Introduction
Breast cancer is one of the most common types of
cancer in women worldwide. Over the last few decades, various
advanced diagnostic methods and treatments, such as novel drugs and
targeted therapies, have been developed; however, patients are
still subject to physical and mental stress (1). Additionally, recurrence and metastasis
remain critical causes of poor prognosis in patients with breast
cancer (2). According to reported
studies, breast cancer stem cells (BCSCs) are closely linked to
these problems as BCSCs can trigger tumor progression, recurrence
and metastasis through their specific characteristics, including
the ability to escape immune surveillance and resistance to drugs
and radiation (3–6). Additionally, BCSCs possess
characteristic stem cell abilities, such as self-renewal and
differentiation into heterogeneous common cancer cells (7). The CD24−/CD44+
population, a representative CSC phenotype in human breast cancer
cells, exhibits enhanced survival, proliferation, clonogenicity,
drug efflux, migration and invasion capacities (8,9). This
population also exhibits gene expression patterns that differ from
those of common cells, not only stemness-associated genes, such as
Oct4, Sox2, c-Myc and Klf transcription factor 4, but also typical
CSC-associated genes are upregulated (10–13).
Hence, the need to find new BCSC targets is urgent to maximize the
potential of current therapies.
The complement cascade is a multifunctional innate
immune mechanism that provides an effective defense system against
pathogens (14). This system can be
activated by three pathways: The classical, mannose-binding lectin
and alternative pathways (15).
Various components produced in the complement activation process,
such as C3a, C5a and C3b, can interact with other immune cells. C3a
and C5a, known as anaphylatoxins, stimulate the inflammatory
response by recruiting neutrophils and monocytes (14). Additionally, C3b and C4b can
inactivate toxic particles by binding the pathogen membrane and
opsonizing pathogens for phagocytosis by macrophages (16). By contrast, complement regulatory
proteins (CRPs) can prevent the activation of immune cells and the
death of pathogens. Among CRPs, complement factor H (CFH) acts as a
major regulator of this process through its inhibitory effects on
multiple steps, including the production of C3Bb, C3b and C3bB
(17,18). CFH is mostly expressed in healthy
liver cells but is also upregulated in several types of cancer
cells, including liver, lung, ovarian and breast cancer cells
(19–23). CFH is mainly present in the
extracellular space and blood, due to its secretion from cells, and
the cytoplasm and cytoplasmic membrane. According to previous
studies, CFH enhances cancer progression and tumorigenesis by
binding C3 in the cytoplasm of lung cancer cells, and
downregulation of CHF in lung cancer cells suppresses tumor growth.
(20,21) CFH also regulates the stemness of
liver cancer cells via late SV40 factor (19). Notably, breast cancer cells exhibit
resistance to complement-mediated lysis based on their high
expression levels of the CRPs, CD55 and CD59 (24). However, little is known about the
direct association between breast cancer cells and CFH. Therefore,
the present study aimed to elucidate the impact of CFH
downregulation on radioresistance and cancer stemness in MDA-MB-231
human breast cancer cells.
Materials and methods
Survival analysis
The Cancer Target Gene Screening database
(http://ctgs.biohackers.net) was used to
analyze the significance between the survival period of patients
with breast cancer and gene expression levels [dataset: Molecular
Taxonomy of Breast Cancer Consortium (METABRIC), http://ctgs.biohackers.net/datasets/].
All primary data were deposited at The European
Genome-phenome-Archive (EGA; accession no. EGAS00001001753) and
were published by Pereira et al (25) in 2016. Data from the METABRIC
dataset, including information for 1,980 patients with breast
cancer, were analyzed. High and low gene expression was determined
by the median. P<0.05 was considered to indicate a statistically
significant difference. The survival and survminer packages in R
(version 4.2; http://www.r-project.org/) were used for conducting
survival analysis, which was validated using the Cox proportional
hazards model.
Cell culture
MDA-MB-231 (human breast adenocarcinoma cell line)
and Hs578T (human breast carcinoma cell line) cells were maintained
in Dulbecco's Modified Eagle's Medium (DMEM; cat. no. 010-013-CV;
Corning, Inc.) supplemented with 10% fetal bovine serum (FBS; cat.
no. 35-015-CV; Corning, Inc.) and 1% Antibiotic-Antimycotic (cat.
no. 15240-062; Gibco; Thermo Fisher Scientific, Inc.) at 37°C in a
humidified incubator with 5% CO2. T47D, ZR751 and BT549
cells (human breast carcinoma cell lines) were maintained in
RPMI-1640 medium (cat. no. 10-040-CV; Corning, Inc.) supplemented
with 10% FBS and 1% Antibiotic-Antimycotic at 37°C in a humidified
incubator with 5% CO2. MCF7 cells (human breast
adenocarcinoma cell line) were maintained in Minimum Essential
Medium Eagle (LM007-54; Welgene, Inc.) supplemented with 0.01 mg/ml
human insulin (cat. no. I9278; MilliporeSigma; Merck KGaA), 10% FBS
and 1% Antibiotic-Antimycotic at 37°C in a humidified incubator
with 5% CO2.
Reverse transcription-quantitative PCR
(RT-qPCR)
RNA was isolated from cells using a Ribospin RNA
purification kit (cat. no. 304-150; GeneAll Biotechnology Co.,
Ltd.), and cDNA was synthesized using a Thermal Cycler Dice PCR
machine (Takara Bio, Inc.) with PrimeScript™ RT Master Mix (cat.
no. RR036A; Takara Bio, Inc.), according to the manufacturer's
instructions. qPCR was performed using a CFX96 Optical Reaction
module (Bio-Rad Laboratories, Inc.) with TB Green Premix (cat. no.
RR420A; Takara Bio, Inc.) according to the manufacturer's
instructions. Quantification was performed by calculating the ΔCq
value of the target gene relative to the Cq value of b-actin, used
as a loading control (26).
Information regarding the primers used for the RT-qPCR is provided
in Table SI.
Short hairpin (sh)RNA knockdown
The vector used for shRNA knockdown was based on
pLKO.1 and utilized the second-generation system. 293T cells were
transfected with a mixture containing non-silencing (NS; negative
control) or CFH-targeting shRNA (6.5 µg), viral packaging DNA (5 µg
of psPAX2 and 2 µg of pMD2G) and 30 µl of X-tremeGENE 9 DNA
Transfection Reagent (cat. no. 06-365-809-001; Roche Diagnostics)
in 500 µl of Opti-MEM (#31985-070, Gibco), for 48 h at 37°C in a
humidified incubator with 5% CO2. The resulting
supernatant was filtered using a 0.45-µm syringe and mixed with
DMEM (2 ml supernatant and 8 ml DMEM) and Polybrene
infection/transfection reagent (cat. no. TR-1003-G; MilliporeSigma;
Merck KGaA). Next, MDA-MB-231 cells were infected with the viral
mixture for 24 h at 37°C in a humidified incubator with 5%
CO2. The infected cells were washed with 1X DPBS and
maintained in complete DMEM. After 48 h, the infected cells were
incubated with selection medium containing 1 µg puromycin in 10 ml
complete DMEM for 72 h at 37°C in a humidified incubator with 5%
CO2. The cells were cultured in complete DMEM with a
concentration of 0.5 µg/ml puromycin for maintenance after
selection. Information regarding the shRNA used for CFH knockdown
is provided in Table SII.
Western blotting
Proteins were extracted from cells using ProEX™ CETi
lysis buffer (cat. no. TLP-121CETi; TransLab) and quantified using
Protein Assay Dye Reagent Concentrate (cat. no. 5000006; Bio-Rad
Laboratories, Inc.). Quantified proteins (20 µg) were mixed with 4X
sample buffer (cat. no. B0007; Thermo Fisher Scientific, Inc.) and
loaded on Bolt™ 4–12% Bis-Tris precast gels (cat. nos. NW04120BOX
and NW04122BOX; Thermo Fisher Scientific, Inc.) for SDS-PAGE and
then transferred onto nitrocellulose membranes (cat. no. 10600002;
GE Healthcare). The membranes were blocked with 5% skim milk (cat.
no. 232100; Difco; BD Biosciences) in TBST (0.05% Tween 20) at room
temperature for 1 h and then incubated with primary antibodies at
4°C overnight. Next, the membranes were washed with TBST three
times and incubated with secondary antibodies at room temperature
for 1 h. After the membranes were washed with TBST a further three
times, they were treated with ECL Select Western blot detection
reagent (cat. no. RPN2235; Cytivia). Target proteins were detected
using a Fusion FX5 image analyzer (Vilber Lourmat). The normalized
relative protein levels (compared with the respective actin bands)
were calculated using ImageJ software (version 1.53k; National
Institutes of Health). Information regarding the antibodies used
for western blotting is provided in Table SIII.
Cytotoxicity assay
Cells were treated with 5 µM
2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein,
acetoxymethyl ester (BCECF, AM; cat. no. B1150; Thermo Fisher
Scientific, Inc.) for 30 min at 37°C and subsequently harvested in
a 1 ml tube. The cells were then washed with sodium veronal buffer
(cat. no. B102; Complement Technology, Inc.) and treated with
normal human serum (NHS; cat. no. NHS; Complement Technology, Inc.)
or 1% SDS (cat. no. 28312; Thermo Fisher Scientific, Inc.) in
veronal buffer (as a positive control) for 30 min at 37°C. Next,
the sample supernatants were transferred to black 96-well plates
and the fluorescence was measured (excitation, 485 nm; emission,
538 nm). The cytotoxicity was calculated using the following
formula: Cytotoxicity=[(A-B)/(C-B)] ×100%. Where A is the BCECF
released from the samples, B is the spontaneous BCECF and C is the
BCECF released from the SDS positive control.
