Introduction
Although the occurrence of liver cancer has declined
for decades and has currently plateaued, it is still considered one
of the most fatal cancers (1).
Cancer statistics showed 906,000 new liver cancer cases and 830,000
deaths worldwide in 2020. The five-year net survival was reported
as 5–30% during 2000–2014 (2).
Metastasis and recurrence present formidable
obstacles to improving the long-term survival of patients with HCC.
Currently, surgical intervention is the most effective approach for
the treatment of HCC. However, when initially diagnosed with HCC, a
considerable number of patients are already at an advanced stage,
rendering them ineligible for curative surgery (3). Consequently, the acquisition of a
comprehensive understanding of the mechanisms underlying HCC
metastasis is of utmost importance for fostering the development of
innovative therapeutic strategies aimed at improving patient
outcomes.
Fatty acids (FAs) are energy storage substances that
cells can utilize as a source of energy for growth and
multiplication (4). To maintain
rapid proliferation, the metabolic level of FA synthesis in
cancerous cells is upregulated to meet the biosynthesis demands of
membranes and signaling molecules (5,6).
Consequently, metabolic dysregulation may contribute to tumor
initiation, development and progression (7,8). Also,
the upregulation of FA metabolism occurs in HCC (9). There is preclinical and clinical
evidence that FA-related genes participate in HCC development and
prognosis (10,11). However, studies concerning the
dysregulation of FA metabolism in HCC metastasis are rare. In
addition to promoting HCC, upregulated FA metabolism has also been
shown to impair the response of HCC to targeted therapies such as
sorafenib (12) and bevacizumab
(13). Furthermore, studies have
suggested that high palmitic acid levels induce the upregulation of
programmed cell death ligand-1 (14), and that the β-oxidation of FAs
(15,16) and short-chain FAs (10,17)
facilitates the activity of Treg cells and M2 macrophages, thereby
impairing their antitumor immune efficacy.
The aim of the present study was to analyze the
association between HCC metastasis and FA metabolism-related genes,
and then to explore the value of the FA genetic signature in
prognosis and as a novel therapeutic target. The findings may
improve our understanding of HCC and help to optimize strategies
for its treatment.
Materials and methods
Data acquisition
The expression arrays and associated clinical
information of patients with HCC from The Cancer Genome Atlas
(TCGA) were obtained via the University of California Santa Cruz
Xena tool (http://xena.ucsc.edu/) (18) for subsequent analysis. After
excluding samples with no clear clinical information and OS <30
days, a total of 362 patients participated. The Human Protein Atlas
portal (https://www.proteinatlas.org/) was
searched to acquire images of HCC and adjacent normal tissue
immunohistochemistry (IHC) to show dysregulation of a key gene at
the protein level. The gene set for FA metabolism was obtained from
the Molecular Signatures Database (MSigDB) (19,20).
After entering ‘fatty’ in the keywords box with the filter of
Homo sapiens, a total of 187 gene sets were obtained. The
target gene set comprised genes from the FA metabolism gene sets
that overlapped with gene sets associated with metastasis and worse
OS risk. Kaplan-Meier survival curve analysis was conducted using
the Gene Expression Profiling Interaction Analysis 2 web
application (http://gepia2.cancer-pku.cn/#index) to explore the
association of specific genes with OS in HCC. The University of
Alabama at Birmingham Cancer Data Analysis (UALCAN) portal
(http://ualcan.path.uab.edu/) was
utilized to analyze the correlation between promoter methylation
level and protein level of the key gene mitochondrial ribosomal
protein L35 (MRPL35) and clinical status. UALCAN
(ualcan.path.uab.edu/analysis-prot.html) was used to analyze the
association between protein level and clinical characteristics from
the Clinical Proteomic Tumor Analysis Consortium (CPTAC). TNMplot
(https://tnmplot.com/analysis/) (21) public database was used to explore
the relationship between MRPL35 and metastatic status in HCC.
Construction of a co-expression
network and exploration of the association between gene modules and
metastatic status
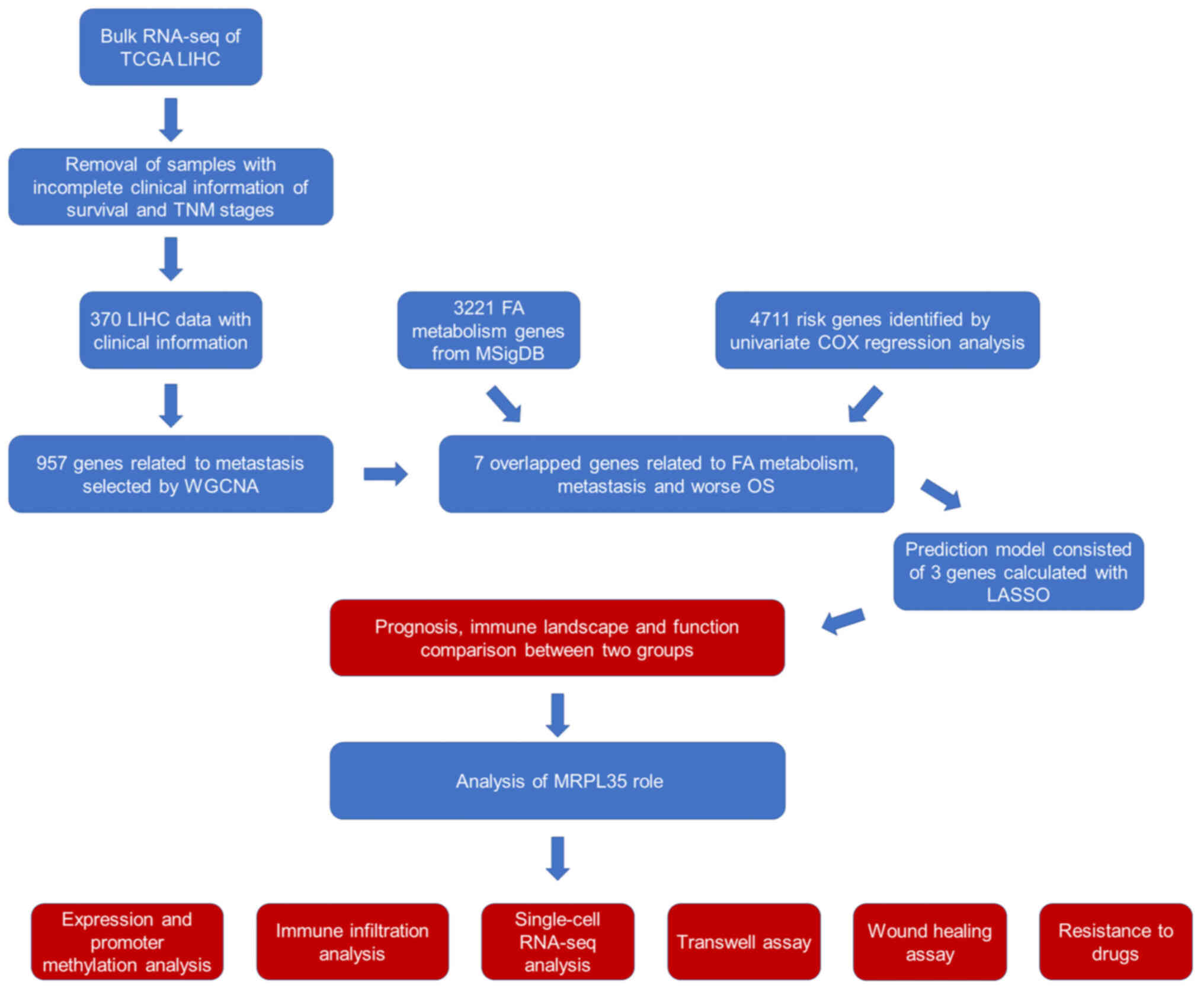
The workflow of the present study is shown in
Fig. 1. Following the acquisition
of the RNA-sequencing (RNA-seq) array and clinical information of
patients with HCC, a weighted correlation network analysis (WGCNA)
(22) was conducted using R
(Vision: 3.6.3; r-project.org/). WGCNA was performed to investigate
the association between gene modules and clinical variables (age, T
stage, N stage and M stage). In this analysis, gene significance
(GS) was determined by taking the logarithm (base 10) of the
P-value resulting from the linear regression analysis conducted on
gene expression and clinical features. Module significance (MS) was
defined as the mean GS across all genes within a module. The module
exhibiting the highest absolute MS among all the modules selected
for a specific clinical feature was considered to be associated
with that feature. To condense the expression patterns of all genes
within a module, a singular representative expression value known
as the module eigengene (ME) was calculated, which served as the
primary component in the principal component analysis. By
evaluating the associations between MEs and clinical features, the
most pertinent gene module for each clinical feature of interest
was identified. Further analysis was conducted on the module
displaying the strongest association with the desired clinical
features.
