Introduction
In recent years, the incidence of esophageal
adenocarcinoma (EAC) has been increasing (1,2). Given
the extremely poor prognosis of EAC, it is crucial to focus on
disease prevention. Barrett's esophagus (BE) is considered a
precursor lesion of EAC and is believed to develop as a result of
chronic esophageal reflux (3,4). The
development of these diseases is influenced by the persistent
inflammatory environment caused by the reflux of gastroduodenal
contents (5–7). These process, gastro esophageal reflux
disease (GERD) to EAC, is thought to be involved in the
inflammation-metaplasia-adenocarcinoma (IMA) sequence (8,9).
Chronic reflux of gastroduodenal contents into the esophagus leads
to severe esophagitis and triggers cell proliferation. Prolonged
proliferation progresses from hyperplasia to metaplasia, ultimately
resulting in EAC. Clinical studies have highlighted the importance
of chronic duodenogastroesophageal reflux (DGER) in the development
of BE and EAC (10–12). Our rat models, which mimic human
reflux esophagitis, have demonstrated that DGER can sequentially
induce BE and EAC (13–15). Other researchers have also reported
the impact of DGER in animal models (16,17).
The arachidonic acid (AA) cascade is extensively
studied as a biological regulatory pathway. Metabolites of the AA
cascade play a crucial role in the development and progression of
inflammatory diseases and cancers. This metabolic pathway involves
cyclooxygenases (COX) and lipoxygenases (LOX). While the
administration of COX-2 inhibitors in rat models has been shown to
suppress the inflammatory-metaplasia-adenocarcinoma (IMA) sequence
(18), the role of the LOX pathway
remains unclear.
Leukotrienes, which are potent pro-inflammatory
lipid mediators, are synthesized from AA in various cell types,
including mast cells, eosinophils, neutrophils, basophils, and
macrophages (19). AA stimulates
the production of two groups of leukotrienes under the action of
5-LOX, LTB4 and cysteinyl leukotrienes (CysLTs). CysLTs bind to
cysteinyl leukotriene receptors, primarily CysLT1R and CysLT2R.
These receptors are expressed on various cells, such as mast cells,
macrophages, and monocytes, and contribute to the production of
various inflammatory cytokines (20).
The LOX pathway has been found to have a significant
impact on various inflammatory conditions. It is released by cells
present or recruited to the site of inflammation. LOX metabolites
act as chemoattractants and activators of inflammatory cells,
contributing to inflammation, immune responses, and host defense
against infections (21). Prolonged
production of leukotrienes leads to sterile inflammation in chronic
inflammatory diseases (22).
Leukotrienes are implicated in several inflammatory responses,
including cell proliferation, angiogenesis, and cell survival,
which are fundamental steps in oncogenesis (23).
Numerous studies have highlighted the crucial role
of LOX-derived leukotrienes in carcinogenesis and cancer
progression (24–29). CysLT1R, a receptor involved in the
LOX pathway, is highly expressed in human colon and prostate
cancers and is negatively associated with patient survival
(25,26). It has been demonstrated that
LOX-derived leukotrienes are more numerous in human esophageal
adenocarcinoma compared with those in the normal esophagus
(27–29). Additionally, the administration of a
LOX pathway inhibitor has demonstrated chemopreventive effects in
certain animal models (30,31). These findings provide evidence that
the LOX pathway of AA metabolism plays a critical role in the
development of cancer.
In clinical practice, medications targeting the LOX
pathway have been implicated in the treatment of asthma and
allergic rhinitis. Pranlukast, montelukast, zafirlukast, and
zileuton are routinely used as primary therapeutic agents. The
pharmacological mechanism of pranlukast, montelukast, and
zafirlukast involves the antagonism of CysLT1R. Meanwhile, zileuton
can be used to treat asthma by inhibiting 5-LOX activity. However,
the clinical application of zileuton is limited due to its
hepatotoxicity. Pranlukast, on the other hand, was introduced for
clinical use in Japan in 1995 and has been marketed in several
countries. Additionally, receptor antagonists have shown great
potential in the treatment of asthma and other diseases. They may
be promising candidates for chemopreventive interventions as their
direct inhibition of these enzymes reduces the inflammatory
response that contributes to carcinogenesis.
In this study, we investigated the chemopreventive
effect of pranlukast, a CysLT1R antagonist, on inflammation-induced
esophageal carcinogenesis using our established rat DGER model,
which develops BE and EAC without the administration of any
carcinogens (15).
Materials and methods
Animals and treatment procedures
Eight-week-old male Wistar rats, weighing
approximately 300 g (Charles River Laboratories Japan, Inc.,
Kanazawa, Japan), were used for the experiments. The rats were
housed in cages with three rats per group and kept in a room
maintained at a temperature of 22±3°C and a humidity of 55±5%,
following a 12-h light-dark cycle. They were provided with standard
solid chow (CRF-1, Charles River Laboratories Japan, Inc.) and tap
water ad libitum. No carcinogens were administered throughout the
study period. Prior to the surgical procedure, the rats underwent a
24-h fasting period and were anesthetized with isoflurane
inhalation anesthesia. In isoflurane anesthesia, the initial dose
is 3–5% and is maintained at approximately 1.5–2.5%. An upper
abdominal incision was made, ensuring preservation of the stomach
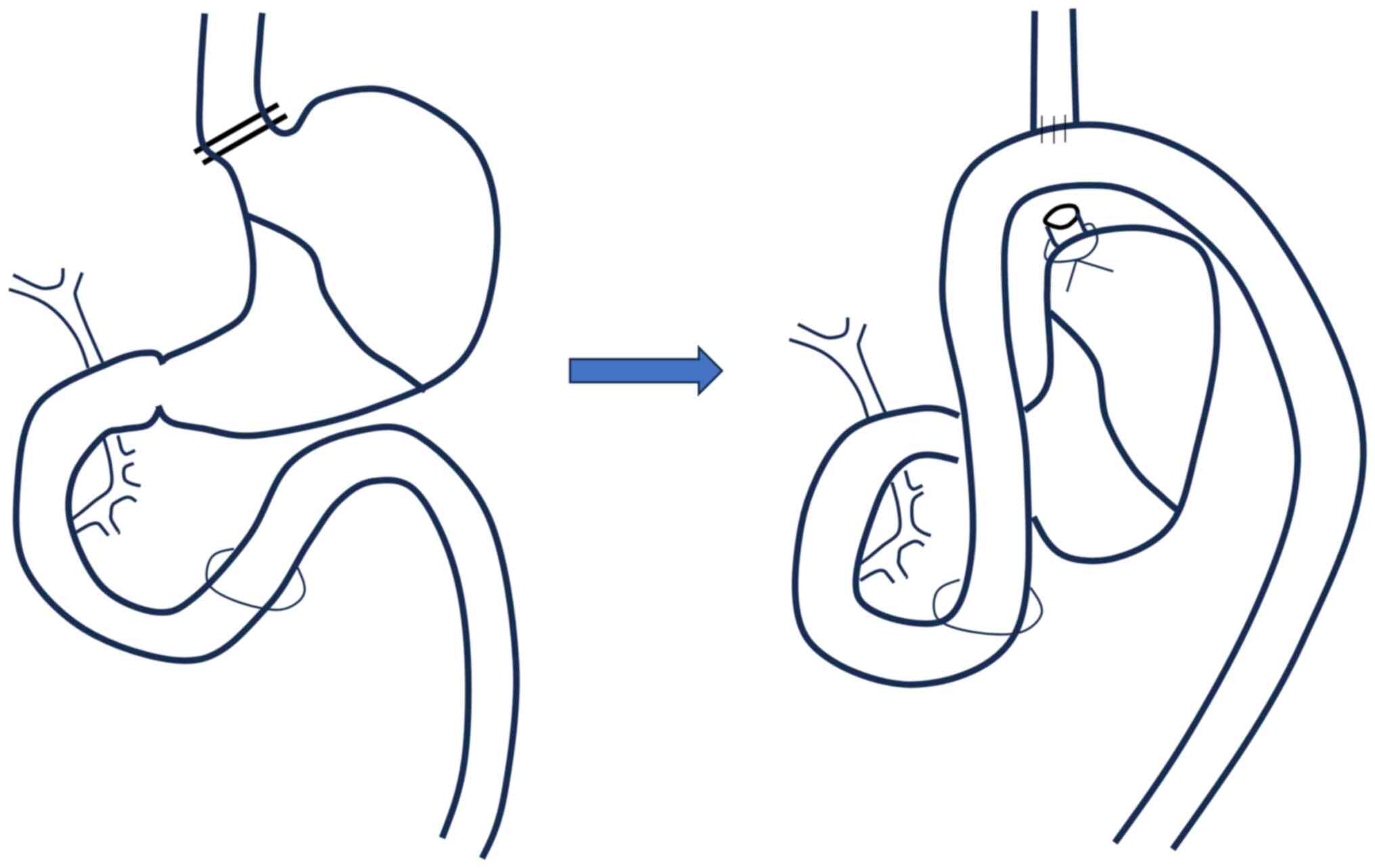
and vagal nerves. The esophagus was excised and ligated to the oral
side of the stomach. A loop in the jejunum, located 4 cm from the
ligament described by Treitz, was identified. An end-to-side
anastomosis was performed between the distal esophagus and jejunum
using interrupted full-thickness stitches with a 7-0 monofilament
suture. This surgical procedure allowed for the direct backflow of
gastric and duodenal juices into the esophagus (Fig. 1). After a 24-h recovery period, the
rats were provided with free access to water and food. In the
non-surgical group, five rats were bred under the same
environmental conditions.