ELISA
ELISAs were performed on the cells using the Human
Complement Component C5a DuoSet ELISA kit (cat. no. DY2037; R&D
Systems, Inc.) according to the manufacturer's protocols. The
optical density of the samples at 450 nm was measured using a
microtiter plate spectrophotometer (Beckman Coulter, Inc.).
Irradiation
Cells were exposed to γ-rays using a
137Cs-ray source (Eckert & Ziegler) at a dose rate
of 2.6 Gy/min. Following irradiation at doses of 0, 2, 4 and 6 Gy,
the cells were incubated for 24 h at 37°C in a humidified incubator
with 5% CO2.
Clonogenic cell survival assay
Cells were irradiated as aforementioned. The cells
were then seeded in 6-well culture plates at a density of 500
cells/well for 10 days at 37°C, in a humidified incubator with 5%
CO2. Following incubation, the colonies were fixed with
2 ml ice-cold methanol for 10 min at 4°C, and were stained with a
1% crystal violet solution in methanol at room temperature for 1 h.
After washing with distilled water, the colonies, which contained
at least 50 cells, were counted manually.
Mammosphere formation assay
Cells were irradiated as aforementioned. Following
exposure to ionizing radiation, the non-irradiated or irradiated
cells were detached from the plate with TrypLE Express (cat. no.
12605-010; Gibco; Thermo Fisher Scientific, Inc.) and reseeded at a
density of 100 cells/well in ultra-low-attachment surface spheroid
96-well microplates (cat. no. 4520; Corning, Inc.) in MammoCult
Human Medium (cat. no. 05620; Stemcell Technologies, Inc.)
supplemented with a heparin solution and hydrocortisone stock
solution (cat. nos. 07980 and 07925, respectively; Stemcell
Technologies, Inc.). The suspended cells were incubated for 7–14
days at 37°C in a humidified incubator with 5% CO2.
Mammosphere formation was monitored, and the sphere size was
measured using the following formula: Sphere size=(long diameter +
short diameter)/2.
Cell migration and invasion
assays
Cells were irradiated as aforementioned. Transwell
inserts (8-µm pore size; cat. no. 353097; Falcon; Corning Life
Sciences) were coated with a collagen solution (cat. no.
C2249-20ML; Sigma-Aldrich; Merck KGaA) or Matrigel (for the
invasion assay; cat. no. 354230; Corning, Inc.) and placed into
24-well microplates containing 800 µl complete medium and 10% FBS.
The cells were suspended in serum-free medium at a density of
5×104 cells/150 µl and seeded on each membrane insert.
After incubation for 20 h at 37°C in a humidified incubator with 5%
CO2, the inserts were washed with PBS and fixed with
ice-cold methanol at 4°C for 10 min. Then, the inserts were stained
with a 1% crystal violet solution in methanol at room temperature
for 1 h. After washing with distilled water, cells that had not
migrated to the upper membrane of the insert were removed using a
cotton swab. The migrated or invaded cells were observed using an
EVOS XL Cell Imaging System (Thermo Fisher Scientific, Inc.) at
×100 magnification.
Flow cytometry
Cells were irradiated as aforementioned. The cells
were then washed with 1X DPBS, harvested by centrifuging at 1,200
rpm (335 × g) for 3 min at 4°C and then fixed with 70% cold ethanol
for 1 h at 4°C. The fixed cells were washed with 1X DPBS and
stained with PI solution for 30 min at 4°C. The stained cells were
then washed and resuspended in 500 µl BD FACS™ Sheath Fluid (cat.
no. 342003; BD biosciences), and the cell cycle was analyzed using
a BD FACSAria Cell Sorter Special Order Research Product (BD
Biosciences) and BD FACSDiva Software (version 6.1.3; BD
Biosciences).
For the Annexin V apoptosis assay, the fixed cells
were washed with 1X DPBS and stained with FITC Annexin V from a
FITC Annexin V Apoptosis Detection Kit I (cat. no. 556547; BD
Biosciences) in 1X binding buffer (from the aforementioned kit).
The cells labeled with FITC Annexin V were stained with a PI
solution and analyzed using BD FACSAria Cell Sorter Special Order
Research Product (BD Biosciences) and BD FACSDiva Software.
Gene expression profiling and function
analysis
Total RNA was isolated from NS and CFH knockdown
MDA-MB-231 cells using a Ribospin RNA purification kit. A total of
six samples (three NS and three CFH knockdown cell lines) were
chosen for microarray analysis to predict the association between
downregulated CFH gene expression and breast cancer. cDNA was
synthesized using the GeneChip WT Amplification kit (cat. no.
902230; Thermo Fisher Scientific, Inc.), as described by the
manufacturer. The Affymetrix GeneChip Human Gene 2.0 ST Array
platform was used for microarray analysis and scanned on a GCS3000
Scanner (Affymetrix; Thermo Fisher Scientific, Inc.). The following
filtering criteria were used to define differentially expressed
genes (DEGs): A change in gene expression by >2-fold and
P<0.01. The QuickGO database was used to classify categories
enriched in the DEGs with comparable functions (GO analysis,
https://www.ebi.ac.uk/QuickGO/annotations; TOPPFUN
analysis, https://toppgene.cchmc.org/enrichment.jsp; DAVID
analysis, http://david.ncifcrf.gov/). The raw
dataset has been uploaded to the ArrayExpress database (accession
no. E-MTAB-13327).
Small interfering RNA (siRNA)
knockdown
MDA-MB-231 cells or shCFH #1 cells in 6-well plates
containing 2 ml Opti-MEM (cat. no. 31985-070; Gibco; Thermo Fisher
Scientific, Inc.) were transfected with a mixture containing 20 nM
universal negative control (NC) siRNA or 20 nM erythrocyte membrane
protein band 4.1-like 3 (EPB41L3)-targeting siRNA (TriFECTa DsiRNA
kit; cat. no. hs.Ri.EPB41L3.13; Integrated DNA Technologies, Inc.)
and 4 µl jetPRIME Transfection Reagent (cat. no. 101000046;
Polyplus-transfection SA) in 200 µl of jetPRIME buffer
(Polyplus-transfection SA) for 24 h at 37°C in a humidified
incubator with 5% CO2. Subsequent experiments were
performed after 24 h incubation. Information regarding the siRNAs
used for EPB41L3 knockdown is provided in Table SIV.
Statistical analysis
All experiments were repeated at least three times.
GraphPad Prism 8 software (Dotmatics) was used for all statistical
analyses. Data are presented as the mean ± standard deviation.
Analyses were performed with unpaired Student's t-test, or one-way
or two-way ANOVA followed by Tukey's test. P<0.05 was considered
to indicate a statistically significant difference.
Results
CFH downregulation sensitizes
MDA-MB-231 cells against complement-dependent cytotoxicity
(CDC)
The CFH expression levels in various breast cancer
cell lines were investigated to evaluate the basal levels in
luminal and triple-negative breast cancer (TNBC) subtypes. Notably,
the TNBC group exhibited significantly higher CFH expression than
the luminal group (Fig. 1A). Next,
stable CFH knockdown cells were established through shRNA viral
transfection into TNBC MDA-MB-231 cells (cells that are highly
aggressive with high metastatic potential). It was confirmed that
CFH expression was downregulated in MDA-MB-231 cells transfected
with shCFH #1 and #5 (Figs. 1B and
S1). To examine the effect of CFH
downregulation on the inhibitory function of NHS containing
complement proteins, cytotoxicity assays and a C5a ELISA were
performed. The cytotoxicity and level of C5a release induced by NHS
were significantly increased in cells transfected with shCFH #1 and
#5 compared with NS cells (Fig. 1C and
D). Therefore, these data demonstrated that CFH knockdown
inhibited the protection against immune attack by complement
proteins in MDA-MB-231 cells.