Functional enrichment analysis
A combined WGCNA and FA (WF) score was calculated by
multiplying the expression value of each gene by the sum of its
respective coefficients. The coefficients calculated by Least
Absolute Shrinkage and Selection Operator (LASSO) (23) were as follows: CYP2S1, 0.043;
MRPL35, 0.178; and PLCD3, 0.061. The HCC cohort was divided into
high and low WF score groups according to the median WF score. Gene
Set Enrichment Analysis (GSEA; gsea-msigdb.org/gsea/index.jsp)
(19) was utilized to investigate
the biological functions that differed between the two groups. The
GSEA employed a cut-off threshold of nominal P<0.05 and a false
discovery rate (FDR) <0.25. The LinkedOmics (https://www.linkedomics.org/admin.php)
(24) online tool was used to
analyze the potential function of MRPL35 in HCC.
LASSO Cox regression modeling and
survival analysis
Cox regression modeling using LASSO was performed
with the LASSO-Cox regression tool available in Sangerbox (25) to select the most relevant genes.
LASSO regression is characterized by variable selection and
complexity regularization while fitting a generalized linear model.
Therefore, regardless of whether the target dependent variable is
continuous or discrete, LASSO regression is used to model and
predict. COX survival analysis was conducted by glmnet, survival
and survminer packages of R based on clinical information.
Analysis of immune cell
infiltration
The differential infiltration of immune cells was
assessed using the CIBERSORT algorithm (https://cibersortx.stanford.edu) (26). In this analysis, the default
signature matrix comprising 1,000 permutations was employed. To
enhance the reliability of the findings, only data with
deconvolution P<0.05 were retained. Wilcoxon rank-sum test and
signed-rank test were utilized appropriately to analyze the
differences between clusters and in immune cell infiltration
between the high and low WF score groups.
To assess the association between MRPL35 expression
and immune infiltration, the TIMER2.0 (timer.comp-genomics.org/)
(27) systematic platform was used
to examine immunological attributes of cancer in TCGA data.
Validation of single-cell RNAseq
(scRNA-seq)
Tumor Immune Single-Cell Hub
(tisch.comp-genomics.org/home/) (TISCH) is a public source of
scRNA-seq data on the tumor microenvironment (TME). TISCH collected
a total of 76 sets of high-quality tumor single-cell transcriptome
data from GEO and ArrayExpress and corresponding patient
information, covering 27 cancer types. It provides specific
annotation of cell type at a single-cell level and allows gene
expression in the TME to be investigated (28).
Cell line and culture
The HepG2 liver cancer cell line (identified by
Short Tandem repeat) was purchased from The Cell Bank of Type
Culture Collection of The Chinese Academy of Sciences. The cells
were cultured at 37°C in a humid atmosphere with 5% CO2
and maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine
serum (FBS; Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml
penicillin and 100 µg/ml streptomycin (Beijing Solarbio Science
& Technology Co., Ltd.).
Small interfering RNA (siRNA) and
reverse transcription-quantitative PCR (RT-qPCR)
Two siRNAs targeting MRPL35 (siMRPL35-1 and
siMRPL35-2) and a control siRNA (siCtrl) were synthesized and
designed by Sangon Biotech Co., Ltd. to suppress the expression of
MRPL35 in HepG2 cells. To initiate transfection, cells were plated
in 6-well plates at a density of 1×105 cells/well and
incubated at 37°C until 70% confluency was reached. Subsequently,
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) was used to transfect the cells with siRNAs or
siCtrl (<60 nM, following the protocols provided by the
manufacturer at room temperature for 20 min. The antisense
sequences of the three siRNAs were as follows: siMRPL35-1,
TAGTCATATTCAGACACCAGTTGTT; siMRPL35-2, CAACTGTGTCAAGAATGCCTCTCTT;
and siCtrl, TAGATACTTAGACACGACTTTCGTT.
To confirm the efficacy of siRNA transfection,
RT-qPCR was utilized to detect the level of MRPL35 after
transfection. Following 36 h incubation under 37°C, total RNA was
isolated from cells using TRIzol® (Invitrogen; Thermo
Fisher Scientific, Inc.). Subsequently, cDNA synthesis was
performed using a RevertAid First Strand cDNA Synthesis kit (Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocols.
RT-qPCR was performed using iQ™ SYBR Green Supermix
(Bio-Rad Laboratories, Inc.). The following primers were employed
for the qPCR analysis: MRPL35 forward, 5′-TTGGCATCTTCAACCTACCGC-3′
and reverse, 5′-GGAGGAAACAACTGGTGTCTGA-3′; GAPDH forward,
5′-ACAACTTTGGTATCGTGGAAGG-3′ and reverse, 5′-GCCATCACGCCACAGTTTC-3′
and the thermocycling condition was 94°C 60sec, 37°C 60 sec, 72°C
for 120 sec and 20–30 cycles. To calculate the relative expression
level of the target gene, the 2−ΔΔCq method was employed
(29).
Wound healing assay
In the wound healing assay, HepG2 cells were seeded
into a 6-well plate and allowed to reach 80% confluence. Under the
deprivation of serum the cell monolayer was then scratched using a
sterile micropipette tip. The floating cells were washed away with
PBS to clear away any debris. Time points of 0, 12 and 24 h were
chosen to observe the progression of wound healing within the gap
with light microscope. Triplicate wells were examined for each
condition to ensure reliability and reproducibility. The migration
and closure of the wound were assessed by measuring the width of
the wound at the two time points with those at 0 h.
Transwell assay
The Transwell assay, also known as the Boyden
chamber assay, is a widely used technique for the study of cell
migration and invasion. In this assay, Transwell chambers were
prepared, each comprising an insert with 8-µm pores. The upper side
of the membrane in the Transwell chamber was coated with a diluted
Matrigel matrix in a 37°C incubator for 30 min. Prior to the assay,
the HepG2 cells were cultured in 2% FBS-DMEM for 12 h to induce
starvation. The cells were then suspended in FBS-free DMEM and
added to the upper chamber of the Transwell chamber at a
concentration of 1×105 cells/chamber. Simultaneously,
DMEM supplemented with 10% FBS was added to the lower compartment.
The Transwell chambers were then incubated for 48 h at 37°C. Cells
that had migrated to the lower surface of the filter membrane were
fixed with 4% paraformaldehyde (20 min at room temperature) and
stained with 0.5% crystal violet (10 min at room temperature).
Cells remaining on the upper surface of the filter membrane were
gently scraped off with a cotton swab. The lower surfaces of the
membrane were then observed and images captured using an inverted
light microscope, and the counting process was repeated three
times.
MRPL35 and drug response
Drug Response CellMiner (https://discover.nci.nih.gov/cellminer/) is as an
integration platform for both molecular and pharmacological data
concerning the NCI-60 cancer cell lines (30). This resource was employed to explore
the correlation between MRPL35 expression and drug response.
Statistical analysis
Statistical analyses were performed using the
aforementioned online databases and R software (version 3.6.3).
Data are presented as mean ± SD. For the statistical analysis of
experimental data, GraphPad Prism 9.0 (GraphPad Software;
Dotmatics) was employed. Unpaired Student's t and Wilcoxon rank-sum
test and one-way ANOVA tests (with LSD as post hoc test) were used
to calculate the significance of differences in data between and
among groups. P<0.05 was considered to indicate a statistically
significant result.
Results
Identification of the core gene set
associated with HCC progression
Following the extraction of gene expression data and
clinical information associated with HCC from TGCA, data on 362
cancerous samples with total prognostic information and 50 normal
samples were obtained. These data were subjected to pre-processing
cleanup and quality control prior to being subjected to WGCNA to
investigate the associations between gene clusters and clinical
characteristics associated with HCC. Based on scale-free topology,
a soft-thresholding power of β=4 was chosen, indicating the
reliability of the chosen power value. The rationality test yielded
positive results as shown in Fig.
S1A-C and following the identification of 44 gene modules
(Fig. S1D), the skyblue3 and
purple modules were found to exhibit significantly positive
connections with M stage (cor=0.86, P<0.05) and N stage
(cor=0.89, P<0.05) (Fig. S1E and
F). As a result, the two modules, consisting of 957 genes, were
recognized as the modules most relevant to tumor-distant metastasis
and lymph node metastasis in HCC.