The operated rats were randomly divided into two
groups: the control group (n=30) received commercial chow (CRF-1),
while the pranlukast group (n=30) received experimental chow that
was mixed with pranlukast (50 ppm, 3.3 mg/kg body wt/day). The
dosage of pranlukast was determined based on a previous study that
reported its inhibitory effect on tumor metastasis (32). Pranlukast was obtained from Cayman
Chemical (USA). The body weight of the rats was measured before
surgery and monthly thereafter.
Pathological evaluation
The rats were euthanized by exsanguination under
isoflurane anesthesia at 10, 20, 30, and 40 weeks after surgery.
The rats in the non-surgical group were euthanized after 40 weeks
of breeding. Euthanasia was confirmed by observing that the rats
were not breathing and by palpation for the absence of a heartbeat.
Following euthanasia, the entire esophagus and jejunum (including
the anastomosis) were surgically resected. The resected tissues
were fixed in a 10% formalin solution for 24 h. Subsequently, the
esophagus was sectioned at 3-mm intervals along its length and
embedded in paraffin. Thin sections of 5 µm thickness were prepared
from each paraffin block for histological evaluation, which
included hematoxylin and eosin staining as well as
immunohistochemistry.
Definition of pathological
findings
The pathological changes due to DGER were defined as
follows: regenerative thickening (RT), epithelial thickening to
more than double the normal thickness, together with acanthosis, an
abnormal extension of papillae toward the mucosal surface, and
parakeratosis; basal-cell hyperplasia (BCH), where the basal layer
in the squamous epithelium thickened and occupied >15% of the
epithelial layer; erosion, a lack of epithelium with cellular
infiltration; BE, esophageal squamous epithelium replaced with
columnar-lined epithelium comprising absorptive cells with brush
borders and goblet cells; adenocarcinoma, an epithelial growth with
atypical cells and structure and invasion of the submucosal
layer.
Immunohistochemistry and the TUNEL
method
Immunohistochemical staining and the TUNEL assay
were performed to assess inflammatory cell infiltration and
cytokine production in the esophageal epithelium. For
immunohistochemical staining, the Envision System (Dako, Denmark)
was utilized, with autoclave acceleration. 5-µm sections of
formalin-fixed, paraffin-embedded blocks were deparaffinized and
then treated with absolute methanol containing 0.3% hydrogen
peroxidase. Subsequently, the sections were incubated with normal
goat serum (1:30) and left overnight at 4°C with the primary
antibody. The primary antibodies used in this study were as
follows: polyclonal rabbit anti-rat 5-LOX (Abbiotec, USA),
monoclonal mouse anti-rat CD68 (Bio-Rad Laboratories, USA),
polyclonal rabbit anti-rat IL8 (Abcam, UK), and monoclonal anti-rat
VEGF (Abcam, UK). Afterward, the sections were treated with a
labeled polymer (Dako) for 2 h. The reaction products were
developed by immersing the sections in 3,3-diaminobenzidine, and
the slides were lightly counterstained with hematoxylin.
Cells that exhibited strong staining were considered
immunohistochemically positive. The expression intensity of 5-LOX,
CD68, IL-8, and VEGF was evaluated by counting labeled cells per
high-power field (HPF) in the esophageal epithelium located 5 mm
from the oral side of the anastomosis.
To assess the proliferative activity of the
esophageal epithelium, immunohistochemical staining for the Ki-67
protein was performed. The primary antibody used for Ki-67
immunohistochemical staining was monoclonal mouse anti-rat Ki-67
antigen (Dako), following the procedure described earlier. The
number of Ki-67-labeled cells was counted per 1000 epithelial basal
cells (Ki-67 labeling index) in the esophageal epithelium, located
5 mm from the oral side of the anastomosis.