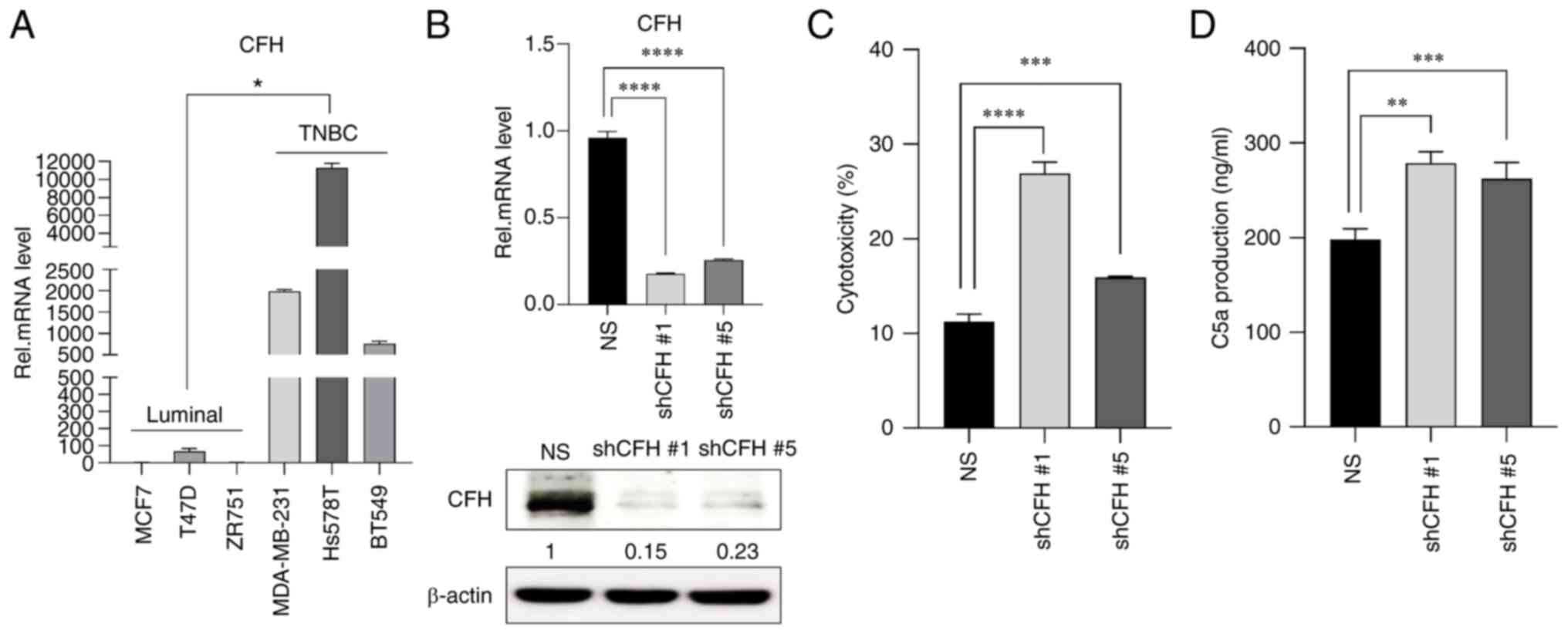 | Figure 1.CFH knockdown in MDA-MB-231 reduces
the ability to defend against CDC. (A) CFH mRNA expression levels
in luminal (MCF7, T47D and ZR751) and TNBC (MDA-MB-231, Hs578T and
BT549) human breast cancer cell lines were analyzed by RT-qPCR. The
results are presented as the mean ± SD. *P<0.05 luminal vs.
TNBC, using the unpaired Student's t test. (B) MDA-MB-231 cells
were transfected with NS, shCFH #1 or shCFH #5, and downregulated
expression was measured by RT-qPCR and western blotting. The
expression levels were semi-quantified and defined beneath each
protein band. The expression levels were calculated as the
target/b-actin ratio for each lane and the ratio values for the
experimental group were normalized by setting the ratio value of
the negative control group to 1. (C) A CDC assay was performed by
measuring 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein,
acetoxymethyl ester release induced by normal human serum from NS
and shCFH transfected cells. (D) C5a production was measured using
a commercially available C5a detection ELISA kit. The results are
presented as the mean ± SD. *P<0.05, **P<0.01, ***P<0.001,
****P<0.0001, using one-way ANOVA and Tukey's test. CDC,
complement-dependent cytotoxicity; CFH, complement factor H; NS,
non-silencing shRNA; Rel, relative; RT-qPCR, reverse
transcription-quantitative PCR; sh(RNA), short hairpin (RNA); TNBC,
triple-negative breast cancer. |
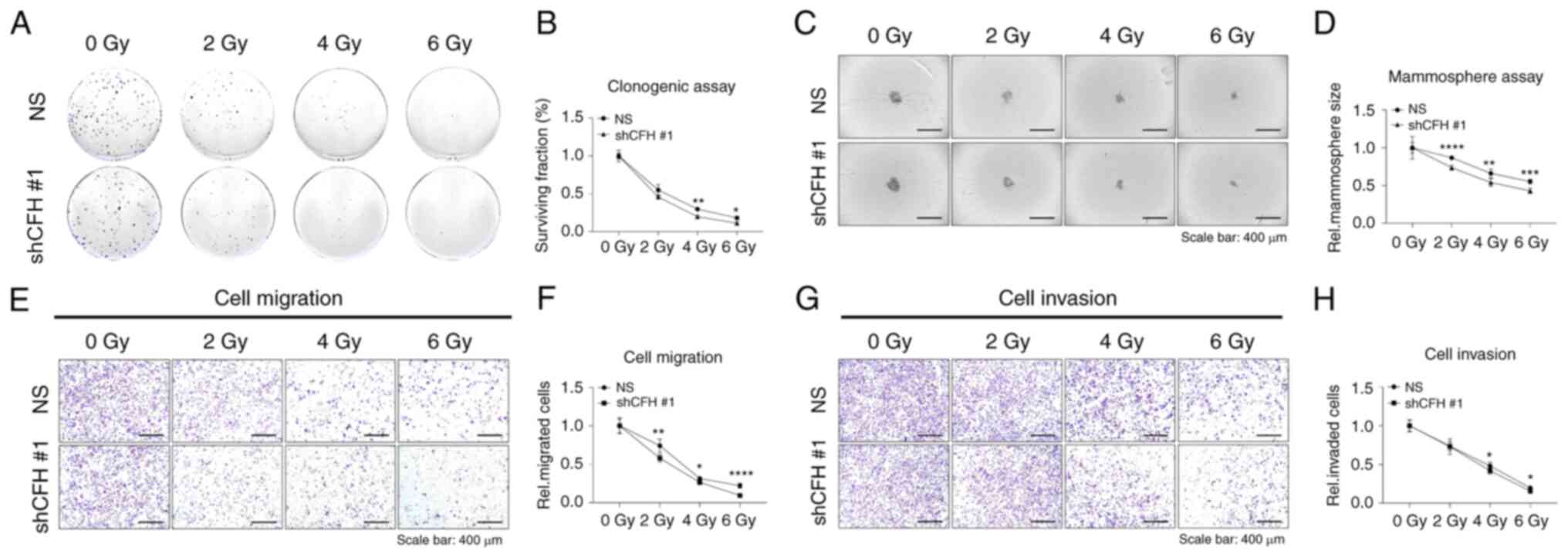
CFH downregulation decreases cell
survival and suppresses motility by decreasing radioresistance and
promotes apoptosis through G2/M phase cell cycle arrest
Experiments were performed to investigate whether
CFH downregulation could decrease the radioresistance and survival
of MDA-MB-231 cells. Irradiated cells were seeded onto 6-well
plates and incubated for 7–10 days. Notably, the number of shCFH #1
cell colonies following irradiation was significantly reduced
compared with that of NS cells in a dose-dependent manner (Fig. 2A and B). Next, to examine whether
CFH could affect anchorage-independent properties, irradiated cells
were cultured on 96-well plates for 7–10 days to develop
mammospheres. The size of the CFH mammospheres was decreased
compared with that of the NS cell mammospheres (Fig. 2C and D). Furthermore, the effect of
CFH knockdown on cell motility was evaluated through migration and
invasion assays. Notably, the relative migration and invasion of
irradiated shCFH #1 cells were decreased compared with those of
irradiated NS cells (Fig. 2E-H).
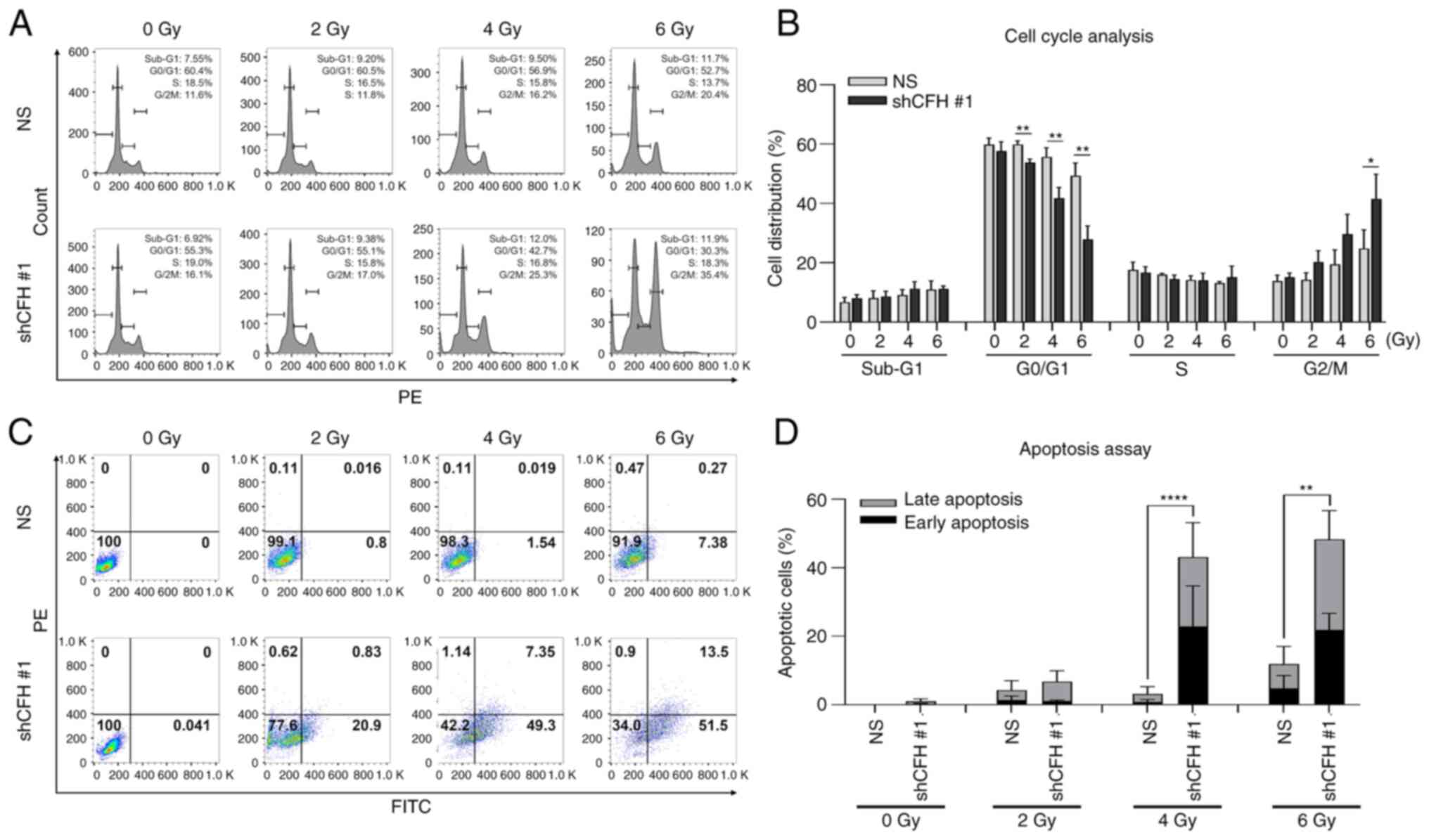
Additionally, cell cycle analysis and an apoptosis assay using flow
cytometry were performed following cell irradiation. The ratio of
shCFH #1 cells arrested in G2/M phase increased with irradiation
(Fig. 3A and B), and these cells
showed a significantly increased apoptotic cell ratio compared with
that of the NS cells (Fig. 3C and
D). These results indicated that CFH downregulation suppressed
cell survival and motility and promoted apoptosis through G2/M
phase cell cycle arrest by sensitizing breast cancer cells to
radiation.