Consolidation of the gene set for
prognosis prediction
There were seven genes in the area of overlap of the
metastasis-associated WGCNA genes with the FA metabolism-related
genes from MSigDB, and genes identified to be associated with the
risk of HCC progression and OS by univariate Cox regression
analysis (Fig. 2A). The LASSO-Cox
regression model was used to further screen the seven core genes in
HCC prognosis, and three genes were identified as preferable
parameters (Fig. 2B and C). The
relationship between the levels of the three genes, namely
phospholipase C d3 (PLCD3), MRPL35 and cytochrome P450 family 2
subfamily S member 1 (CYP2S1), and outcomes is shown in Fig. 2D. A significant difference in OS was
observed between the high and low WF score groups by Kaplan-Meier
analysis (P=0.009; Fig. 2E).
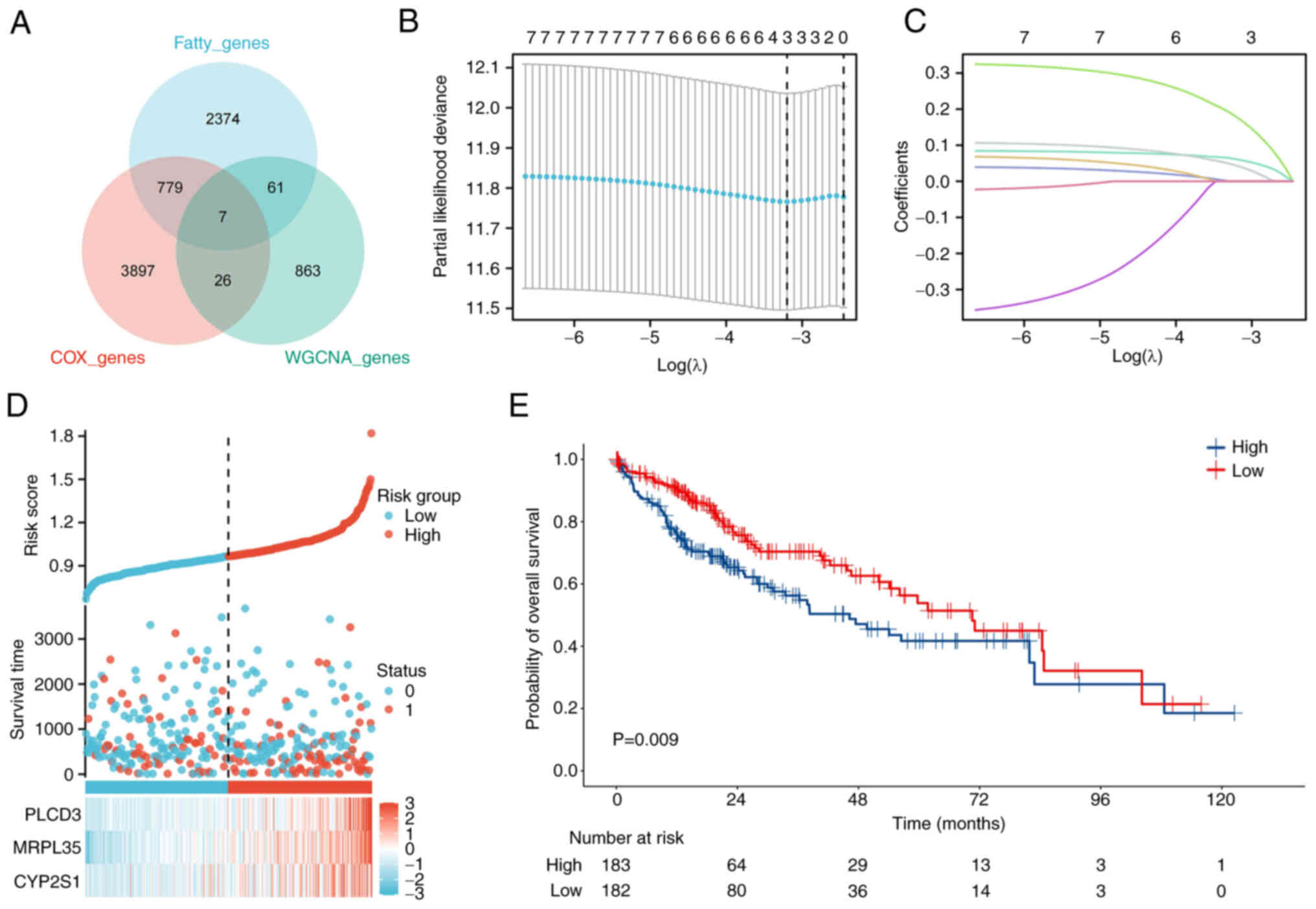 | Figure 2.Consolidation of the predictive
model. (A) Seven genes from the WGCNA, fatty acid
metabolism-related and OS risk gene datasets overlapped in HCC. (B)
LASSO analysis was conducted based on the seven genes, and three
key genes were identified. (C) Dynamic screening process of
variable coefficients. (D) Relationship between the expression
levels of the three genes and the clinical status of patients with
HCC. (E) Based on the WF scores calculated by LASSO, a worse
outcome was observed in the group with high WF scores. WGCNA,
weighted correlation network analysis; OS, overall survival; HCC,
hepatocellular carcinoma; LASSO, Least Absolute Shrinkage and
Selection Operator; WF, WGCNA and fatty acid; PLCD3, phospholipase
C d3; MRPL35, mitochondrial ribosomal protein L35; CYP2S1,
cytochrome P450 family 2 subfamily S member 1. |
The univariate Cox regression analysis of the seven
genes is shown in Fig. 3A, each
with P<0.05 and hazard ratio (HR)>1 for OS. MRPL35 had the
highest HR among the three genes of particular interest (HR=1.461,
P<0.05). Survival analyses were also conducted for the three
preferred genes, CYP2S1 (HR=1.5, P=0.024, Fig. 3B), PLCD3 (HR=1.4, P=0.042, Fig. 3C and MRPL35 (HR=1.9, P<0.001,
Fig. 3D). Subsequent expression
analysis further indicated that PLCD3 and MRPL35 were significantly
upregulated in cancerous samples compared with normal tissue
(P<0.05; Fig. 3E-G).
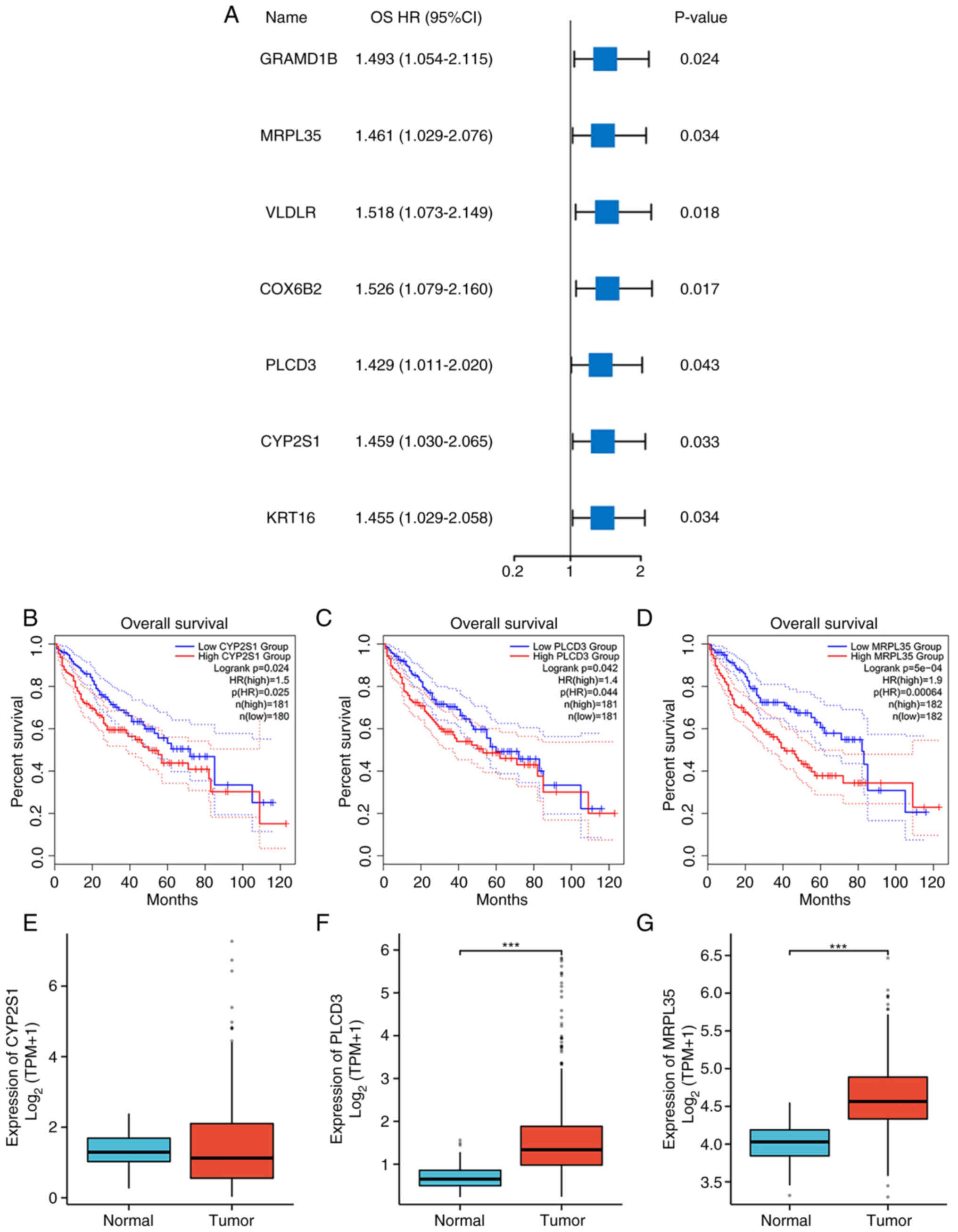 | Figure 3.Survival and expression analysis. (A)
Forest plot showing the OS HR and P-value of seven genes. OS
analysis of (B) CYP2S1, (C) PLCD3 and (D) MRPL35 in HCC. Expression
levels of (E) CYP2S1, (F) PLCD3 and (G) MRPL35 in normal and HCC
tumor samples. ***P<0.001. OS, overall survival; HR, hazard
ratio; CYP2S1, cytochrome P450 family 2 subfamily S member 1;
PLCD3, phospholipase C d3; MRPL35, mitochondrial ribosomal protein
L35; HCC, hepatocellular carcinoma; GRAMD1B, GRAM domain containing
1B; VLDLR, very low density lipoprotein; COX6B2, cytochrome C
oxidase subunit 6B2; KRT16, keratin 16; TPM, transcripts per
million. |
These results indicate that the analysis of samples
based on PLCD3, MRPL35 and CYP2S1 expression may be utilized to
predict the OS of HCC. Therefore, the mechanism underlying the
difference is worthy of exploration.
