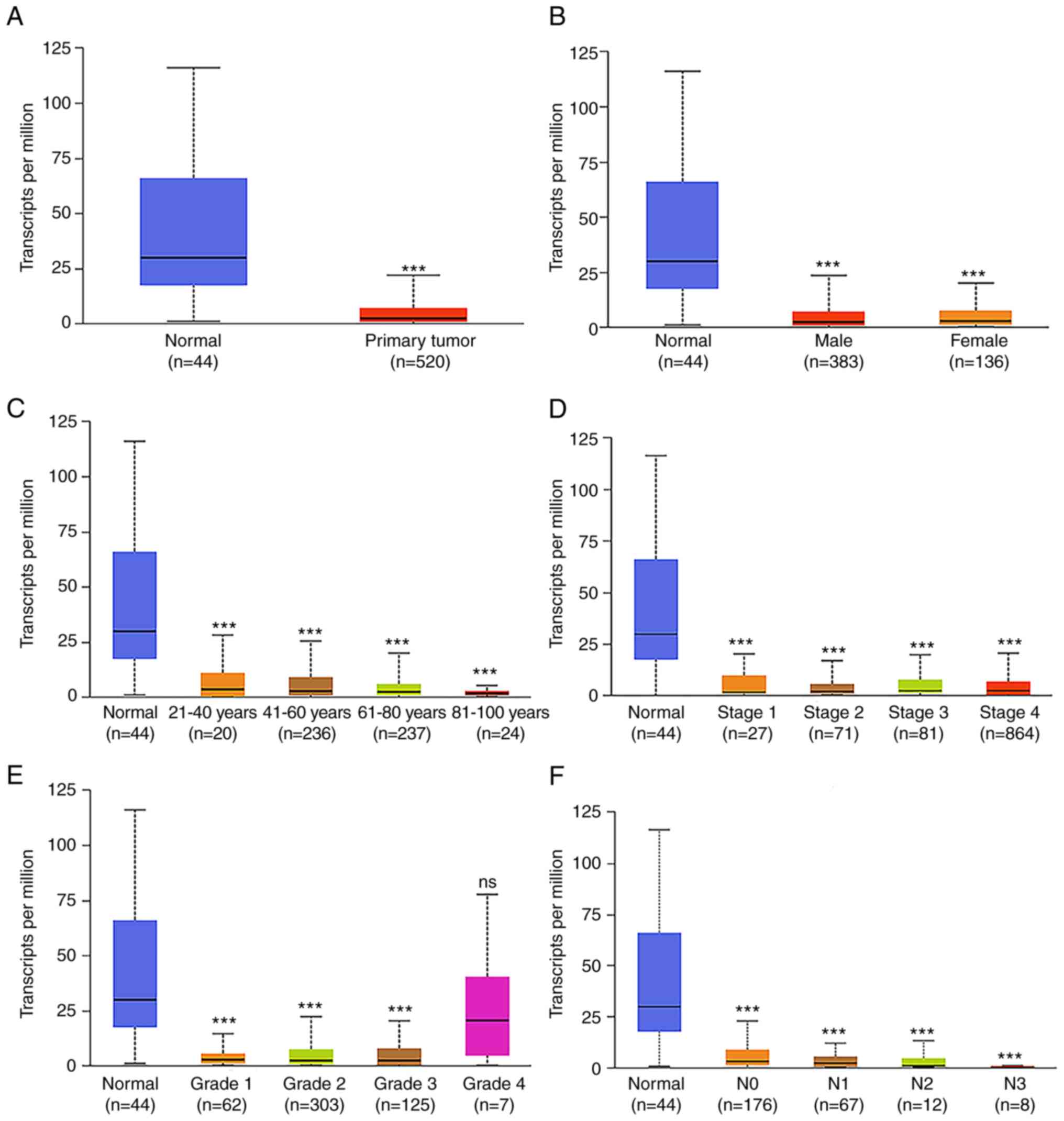
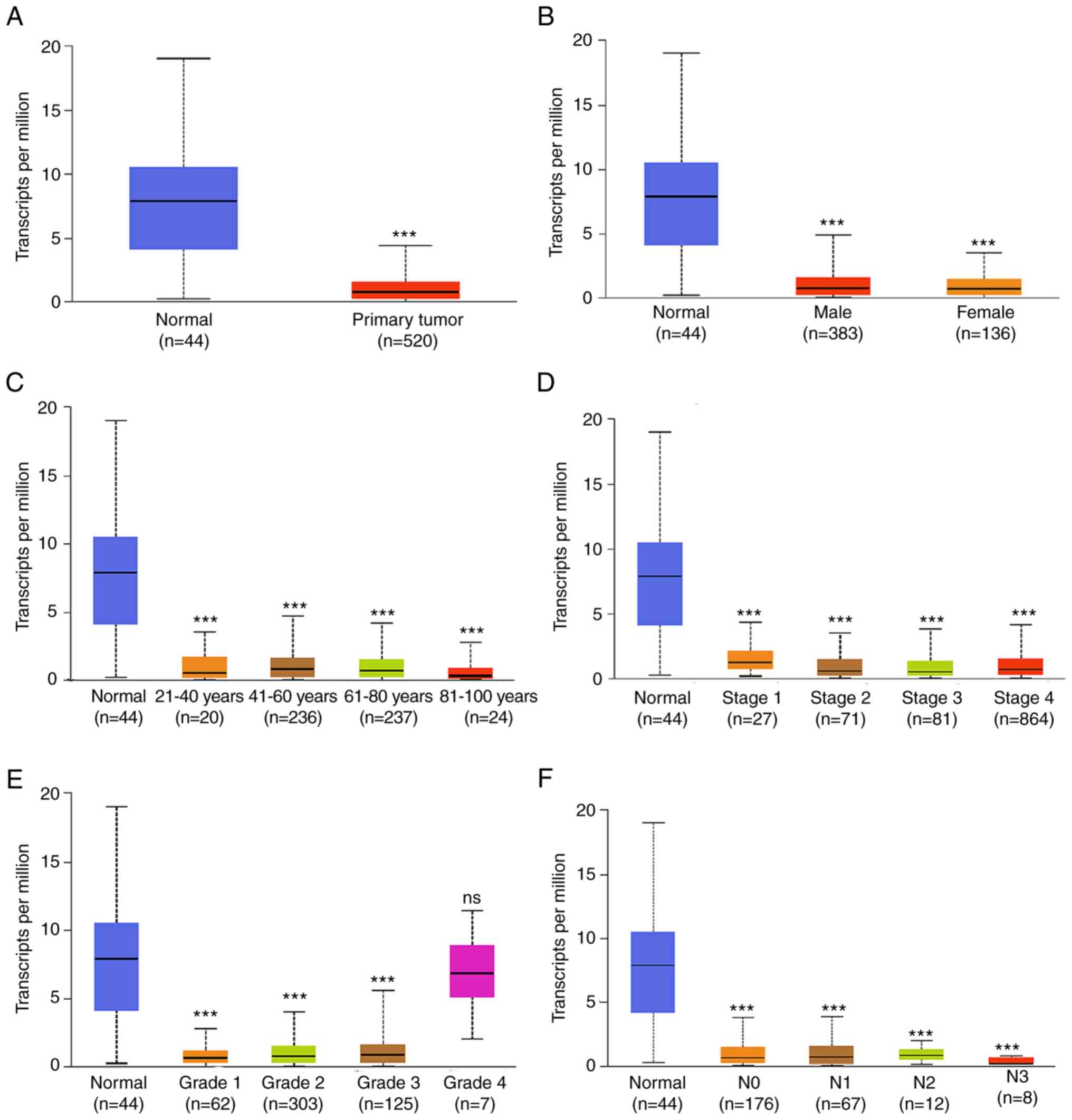
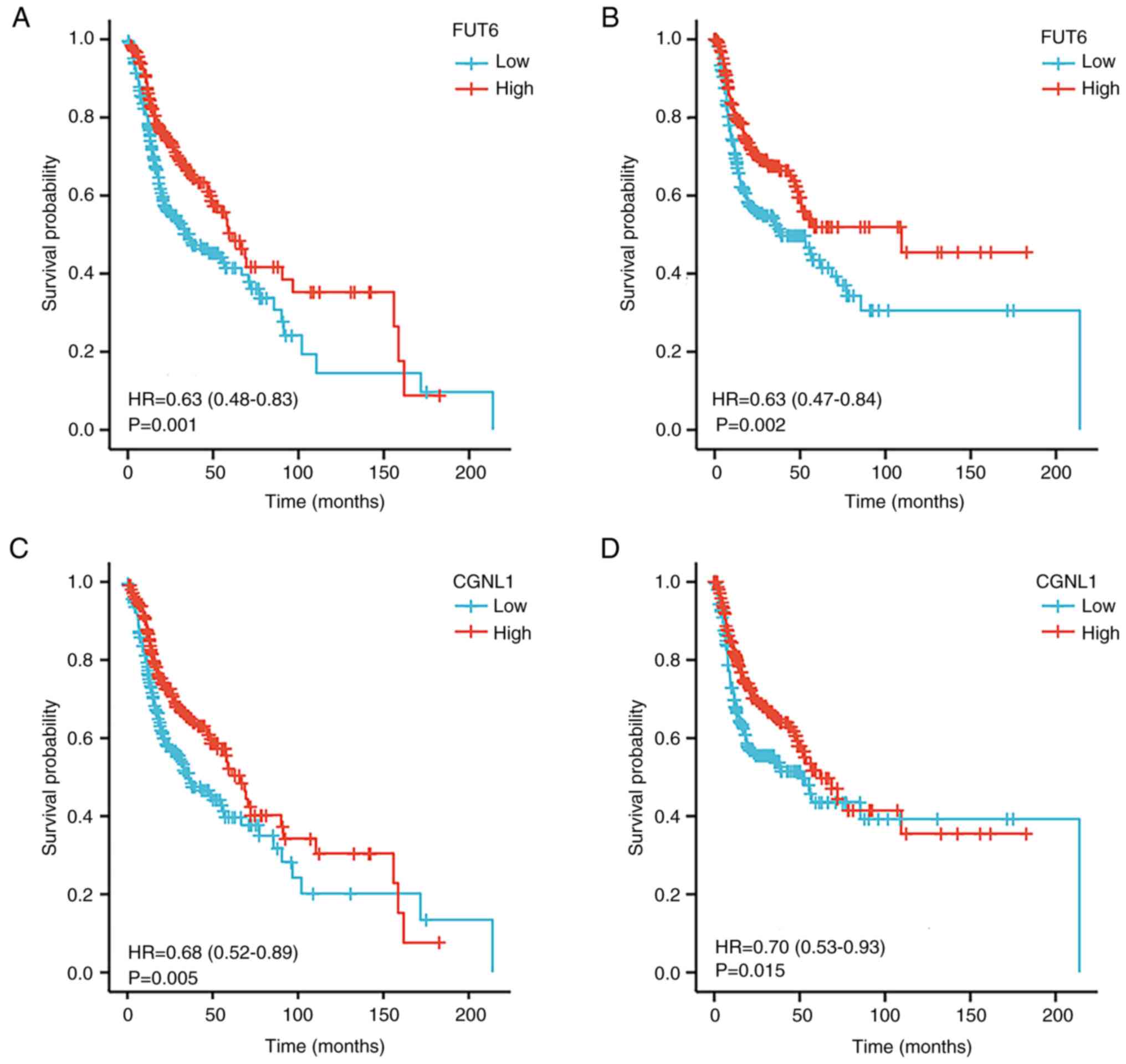
Association of FUT6 and CGNL1
expression with clinical characteristics and survival
The subgroup analysis of pathological
characteristics of HNSCC samples using UALCAN database demonstrated
a decrease in FUT6 and CGNL1 transcription levels. Subgroup
analysis of sex, age, disease stage and lymph node metastasis
demonstrated that expression levels of FUT6 and CGNL were
significantly lower in patients with HNSCC compared with controls
(Figs. 6 and 7). Logistic regression analysis of TCGA
data indicated that FUT6 expression was lower in T3 and T4 than in
T1 and in stages II and IV than at stage I. Expression of CGNL1 was
lower in T3 and T4 than in T1 (Table
VI). In addition, increased FUT6 and CGNL1 expression was
independent of sex, age and lymph node and distant metastasis
(Tables VI and VII). To analyze clinical significance of
FUT6 and CGNL1, prognostic analysis was performed using TCGA.
Increased FUT6 and CGNL1 expression was associated with longer OS
