Introduction
Sarcoma is a mesenchymal-derived tumor that accounts
for ~1% of all cancers and has a particularly high incidence in
children, accounting for ~20% of pediatric solid cancers (1,2). The
World Health Organization classifies sarcomas into soft tissue and
bone tumors, and numerous subtypes have been identified (3). Sarcomas can occur in various
locations, including the abdominal cavity, arms, legs, and head and
neck. The prognoses of sarcomas vary depending on their location
and subtype. The 5-year survival rate for sarcomas is >50%
(4,5).
The number of cancer survivors is increasing,
leading to an increased occurrence of secondary malignancies
(6,7). Secondary malignancy is defined as the
development of a new primary tumor unrelated to the recurrence or
metastasis of the initial tumor (8). Owing to the growing interest in
secondary malignancies, their incidence, risk factors and screening
have been studied (9–11). Previous studies suggested that age
and treatment dose as well as the well-known chemotherapy and
radiotherapy (RT) were associated with the development of secondary
hematological malignancies (SHMs) (12–15).
Regarding the epidemiology of sarcoma, young adults
and adolescents comprise 30–50% of the patient population, in which
long-term survival has been reported (1). However, the common treatment methods,
cytotoxic chemotherapy and radiation therapy, can increase the risk
of secondary malignancies; thus, the incidence of these
malignancies in patients with sarcoma is expected to increase
(16–19). Nevertheless, research on SHMs in
sarcoma, including the clinical characteristics, SHM types, and
prognosis of affected patients, is scarce (20–22).
Furthermore, previous studies have focused only on specific
subgroups including young patients, subtypes of sarcoma, or
therapy-related acute myeloid leukemia (t-AML) alone (20,22,23).
Therefore, the present retrospective study
investigated the clinical characteristics and prognoses of all SHMs
in patients with sarcoma.
Materials and methods
Study design and patients
A retrospective analysis of the data of patients
diagnosed with SHMs after receiving treatment for sarcoma was
performed at the Korea Cancer Center Hospital (Seoul, South Korea)
between January 2000 and May 2023 (Seoul, Korea). The inclusion
criteria were: (i) Pathological diagnosis of sarcoma, ii) sarcoma
treatment history, and iii) SHMs diagnosis after sarcoma treatment.
The exclusion criterion was sarcoma, identified as a secondary
malignancy that developed after a primary cancer diagnosis. The
present study was approved (approval no. 2023-05-004) by the
Institutional Review Board of Korea Cancer Center Hospital. Due to
the retrospective nature of the current analysis, informed consent
was not required for the present study, which was conducted in
accordance with the tenets of the 2013 Declaration of Helsinki.
Data collection
Clinical characteristics were obtained from the
patients' medical records. The following variables were analyzed:
Age, sex, date of diagnosis of sarcoma, pathological diagnosis of
the sarcoma, stage of the sarcoma, primary site of the sarcoma,
whether treatment was performed, chemotherapy regimen, number of
chemotherapy cycles, whether radiation therapy was performed, date
of SHM diagnosis, SHM type, sarcoma status at the time of its
diagnosis, latency between the dates of sarcoma and SHM diagnoses,
SHM treatment, and date of patient's death.
Statistical analysis
Categorical variables are presented as numbers and
proportions, while continuous variables are described as medians
(ranges). Latency was defined as the time interval between the
diagnosis of sarcoma and the occurrence of SHMs. Overall survival
(OS) indicated the period from SHM diagnosis to death, regardless
of the cause. Statistical analyses were performed using IBM SPSS
Statistics for Windows, version 26.0 (IBM Corp.).
Results
From January 2020 to May 2023, 2,953 patients were
diagnosed with sarcoma and received treatment, including
chemotherapy, in 1,658 patients. Among the patients diagnosed with
sarcoma, 18 who developed SHMs were ultimately enrolled in the
present study. The patient details are summarized in Fig. 1.
Patient characteristics
The clinical features of the 18 patients included in
the present study are included in Table
I. Their median age was 39.5 (range, 9–72) years, with a
predominance of men (n=14, 77.7%). Osteosarcoma accounted for half
of the cases (n=9, 50%), followed by undifferentiated pleomorphic
sarcoma (n=4, 22.2%) and Ewing's sarcoma (n=2, 11.1%). Regarding
the initial disease stage, most patients had localized disease
(n=14, 77.7%), whereas a small proportion had metastatic disease
(n=4, 22.2%). The most frequent primary tumor sites were the femur
and thigh (n=6; 33.3%), followed by the tibia (n=3; 16.5%) and
buttocks (n=1; 5.5%). All patients (n=18, 100%) underwent surgery
as part of their treatment and did not receive RT. Most patients
(n=16, 88.8%) received chemotherapy.
 | Table I.Baseline patient characteristics
(n=18). |
Table I.