Apoptosis was evaluated using the TUNEL method
(Apoptosis In Situ Detection Kit; Wako, Japan) according to
the manufacturer's instructions. The number of apoptotic cells was
expressed as the number of apoptotic cells per 1000 epithelial
cells in the esophageal epithelium, located 5 mm from the oral side
of the anastomosis.
Statistical analysis
The statistical analysis of the incidence of
pathological findings was performed using Fisher's exact test. The
expression of 5-LOX, CD68, IL-8, and VEGF, as well as the
proliferative activity and apoptosis, were presented as the mean
value ± SD. Comparisons between groups were conducted using the
Mann-Whitney U test. Differences were considered statistically
significant when the P-value was <0.05.
Results
General observations
Fifty-six of the 60 rats were included in this
study. Four rats (two from the control group and two from the
pranlukast group) died due to complications, including
malnutrition, pneumonia, and unknown causes. The effective number
of rats examined in each group at different time points were as
follows: 5 rats at weeks 10, 20, and 30, and 13 rats at week 40
after surgery (Table I). There was
no significant difference in mortality observed between the two
groups. Body weights were comparable between the control and
pranlukast groups at 0, 10, 20, 30, and 40 weeks. Microscopic
examination of the lungs, livers, and kidneys of the pranlukast
group at 40 weeks did not reveal any pathological changes,
suggesting no adverse effects of pranlukast.
 | Table I.Incidence of pathological
findings. |
Table I.
Incidence of pathological
findings.
|
|
|
| Incidence of
pathological findings (%) |
|---|
|
|
|
|
|
|---|
| Post-operative
week | Group | n | RT | BCH | Erosion | BE | EAC |
|---|
| 10 | Control | 5 | 5 (100) | 5 (100) | 5 (100) | 0 (0) | 0 (0) |
|
| Pranlukast | 5 | 5 (100) | 5 (100) | 4 (80) | 0 (0) | 0 (0) |
| 20 | Control | 5 | 5 (100) | 5 (100) | 5 (100) | 3 (60) | 1 (20) |
|
| Pranlukast | 5 | 5 (100) | 5 (100) | 5 (100) | 1 (20) | 0 (0) |
| 30 | Control | 5 | 5 (100) | 5 (100) | 5 (100) | 4 (80) | 2 (40) |
|
| Pranlukast | 5 | 5 (100) | 4 (80) | 4 (80) | 2 (40) | 0 (0) |
| 40 | Non-surgical | 5 | 2 (40) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
|
| Control | 13 | 13 (100) | 13 (100) | 13 (100) | 13
(100)a | 9 (69)a |
|
| Pranlukast | 13 | 13 (100) | 13 (100) | 12 (92) | 8 (62)a | 2 (15)a |
Histopathological findings
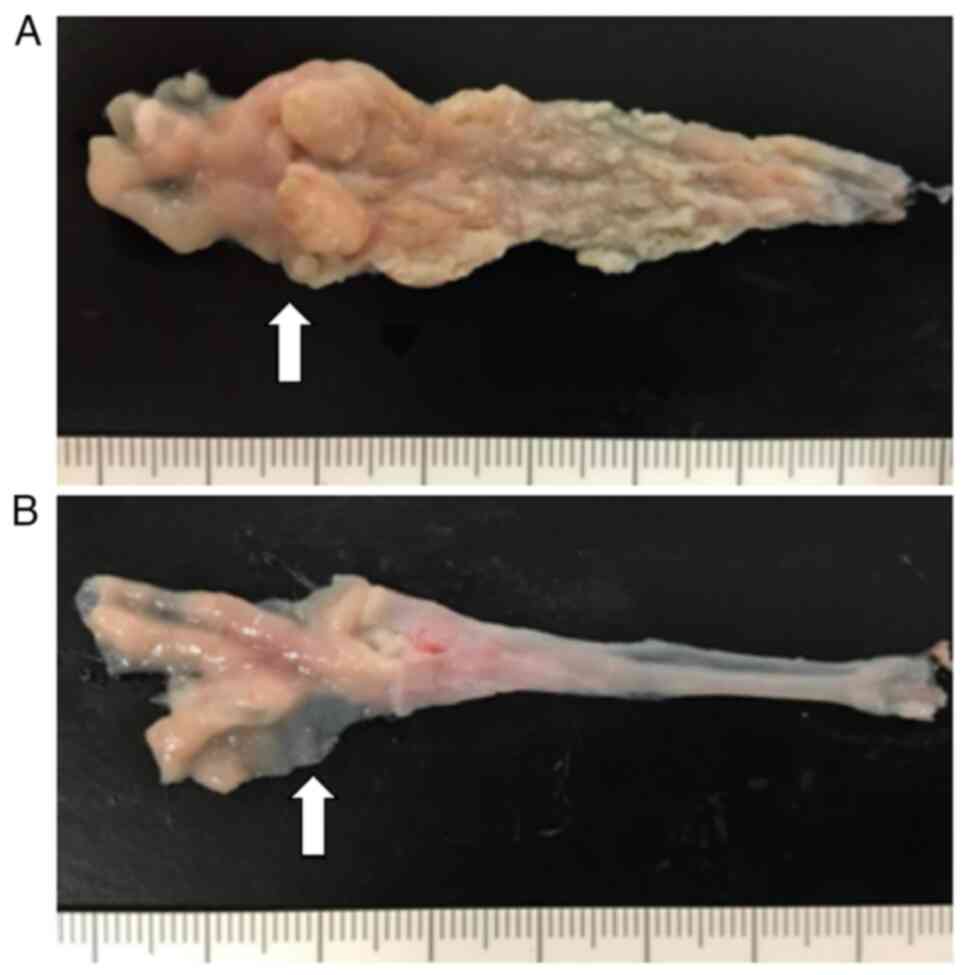
In the control group, the distal portion of the
esophagus exhibited macroscopic thickening and irregularity. Some
areas of the rough epithelium showed small nodular elevations
(Fig. 2A). Severe squamous
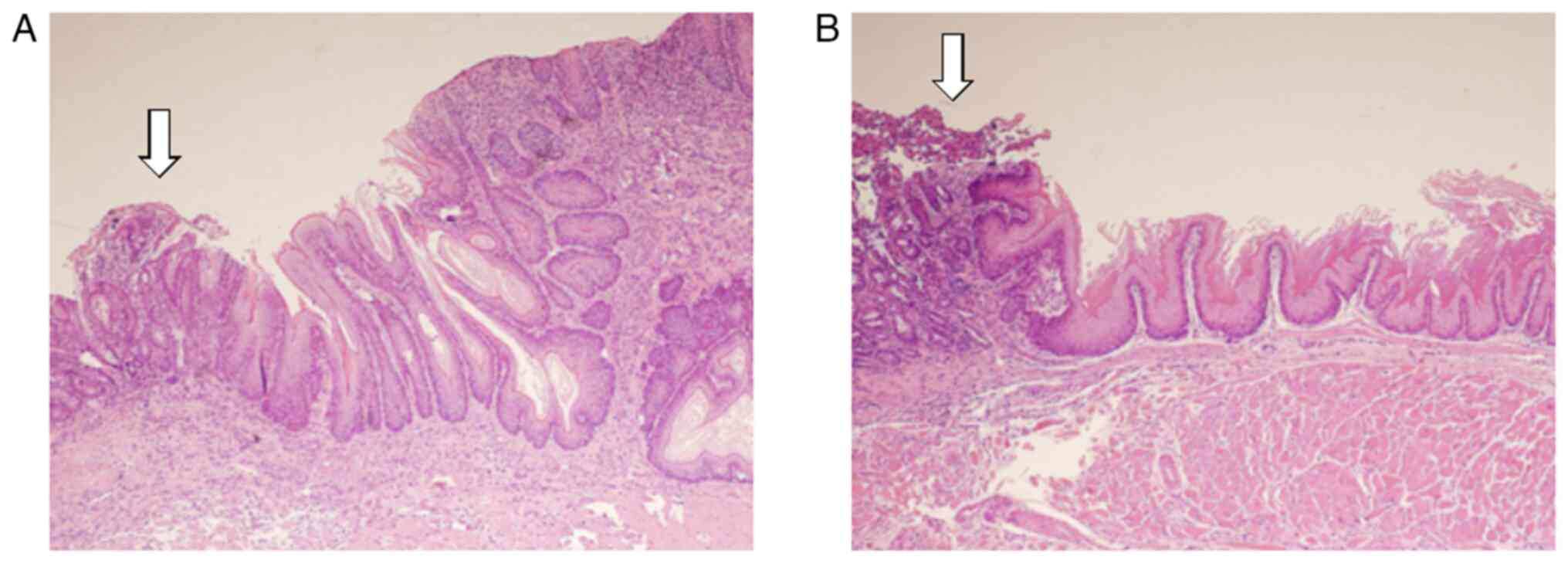
esophagitis was observed in the distal portion of the esophagus
(Fig. 3A). In the presence of
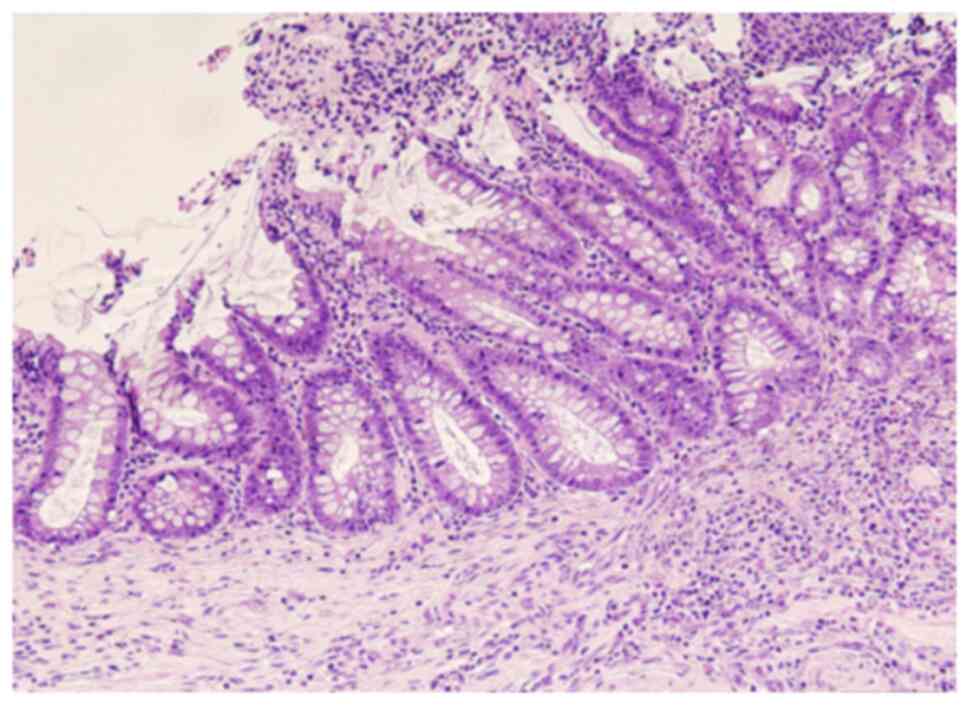
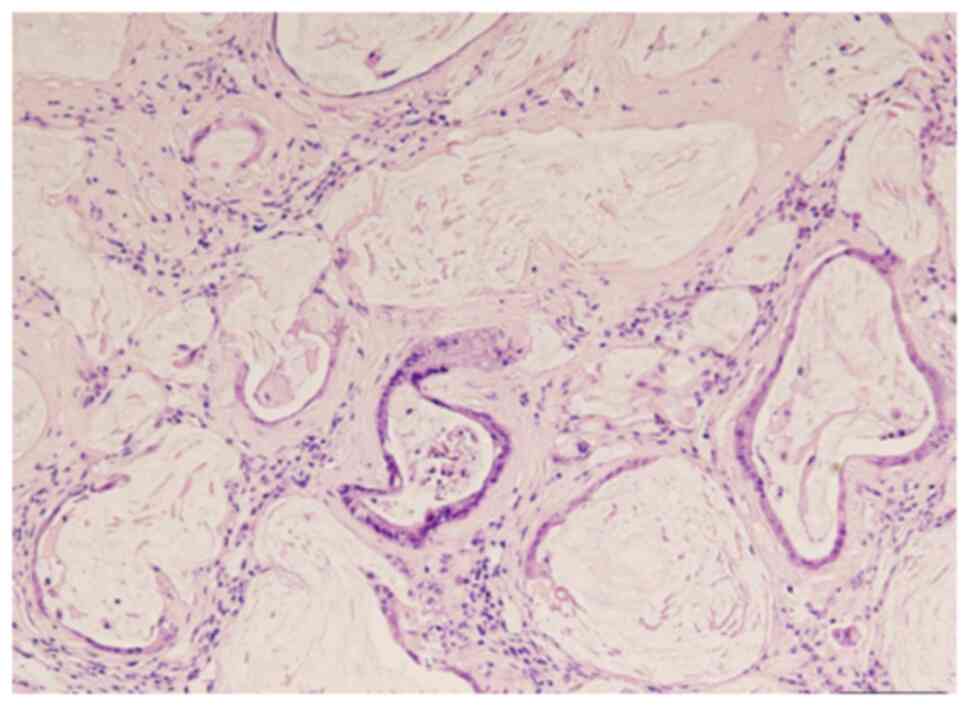
severe esophagitis, BE and EAC developed in the surrounding areas
(Fig. 4 and Fig. 5). BE was observed starting from the
20th week (60%) and progressively increased to 100% by the 40th
week (Table I). EAC was observed
starting from the 20th week (20%) and progressively increased to
69% by the 40th week (Table I).
In the pranlukast group, the distal portions of the
esophagus appeared relatively smooth, and the degree of thickening
was mild (Fig. 2B). Squamous
esophagitis was milder compared to that observed in the control
group (Fig. 3B). RT, BCH, and
erosion were observed, but to a lesser extent. The incidences of BE
and EAC at 40 weeks after surgery were significantly lower in the
pranlukast group compared to the control group (P<0.05)
(Table I). The length of Barrett's
esophagus tended to be shorter in the pranlukast group than in the
control group, but there was no significant difference (Table II).
| Table II.Length of Barrett's esophagus. |
Table II.