Immune infiltration and functional
enrichment analysis
The effective infiltration of lymphocytes and
ability of tumor cells to escape them affects the antitumor
efficacy of immunity and the outcome of patients. With the
CIBERSORT algorithm, the immune cells in the microenvironment of
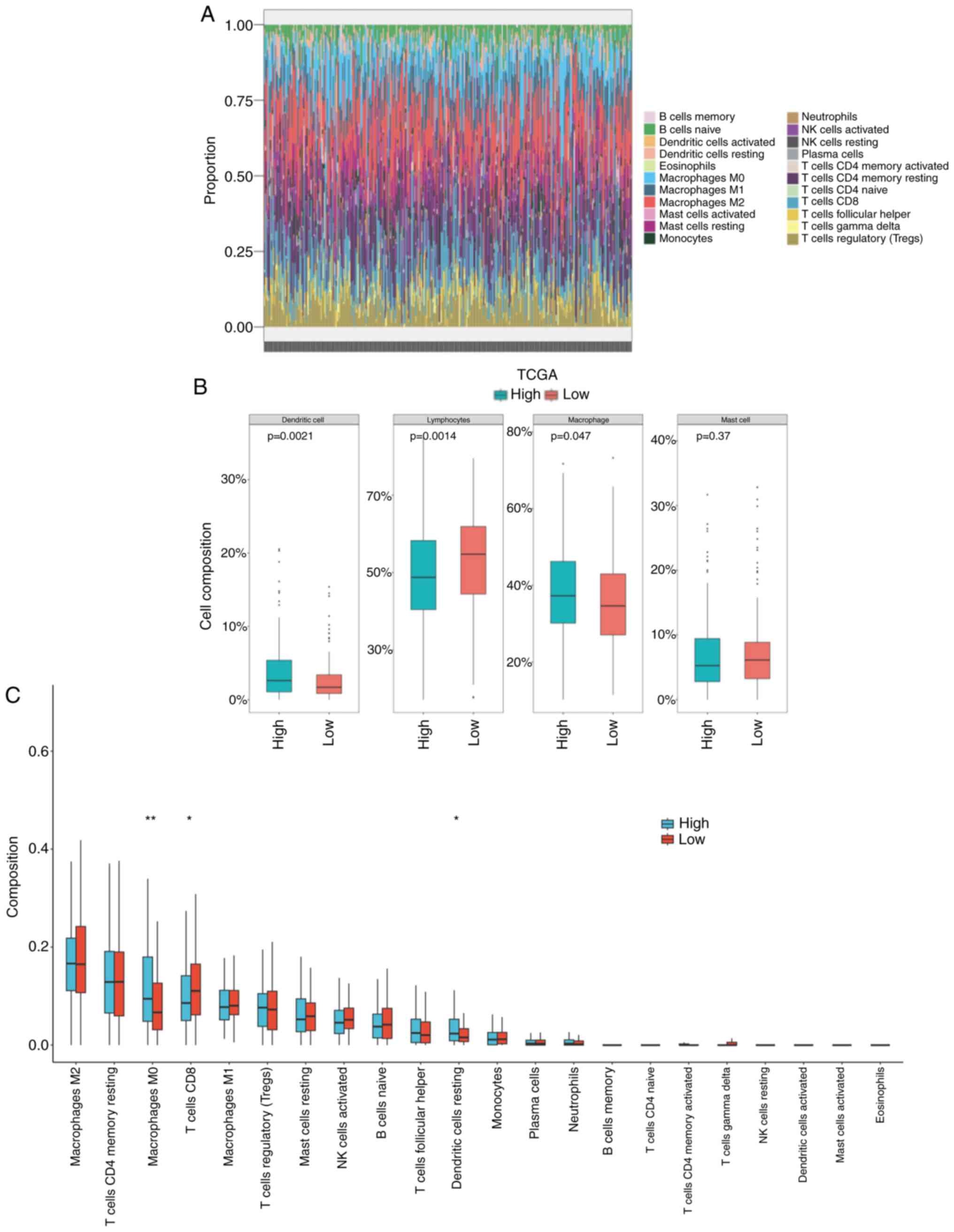
each sample were analyzed and predicted (Fig. 4A). In the high WF score group, it
was predicted that the infiltration of lymphocytes was lower
(P=0.0014) and the infiltration of macrophages was higher (P=0.047)
than that in the low WF score group (Fig. 4B). The specific types of immune cell
were analyzed, which indicated the presence of more M0 macrophages
and fewer CD8+ T cells in the high WF score group
compared with the low WF score group (P<0.05; Fig. 4C). The impaired immune efficacy
might be a reason for the worse outcome of patients with a high WF
score.