CFH downregulation alters the
expression levels of multiple genes associated with BCSCs
The specific characteristics of CSCs might be
maintained through their enhanced survival, motility and
radioresistance through gene expression regulation. To explore
whether CFH can regulate the expression of various genes associated
with BCSCs, RT-qPCR was performed to measure the levels of BCSC
marker genes. Compared with the NS cells, the shCFH #1 cells showed
increased CD24 expression and decreased CD44 and CD133 expression,
indicating the loss of the typical phenotype for BSCS cells, such
as having a CD24low/CD44high population
(Fig. 4A). Additionally, the
expression levels of Oct4, Sox2, c-Myc and epithelial cellular
adhesion molecule (EpCAM) were decreased in shCFH #1 cells compared
with NS cells. Moreover, it was found that the differences in
protein levels of these genes were similar to the changes in mRNA
levels (Figs. 4B and S2). These results demonstrated that CFH
knockdown decreased the CSC properties related to radioresistance
by regulating gene expression in breast cancer cells.
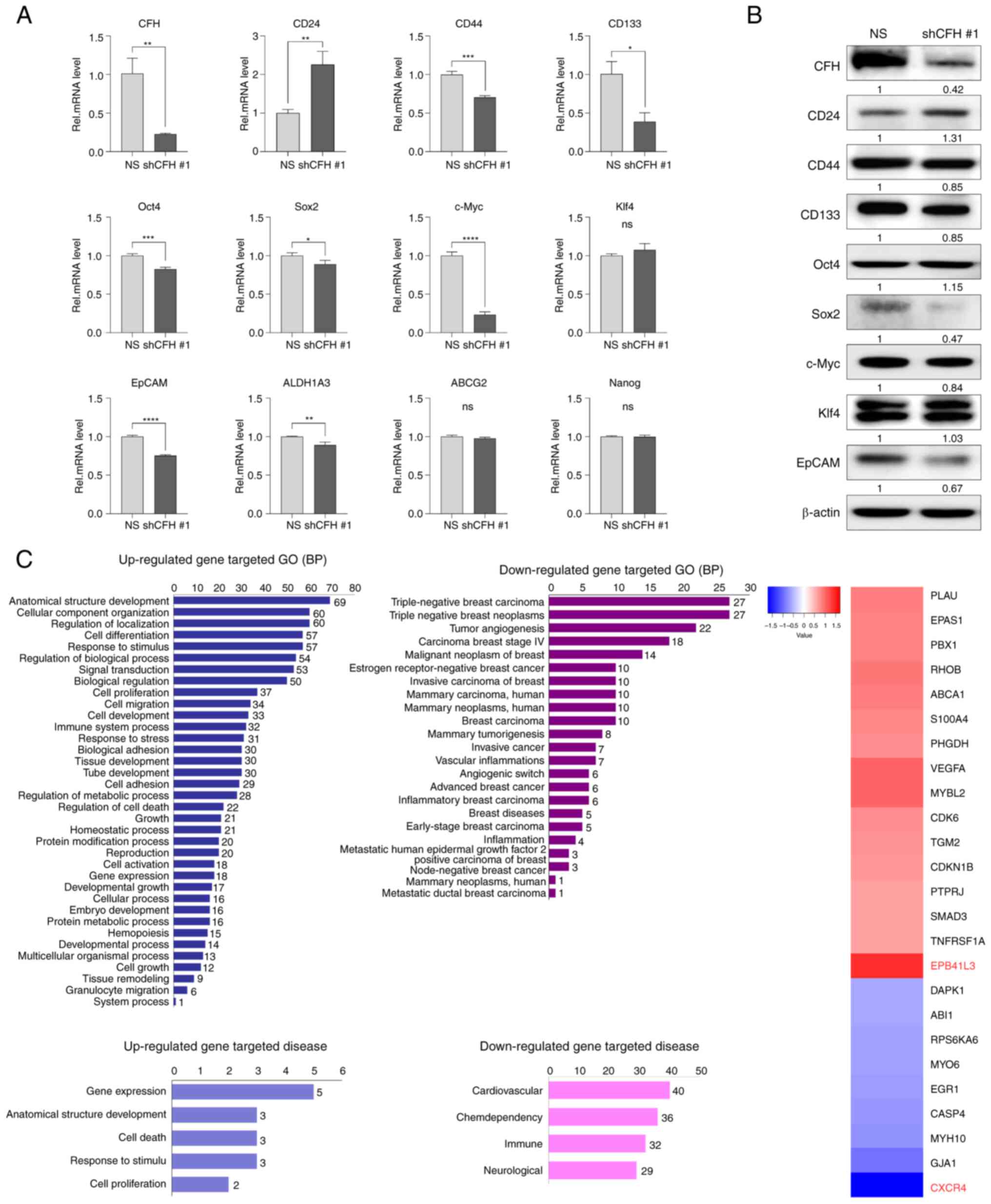 | Figure 4.CFH knockdown controls the expression
patterns of BCSC-associated genes. The expression levels of
BCSC-specific and stemness-related genes were analyzed by (A)
reverse transcription-quantitative PCR and (B) western blotting,
following irradiation and shRNA transfection. The expression levels
were semi-quantified and defined beneath each protein band. The
expression levels were calculated as the target/b-actin ratio for
each lane and the ratio values for the experimental group were
normalized by setting the ratio value of the negative control group
to 1. (C) Gene expression profiling identified several pro- or
antitumor genes as major candidate downstream factors of the CFH
signaling mechanism, through BP, disease and heatmap analyses using
bioinformatics tools. The results are presented as the mean ± SD.
*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, using an
unpaired Student's t test. ABCG2, ATP biding cassette subfamily G
member 2; ALDH1A3, aldehyde dehydrogenase 1 family member A3; BCSC,
breast cancer stem cell; BP, biological process; CFH, complement
factor H; EpCAM, epithelial cellular adhesion molecule; GO, Gene
Ontology; Klf4, Klf transcription factor 4; NS (uppercase),
non-silencing shRNA; ns (lowercase), not significant; Rel,
relative; sh(RNA), short hairpin (RNA). |
Next, microarray expression profiling was performed
to identify genes regulated by decreased CFH expression (Fig. 4C). It was found that the DEGs were
associated with motility, growth, signal transduction and, in
particular, breast cancer. Based on these analyses, 25 genes were
selected as candidate hub genes that may interact with numerous
other genes. As such, the biological functions, regulatory
mechanisms and signaling pathways of the 25 genes in cancer cells
were further investigated and the diverse characteristics of the 25
genes in cancer were verified through bioinformatic analysis. Then,
seven genes that had a high correlation with survivability,
motility, radioresistance and cancer stemness were selected. The
selected seven genes were endothelial PAS domain protein 1,
cyclin-dependent kinase inhibitor 1B, protein tyrosine phosphatase
receptor type J and EPB41L3, which showed increased expression due
to CFH downregulation, and myosin VI, gap junction protein α1 and
C-X-C motif chemokine receptor 4 (CXCR4), which exhibited decreased
expression. Taken together, these microarray findings indicated
that several genes were regulated by CFH knockdown and allowed for
the selection of target genes. A list of gene ontology terms is
provided in Table SV, Table SVI, Table VII, Table VIII.
CFH downregulation attenuates CSC
characteristics via the regulation of MAPK signaling by
upregulating EPB41L3 in MDA-MB-231 breast cancer cells
A total of seven notable genes that were
differentially expressed in NS and shCFH #1 cells were identified
(Fig. 5A). Of these genes, CXCR4 is
a tumor promoter and EPB41L3 plays a role in tumor suppression.
CXCR4, a widely expressed oncogenic factor in malignant solid
tumors, is correlated with cancer progression, invasion, DNA
repair, radioresistance and CSC properties, and is involved in
survival or oncogenic signaling pathways, including STAT3, MAPK,
PI3K/AKT and sonic hedgehog (27,28).
By contrast, EPB41L3 inhibits invasion, metastasis and tumor
development in various cancer cells, such as breast, ovarian,
prostate and gastric cancer cells, and induces apoptosis (29,30).