and progression-free interval (PFI) of patients with HNSCC
(Fig. 8).
| Table VI.Logistic regression analysis of
fucosyltransferase 6. |
Table VI.
Logistic regression analysis of
fucosyltransferase 6.
| Characteristic | n | Odds ratio (95%
CI) | P-value |
|---|
| T2 vs. T1 | 177 | 0.543
(0.238–1.181) | 0.132 |
| T3 vs. T1 | 164 | 0.449
(0.196–0.983) | 0.050a |
| T4 vs. T1 | 212 | 0.432
(0.192–0.927) | 0.035a |
| N1 vs. N0 | 319 | 0.650
(0.386–1.083) | 0.101 |
| N2 vs. N0 | 393 | 1.054
(0.703–1.582) | 0.798 |
| N3 vs. N0 | 246 | 0.731
(0.142–3.385) | 0.686 |
| M1 vs. M0 | 477 | 0.672
(0.088–4.093) | 0.665 |
| Stage II vs. I | 114 | 0.295
(0.090–0.840) | 0.029a |
| Stage III vs.
I | 121 | 0.386
(0.118–1.092) | 0.088 |
| Stage IV vs. I | 291 | 0.342
(0.108–0.920) | 0.045a |
| Male vs.
female | 502 | 0.751
(0.504–1.117) | 0.158 |
| Age >60 vs. ≤60
years | 501 | 0.873
(0.614–1.239) | 0.447 |
| Table VII.Logistic regression analysis of
cingulin-like 1. |
Table VII.
Logistic regression analysis of
cingulin-like 1.
| Characteristic | n | Odds ratio (95%
CI) | P-value |
|---|
| T2 vs. T1 | 177 | 0.720