Length of Barrett's esophagus.
| Post-operative
week | Group | n | Length of BE,
mm |
|---|
| 20 | Control | 3 | 9.6±4.7 |
|
| Pranlukast | 1 | 3.0±0.0 |
| 30 | Control | 4 | 15.5±5.4 |
|
| Pranlukast | 2 | 9.5±2.1 |
| 40 | Control | 13 | 16.2±8.4 |
|
| Pranlukast | 8 | 10.8±4.8 |
Immunohistochemistry of 5-LOX, CD68,
IL-8, and VEGF
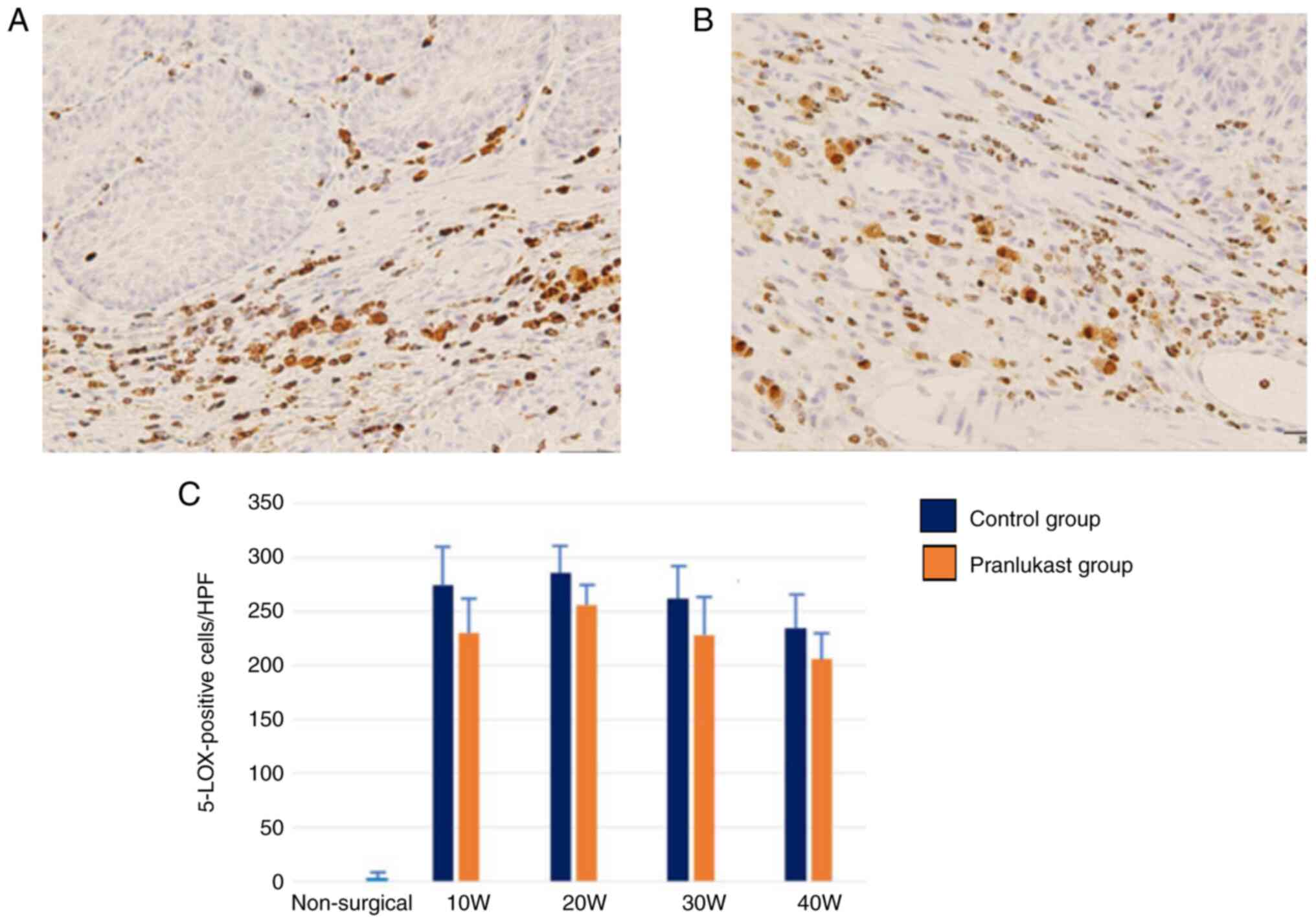
Both the control and pranlukast groups showed 5-LOX
immunoreactivity throughout the study. The primary expression of
5-LOX was observed in infiltrating macrophages, with additional
expression observed in other infiltrating leukocytes such as
neutrophils, mast cells, and eosinophils. A few squamous epithelial
cells also exhibited positive staining for the 5-LOX protein. There
was no significant difference in 5-LOX expression in the submucosal
tissue of the lower esophagus between the control and pranlukast
groups (Fig. 6). In the lower
esophagus of rats in the non-surgical group, 5-LOX immunoreactivity
was barely observed.
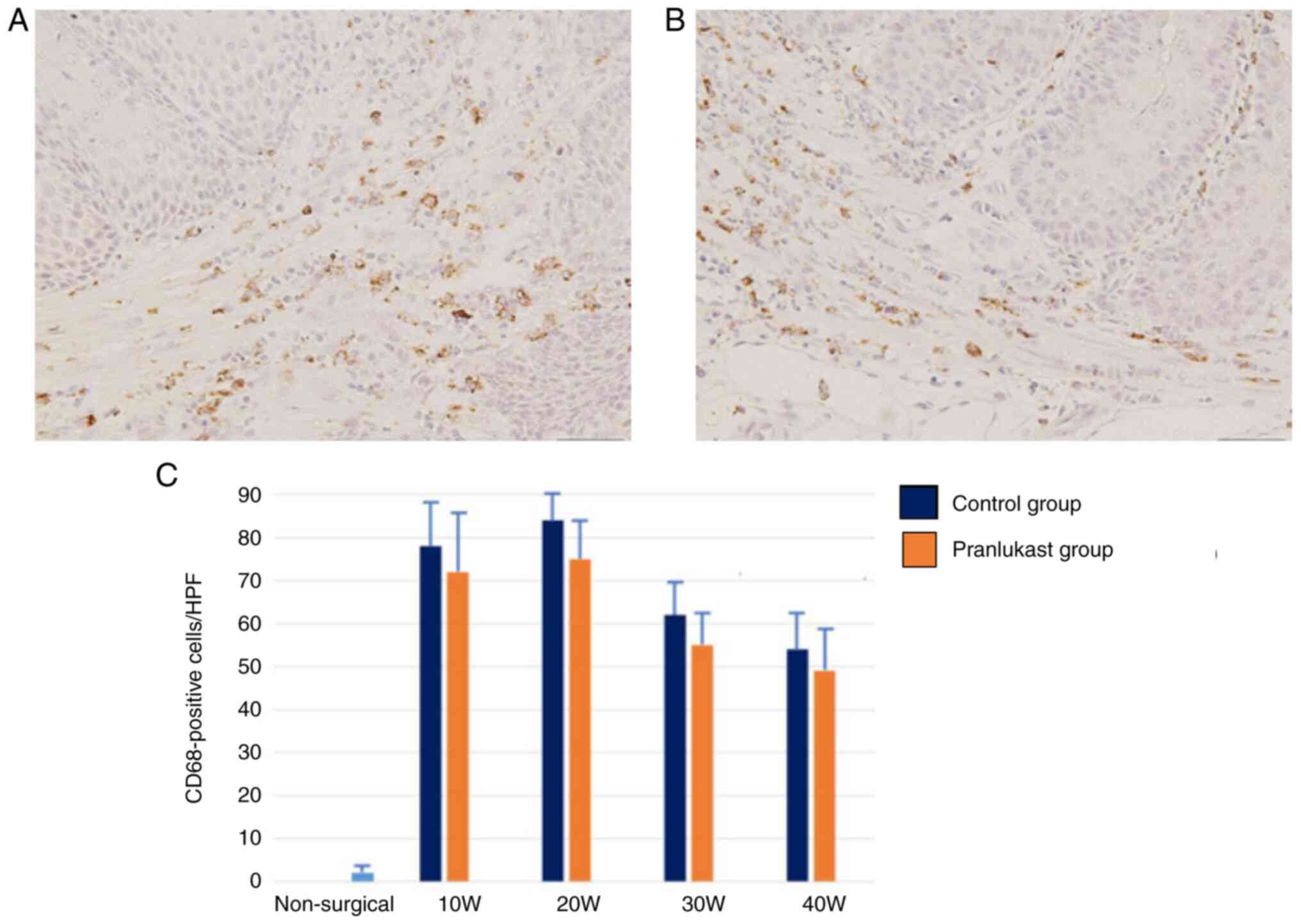
CD68 immunoreactivity indicated the presence of
macrophages in both groups throughout the study. There was no
significant difference in CD68 expression in the submucosal tissue
of the lower esophagus between the control and pranlukast groups
(Fig. 7). In the lower esophagus of
rats in the non-surgical group, CD68 immunoreactivity was barely
observed.