Therefore, CXCR4 and EPB41L3, which exhibited significant
expression changes following CFH knockdown, were selected as
candidate factors for further validation.
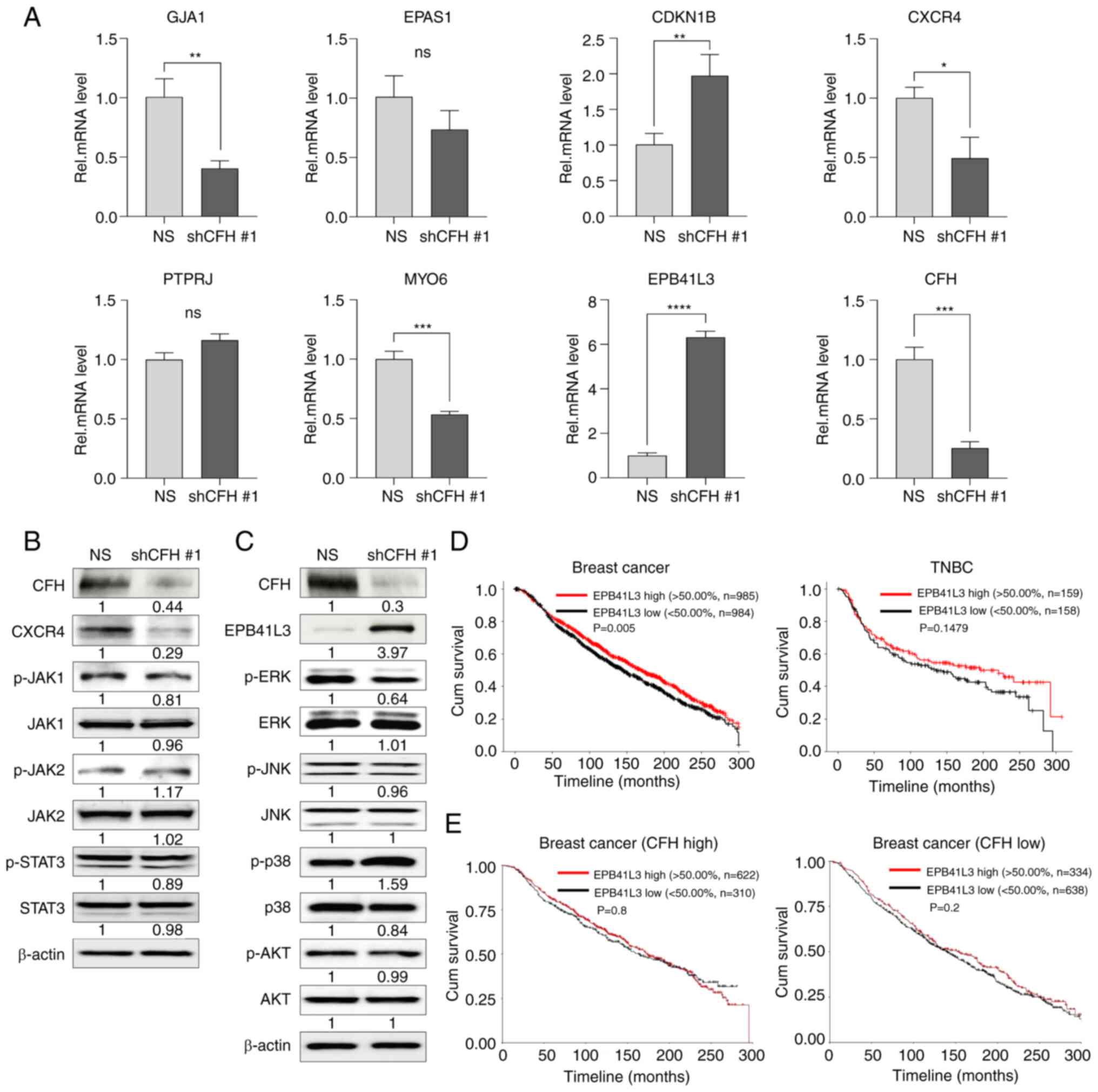 | Figure 5.CFH knockdown regulates the MAPK
signaling pathway by increasing EPB41L3 expression. (A) The mRNA
expression levels of seven major candidate genes were measured by
reverse transcription-quantitative PCR. The results are presented
as the mean ± SD. *P<0.05, **P<0.01, ***P<0.001,
****P<0.0001, using an unpaired Student's t test. The expression
levels of (B) CXCR4 and (C) EPB41L3 signaling pathway proteins were
analyzed by western blotting. The expression levels were
semi-quantified and defined beneath each protein band. The
expression levels were calculated as the target/b-actin ratio for
each lane and the ratio values for the experimental group were
normalized by setting the ratio value of the negative control group
to 1. (D) Correlation between the EPB41L3 expression level and the
survival rate of patients with all types of breast cancer or TNBC
was analyzed using METABRIC data using the log-rank test. The
results were validated using the Cox proportional hazards model.
(E) Correlation of the survival rates of patients with breast
cancer according to the level of EPB41L3 expression in the CFH high
or low expression cohorts. This analysis utilized the survival and
survminer packages in R for conducting survival analysis, and was
validated using the Cox proportional hazards model. CDKN1B,
cyclin-dependent kinase inhibitor 1B; CFH, complement factor H;
CXCR4, C-X-C motif chemokine receptor 4; EPAS1, endothelial PAS
domain protein 1; EPB41L3, erythrocyte membrane protein band
4.1-like 3; GJA1, gap junction protein α1; JAK, Janus kinase;
METABRIC, Molecular Taxonomy of Breast Cancer International
Consortium; MYO6, myosin VI; NS (uppercase), non-silencing; ns
(lowercase), not significant; PTPRJ, protein tyrosine phosphatase
receptor type J; shRNA; Rel, relative; sh(RNA), short hairpin
(RNA); TNBC, triple-negative breast cancer. |
To investigate whether CFH knockdown affected the
CXCR4 or EPB41L3 signaling cascade, CXCR4- or EPB41L3-associated
protein phosphorylation were examined using western blotting.
First, the CXCR4/ Janus kinase (JAK)/STAT3 signaling pathway, a
major oncogenic cascade in CSCs (27,28),
was analyzed. However, the phosphorylation levels of JAK1, JAK2 and
STAT3 were not affected by CFH knockdown (Figs. 5B and S3). Next, the tumor suppressor protein,
EPB41L3, which is involved in the PI3K/AKT and MAPK pathways
(29,30), was analyzed. For this, whether
increased EPB41L3 expression could regulate the phosphorylation
levels of AKT, ERK, JNK and p38 MAPK was explored. Notably,
phosphorylated-ERK (p-ERK) levels were reduced and p-p38 levels
were significantly increased in shCFH #1 cells, which contained
upregulated EPB41L3 expression following CFH knockdown, compared
with NS cells (Figs. 5C and
S4).
Moreover, after analyzing the METABRIC breast cancer
and TNBC data, it was demonstrated that the patient groups with
high EPB41L3 expression had an improved survival rate compared with
the low expression groups (Fig.
5D). However, the results of the analysis of the TNBC data were
not statistically significant. In addition, the survival rates of
patients with breast cancer were further analyzed according to the
levels of EPB41L3 expression in the CFH high and CFH low expression
cohorts (Fig. 5E). In the CFH low
expression cohort, the EPB41L3 high expression cohort had a
slightly higher survival rate than the EPB41L3 low expression
cohort, but this result was not statistically significant. These
results indicated that the EPB41L3 expression level may influence
the survival rates of patients with breast cancer. However, the
correlation with CFH was not statistically significant.
EPB41L3 downregulation restores cell
survival, migration and invasion abilities through the enhancement
of radioresistance via the regulation of MAPK signaling in
MDA-MB-231 breast cancer cells
It was next investigated whether the
re-downregulation of EPB41L3, which was upregulated by CFH
suppression, could rescue the CFH suppression effect. To confirm
the downregulation of EPB41L3 siRNA, MDA-MB-231 cells were
transfected with universal NC siRNA or siEPB41L3-#1, -#2 and -#3,
and RT-qPCR and western blotting were performed (Figs. 6A and S5). Moreover, EPB41L3 downregulation by
siEPB41L3 #2 in shCFH #1 cells induced an increase in p-ERK and a
decrease in p-p38 levels compared with shCFH #1 or NC cells
(Figs. 6B and S6). In accordance with these results, it
was next examined whether reactivation of the signaling pathways
induced by downregulation of EPB41L3 could transform cancer cells
to aggressive phenotypes. shCFH #1 cells were irradiated following
transfection with NC or EPB41L3 #2 siRNA, and then clonogenic,
migration and invasion assays were performed. Compared with shCFH
#1 or NC cells, the shCFH #1 + siEPB41L3 #2 cells showed increased
survival and motility upon radiation treatment (Fig. 6C-E). These data demonstrated that
upregulation of the EPB41L3 tumor suppressor by CFH knockdown
increased the radiotherapy sensitivity of MDA-MB-231 cells through
the decrease of radioresistance via MAPK regulation. Consequently,
CFH downregulation suppressed the expression of CSC-associated
genes via the regulation of ERK and p38 MAPK signaling by
increasing EPB41L3 protein expression.