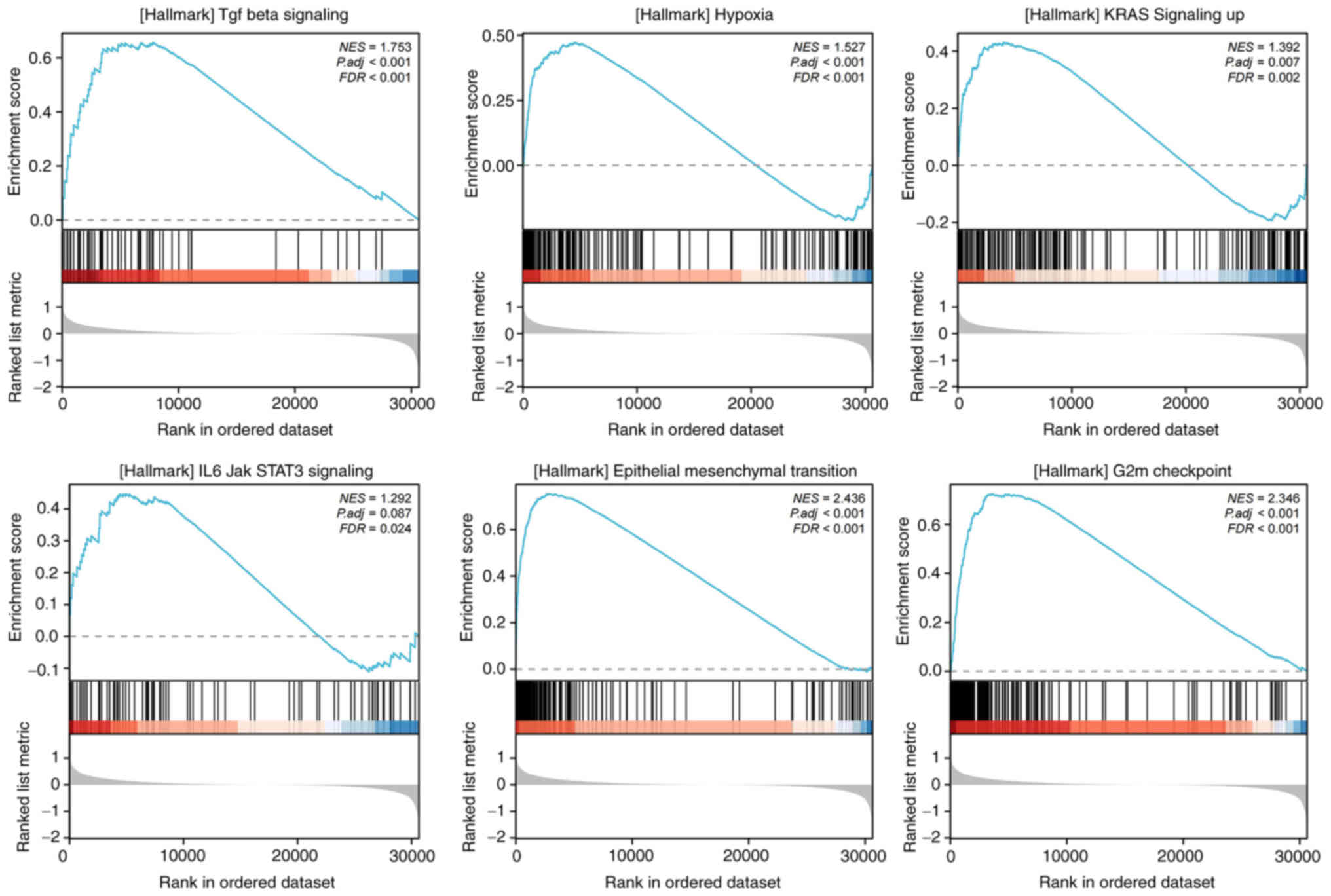
To investigate the downstream pathways and
characteristics, GSEA was performed for the high and low WF score
groups, which demonstrated that several tumor promotive pathways
were evidently activated, namely TGF-β signaling, hypoxia, KRAS
signaling, IL6/JAK/STAT3 signaling, epithelial-mesenchymal
transition (EMT) and G2M checkpoint pathways, each with FDR
<0.25 and normalized enrichment score >0 (Fig. 5). These pathways are known to
promote tumor development and immune dysfunction.
Dysregulation of MRPL35 in HCC
Given the clinical value and potential mechanism
based on the WF score, focus was given to the MRPL35 gene, which
had the highest HR value, based on the assumption that it is the
main factor facilitating tumor progression. In agreement with its
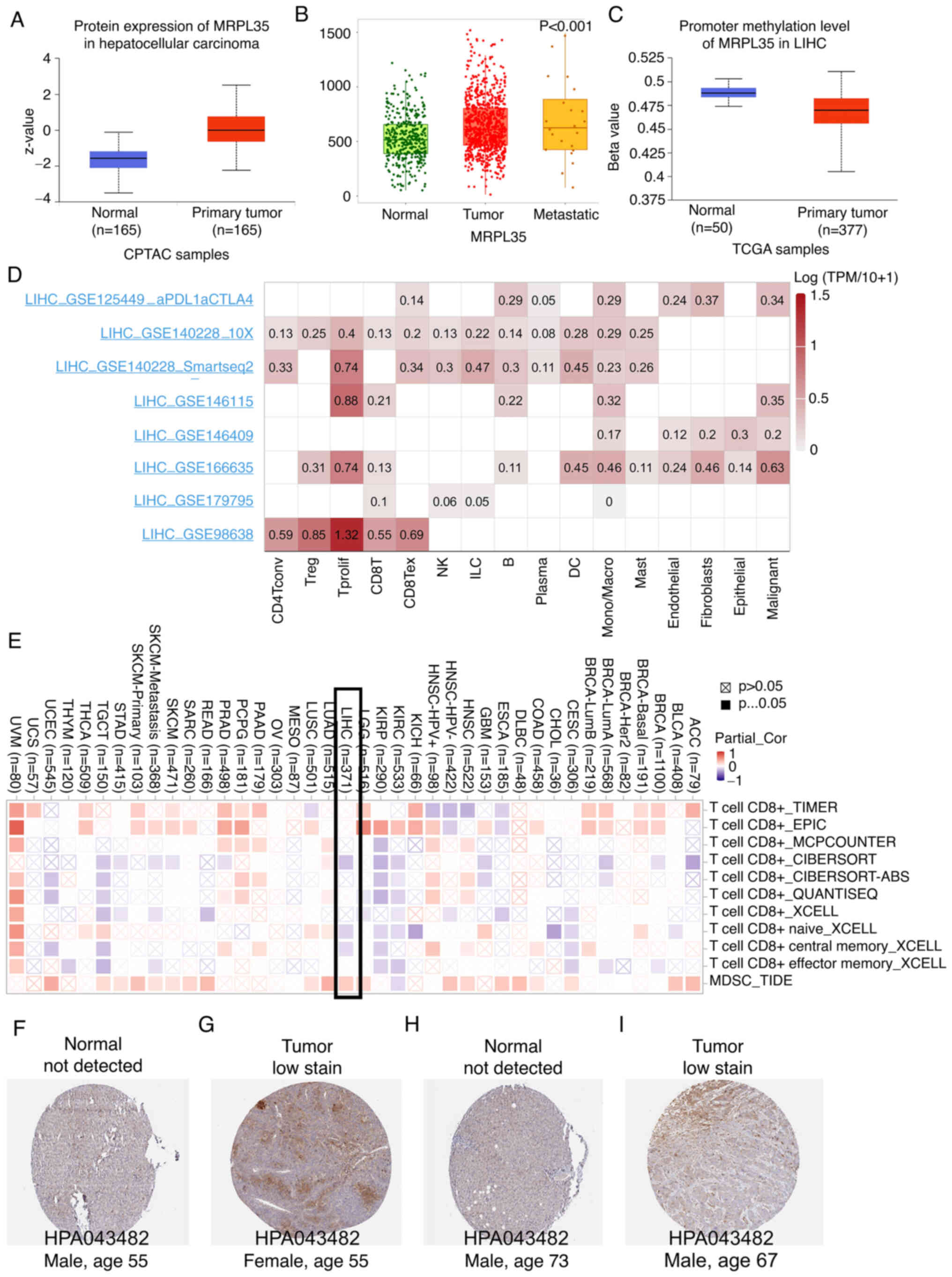
mRNA level, the protein level of MRPL35 in HCC tumor tissue was
higher than that in normal tissue (Fig.
6A), and a significant association with metastasis status was
identified using TNMplot (P<0.05, Fig. 6B). Hypermethylation of a gene
promoter region is known to regulate transcriptional silencing, and
promoter hypomethylation can result in the upregulation of
downstream genes. The analysis performed in the present study found
that the promoter methylation levels of MRPL35 in HCC were
significantly lower compared with those in tumor samples
(P<0.05, Fig. 6C).
Downstream function and association
with clinical characteristics of MRPL35 in HCC
LinkedOmics analysis was performed to evaluate the
potential biological function of elevated MRPL35, and the results
included activation of ‘citrate cycle (TCA cycle)’ and ‘fatty acid
metabolism’ (Fig. S2A and B).
The associations of MRPL35 with clinical
characteristics are shown in Fig.
S2C-G. The results indicate that the level of MRPL35 was
significantly higher in cases with residual tumor status R1 (vs.
R0; Fig. S2E) and female patients
(Fig. S2G), both P<0.05.
Validation of scRNA-seq and
infiltration analysis
When considering whether a gene possesses value as a
therapeutic target or predictive parameter, the precise location of
the gene at the cell level is important. The scRNA-seq method
allows the expression level of a gene to be explored among
different types of cells. In the microenvironment of HCC, MRPL35
was found to be mainly expressed by proliferating T cells and
malignant cells (Fig. 6D).
Similarly, a negative association with CD8+ T cells and
positive association with myeloid-derived suppressor cells (MDSCs)
were observed in HCC (Fig. 6E).
Furthermore, the comparison of MRPL35 IHC between tumor and normal
human tissues supported the oncogenic role of MRPL3 (Fig. 6F-I).
The aforementioned findings suggest that inhibiting
the expression of MRPL35 in tumor cells may impair their
proliferation, migration and immune-escape abilities.