(0.315–1.570) | 0.419 |
| T3 vs. T1 | 164 | 0.436
(0.190–0.953) | 0.042a |
| T4 vs. T1 | 212 | 0.386
(0.171–0.828) | 0.017a |
| N1 vs. N0 | 319 | 0.721
(0.430–1.198) | 0.209 |
| N2 vs. N0 | 393 | 1.170
(0.780–1.759) | 0.448 |
| N3 vs. N0 | 246 | 1.300
(0.281–6.718) | 0.735 |
| M1 vs. M0 | 477 | 0.244
(0.012–1.662) | 0.208 |
| Stage II vs. I | 114 | 0.535
(0.175–1.475) | 0.242 |
| Stage III vs.
I | 121 | 0.379
(0.125–1.039) | 0.068 |
| Stage IV vs. I | 291 | 0.462
(0.158–1.204) | 0.128 |
| Male vs.
female | 502 | 1.085
(0.730–1.613) | 0.687 |
| Age >60 vs. ≤60
years | 501 | 0.901
(0.634–1.279) | 0.561 |
Validation of differential expression
of hsa_circ_0035431, hsa-miR-940, FUT6 and CGNL1 in HNSCC
RT-qPCR was performed using the same RNA samples
used in sequencing analysis. Results demonstrated that the
expression levels of hsa_circ_0035431, FUT6 and CGNL1 were
decreased and hsa-miR-940 expression levels were elevated in HNSCC
compared with the HOK and adjacent groups (Fig. 9A-D). Notably, these results were
consistent with those obtained using transcriptome sequencing
(Fig. 9E-H).