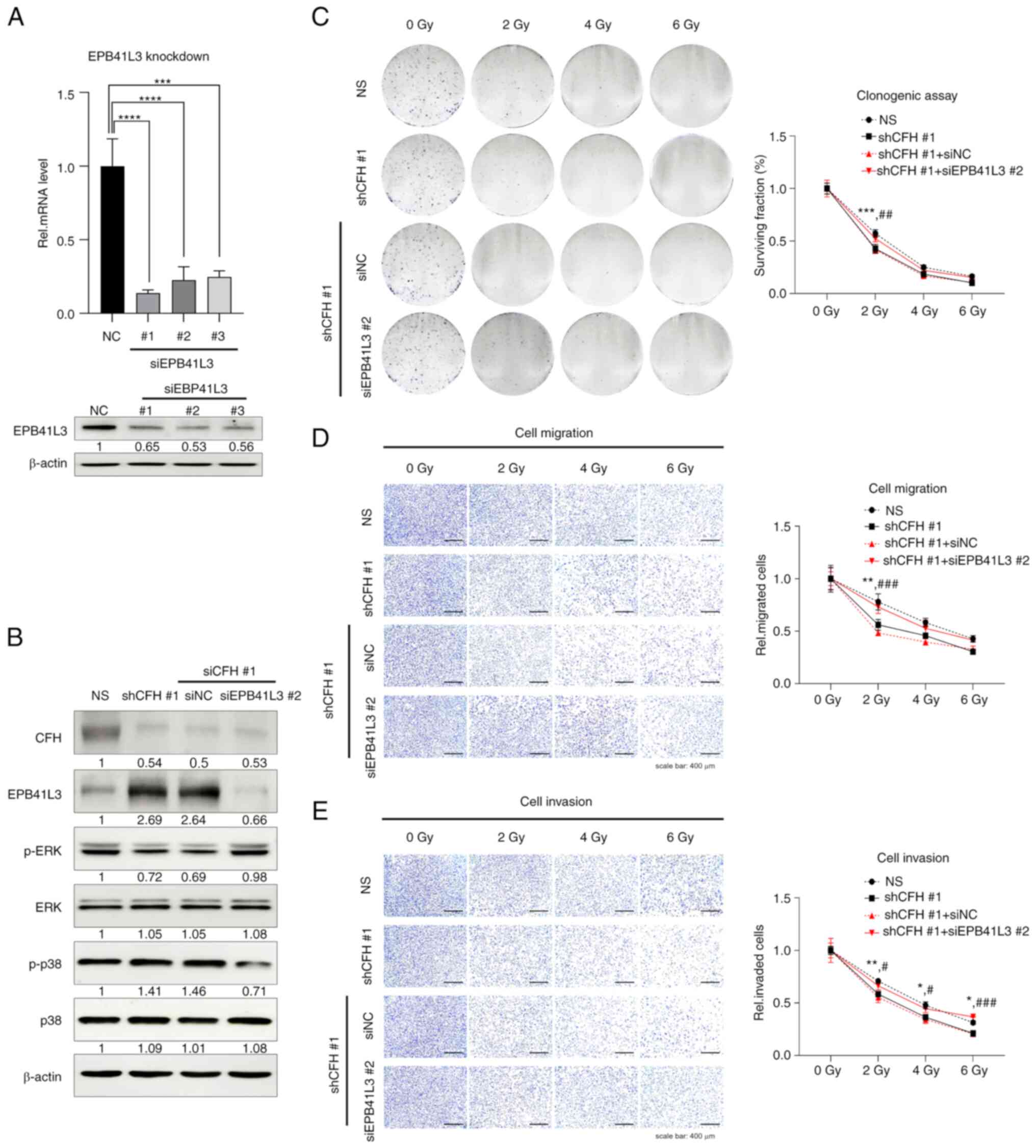 | Figure 6.EPB41L3 knockdown may eliminate the
effect of CFH suppression on cell survival, migration and invasion
abilities. (A) MDA-MB-231 cells were transfected with NC, EPB41L3
#1, #2 or #3. The mRNA and protein expression levels of EPB41L3
were measured by reverse transcription PCR and western blotting.
The results are presented as the mean ± SD. ***P<0.001,
****P<0.0001, using one-way ANOVA and Tukey's test. (B) The
phosphorylation levels of ERK and p38 MAPK were confirmed by
western blotting following NC or siEPB41L3 #2 transfection. The
expression levels were semi-quantified and defined beneath each
protein band. The expression levels were calculated as the
target/b-actin ratio for each lane and the ratio values for the
experimental group were normalized by setting the ratio value of
the negative control group to 1. (C) A clonogenic assay was
performed using a crystal violet solution after NC or siEPB41L3 #2
transfection and irradiation. The surviving fraction was measured
by counting the colonies. (D) Cell migration and (E) invasion
assays were performed using Transwell inserts with an 8-µm pore
size coated with collagen or Matrigel, and cells were stained with
a crystal violet solution. NC or siEPB41L3 #2 transfected cells
were irradiated. The results are presented as the mean ± SD.
*P<0.05, **P<0.01, ***P<0.001 NS vs. shCFH #1;
#P<0.05, ##P<0.01,
###P<0.001 shCFH #1 + siNC vs. shCFH #1 + siEPB41L3
#2, using two-way ANOVA and Tukey's test (the two variables were
presence of gene regulation and the radiation dose). CFH,
complement factor H; EPB41L3, erythrocyte membrane protein band
4.1-like 3; EpCAM, epithelial cellular adhesion molecule; NC,
negative control; NS, non-silencing; p-, phosphorylated; Rel,
relative; sh(RNA), short hairpin (RNA); si(RNA), small interfering
(RNA). |
Discussion
The complement cascade protects the body against
diverse pathogens. This process is accompanied by the release of
inflammatory factors, such as interleukins, IFN-γ, TNF-α and TGF-β,
from mast cells, monocytes, macrophages, peripheral blood
mononuclear cells, antigen-presenting cells and T cells. These
cells and cytokines control the immune response by interacting and
communicating with each other. However, if this process is not
terminated and instead maintained continuously, it can generate
chronic inflammation and a favorable environment for tumors. In
fact, the sublytic pore of the cell membrane is known to trigger
tumor progression by promoting chronic inflammation (14,31).
Therefore, cancer cell death should be induced through an effective
immune response to prevent a chronic inflammatory environment.
Human breast cancer cells are classified into
several types, such as luminal A and B, HER2+ and TNBC
cells. TNBC cells are difficult to completely eliminate despite
advanced drug and radiotherapy techniques due to their high
capacities for survival, invasion and radioresistance, which lead
to metastasis and recurrence (32).
BCSCs can exhibit various phenotypes by maintaining high expression
levels of CD44 and CD133, which enhance stemness, cell survival,
growth, migration, invasion, chemoresistance and radioresistance
through signaling pathways such as NF-κB, cAMP response
element-binding protein/TGF-β2, β-catenin and IL-6/Notch3 (33–36).
For these reasons, the discovery of targets to suppress cancer
stemness is crucial to prevent recurrence and metastasis and
improve therapeutic efficacy. According to a report, suppressor of
cytokine signaling-1 and −3/JAK/STAT4, an oncogenic transcription
factor pathway, increases the expression of CFH in A549 lung cancer
cells (37). Moreover,
intracellular CFH overexpression promotes proliferation, migration
and survival independent of the complement cascade in A498 renal
cancer cells and A549 cells (20).
Additionally, CFH uptake controls intracellular C3 activation in
apoptotic cells and represses the inflammatory potential of
released nucleosomes (38).
However, to the best of our knowledge, the distinct role of
intracellular CFH in cancer cells is not known. According to a
recent study, CFH in exosomes released from metastatic
hepatocellular carcinoma cells induced tumorigenesis and metastasis
by increasing cell growth, migration and invasiveness (39). More notably, CFH expressed by breast
cancer cells promotes the differentiation of CD14+
monocytes into immunosuppressive macrophages (23). These studies suggest that CFH may
affect tumor progression and the maintenance of stemness potential
through direct regulation of specific genes and indirect
interaction with their surrounding microenvironments, including
various immune cells (23,38,39).
Therefore, we hypothesize that CFH is a target factor to enhance
the effectiveness of drugs and radiotherapy by suppressing stemness
and aggressive phenotypes.
Indeed, the microarray results of the present study
demonstrated that numerous genes regulated by CFH knockdown were
involved in breast cancer. These genes were associated with
essential biological processes for survival, including
proliferation, growth, migration and signal transduction. CXCR4, a
major pro-tumor receptor that promotes signal transduction by
binding the CXCL12 ligand, is upregulated in a number of cancer
types. The upregulation of CXCR4 in the tumors of patients with
TNBC is known to promote tumor growth and metastasis, and
upregulated CXCR4 is also associated with proliferation, motility
and metastasis in colon and gastric cancer (40–42).
In the present study, CXCR4 expression was suppressed by CFH
knockdown but this did not affect the downstream signaling of
CXCR4. The CXCR4 downstream pathway, JAK/STAT3, is likely to be
activated by alternative ligands including EGF, IL-10, IL-6 and
IL-11. (43) By contrast, EPB41L3
is a potential antitumor gene that plays a key role in cytoskeletal
organization and remodeling and participates in diverse biological
processes through its interaction with cytoplasmic or membrane
proteins via the four one-ezrin-radixin-moesin domain (29). According to a previous study,
EPB41L3 overexpression represses proliferation and induces
apoptosis in esophageal squamous cell carcinoma cells through G2/M
arrest (44). In addition, EPB41L3
inhibited osteosarcoma cell invasion through the suppression of
epithelial-mesenchymal transition induced by snail-1 (30). The results of the present study
corroborate the antitumor role of EPB41L3 and its related signaling
pathway.
The activation of ERK plays a radioprotective
function by enhancing Bcl-2 and p65 expression while repressing Bax
and p53 expression (45).