Validation via in vitro
experiments
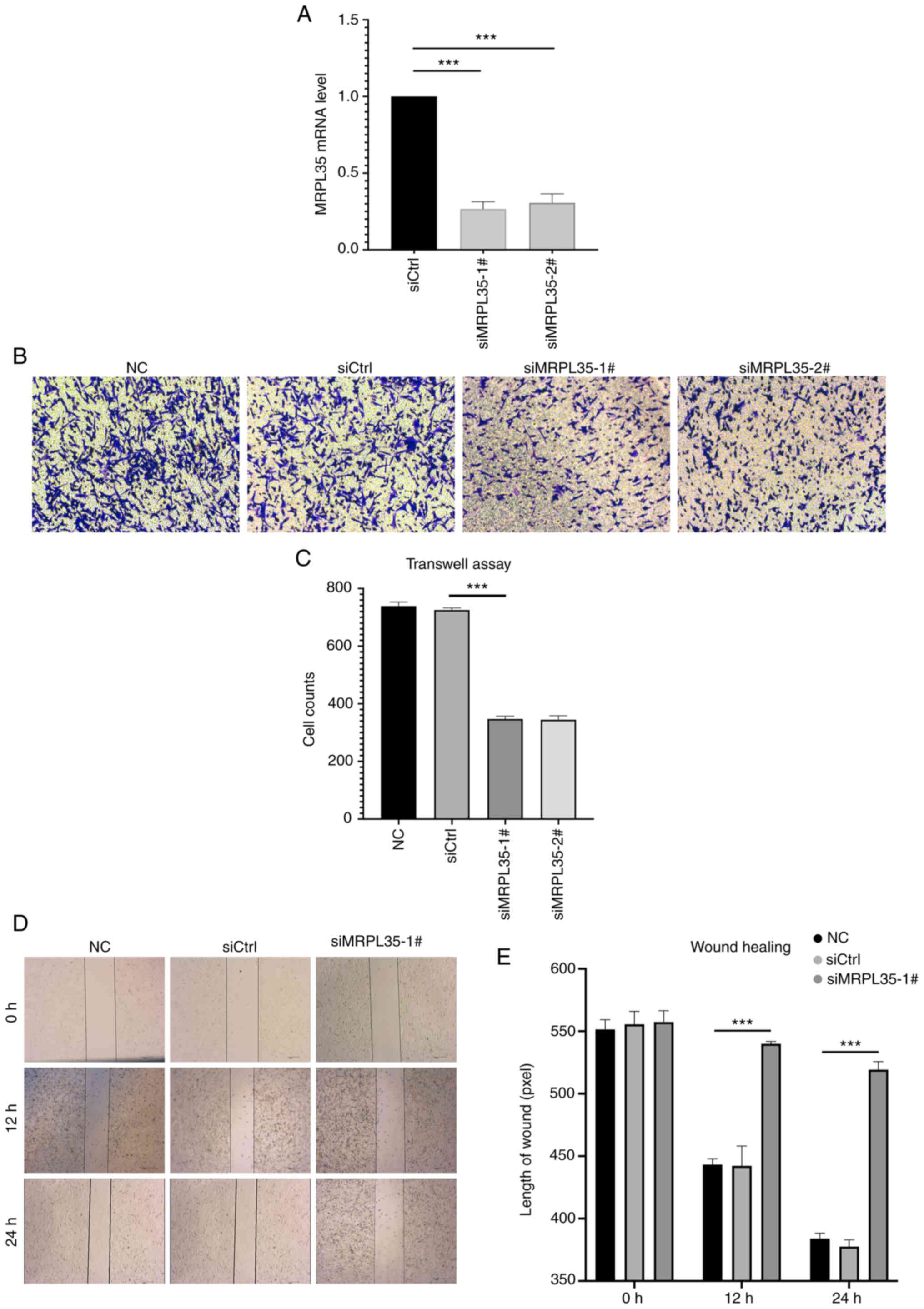
To validate the role of MRPL35 as a risk factor and
its potential as a novel therapeutic target, assays were conducted
to evaluate the effect of its knockdown on the invasion and
migration competence of HepG2 cells. Following the confirmation of
the efficacy of knockdown by the MRPL35 siRNA compared with the
siCtrl (P<0.001; Fig. 7A),
negative control (NC), siCtrl and MRPL35 siRNA groups were
evaluated in vitro. In the Transwell assay, the two siRNAs
against MRPL35 showed a significant inhibitory effect on invasion
after 48 h compared with siCtrl (P<0.001; Fig. 7B and C). In addition, the wound
healing assay indicated that liver cancer cells with MRPL35
knockdown possessed significantly impaired ability to migrate
compared with the controls (P<0.001; Fig. 7D and E). These experimental results
suggest that MRPL35 may contribute to liver cancer metastasis and
the blockade of MRPL35 could be a promising therapeutic target.
Correlation between the response to
drugs and MRPL35 levels
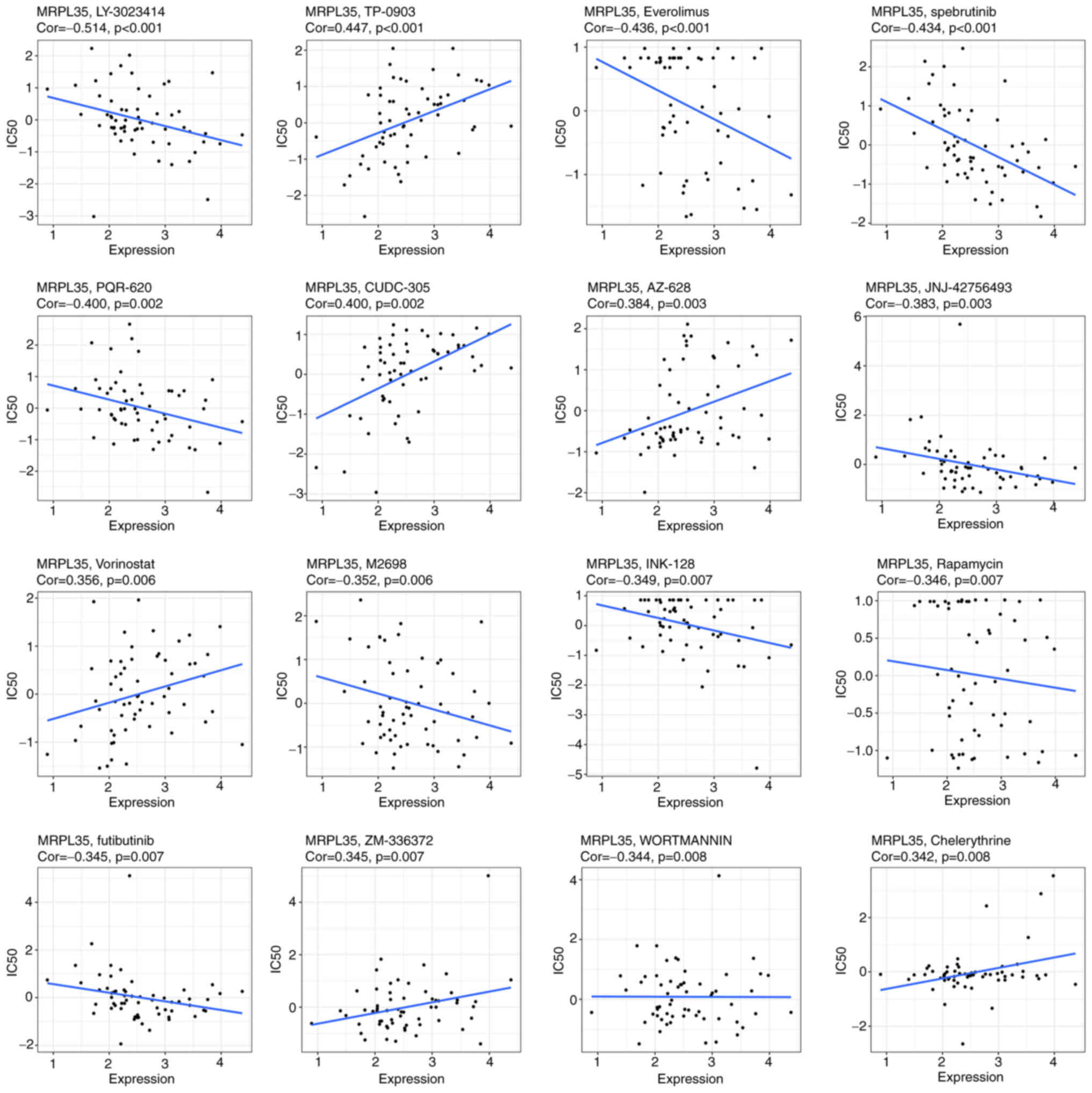
To investigate the potential value of MRPL35 in drug
treatment, an analysis of the correlation between the expression of
MRPL35 and the response to the drug in 60 cell lines was conducted.