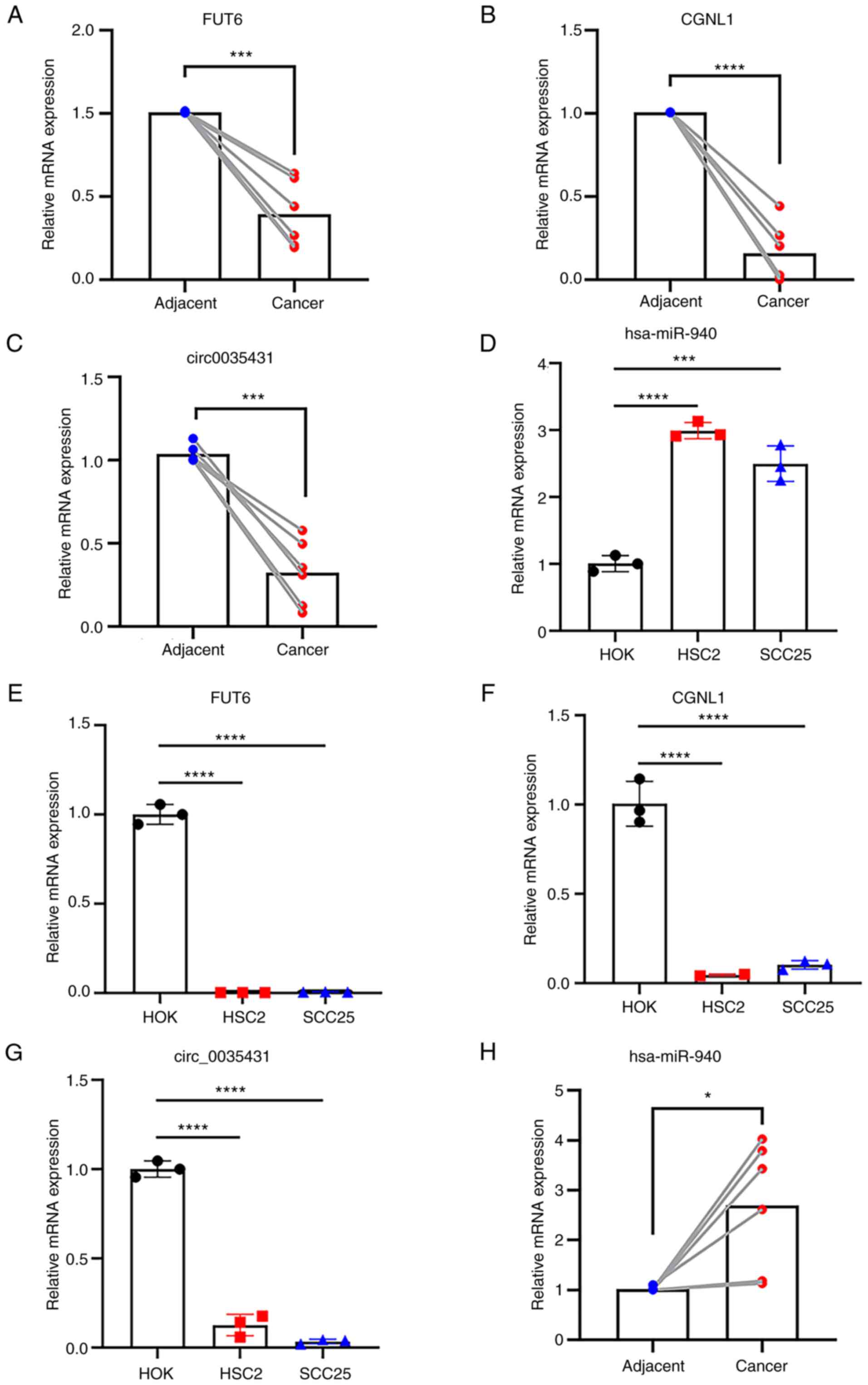 | Figure 9.Reverse transcription-quantitative
PCR validation of expression levels of hsa_circ_0035431,
hsa-miR-940, FUT6 and CGNL1 in HNSCC tissues and cells.
Transcription levels of (A) FUT6, (B) CGNL1, (C) hsa_circ_0035431
and (D) hsa_miR-940 in tissue. Transcription levels of (E) FUT6,
(F) CGNL1, (G) hsa_circ_003543 and (H) hsa_miR-940 in HNSCC cell
lines. *P<0.05, ***P<0.001, ****P<0.0001. FUT6,
fucosyltransferase 6; CGNL1, cingulin-like 1; HNSCC, head and neck
squamous cell carcinoma; circ, circular; miR, microRNA. |
Discussion
HNSCC is the seventh most common type of cancer in
the world; however, treatment is complex and options include
surgery, radiotherapy and chemotherapy (1). Determining the underlying pathogenic
mechanisms may aid discovery of effective treatment options to
improve the survival rate of patients.