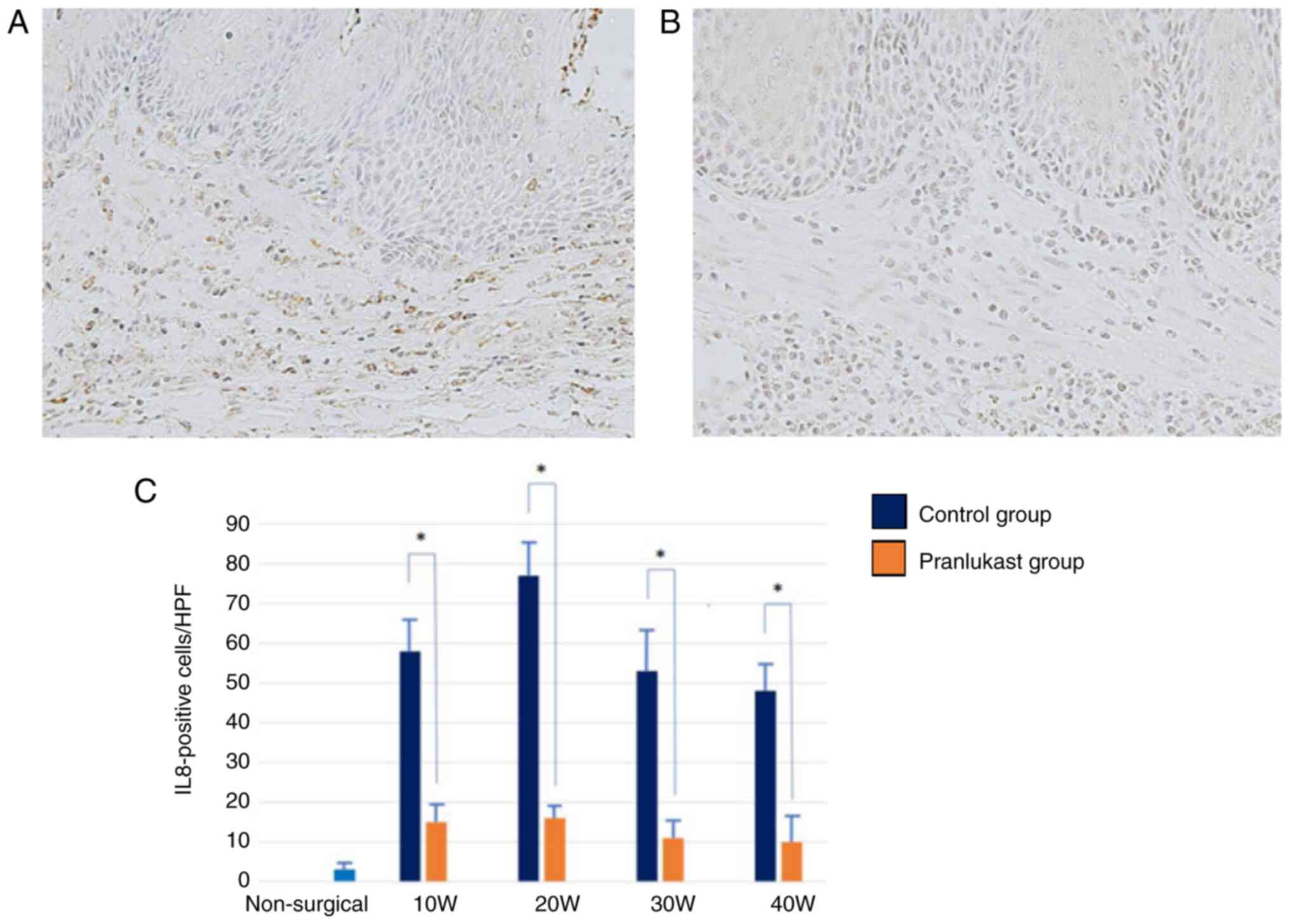
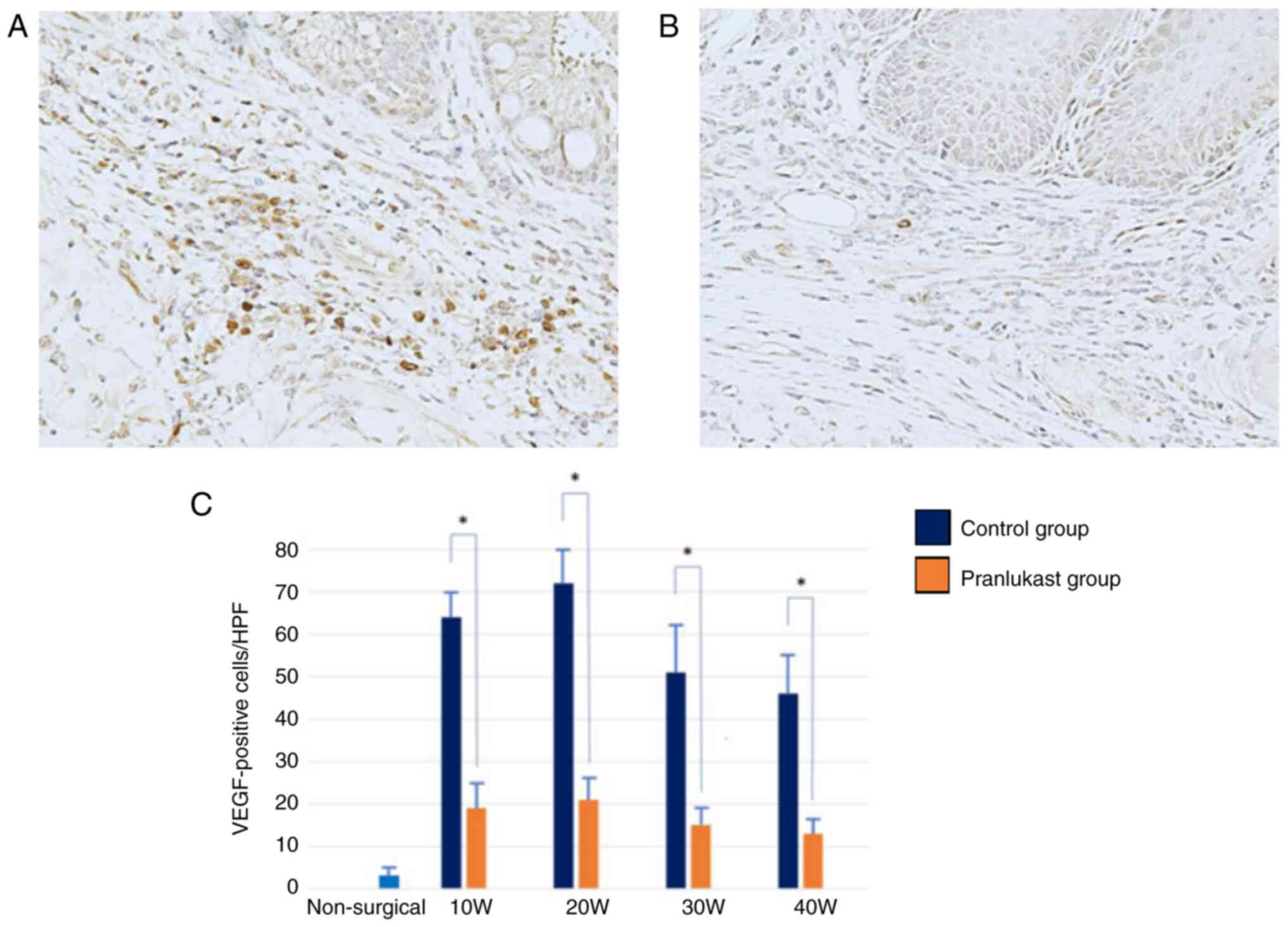
Immunoreactivity for IL-8 and VEGF was observed in
both groups throughout the study. IL-8 and VEGF are primarily
expressed in macrophages. The expression of IL-8 and VEGF increased
in weeks 10 and 20, and then decreased sequentially in weeks 30 and
40 (Fig. 8 and Fig. 9). They were significantly suppressed
in the pranlukast group compared to the control (P <0.05)
(Fig. 8 and Fig. 9). In the lower esophagus of rats in
the non-surgical group, IL-8 and VEGF immunoreactivity was hardly
observed.