The results indicated that MRPL35 was negatively correlated with
the IC50 of 10 in 16 drugs: LY-3023414, Everolimus,
spebrutinib, PQR-620, JNJ-42756493, M2698, INK-128, Rapamycin,
futibutinib and WORTMANNIN, all with a correlation coefficient
<0 and P<0.05 (Fig. 8). This
provides preliminary guidance for strategies that may be applicable
to patients with high MRPL35 levels.
Discussion
The occurrence of metastasis is a fatal sign for
patients with HCC as it indicates the loss of opportunity to
receive surgical treatment (3).
Therefore, studies of biomarkers and promising therapeutic target
are required.
Given the tight physiological and pathological
relationship between FA metabolism and the liver (31), the clinical value of FA
metabolism-related genes in HCC was explored in the present study.
WGCNA identified 957 genes that may be promotive for HCC
metastasis, from which seven were identified to have an association
with FA metabolism and OS. After filtering using the LASSO
algorithm, three core genes remained: PLCD3, MRPL35 and CYP2S1.
Studies have reported the tumor-promotive role of PLCD3 in thyroid
cancer (32) and osteosarcoma
(33), the role of MRPL35 in
gastric (34) and colorectal
(35) cancer, and the role of
CYP2S1 in breast (36) and lung
(37) cancer. Consistent with this,
the group with high WF scores, calculated for the three genes,
showed significantly worse outcomes compared with those with low WF
scores. Moreover, a relatively repressed immune function was
observed in the low WF group, with lower infiltration of
CD8+ cells and higher infiltration of M0 macrophages and
dendritic resting cells. The relationship between FA metabolism and
dysfunctional immunity has been comprehensively reported by Zhang
et al (38), who indicated
that in breast cancer, obesity remodeled the tumor immune
microenvironment by promoting the migration of CD8+ T
cells through a metabolic shift, thereby suppressing immunity. This
metabolic shift was characterized by enhanced FA signaling, leading
to STAT3 activation and the subsequent promotion of FA β oxidation
and concurrent inhibition of glycolysis due to glucose deprivation.
As a result, the ability of CD8+ T cells to restrict
tumor proliferation was impaired. In further investigation of the
downstream mechanisms in the present study, the results of GSEA
indicated that in the high WF group, the TGF-β, hypoxia and
IL6/JAK/STAT3 pathways were significantly activated. The activation
of each of these signaling pathways has been considered as a strong
etiology for tumor development and immune escape in previous
studies (39–41); notably, the IL6/JAK/STAT3 signaling
pathway has been reported to contribute to immunotherapy resistance
(42). These results concur with
the conclusions of Fang et al (43), Povero et al (44) and Zhang et al (38). Furthermore, regarding tumor growth,
the KRAS, G2M checkpoint and EMT pathways, which contribute to
tumor development and metastasis, were also found to be activated
in the high WF score group. Consequently, exploration of the
dysregulation of FA metabolism-related genes in HCC has high
predictive and therapeutic value.
Due to MRPL35 having the highest HR value among the
three genes obtained by the LASSO model, it was chosen for further
bioinformatic analysis and validation experiments. The upregulation
of MRPL35 at the mRNA and protein levels was observed in cancerous
samples compared with normal tissues and in tumors with metastatic
status. Since studies have confirmed that methylation of the
promoter region of a gene can lead to oncogene activation and the
advancement of tumors (45,46), the promoter methylation level of
MRPL35 was compared between normal and tumor samples, which showed
a significantly lower promoter methylation level in tumors. A
relatively high MRPL35 was also be observed in patients with
residual tumor status R1 and in female patients, which is of value
for clinical guidance. scRNA-seq analysis further confirmed that
MRPL35 expression was located in tumor and proliferating T cells.
With regard to the immune microenvironment, MRPL35 was found to
have a negative correlation with CD8+ T cells and a
positive correlation with MDSCs, suggesting the dysfunctional
immunity in the high WF score group was probably induced via
MRPL35-related pathways. Similarly, Yuan et al (34) found that in gastric carcinoma the
knockdown of MRPL35 inhibited cell proliferation and colony
formation, and induced apoptosis. Furthermore, Zhang et al
(35) and Alshabi et al
(47) proposed that MRPL35 plays a
promotive role in colorectal cancer and glioblastoma. The present
analysis investigated MRPL35-related functions in HCC, which
indicated that tumors with a high level of MRPL35 may activate the
energy supply process.
In vitro experiments were performed to verify
the promotive role of MRPL35 in liver cancer cell migration and
invasion, which indicated that after MRPL35 knockdown, these
abilities of HepG2 cells were significantly impaired. These results
not only confirm the risk role of MRPL35 in metastasis but also
support the potential value of MRPL35 as a therapeutic target.
Although the association between the dysregulation
of genes associated with FA metabolism was analyzed and validated
with in vitro experiments, this study has several
limitations. First, despite bioinformatics analysis providing a
preliminary insight into the crucial role of MRPL35 in HCC
progression and drug resistance, future biological experiments
in vivo are necessary to explore this further. Secondly, the
exploration of MRPL35 role in special isotypes of patients also
needs conducted, such as patients with certain genetic mutation and
with resistance to target/immunotherapy drugs.
In conclusion, the present study uncovered the
predictive and therapeutic value of FA metabolism genes in HCC and
suggests the potential of targeting these genes to prevent tumor
initiation, progression and resistance to drugs.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZZ, JS, LW and GT were responsible for study
conception and design. JS and LW developed the methodology. LW and
ZZ performed data collection. JS, LZ and CJ analyzed and
interpreted the data. ZZ and JS wrote, reviewed and revised the
manuscript. GT and LW confirm the authenticity of all the raw data.
All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2019. CA Cancer J Clin. 69:7–34. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Allemani C, Matsuda T, Di Carlo V,
Harewood R, Matz M, Nikšić M, Bonaventure A, Valkov M, Johnson CJ,
Estève J, et al: Global surveillance of trends in cancer survival
2000-14 (CONCORD-3): Analysis of individual records for 37 513 025
patients diagnosed with one of 18 cancers from 322 population-based
registries in 71 countries. Lancet. 391:1023–1075. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Reig M, Forner A, Rimola J, Ferrer-Fàbrega
J, Burrel M, Garcia-Criado Á, Kelley RK, Galle PR, Mazzaferro V,
Salem R, et al: BCLC strategy for prognosis prediction and
treatment recommendation: The 2022 update. J Hepatol. 76:681–693.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Swinnen JV, Brusselmans K and Verhoeven G:
Increased lipogenesis in cancer cells: New players, novel targets.
Curr Opin Clin Nutr Metab Care. 9:358–365. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Santos CR and Schulze A: Lipid metabolism
in cancer. FEBS J. 279:2610–2623. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Medes G, Thomas A and Weinhouse S:
Metabolism of neoplastic tissue. IV. A study of lipid synthesis in
neoplastic tissue slices in vitro. Cancer Res. 13:27–29.
1953.PubMed/NCBI
|
|
7
|
Alannan M, Fayyad-Kazan H, Trézéguet V and
Merched A: Targeting lipid metabolism in liver cancer.
Biochemistry. 59:3951–3964. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Budhu A, Roessler S, Zhao X, Yu Z, Forgues
M, Ji J, Karoly E, Qin LX, Ye QH, Jia HL, et al: Integrated
metabolite and gene expression profiles identify lipid biomarkers
associated with progression of hepatocellular carcinoma and patient
outcomes. Gastroenterology. 144:1066–1075.e1. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sangineto M, Villani R, Cavallone F,
Romano A, Loizzi D and Serviddio G: Lipid metabolism in development
and progression of hepatocellular carcinoma. Cancers (Basel).
12:14192020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kumagai S, Togashi Y, Kamada T, Sugiyama
E, Nishinakamura H, Takeuchi Y, Vitaly K, Itahashi K, Maeda Y,
Matsui S, et al: The PD-1 expression balance between effector and
regulatory T cells predicts the clinical efficacy of PD-1 blockade
therapies. Nat Immunol. 21:1346–1358. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou L, Xia S, Liu Y, Ji Q, Li L, Gao X,
Guo X, Yi X and Chen F: A lipid metabolism-based prognostic risk
model for HBV-related hepatocellular carcinoma. Lipids Health Dis.