A total of 1–2% of transcribed genes encode proteins
in eukaryotic cells and the majority of remaining genes are
transcribed into non-coding RNAs (ncRNAs) (18). ncRNAs are divided according to their
length as small (18–200 nucleotides) and long ncRNAs (>200
nucleotides). miRNAs are the most commonly studied type of ncRNA
(19,20). circRNAs, another type of ncRNA with
an average length of 1 kb, serve key roles in tumors through acting
as molecular sponges for miRNAs (21). Although circRNAs were discovered in
eukaryotes as early as 1991, the circRNA transcriptome has only
been studied in detail in recent years (22). A previous study demonstrated that
circRNAs serve as endogenous competitive RNAs, thereby regulating
proliferation, invasion and other physiological activities of tumor
cells (23). High-throughput
sequencing is widely used in circRNA studies to identify associated
functions (24,25). BPs, MFs and signaling pathways may
aid in determining the mechanisms of HNSCC onset and progression.
Numerous circRNAs serve a regulatory role in colorectal cancer
progression (26). Microtubule
cross-linking factor 1 circRNA promotes progression of advanced
laryngeal squamous carcinoma by inhibiting C1q-binding protein
ubiquitin degradation and mediating β-catenin activation,
demonstrating the pro-carcinogenic role of circRNA (27). circ_0109291 is highly expressed in
cisplatin-resistant oral squamous cell carcinoma tissue and
promotes cisplatin resistance by sponging miR-188-3p, suggesting
the therapeutic efficacy of circRNA (28). Therefore, clarifying the regulatory
role of RNAs in HNSCC may provide a novel theoretical basis for
disease progression and treatment.
circRNAs regulate gene expression through
competitive binding to miRNA (29).
miRNAs block protein translation and regulate mRNA stability at the
post-transcriptional level by binding to miRNA recognition elements
(30). circRNA/miRNA/mRNA
interactions have been identified in progression and prognosis of
non-small cell lung (31) and
gastric cancer (32) and
nasopharyngeal carcinoma (33).
Although circRNA/miRNA/mRNA interactions serve a role in
tumorigenesis and progression, the pathogenesis and potential
treatment of HNSCC are poorly understood and further investigation
is required.
In the present study, the whole genome of six pairs
of HNSCC and adjacent healthy tissue were sequenced and the
abnormal expression of circRNA, miRNA and mRNA were screened. In
addition, key target genes, FUT6 and CGNL1 were determined by
targeted prediction and online databases, yielding
hsa_circ_0035431/hsa-miR-940/FUT6 and
hsa_circ_0035431/hsa-miR-941/CGNL1 axes, which were associated with
the occurrence, development and prognosis of HNSCC.
Results of a previous study demonstrated that
hsa_circ_0035431 is the most differentially altered circRNA in
patients with gastric cancer, suggesting that it may be involved in
the pathological process of gastric cancer progression (34). However, the role of circRNA in HNSCC
is yet to be fully elucidated. A previous study confirmed the role
of miRNA-940 in tumor progression: miRNA-940 promotes gastric
cancer cell proliferation and migration by regulating programmed
death ligand 1 (35) and promotes
invasion and metastasis through downregulation of zinc finger
transcription factor 24 (36). Su
et al (37) demonstrated
that miR-940 overexpression promotes progression of human cervical
cancer through inhibiting p27 and PTEN, highlighting that miRNA-940
inhibition may be a target for treatment of cervical cancer. In the
present study, the Oncomir database and circRNA targeting
prediction were used to determine whether hsa-miR-940 was
associated with development and prognosis of HNSCC. An association
between hsa_circ_0035431 and hsa-miR-940 was established,
suggesting that the hsa_circ_0035431/hsa-miR-940 axis may be
involved in progression and prognosis of HNSCC. In addition, the
present study demonstrated FUT6 and CGNL1 were the target genes of
hsa-miR-940 through target gene prediction.
FUT6 is a member of the FUT family and is involved
in the synthesis of α-1,3-fucosyl bonds (38). A previous study demonstrated that
high FUT6 expression is associated with lower event-free survival
and OS; thus, FUT6 may serve as an independent adverse prognostic
factor in patients with acute myeloid leukemia (39). Based on TCGA HNSCC cohort, Mai et
al (40) revealed a robust
differentially expressed metabolic enzyme-based prognostic
signature, which included FUT6, for predicting clinical outcomes of
HNSCC. Moreover, a novel 19-gene risk predictive score model, which
included FUT6, was developed based on genes associated with lipid
metabolism. The aforementioned study demonstrated that FUT6 may
serve as a prognostic indicator and therapeutic target of gastric
cancer (41). By contrast, Li et
al (42) demonstrated that FUT6
overexpression may decrease the proliferation, migration and
invasion of human breast cancer cells. Thus, it was hypothesized
that FUT6 may be involved in different biological processes in
numerous types of cancer.