Lapatinib, an EGFR/HER2 kinase inhibitor, induces
radiosensitization through the inhibition of ERK (46). Furthermore, the p38 signaling
pathway enhances radiosensitivity by increasing G2/M phase arrest
and apoptosis through the suppression of cyclin B1, CDK1 and Bcl-2
(47). Activation of the p38
signaling pathway promotes apoptosis by regulating the
Bax/Bcl-2/caspase-3, −7 and −9 cascade and induces cell cycle
arrest through the suppression of CDK2, CDK4, cyclin D1 and cyclin
E1 (48,49). As is well-known, the ERK and p38
signaling pathways play a crucial role as key mediators in
regulating multiple phenotypes of cancer cells. The results of the
present study revealed that CFH could enhance stemness, which is
associated with malignant phenotypes and chemoresistance and
radioresistance, through the regulation of the EPB41L3/ERK/p38
signaling pathway. It was demonstrated in the present study that
downregulation of CFH, which regulates the activity of MAPKs and
suppresses stemness-related phenotypes (such as
CD24−/CD44+, CD133+ and
EpCAM+), enhanced radiosensitivity, thereby reducing
cell survival and motility due to irradiation-induced damage.
Additionally, CFH-knock down cells with increased radiosensitivity
underwent cell cycle arrest upon irradiation, leading to a higher
level of apoptotic signals. Collectively, these results
demonstrated that downregulation of CFH in MDA-MB-231 cells
increased the expression of the EPB41L3 tumor suppressor,
suppressed ERK activation, promoted p38 activation, suppressed
cancer stemness and enhanced irradiation-induced damage.
A future study stemming from the present study would
be to analyze the level of complement activation and CDC as well as
the tumor suppression effect of CFH downregulation through tumor
xenograft experiments. However, it is difficult to select a
suitable mouse model with a normal immune system that can be used
for xenografting as the structural differences in complement
factors between humans and mice might lead to inaccurate
experimental data related to abnormal activation of complement
responses, such as hyperactivation or inactivation. Therefore, ways
to develop a proper mouse model through CRISPR-Cas technology and
humanized mouse models are being studied. In the future, the
correlation of the complement system and human breast cancer will
be studied using a mouse model that is currently in
development.
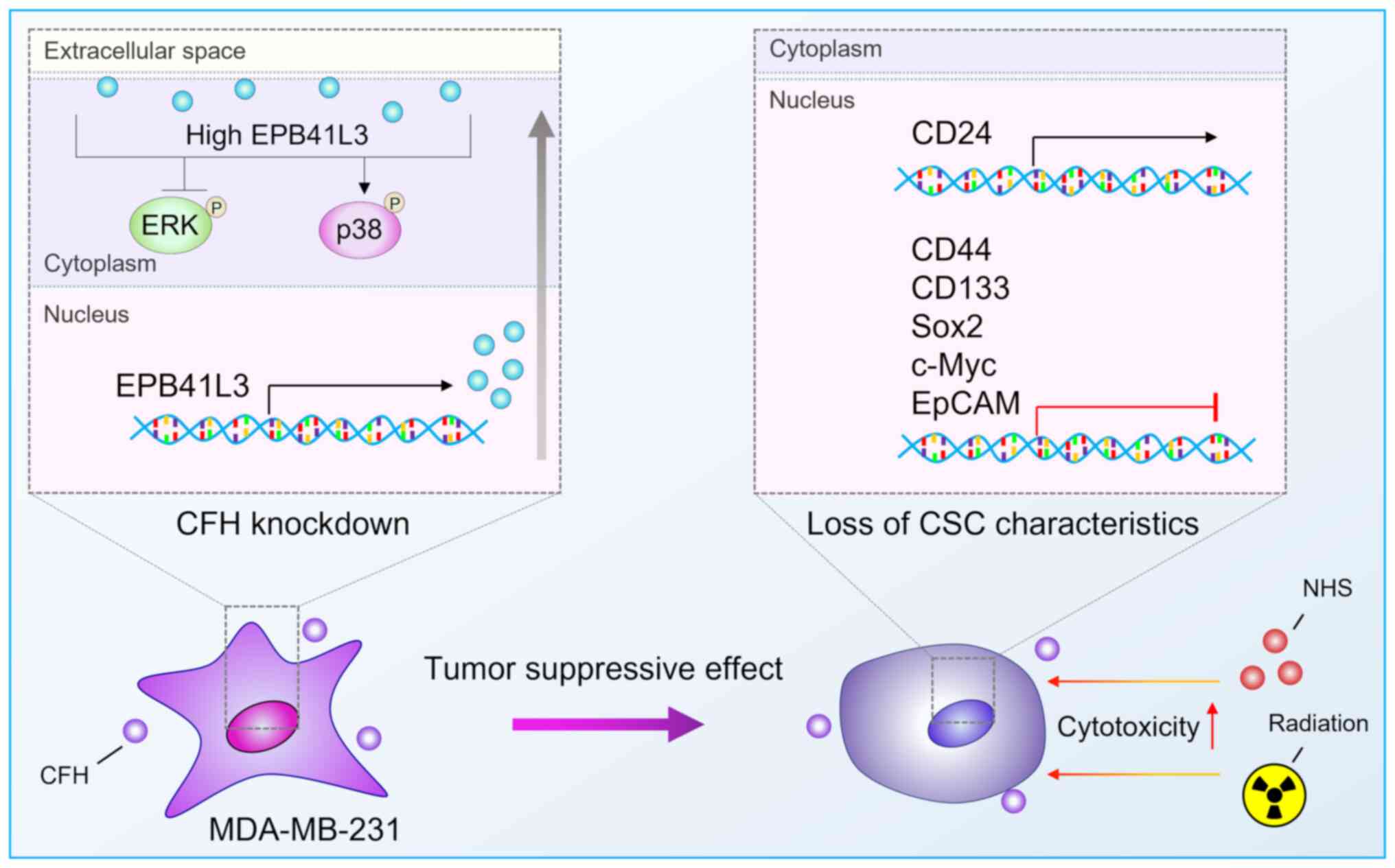
In conclusion, CFH downregulation suppressed the
expression of BCSC-specific genes by promoting EPB41L3 expression
via regulation of the MAPK signaling pathway in MDA-MB-231 breast
cancer cells (Fig. 7).
Additionally, since CFH downregulation promoted the activation of
CDC, it might contribute to overcoming many side effects of
anticancer therapies by improving therapeutic efficiency.
Therefore, CFH is a potential target for efficient radiotherapy for
human breast cancer treatment.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by The Dongnam Institute of Radiological
& Medical Sciences grant funded by The Korean Government (grant
no. 50591-2023).
Availability of data and materials
The datasets generated by microarray are available
in the ArrayExpress database (https://www.ebi.ac.uk/biostudies/arrayexpress/studies/E-MTAB-13327?query=E-MTAB-13327).
All other datasets used and/or analyzed during the current study
are available from the corresponding author on reasonable
request.
Authors' contributions
SYP, DYE and YJC designed the study. SYP, DYE, YJ
and JWS performed the experiments. CYL analyzed the microarray
data. SHC, SJP and KH analyzed and interpreted the data. SYP and
YJC wrote the paper. All authors read and approved the final
version of the manuscript. SYP and YJC confirm the authenticity of
all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
BCSCs
|
breast cancer stem cells
|
|
CFH
|
complement factor H
|
|
CRPs
|
complement regulatory proteins
|
|
CXCR4
|
C-X-C motif chemokine receptor 4
|
|
EPB41L3
|
erythrocyte membrane protein band
4.1-like 3
|
|
NHS
|
normal human serum
|
|
TNBC
|
triple-negative breast cancer
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics. CA Cancer J Clin. 69:7–34. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Weigelt B, Peterse JL and van ‘t Veer LJ:
Breast cancer metastasis: Markers and models. Nat Rev Cancer.
5:591–602. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bai X, Ni J, Beretov J, Graham P and Li Y:
Cancer stem cell in breast cancer therapeutic resistance. Cancer
Treat Rev. 69:152–163. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brooks MD, Burness ML and Wicha MS:
Therapeutic implications of cellular heterogeneity and plasticity
in breast cancer. Cell Stem Cell. 17:260–271. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
El-Sahli S and Wang L: Cancer stem
cell-associated pathway in the metabolic reprogramming of breast
cancer. Int J Mol Sci. 21:91252020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Badve S and Nakshtri H: Breast-cancer stem
cells-beyond semantics. Lancet Oncol. 13:e43–e48. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Walcher L, Kistenmacher AK, Suo H, Kitte
R, Dluczek S, Strauß A, Blaudszun AR, Yevsa T, Fricke S and
Kossatz-Boehlert U: Cancer stem cell-Origins and biomarkers:
Perspectives for targeted personalized therapies. Front Immunol.
11:12802020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fultang N, Chakraborty M and Peethambaran
B: Regulation of cancer stem cells in triple negative breast
cancer. Cancer Drug Resist. 4:321–342. 2021.PubMed/NCBI
|
|
9
|
J O'Conor C, Chen T, González I, Cao D and
Peng Y: Cancer stem cells in triple-negative breast cancer: a
potential target and prognostic marker. Biomark Med. 12:813–820.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Barzaman K, Karami J, Zarei Z,
Hosseinzadeh A, Kazemi MH, Moradi-Kalbolandi S, Safari E and
Farahmand L: Breast cancer: Biology, biomarkers, and treatments.