22:462023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Feng T, Wu T, Zhang Y, Zhou L, Liu S, Li
L, Li M, Hu E, Wang Q, Fu X, et al: Stemness analysis uncovers that
the peroxisome proliferator-activated receptor signaling pathway
can mediate fatty acid homeostasis in sorafenib-resistant
hepatocellular carcinoma cells. Front Oncol. 12:9126942022.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bensaad K, Favaro E, Lewis CA, Peck B,
Lord S, Collins JM, Pinnick KE, Wigfield S, Buffa FM, Li JL, et al:
Fatty acid uptake and lipid storage induced by HIF-1α contribute to
cell growth and survival after hypoxia-reoxygenation. Cell Rep.
9:349–365. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Murai H, Kodama T, Maesaka K, Tange S,
Motooka D, Suzuki Y, Shigematsu Y, Inamura K, Mise Y, Saiura A, et
al: Multiomics identifies the link between intratumor steatosis and
the exhausted tumor immune microenvironment in hepatocellular
carcinoma. Hepatology. 77:77–91. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gualdoni GA, Mayer KA, Göschl L, Boucheron
N, Ellmeier W and Zlabinger GJ: The AMP analog AICAR modulates the
Treg/Th17 axis through enhancement of fatty acid oxidation. FASEB
J. 30:3800–3809. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen L, Zhou Q, Liu J and Zhang W: CTNNB1
alternation is a potential biomarker for immunotherapy prognosis in
patients with hepatocellular carcinoma. Front Immunol.
12:7595652021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shen S, Khatiwada S, Behary J, Kim R and
Zekry A: Modulation of the gut microbiome to improve clinical
outcomes in hepatocellular carcinoma. Cancers (Basel). 14:20992022.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Goldman MJ, Craft B, Hastie M, Repečka K,
McDade F, Kamath A, Banerjee A, Luo Y, Rogers D, Brooks AN, et al:
Visualizing and interpreting cancer genomics data via the Xena
platform. Nat Biotechnol. 38:675–678. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mootha VK, Lindgren CM, Eriksson KF,
Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E,
Ridderstråle M, Laurila E, et al: PGC-1alpha-responsive genes
involved in oxidative phosphorylation are coordinately
downregulated in human diabetes. Nat Genet. 34:267–273. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bartha Á and Győrffy B: TNMplot.com: A web
tool for the comparison of gene expression in normal, tumor and
metastatic tissues. Int J Mol Sci. 22:26222021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ranstam J and Cook JA: Statistical nugget
LASSO regression. Br J Surg. 105:13482018. View Article : Google Scholar
|
|
24
|
Vasaikar SV, Straub P, Wang J and Zhang B:
LinkedOmics: Analyzing multi-omics data within and across 32 cancer
types. Nucleic Acids Res. 46(D1): D956–D963. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shen W, Song Z, Zhong X, Huang M, Shen D,
Gao P, Qian X, Wang M, He X, Wang T, et al: Sangerbox: A
comprehensive, interaction-friendly clinical bioinformatics
analysis platform. iMeta. 1:e362022. View Article : Google Scholar
|
|
26
|
Newman AM, Liu CL, Green MR, Gentles AJ,
Feng W, Xu Y, Hoang CD, Diehn M and Alizadeh AA: Robust enumeration
of cell subsets from tissue expression profiles. Nat Methods.
12:453–457. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q,
Li B and Liu XS: TIMER2.0 for analysis of tumor-infiltrating immune
cells. Nucleic Acids Res. 48((W1)): W509–W514. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun D, Wang J, Han Y, Dong X, Ge J, Zheng
R, Shi X, Wang B, Li Z, Ren P, et al: TISCH: A comprehensive web
resource enabling interactive single-cell transcriptome
visualization of tumor microenvironment. Nucleic Acids Res. 49(D1):
D1420–D1430. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Reinhold WC, Sunshine M, Liu H, Varma S,
Kohn KW, Morris J, Doroshow J and Pommier Y: CellMiner: A web-based
suite of genomic and pharmacologic tools to explore transcript and
drug patterns in the NCI-60 cell line set. Cancer Res.
72:3499–3511. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jeon YG, Kim YY, Lee G and Kim JB:
Physiological and pathological roles of lipogenesis. Nat Metab.
5:735–759. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lin L, Wen J, Lin B, Chen H, Bhandari A,
Qi Y, Zheng D and Wang O: Phospholipase C Delta 3 inhibits
apoptosis and promotes proliferation, migration, and invasion of
thyroid cancer cells via Hippo pathway. Acta Biochim Biophys Sin
(Shanghai). 53:481–491. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hu H, Yin Y, Jiang B, Feng Z, Cai T and Wu
S: Cuproptosis signature and PLCD3 predicts immune infiltration and
drug responses in osteosarcoma. Front Oncol. 13:11564552023.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yuan L, Li JX, Yang Y, Chen Y, Ma TT,
Liang S, Bu Y, Yu L and Nan Y: Depletion of MRPL35 inhibits gastric
carcinoma cell proliferation by regulating downstream signaling
proteins. World J Gastroenterol. 27:1785–1804. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang L, Lu P, Yan L, Yang L, Wang Y, Chen
J, Dai J, Li Y, Kang Z, Bai T, et al: MRPL35 is up-regulated in
colorectal cancer and regulates colorectal cancer cell growth and
apoptosis. Am J Pathol. 189:1105–1120. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Aiyappa-Maudsley R, Storr SJ, Rakha EA,
Green AR, Ellis IO and Martin SG: CYP2S1 and CYP2W1 expression is
associated with patient survival in breast cancer. J Pathol Clin
Res. 8:550–566. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Guo H, Zeng B, Wang L, Ge C, Zuo X, Li Y,
Ding W, Deng L, Zhang J, Qian X, et al: Knockdown CYP2S1 inhibits
lung cancer cells proliferation and migration. Cancer Biomark.
32:531–539. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang C, Yue C, Herrmann A, Song J,
Egelston C, Wang T, Zhang Z, Li W, Lee H, Aftabizadeh M, et al:
STAT3 activation-induced fatty acid oxidation in CD8+ T
effector cells is critical for obesity-promoted breast tumor
growth. Cell Metab. 31:148–161.e5. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Datta J, Dai X, Bianchi A, De Castro Silva
I, Mehra S, Garrido VT, Lamichhane P, Singh SP, Zhou Z, Dosch AR,
et al: Combined MEK and STAT3 inhibition uncovers stromal
plasticity by enriching for cancer-associated fibroblasts with
mesenchymal stem cell-like features to overcome immunotherapy
resistance in pancreatic cancer. Gastroenterology. 163:1593–1612.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xie F, Zhou X, Su P, Li H, Tu Y, Du J, Pan
C, Wei X, Zheng M, Jin K, et al: Breast cancer cell-derived
extracellular vesicles promote CD8+ T cell exhaustion
via TGF-β type II receptor signaling. Nat Commun. 13:44612022.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bourayou E and Golub R: Signaling pathways
tuning innate lymphoid cell response to hepatocellular carcinoma.
Front Immunol. 13:8469232022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Johnson DE, O'Keefe RA and Grandis JR:
Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev
Clin Oncol. 15:234–248. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fang Y, Zhang Q, Lv C, Guo Y, He Y, Guo P,
Wei Z, Xia Y and Dai Y: Mitochondrial fusion induced by
transforming growth factor-β1 serves as a switch that governs the
metabolic reprogramming during differentiation of regulatory T
cells. Redox Biol. 62:1027092023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Povero D, Chen Y, Johnson SM, McMahon CE,
Pan M, Bao H, Petterson XT, Blake E, Lauer KP, O'Brien DR, et al:
HILPDA promotes NASH-driven HCC development by restraining
intracellular fatty acid flux in hypoxia. J Hepatol. 79:378–393.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Piao GH, Piao WH, He Y, Zhang HH, Wang GQ
and Piao Z: Hyper-methylation of RIZ1 tumor suppressor gene is
involved in the early tumorigenesis of hepatocellular carcinoma.
Histol Histopathol. 23:1171–1175. 2008.PubMed/NCBI
|
|
46
|
Chen G, Fan X, Li Y, He L, Wang S, Dai Y,
Bin C, Zhou D and Lin H: Promoter aberrant methylation status of
ADRA1A is associated with hepatocellular carcinoma. Epigenetics.
15:684–701. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Alshabi AM, Vastrad B, Shaikh IA and
Vastrad C: Identification of crucial candidate genes and pathways
in glioblastoma multiform by bioinformatics analysis. Biomolecules.
9:2012019. View Article : Google Scholar : PubMed/NCBI
|