CGNL1, also known as paracingulin, is an endothelial
junctional complex protein localized to adhesion and tight
junctions. CGNL1 serves important roles in regulating vascular
growth in embryonic development and adult vascular-associated
diseases (43). Low expression of
CGNL1, the target gene of miR-149-3p, is associated with a poor
prognosis in patients with uterine corpus endometrial carcinoma
(44). Notably, CGNL1 may be
involved in processes associated with progesterone resistance
(45). Moreover, CGNL1 is a
prognostic gene for carriers of the HNSCC TP53 mutation (46). A previous study indicated that CGNL1
may act as a urine biomarker of high-grade bladder urothelial
carcinoma (HGBC), thus exhibiting potential as a diagnostic
indicator, prognostic predictor and treatment target for HGBC.
Thus, CGNL1 may improve prognosis of patients with HGBC (47).
Through data mining, the expression levels of FUT6
and CGNL1 were investigated in HNSCC in the present study. Results
from the GEPIA database demonstrated that FUT6 and CGNL1 expression
were lower in tumor tissues of patients with HNSCC compared with
adjacent healthy tissue. Subsequently, the expression levels of
FUT6 and CGNL1 were compared using UALCAN and TCGA databases. FUT6
and CGNL1 expression levels were associated with the staging of
patients with HNSCC. In addition, patients with low FUT6 and CGNL1
expression levels exhibited decreased OS and PFI compared with the
those with high expression levels. Collectively, the present study
demonstrated that FUT6 and CGNL1 may serve key roles in the
development and prognosis of HNSCC.
RT-qPCR was performed to validate the accuracy of
the microarray. miRNA-940 was highly expressed in HNSCC and FUT6
and CGNL1 were expressed at low levels. These results supported the
hypothesis that hsa_circ_0035431 may act as a potential prognostic
marker targeting and regulating the expression of FUT6 and CGNL1
through hsa-miR-940, thus affecting HNSCC progression.
However, the present study had numerous limitations.
First, only six pairs of tumor tissue samples were used to validate
circRNA/miRNA/mRNA interaction. Expression levels were only
verified at the genetic level and further investigations into
protein expression levels are required. In addition, western blot,
immunohistochemistry, transfection, Transwell and wound healing
assay and in vivo experiments are further required to
validate the dysregulation of circRNA/miRNA/mRNA expression in
HNSCC. Moreover, human papillomavirus infection status was not
considered in the present study and may impact the progression and
prognosis of HNSCC (48).
In conclusion, bioinformatics analysis revealed the
potential regulatory mechanisms of the
hsa_circ_0035431/hsa-miR-940/FUT6 and
hsa_circ_0035431/hsa-miR-940/CGNL1 axes in development and
prognosis of HNSCC. Notably, hsa_circ_0035431 may regulate
expression of FUT6 and CGNL1 via sponging hsa-miR-940, thus
impacting the progression and prognosis of HNSCC, and may exhibit
potential as a drug target for the treatment of HNSCC.
Acknowledgements
Not applicable.
Funding
The present study was supported by National Natural Science
Foundation of China (grant nos. 31900441 and 81903102), Natural
Science Foundation of Shandong Province (grant nos. ZR2018BH026 and
ZR2019PH075), Projects of Medical and Health Technology Development
Program in Shandong Province (grant nos. 2017WS231 and
202008020682), Youth Project of Traditional Chinese Medicine
Science and Technology in Shandong Province (grant no. 2020Q062)
and Qilu Outstanding Young Talents in Health Project, Taishan
Scholarship and Double-Hundred Foreign Talent Plan (grant no.
WSG2018019).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the Gene Expression Omnibus
repository (accession no. GSE190224).
Authors' contributions
XYL and LLZ confirm the authenticity of all the raw
data. XRM conceived the project. XYL, LLZ, and WLW designed the
experiments and analyzed data. XYL and LLZ performed experiments.
WLW, ZPL, FW, YW and SYZ were responsible for the collection of
clinical samples and data. JD and XRM performed bioinformatics.
XYL, LLZ and ZPL wrote the manuscript. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
The experimental procedures were approved by The
Ethics Committee of Binzhou Medical University Hospital (Binzhou,
China; approval no. 2018-G010-01) and The West China Hospital of
Stomatology, Sichuan University (Chengdu, China; approval no.
WCHSIRB-CT-2021-022). All patients provided written informed
consent.
Patient consent for publication
Not applicable.
Authors' information
LX, ORCID ID: 0000-0002-7288-7531. ZL, ORCID ID:
0000-0002-4454-4929. WW, ORCID ID: 0000-0001-7014-2985.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chow L: Head and neck cancer. N Engl J
Med. 382:60–72. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang J, Chen X, Tian Y, Zhu G, Qin Y, Chen
X, Pi L, Wei M, Liu G, Li Z, et al: Six-gene signature for
predicting survival in patients with head and neck squamous cell
carcinoma. Aging (Albany NY). 12:767–783. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Garbo S, Maione R, Tripodi M and
Battistelli C: Next RNA therapeutics: The mine of Non-coding. Int J
Mol Sci. 23:74712022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tang X, Ren H, Guo M, Qian J, Yang Y and
Gu C: Review on circular RNAs and new insights into their roles in
cancer. Comput Struct Biotechnol J. 19:910–928. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Qiu S, Li B, Xia Y, Xuan Z, Li Z, Xie L,
Gu C, Lv J, Lu C, Jiang T, et al: CircTHBS1 drives gastric cancer
progression by increasing INHBA mRNA expression and stability in a
ceRNA- and RBP-dependent manner. Cell Death Dis. 13:2662022.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang J, Qi M, Fei X, Wang X and Wang K:
Hsa_circRNA_0088036 acts as a ceRNA to promote bladder cancer
progression by sponging miR-140-3p. Cell Death Dis. 13:3222022.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mo M, Liu B, Luo Y, Tan J, Zeng X, Zeng X,
Huang D, Li C, Liu S and Qiu X: Construction and comprehensive
analysis of a circRNA-miRNA-mRNA regulatory network to reveal the
pathogenesis of hepatocellular carcinoma. Front Mol Biosci.