Proliferative activity and
apoptosis
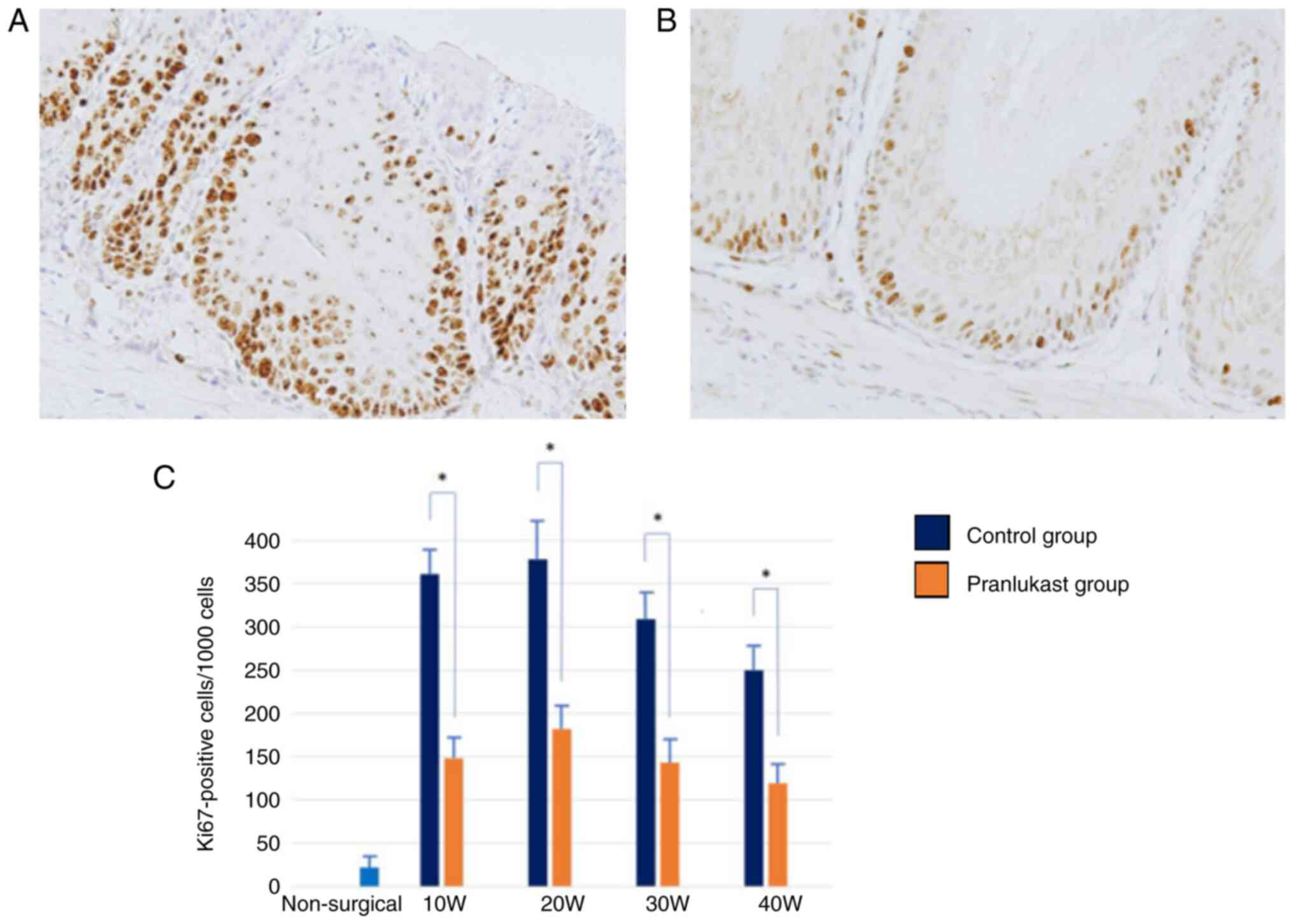
Ki-67 immunoreactivity was observed in the basal
cells of the esophageal epithelium. The prevalence of
Ki-67-positive cells was higher in the esophageal epithelium of
rats with DGER compared to the epithelium of non-surgical rats
(P<0.01). Proliferative activity increased at weeks 10 and 20
and then decreased sequentially at weeks 30 and 40, similar to IL-8
and VEGF expression. Pranlukast significantly suppressed the
proportion of Ki-67 positive cells at weeks 10, 20, 30, and 40
(P<0.05) (Fig. 10).
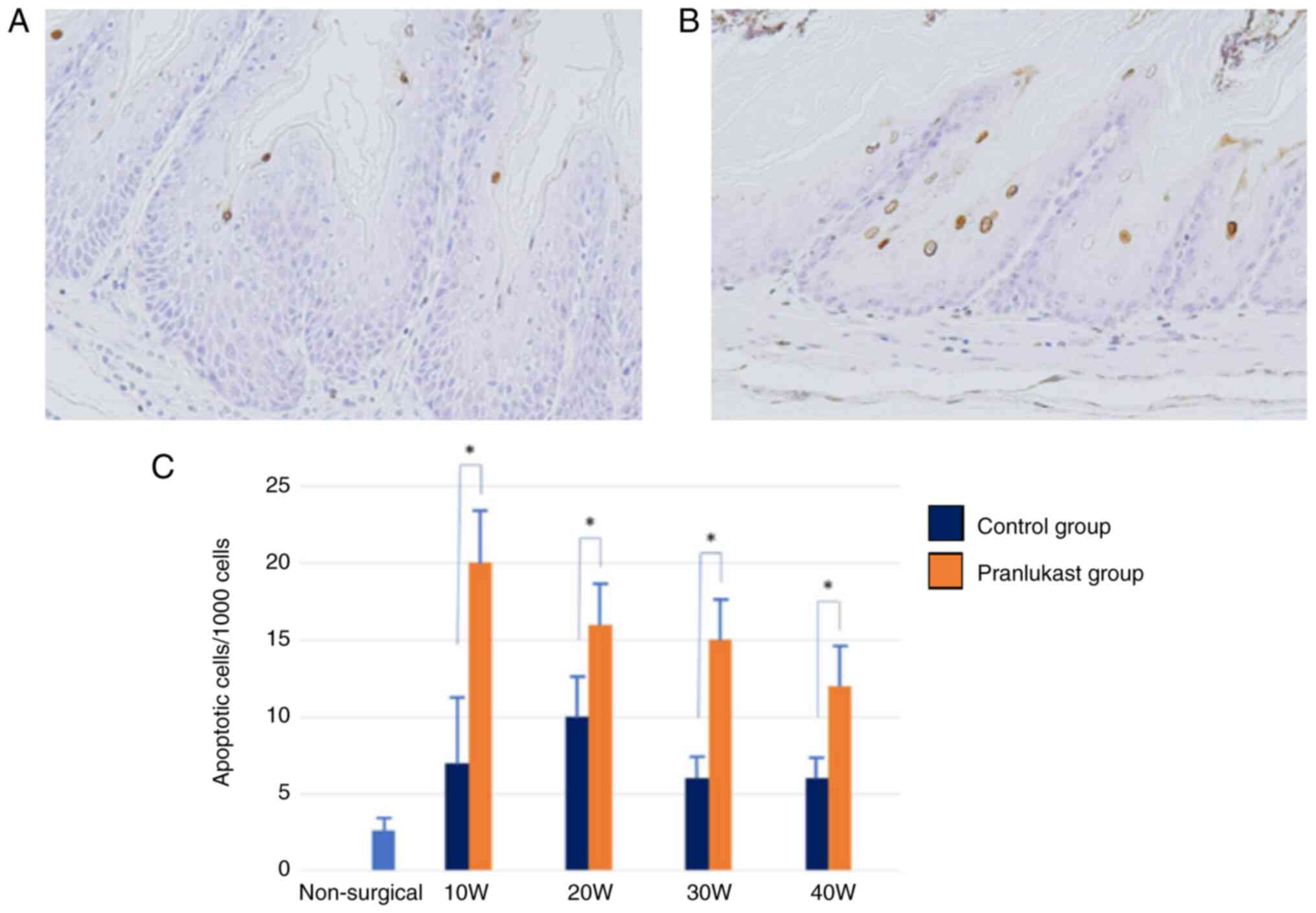
Apoptosis, calculated using the TUNEL method, was
more prevalent in the esophageal epithelium of rats with DGER
compared to the epithelium of non-surgical rats (P<0.05).
Pranlukast significantly increased the proportion of apoptotic
cells at weeks 10, 20, 30, and 40 (P<0.05) (Fig. 11).
Discussion
The present study provides evidence that chronic
reflux of gastric and duodenal contents leads to the development of
esophagitis, BE, and EAC in rats. This process is accompanied by
the upregulation of IL-8 and VEGF, as well as the induction of
5-LOX by infiltrated macrophages. Consequently, increased cell
proliferation and survival occur in the esophageal epithelium. The
CysLT1R antagonist pranlukast effectively suppresses the
upregulation of IL-8 and VEGF, resulting in decreased cell
proliferation and a reduction in the development of BE and EAC.
The 5-LOX pathway plays a crucial role in
inflammation, particularly in inflammatory diseases where it
contributes to cellular damage by catalyzing the production of
leukotrienes. Studies have shown a significant increase in
leukotriene levels in human esophageal biopsy samples from patients
with reflux esophagitis and BE (33). LOX metabolites are thought to act as
important mediators of inflammation and inflammation-associated
esophageal carcinogenesis. In tissues exposed to chronic
inflammation, the continuous activation of the LOX pathway promotes
epithelial cell proliferation.
The observed changes in this study are primarily
triggered by inflammatory infiltrates, with 5-LOX and cytokine
expression predominantly observed in the infiltrated macrophages.