Int Immunopharmacol. 84:1065352020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Crabtree JS and Miele L: Breast cancer
stem cells. Biomedicines. 6:772018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang T, Song X, Xu D, Tiek D, Goenka A,
Wu B, Sastry N, Hu B and Cheng SY: Stem cell programs in cancer
initiation, progression, and therapy resistance. Theranostics.
10:8721–8743. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Franco SS, Szczesna K, Iliou MS,
Al-Qahtani M, Mobasheri A, Kobolák J and Dinnyés A: In vitro models
of cancer stem cells and clinical applications. BMC Cancer.
16:7382016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Afshar-Kharghan V: The role of the
complement system in cancer. J Clin Invest. 127:780–789. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bajic G, Degn SE, Thiel S and Andersen GR:
Complement activation, regulation, and molecular basis for
complement-related diseases. EMBO J. 34:2735–2757. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lewis LA, Ram S, Prasad A, Gulatin S,
Getzlaff S, Blom AM, Vogel U and Rice PA: Defining targets for
complement components C4b and C3b on the pathogenic neisseriae.
Infect Immun. 76:339–350. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Parente R, Clark SJ, Inforzato A and Day
AJ: Complement factor H in host defense and immune evasion. Cell
Mol Life Sci. 74:1605–1624. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cserhalmi M, Papp A, Brandus B, Uzonyi B
and Jozsi M: Regulation of regulators: Role of the complement
factor H-related proteins. Semin Immunol. 45:1013412019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Seol HS, Lee SE, Song JS, Rhee JK, Singh
SR, Chang S and Jang SJ: Complement proteins C7 and CFH control the
stemness of liver cancer cells via LSF-1. Cancer Lett. 372:24–35.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Daugan MV, Revel M, Thouenon R,
Dargon-Durey MA, Robe-Rybkine T, Torset C, Merle NS, Noé R,
Verkarre V, Oudard SM, et al: Intracellular factor H drives tumor
progression independently of the complement cascade. Cancer Immunol
Res. 9:909–925. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ajona D, Hsu YF, Corrales L, Montuenga LM
and Pio R: Down-regulation of human complement factor H sensitizes
non-small cell lung cancer cells to complement attack and reduces
in vivo tumor growth. J Immunol. 178:5991–5998. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Junnikkala S, Hakulinen J, Jarva H,
Manuelian T, Bjørge L, Bützow R, Zipfel PF and Meri S: Secretion of
soluble complement inhibitors factor H and factor H-like protein
(FHL-1) by ovarian tumour cells. Br J Cancer. 87:1119–1127. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Smolag KI, Mueni CM, Leandersson K,
Jirström K, Hagerling C, Mörgelin M, Barlow PN, Martin M and Blom
AM: Complement inhibitor factor H expressed by breast cancer cells
differentiates CD14+ human monocytes into
immunosuppressive macrophages. Oncoimmunology. 9:17311352020.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu M, Yang YJ, Zheng H, Zhong XR, Wang Y,
Wang Z, Wang YG and Wang YP: Membrane-bound complement regulatory
proteins are prognostic factors of operable breast cancer treated
with adjuvant trastuzumab: A retrospective study. Oncol Rep.
32:2619–2627. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pereira B, Chin SF, Rueda OM, Vollan HKM,
Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ,
et al: The somatic mutation profiles of 2,433 breast cancers
refines their genomic and transcriptomic ladnscapes. Nat Commun.
7:114792016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu X, Xiao Q, Bai X, Yu Z, Sun M, Zhao H,
Mi X, Wang E, Yao W, Jin F, et al: Activation of STAT3 is involved
in malignancy mediated by CXCL12-CXCR4 signaling in human breast
cancer. Oncol Rep. 32:2760–2768. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Trautmann F, Cojoc M, Kurth I, Melin N,
Bouchez LC, Dubrovska A and Peitzsch C: CXCR4 as biomarker for
radioresistant cancer stem cells. Int J Radiat Biol. 90:687–699.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yuan X, Piao L, Wang L, Han X, Zhuang M
and Liu Z: Pivotal roles of protein 4.1B/DAL-1, a FERM-domain
containing protein, in tumor progression. Int J Oncol. 55:979–987.
2019.PubMed/NCBI
|
|
30
|
Yuan X, Piao L, Wang L, Han X, Tong L,
Shao S, Xu X, Zhuang M and Liu Z: Erythrocyte membrane protein band
4.1-like 3 inhibits osteosarcoma cell invasion through regulation
of Snai1-induced epithelial-to-mesenchymal transition. Aging.
13:1947–1961. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vlaicu SI, Tatomir A, Rus V and Rus H:
Role of C5b-9 and RGC-32 in cancer. Front Immunol. 10:10542019.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lagadec C, Vlashi E, Donna LD, Dekmezian C
and Pajonk F: Radiation-induced reprogramming of breast cancer
cells. Stem Cells. 30:833–844. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kola P, Nagesh PKB, Roy PK, Deepak K, Reis
RL, Kundu SC and Mandal M: Innovative nanotheranostics: Smart
nanoparticles based approach to overcome breast cancer stem cells
mediated chemo- and radioresistances. Wiley Interdiscip Rev Nanomed
Nanobiotechnol. 15:e18762023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jain V, Kumar H, Anod HV, Chand P, Gupta
NV, Dey S and Kesharwani SS: A review of nanotechnology-based
approaches for breast cancer and triple-negative breast cancer. J
Control Release. 326:628–647. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Song K and Faraneh M: Signaling pathways
governing breast cancer stem cells behavior. Stem Cell Res Ther.
12:2452021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zheng Q, Zhang M, Zhou F, Zhang L and Meng
X: The breast cancer stem cells traits and drug resistance. Front
Pharmacol. 11:5999652021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yoon YH, Hwang HJ, Sung HJ, Heo SH, Kim
DS, Hong SH, Lee KH and Cho JY: Upregulation of complement factor H
by SOCX-1/3-STAT4 in lung cancer. Cancers (Basal). 11:4712019.
View Article : Google Scholar
|
|
38
|
Martin M, Leffler J, Smolag KI, Mytych J,
Björk A, Chaves LD, Alexander JJ, Quigg RJ and Blom AM: Factor H
uptake regulates intracellular C3 activation during apoptosis and
decreases the inflammatory potential of nucleosomes. Cell Death
Differ. 23:903–911. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mao X, Zhou L, Tey SK, Ma APY, Yeung CLS,
Ng TH, Wong SWK, Liu BHM, Fung YME, Patz EF Jr, et al: Tumour
extracellular vesicle-derived complement factor H promotes
tumorigenesis and metastasis by inhibiting complement-dependent
cytotoxicity of tumour cells. J Extracell Vesicels. 10:e120312020.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gupta N, Mohan CD, Shanmugam MK, Jung YY,
Chinnathambi A, Alharbi SA, Ashrafizadeh M, Mahale M, Bender A,
Kumar AP, et al: CXCR4 expression is elevated in TNBC patient
derived samples and Z-guggulsterone abrogates tumor progression by
targeting CXCL12/CXCR4 siganling axis in preclinical breast cancer
model. Environ Res. 232:1163352023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Alsaab HO and Almalki AH: Anti-HSP70
alleviates cell migration and proliferation in colorectal cancer
cells (CRC) by targeting CXCR4 (in vitro study). Med Oncol.
40:2562023. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhao H, Jiang R, Zhang C, Feng Z and Wang
X: The regulatory role of cancer stem cell marker gene CXCR4 in the
growth and metastasis of gastric cancer. NPJ Precis Oncol.
7:862023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
To SQ, Dmello RS, Richards AK, Ernst M and
Chand AL: STAT3 signaling in breast cancer: Multicellular actions
and therapeutic potential. Cancers (Basel). 14:4292022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zeng R, Liu Y, Jiang ZJ, Huang JP, Wang Y,
Li XF, Xiong WB, Wu XC, Zhang JR, Wang QE and Zheng YF: EPB41L3 is
a potential tumor suppressor gene and prognostic indicator in
esophageal squamous cell carcinoma. Int J Oncol. 52:1443–1454.
2018.PubMed/NCBI
|
|
45
|
Lu Y, Liu B, Liu Y, Yu X and Cheng G: Dual
effects of active ERK in cancer: A potential target for enhancing
radiosensitivity. Oncol Lett. 20:993–1000. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sambade M, Camp JT, Kimple RJ, Sartor CI
and Shields JM: Mechanism of lapatinib-mediated radiosensitization
of breast cancer cells is primarily by inhibition of the
Raf>MEK>ERK mitogen-activated protein kinase cascade and
radiosensitization of lapatinib-resistant cells restored by direct
inhibition of MEK. Radiother Oncol. 93:639–644. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
He H, Lin K, Zou C, Pan J, Fu W, Zhou Y,
Lin H, Chen C and Su Y: Knockdown of annexin A2 enhances
radiosensitivity by increasing G2/M-phase arrest, apoptosis and
activating the p38 MAPK-HSP27 pathway in nasopharyngeal carcinoma.
Front Oncol. 12:7695442022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhou Y, Zhao W, Xie G, Huang M, Hu M,
Jiang X, Zeng D, Liu J, Zhou H, Chen H, et al: Induction of
Nur77-dependent apoptotic pathway by a coumarin derivative through
activation of JNK and p38 MAPK. Carcinogenesis. 35:2660–2669. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tang J, Wu W, Yang F, Liu L, Yang Z, Liu
L, Tang W, Sun F and Lin H: Marine sponge-derived smenospongine
preferentially eliminates breast cancer stem-like cells via
p38/AMPKa pathways. Cancer Med. 7:3965–3976. 2018. View Article : Google Scholar : PubMed/NCBI
|