9:8014782022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nath M, Roy D and Choudhury Y: Circular
RNAs are potential prognostic markers of head and neck squamous
cell carcinoma: Findings of a Meta-analysis study. Front Oncol.
12:7824392022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu Y, Zhang Y, Zheng X, Dai F, Lu Y, Dai
L, Niu M, Guo H, Li W, Xue X, et al: Circular RNA circCORO1C
promotes laryngeal squamous cell carcinoma progression by
modulating the let-7c-5p/PBX3 axis. Mol Cancer. 19:992020.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim D, Langmead B and Salzberg SL: HISAT:
A fast spliced aligner with low memory requirements. Nat Methods.
12:357–360. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Robinson MD, Mccarthy DJ and Smyth GK:
edgeR: A Bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Young MD, Wakefield MJ, Smyth GK and
Oshlack A: Gene ontology analysis for RNA-seq: Accounting for
selection bias. Genome Biol. 11:R142010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ke MJ, Ji LD and Li YX: Explore prognostic
marker of colorectal cancer based on ceRNA network. J Cell Biochem.
120:19358–19370. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Enright AJ, John B, Gaul U, Tuschl T,
Sander C and Marks DS: MicroRNA targets in drosophila. Genome Biol.
5:R12003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kruger J and Rehmsmeier M: RNAhybrid:
microRNA target prediction easy, fast and flexible. Nucleic Acids
Res. 34:W451–W454. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Knapek KJ, Georges HM, Van Campen H,
Bishop JV, Bielefeldt-Ohmann H, Smirnova NP and Hansen TR: Fetal
lymphoid organ immune responses to transient and persistent
infection with bovine viral diarrhea virus. Viruses. 12:8162020.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Garcia-Hidalgo MC, Gonzalez J, Benitez ID,
Carmona P, Santisteve S, Perez-Pons M, Moncusi-Moix A,
Gort-Paniello C, Rodriguez-Jara F, Molinero M, et al:
Identification of circulating microRNA profiles associated with
pulmonary function and radiologic features in survivors of
SARS-CoV-2-induced ARDS. Emerg Microbes Infect. 11:1537–1549. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Djebali S, Davis CA, Merkel A, Dobin A,
Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F,
et al: Landscape of transcription in human cells. Nature.
489:101–108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Z, Zhang J, Diao L and Han L: Small
non-coding RNAs in human cancer: Function, clinical utility, and
characterization. Oncogene. 40:1570–1577. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yousefi H, Maheronnaghsh M, Molaei F,
Mashouri L, Reza AA, Momeny M and Alahari SK: Long noncoding RNAs
and exosomal lncRNAs: Classification, and mechanisms in breast
cancer metastasis and drug resistance. Oncogene. 39:953–974. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guo L, Jia L, Luo L, Xu X, Xiang Y, Ren Y,
Ren D, Shen L and Liang T: Critical roles of circular RNA in tumor
metastasis via acting as a sponge of miRNA/isomiR. Int J Mol Sci.
23:70242022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Prats AC, David F, Diallo LH, Roussel E,
Tatin F, Garmy-Susini B and Lacazette E: Circular RNA, the key for
translation. Int J Mol Sci. 21:85912020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou R, Wu Y, Wang W, Su W, Liu Y, Wang Y,
Fan C, Li X, Li G, Li Y, et al: CircularRNAs (circRNAs) incancer.
Cancer Lett. 425:134–142. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Derks KW, Misovic B, van den Hout MC,
Kockx CE, Gomez CP, Brouwer RW, Vrieling H, Hoeijmakers JH, van
Ijcken WF and Pothof J: Deciphering the RNA landscape by RNAome
sequencing. RNA Biol. 12:30–42. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Szabo L and Salzman J: Detecting circular
RNAs: Bioinformatic and experimental challenges. Nat Rev Genet.
17:679–692. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zheng R, Zhang K, Tan S, Gao F, Zhang Y,
Xu W, Wang H, Gu D, Zhu L, Li S, et al: Exosomal circLPAR1
functions in colorectal cancer diagnosis and tumorigenesis through
suppressing BRD4 via METTL3-eIF3h interaction. Mol Cancer.
21:492022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang Z, Sun A, Yan A, Yao J, Huang H, Gao
Z, Han T, Gu J, Li N, Wu H and Li K: Circular RNA MTCL1 promotes
advanced laryngeal squamous cell carcinoma progression by
inhibiting C1QBP ubiquitin degradation and mediating beta-catenin
activation. Mol Cancer. 21:922022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gao F, Han J, Wang Y, Jia L, Luo W and
Zeng Y: Circ_0109291 promotes cisplatin resistance of oral squamous
cell carcinoma by sponging miR-188-3p to increase ABCB1 expression.