Similar findings have been reported in studies examining chronic
kidney disease models, where macrophages were identified as the
primary source of 5-LOX (34).
Another study reported that macrophages mediate epithelial damage
by producing 5-LOX (35). In the
esophageal epithelium affected by inflammation, 5-LOX metabolites
were strongly detected in the infiltrating inflammatory cells
within the stroma, while they were not detected in the squamous
epithelium (36).
Macrophages are the main sources of IL-8 and VEGF
(37,38). In this study, macrophage
infiltration was not influenced by the CysLT1R antagonist despite
the significant suppression of IL-8 and VEGF expression. The
current study reported that the inhibition of 5-LOX did not appear
to change the infiltration of leukocytes, which is similar to the
results of this our study (34).
Although inflammatory cell infiltration was not influenced by
pranlukast, it inhibited the production of inflammatory cytokines
such as IL-8 and VEGF by suppressing the activity of infiltrating
cells.
The interaction between CysLTs and CysLT1R in innate
immune cells leads to the release of various inflammatory
mediators, including IL-8 and VEGF (20). Several studies have investigated the
role of IL-8 in esophageal carcinogenesis. Fitzgerald et al
found increased IL-8 expression in patients with reflux
esophagitis, particularly at the squamous-columnar junction where
inflammation is most pronounced (39). Nguyen et al reported a
correlation between elevated IL-8 expression and poor prognosis in
esophageal cancer, indicating the pro-tumor role of IL-8 (40). In an in vitro study, IL-8 was
significantly upregulated during esophageal carcinogenesis, and
inhibiting the IL-8 receptor led to reduced invasiveness of
esophageal adenocarcinoma (41).
VEGF is also associated with esophageal carcinogenesis. Möbius
et al observed increased VEGF expression in Barrett's
esophagus and esophageal adenocarcinoma (42). LTD4, the CysLTs, induced VEGF
production and enhanced VEGF release through CysLT1R activation in
human monocytes/macrophages, and these effects were completely
inhibited by pranlukast (43). In
the present study, IL-8 and VEGF were found to be overexpressed
under chronic inflammatory conditions and were effectively
suppressed by pranlukast, consistent with previous findings. This
strongly suggests that IL-8 and VEGF play a significant role in
inflammation-induced carcinogenesis.
The chemopreventive effects of CysLT1R antagonists
have been demonstrated in large retrospective cohort studies
(44,45). Tsai et al showed that the use
of a CysLT1R antagonist reduced cancer risk in a dose-dependent
manner in patients with asthma compared to non-users of CysLT1R
antagonists (44). In the present
study, an increase in apoptosis cells was observed in the Barrett
epithelium in the pranlukast group from 10 to 40 weeks
postoperatively. These results suggest that pranrukast induces
apoptosis not only in esophageal adenocarcinoma cells, but also in
metaplastic cells of Barrett's esophagus or its progenitor atypical
cells in a background of chronic inflammation. Previously,
Hormi-Carver et al reported that all trans-retinoic acid
induces via p38 and caspase pathway in a non-neoplastic,
metaplastic Barret's cell line, thus retinoid treatment inhibits
carcinogenesis (46). Although
further studies are needed to elucidate the mechanism by which
pranlukast induces apoptosis of non-neoplastic metaplastic cells
via inhibition of the 5-LOX pathway, the present findings indicate
the potential use of CysLT1R antagonists for chemoprevention in
Barrett's esophagus. Clinical studies on colorectal, pancreatic,
urological, and breast cancers have reported upregulation of
CysLT1R expression in cancer tissues compared to normal tissues,
and this upregulation is associated with poor prognosis (25,26,47,48).
CysLT1R expression has also been detected in human colon cancer
cell lines, and its overexpression enhances cell viability
(25). Furthermore, Bellamkonda
et al found that LTD4 promoted tumor growth induced by
cancer-initiating cells in a xenograft model of nude mice injected
with human colon cancer cells (49). The LOX pathway not only influences
oncogenesis but also cancer progression through the interaction
between cancer cells and stromal cells. While this study did not
specifically assess CysLT1R-expressing cells, the CysLT1R
antagonist is expected to have a dual antitumor effect by targeting
both macrophages and cancer cells.
The dose of pranrukast administered to the DGER
model in this study (3.3 mg/kg/day) was less than the human dose
given in the treatment of asthma and allergic rhinitis (around 9
mg/kg/day). Also, given that the toxic dose in rats is considered
to be 1000 mg/kg/day or higher, the dose used in our study is
considered to be safe enough to be clinically applicable to
humans.
In this study, we investigated the anti-carcinogenic
effects of pranlukast in a rat model. These results indicate that
the administration of a CysLT1R antagonist may suppress esophageal
carcinogenesis associated with the IMA sequence. However, this
topic has not yet been fully elucidated. Further studies are
required to completely understand the relationship between
esophageal carcinogenesis and the LOX pathway.
Acknowledgements
The authors would like to thank Ms. Futakuchi
(Department of Gastrointestinal Surgery Kanazawa University,
Kanazawa, Japan) for their help with the immunohistochemistry.
Funding
Funding: No funding was received.
Availability of data and materials
All data generated or analyzed during the present
study are included in the published article.
Authors' contributions
TK, JK, KO, HS, MS, TT, DY, HM, NI and TO
contributed to the study conception and design. Material
preparation, data collection was performed by TK, KO, HS, and MS.
Data analysis were performed by JK, TT, DY and HM. TK and KO
confirm the authenticity of all raw data. The first draft of the
manuscript was written by TK, NI and TO, and all authors commented
on previous versions of the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All animal studies were reviewed and approved by The
Ethics Committee of Kanazawa University (Kanazawa, Japan; approval
no. 153650). All animal procedures were performed according to
approved protocols from the Institutional Animal Care and Use
Committee (IACUC) of Kanazawa University.
Patient consent for publication
Not applicable.
Authors' information
Professor Jun Kinoshita, ORCID:
0000-0002-2871-9549.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pera M: Epidemiology of esophageal cancer,
especially adenocarcinoma of the esophagus and esophagogastric
junction. Recent Results Cancer Res. 115:1–14. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pohl H and Welch HG: The role of
overdiagnosis and reclassification in the marked increase of
esophageal adenocarcinoma incidence. J Natl Cancer Inst.
97:142–146. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Winters C Jr, Spurling TJ, Chobanian SJ,
Curtis DJ, Esposito RL, Hacker JF III, Johnson DA, Cruess DF,
Cotelingam JD, Gurney MS, et al: Barrett's esophagus. A prevalent,
occult complication of gastroesophageal reflux disease.
Gastroenterology. 92:118–124. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Reid BJ: Barrett's esophagus and
esophageal adenocarcinoma. Gastroenterol Clin North Am. 20:817–834.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gillen P, Keeling P, Byrne PJ, Healy M,
O'Moore RR and Hennessy TP: Implication of duodenogastric reflux in
the pathogenesis of Barrett's oesophagus. Br J Surg. 75:540–543.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Falk GW: Barrett's esophagus.
Gastroenterology. 122:1569–1591. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kauer WK, Peters JH, DeMeester TR, Ireland
AP, Bremner CG and Hagen JA: Mixed reflux of gastric and duodenal
juices is more harmful to the esophagus than gastric juice alone.
The need for surgical therapy re-emphasized. Ann Surg. 222:525–533.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fujimura T, Oyama K, Sasaki S, Nishijima
K, Miyashita T, Ohta T, Miwa K and Hattori T: Inflammation-related
carcinogenesis and prevention in esophageal adenocarcinoma using
rat duodenoesophageal reflux models. Cancers (Basel). 3:3206–3224.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jankowski JA, Harrison RF, Perry I,
Balkwill F and Tselepis C: Barrett's metaplasia. Lancet.
356:2079–2085. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Stein HJ, Feussner H, Kauer W, DeMeester
TR and Siewert JR: Alkaline gastroesophageal reflux: Assessment by
ambulatory esophageal aspiration and pH monitoring. Am J Surg.
167:163–168. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Iftikhar SY, Ledingham S, Steele RJ, Evans
DF, Lendrum K, Atkinson M and Hardcastleet JD: Bile reflux in
columnar-lined Barrett's oesophagus. Ann R Coll Surg Engl.
75:411–416. 1993.PubMed/NCBI
|
|
12
|
Lagergren J, Bergström R, Lindgren A and
Nyrén O: Symptomatic gastroesophageal reflux as a risk factor for
esophageal adenocarcinoma. N Engl J Med. 340:825–831. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Miwa K, Segawa M, Takano Y, Matsumoto H,
Sahara H, Yagi M, Miyazaki I and Hattori T: Induction of
oesophageal and forestomach carcinomas in rats by reflux of
duodenal contents. Br J Cancer. 70:185–189. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Miwa K, Sahara H, Segawa M, Kinami S, Sato
T, Miyazaki I and Hattori T: Reflux of duodenal or gastro-duodenal
contents induces esophageal carcinoma in rats. Int J Cancer.