Cancer Biother Radiopharm. 37:233–245. 2022.PubMed/NCBI
|
|
29
|
Qi X, Zhang DH, Wu N, Xiao JH, Wang X and
Ma W: ceRNA in cancer: Possible functions and clinical
implications. J Med Genet. 52:710–718. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Thomson DW and Dinger ME: Endogenous
microRNA sponges: Evidence and controversy. Nat Rev Genet.
17:272–283. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ge P, Chen X, Liu J, Jing R, Zhang X and
Li H: Hsa_circ_0088036 promotes nonsmall cell lung cancer
progression by regulating miR-1343-3p/Bcl-3 axis through
TGFβ/Smad3/EMT signaling. Mol Carcinog. 62:1073–1085. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shen Y, Zhang N, Chai J, Wang T, Ma C, Han
L and Yang M: CircPDIA4 induces gastric cancer progressionby
promoting ERK1/2 activation and enhancing biogenesisof oncogenic
circRNAs. Cancer Res. 83:538–552. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mo H, Shen J, Zhong Y, Chen Z, Wu T, Lv Y,
Xie Y and Hao Y: CircMAN1A2 promotes vasculogenic mimicry of
nasopharyngeal carcinoma cells through upregulating ERBB2 via
sponging miR-940. Oncol Res. 30:187–199. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dang Y, Ouyang X, Zhang F, Wang K, Lin Y,
Sun B, Wang Y, Wang L and Huang Q: Circular RNAs expression
profiles in human gastric cancer. Sci Rep. 7:90602017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fan Y, Che X, Hou K, Zhang M, Wen T, Qu X
and Liu Y: MiR-940 promotes the proliferation and migration of
gastric cancer cells through up-regulation of programmed death
ligand-1 expression. Exp Cell Res. 373:180–187. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu X, Ge X, Zhang Z, Zhang X, Chang J, Wu
Z, Tang W, Gan L, Sun M and Li J: MicroRNA-940 promotes tumor cell
invasion and metastasis by downregulating ZNF24 in gastric cancer.
Oncotarget. 6:25418–25428. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Su K, Wang C, Zhang Y, Cai Y, Zhang Y and
Zhao Q: miR-940 upregulation contributes to human cervical cancer
progression through p27 and PTEN inhibition. Int J Oncol.
50:1211–1220. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cheng L, Luo S, Jin C, Ma H, Zhou H and
Jia L: FUT family mediates the multidrug resistance of human
hepatocellular carcinoma via the PI3K/Akt signaling pathway. Cell
Death Dis. 4:e9232013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dai Y, Cheng Z, Pang Y, Jiao Y, Qian T,
Quan L, Cui L, Liu Y, Si C, Chen J, et al: Prognostic value of the
FUT family in acute myeloid leukemia. Cancer Gene Ther. 27:70–80.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mai Z, Chen H, Huang M, Zhao X and Cui L:
A robust metabolic enzyme-based prognostic signature for head and
neck squamous cell carcinoma. Front Oncol. 11:7702412022.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wei X, Luo T, Li J, Xue Z, Wang Y, Zhang
Y, Chen Y and Peng C: Development and validation of a prognostic
classifier based on lipid metabolism-related genes in gastric
cancer. Front Mol Biosci. 8:6911432021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li N, Liu Y, Miao Y, Zhao L, Zhou H and
Jia L: MicroRNA-106b targets FUT6 to promote cell migration,
invasion, and proliferation in human breast cancer. IUBMB Life.
68:764–775. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chrifi I, Hermkens D, Brandt MM, van Dijk
C, Burgisser PE, Haasdijk R, Pei J, van de Kamp E, Zhu C, Blonden
L, et al: Cgnl1, an endothelial junction complex protein, regulates
GTPase mediated angiogenesis. Cardiovasc Res. 113:1776–1788. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lu X, Jing L, Liu S, Wang H and Chen B:
miR-149-3p is a potential prognosis biomarker and correlated with
immune infiltrates in uterine corpus endometrial carcinoma. Int J
Endocrinol. 2022:50061232022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li W, Wang S, Qiu C, Liu Z, Zhou Q, Kong
D, Ma X and Jiang J: Comprehensive bioinformatics analysis of
acquired progesterone resistance in endometrial cancer cell line. J
Transl Med. 17:582019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Jin Y and Qin X: Significance of TP53
mutation in treatment and prognosis in head and neck squamous cell
carcinoma. Biomark Med. 15:15–28. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Song Y, Jin D, Ou N, Luo Z, Chen G, Chen
J, Yang Y and Liu X: Gene expression profiles identified novel
urine biomarkers for diagnosis and prognosis of high-grade bladder
urothelial carcinoma. Front Oncol. 10:3942020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kutz LM, Abel J, Schweizer D, Tribius S,
Krull A, Petersen C and Loser A: Quality of life, HPV-status and
phase angle predict survival in head and neck cancer patients under
(chemo)radiotherapy undergoing nutritional intervention: Results
from the prospective randomized HEADNUT-trial. Radiother Oncol.
166:145–153. 2022. View Article : Google Scholar : PubMed/NCBI
|