67:269–274. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sato T, Miwa K, Sahara H, Segawa M and
Hattori T: The sequential model of Barrett's esophagus and
adenocarcinoma induced by duodeno-esophageal reflux without
exogenous carcinogens. Anticancer Res. 22:39–44. 2002.PubMed/NCBI
|
|
16
|
Goldstein SR, Yang GY, Curtis SK, Reuhl
KR, Liu BC, Mirvish SS, Newmark HL and Yanget CS: Development of
esophageal metaplasia and adenocarcinoma in a rat surgical model
without the use of a carcinogen. Carcinogenesis. 18:2265–2270.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fein M, Peters JH, Chandrasoma P, Ireland
AP, Oberg S, Ritter MP, Bremner CG, Hagen JA and DeMeesteret TR:
Duodenoesophageal reflux induces esophageal adenocarcinoma without
exogenous carcinogen. J Gastrointest Surg. 2:260–268. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oyama K, Fujimura T, Ninomiya I, Miyashita
T, Kinami S, Fushida S, Ohta T and Miwa K: A COX-2 inhibitor
prevents the esophageal inflammation-metaplasia-adensocarcinoma
sequence in rats. Carcinogenesis. 26:565–570. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Peters-Golden M, Gleason MM and Togias A:
Cysteinyl leukotrienes: Multi-functional mediators in allergic
rhinitis. Clin Exp Allergy. 36:689–703. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Theron AJ, Steel HC, Tintinger GR, Gravett
CM, Anderson R and Feldman C: Cysteinyl leukotriene receptor-1
antagonists as modulators of innate immune cell function. J Immunol
Res. 2014:6089302014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Byrum RS, Goulet JL, Snouwaert JN,
Griffiths RJ and Koller BH: Determination of the contribution of
cysteinyl leukotrienes and leukotriene B4 in acute inflammatory
responses using 5-lipoxygenase- and leukotriene A4
hydrolase-deficient mice. J Immunol. 163:6810–6819. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Subramanian BC, Majumdar R and Parent CA:
The role of the LTB4-BLT1 axis in chemotactic gradient sensing and
directed leukocyte migration. Semin Immunol. 33:16–29. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tong WG, Ding XZ, Witt RC and Adrian TE:
Lipoxygenase inhibitors attenuate growth of human pancreatic cancer
xenografts and induce apoptosis through the mitochondrial pathway.
Mol Cancer Ther. 1:929–935. 2002.PubMed/NCBI
|
|
24
|
Wang D and Dubois RN: Eicosanoids and
cancer. Nat Rev Cancer. 10:181–193. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ohd JF, Nielsen CK, Campbell J, Landberg
G, Löfberg H and Sjölander A: Expression of the leukotriene D4
receptor CysLT1, COX-2, and other cell survival factors in
colorectal adenocarcinomas. Gastroenterology. 124:57–70. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Matsuyama M, Hayama T, Funao K, Kawahito
Y, Sano H, Takemoto Y, Nakatani T and Yoshimura R: Overexpression
of cysteinyl LT1 receptor in prostate cancer and CysLT1R antagonist
inhibits prostate cancer cell growth through apoptosis. Oncol Rep.
18:99–104. 2007.PubMed/NCBI
|
|
27
|
Moore GY and Pidgeon GP: Cross-talk
between cancer cells and the tumour microenvironment: The role of
the 5-lipoxygenase pathway. Int J Mol Sci. 18:2362017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Boger PC, Shutt JD, Neale JR, Wilson SJ,
Bateman AC, Holloway JW, Patel P and Sampson AP: Increased
expression of the 5-lipoxygenase pathway and its cellular
localization in Barrett's adenocarcinoma. Histopathology.
61:509–517. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shutt JD, Boger P, Neale JR, Patel P and
Sampson AP: Activity of the leukotriene pathway in Barrett's
metaplasia and oesophageal adenocarcinoma. Inflamm Res.
61:1379–1384. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang H, Jia X, Chen X, Yang CS and Li N:
Time-selective chemoprevention of vitamin E and selenium on
esophageal carcinogenesis in rats: The possible role of nuclear
factor kappaB signaling pathway. Int J Cancer. 131:1517–1527. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li N, Sood S, Wang S, Fang M, Wang P, Sun
Z, Yang CS and Chen X: Overexpression of 5-lipoxygenase and
cyclooxygenase 2 in hamster and human oral cancer and
chemopreventive effects of zileuton and celecoxib. Clin Cancer Res.
11:2089–2096. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nozaki M, Yoshikawa M, Ishitani K,
Kobayashi H, Houkin K, Imai K, Ito Y and Muraki T: Cysteinyl
leukotriene receptor antagonists inhibit tumor metastasis by
inhibiting capillary permeability. Keio J Med. 59:10–18. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Triadafilopoulos G, Kaczynska M and Iwane
M: Esophageal mucosal eicosanoids in gastroesophageal reflux
disease and Barrett's esophagus. Am J Gastroenterol. 91:65–74.
1996.PubMed/NCBI
|
|
34
|
Montford JR, Bauer C, Dobrinskikh E, Hopp
K, Levi M, Weiser-Evans M, Nemenoff R and Furgeson SB: Inhibition
of 5-lipoxygenase decreases renal fibrosis and progression of
chronic kidney disease. Am J Physiol Renal Physiol. 316:F732–F742.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Peters-Golden M and Henderson WR Jr:
Leukotrienes. N Engl J Med. 357:1841–1854. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen X, Li N, Wang S, Wu N, Hong J, Jiao
X, Krasna MJ, Beer DG and Yang CS: Leukotriene A4 hydrolase in rat
and human esophageal adenocarcinomas and inhibitory effects of
bestatin. J Natl Cancer Inst. 95:1053–1061. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Arango Duque G and Descoteaux A:
Macrophage cytokines: Involvement in immunity and infectious
diseases. Front Immunol. 5:4912014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Maruyama K, Kidoya H, Takemura N, Sugisawa
E, Takeuchi O, Kondo T, Eid MMA, Tanaka H, Martino MM, Takakura N,
et al: Zinc finger protein St18 protects against septic death by
inhibiting VEGF-A from macrophages. Cell Rep. 32:1079062020.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fitzgerald RC, Onwuegbusi BA,
Bajaj-Elliott M, Saeed IT, Burnham WR and Farthing MJG: Diversity
in the oesophageal phenotypic response to gastro-oesophageal
reflux: Immunological determinants. Gut. 50:451–459. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nguyen GH, Schetter AJ, Chou DB, Bowman
ED, Zhao R, Hawkes JE, Mathé EA, Kumamoto K, Zhao Y, Budhu A, et
al: Inflammatory and microRNA gene expression as prognostic
classifier of Barrett's-associated esophageal adenocarcinoma. Clin
Cancer Res. 16:5824–5834. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shrivastava MS, Hussain Z, Giricz O,
Shenoy N, Polineni R, Maitra A and Verma A: Targeting chemokine
pathways in esophageal adenocarcinoma. Cell Cycle. 13:3320–3327.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Möbius C, Stein HJ, Becker I, Feith M,
Theisen J, Gais P, Jütting U and Siewert JR: The ‘angiogenic
switch’ in the progression from Barrett's metaplasia to esophageal
adenocarcinoma. Eur J Surg Oncol. 29:890–894. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Haneda Y, Hasegawa S, Hirano R, Hashimoto
K, Ohsaki A and Ichiyama T: Leukotriene D4 enhances tumor necrosis
factor-α-induced vascular endothelial growth factor production in
human monocytes/macrophages. Cytokine. 55:24–28. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tsai MJ, Wu PH, Sheu CC, Hsu YL, Chang WA,
Hung JY, Yang CJ, Yang YH, Kuo PL and Huang MS: Cysteinyl
leukotriene receptor antagonists decrease cancer risk in asthma
patients. Sci Rep. 6:239792016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sutton SS, Magagnoli J, Cummings TH and
Hardin JW: Leukotriene inhibition and the risk of lung cancer among
U.S. veterans with asthma. Pulm Pharmacol Ther. 71:1020842021.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hormi-Carver K, Feagins L, Spechler S and
Souza R: All trans-retinoic acid induces apoptosis via p38 and
caspase pathways in metaplastic Barrett's cells. Am J Physiol
Gastrointest Liver Physiol. 292:G18–G27. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kachi K, Kato H, Naiki-Ito A, Komura M,
Nagano-Matsuo A, Naitoh I, Hayashi K, Kataoka H, Inaguma S and
Takahashi S: Anti-allergic drug suppressed pancreatic
carcinogenesis via down-regulation of cellular proliferation. Int J
Mol Sci. 22:74442021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Magnusson C, Liu J, Ehrnström R, Manjer J,
Jirström K, Andersson T and Sjölander A: Cysteinyl leukotriene
receptor expression pattern affects migration of breast cancer
cells and survival of breast cancer patients. Int J Cancer.
129:9–22. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bellamkonda K, Chandrashekar NK, Osman J,
Selvanesan BC, Savari S and Sjölander A: The eicosanoids
leukotriene D4 and prostaglandin E2 promote the tumorigenicity of
colon cancer-initiating cells in a xenograft mouse model. BMC
Cancer. 16:4252016. View Article : Google Scholar : PubMed/NCBI
|