Baseline patient characteristics
(n=18).
| Characteristic | Value |
|---|
| Sex, n (%) |
|
| Male | 14 (77.7) |
|
Female | 4 (22.3) |
| Median age at
diagnosis of sarcoma (range), years | 39.5 (9–72) |
| Sarcoma histology, n
(%) |
|
|
Osteosarcoma | 9 (50.0) |
| Ewing
sarcoma | 2 (11.1) |
|
Chondrosarcoma | 1 (5.5) |
|
Liposarcoma | 1 (5.5) |
|
Undifferentiated pleomorphic
sarcoma | 4 (22.2) |
| Synovial
sarcoma | 1 (5.5) |
| Stage of sarcoma at
initial diagnosis, n (%) |
|
|
Localized | 14 (77.7) |
|
Metastatic | 4 (22.2) |
| Primary site of
tumor, n (%) |
|
|
Femur | 6 (33.3) |
|
Thigh | 6 (33.3) |
|
Tibia | 3 (16.6) |
|
Buttock | 1 (5.5) |
| Chest
wall | 1 (5.5) |
|
Axilla | 1 (5.5) |
| Sarcoma treatment,
n (%) |
|
|
Surgerya |
|
|
Yes | 18 (100.0) |
|
No | 0 (0.0) |
|
Chemotherapya |
|
|
Yes | 16 (88.8) |
| No | 2 (11.1) |
| Radiotherapy |
|
|
Yes | 0 (0.0) |
| No | 18 (100.0) |
| SHM type, n
(%) |
|
|
Myelodysplastic syndrome | 5 (27.7) |
| Acute
myelogenous leukemia | 6 (33.3) |
| Acute
lymphocytic leukemia | 3 (16.6) |
|
Aplastic anemia | 1 (5.5) |
| Chronic
myeloid leukemia | 1 (5.5) |
|
Follicular lymphoma | 1 (5.5) |
|
Multiple myeloma | 1 (5.5) |
| Median age at SHM
diagnosis (range), years | 44 (10–77) |
| Median time from
sarcoma diagnosis to | 30 (11–121) |
| SHM diagnosis
(range), months |
|
| SHM treatment, n
(%) |
|
|
Yes | 13 (72.2) |
| No | 5 (27.7) |
| Sarcoma status at
SHM diagnosis, n (%) |
|
|
NED | 9 (50.0) |
| Local
recurrence | 1 (5.5) |
| Distant
recurrence | 8 (44.4) |
Chemotherapy
The data of the 16 patients who received
chemotherapy for sarcoma treatment are presented in Table II. A median of 8.5 chemotherapy
cycles was administered (range, 3–21 cycles). Among these patients,
six received more than third-line chemotherapy. Each patient
received a median of five chemotherapy agents (range, 2–8). The
most commonly used chemotherapeutic agent was
doxorubicin/adriamycin (15 patients, 83.3%), followed by ifosfamide
and cisplatin [13 (72.2%) and 11 (61.1%) patients, respectively].
Among topoisomerase II inhibitors, doxorubicin/adriamycin (15
patients, 83.3%) and etoposide (eight patients, 44.4%) were
commonly administered. Among alkylating agents, ifosfamide,
cisplatin, carboplatin and dacarbazine were administered to 13
(72.2%), 11 (61.1%), eight (44.4%), and four (22.2%) patients,
respectively. Additionally, seven (38.8%) and one (5.5%) patients
received methotrexate and gemcitabine, respectively. A detailed
list of the chemotherapeutic agents administered is provided in
Table SI.
| Table II.Clinical and therapeutic details and
outcomes of the study patients. |
Table II.
Clinical and therapeutic details and
outcomes of the study patients.
| Patient no. | Age at sarcoma
diagnosis (years) | Sex | Histology | Chemotherapy
regimen | Sarcoma status at
the time of SHM diagnosis | SHM | Cytogenetics | Latency
(months) | SHM treatment | Treatment
regimen | Current status | Median OS time
after SHM diagnosis (months) |
|---|
|
|---|
| Chromosome | FISH |
|---|
| 1 | 24 | M | Osteosarcoma | MMCA (6
cycles) | Distant | MDS |
| del (7q) | 59.3 | N | Not treated | Dead | 3.3 |
|
|
|
|
| MMIB (6
cycles) | recurrence |
|
|
|
|
|
|
|
|
| 2 | 57 | F | Synovial
sarcoma | MAID (1 cycle) | Distant | MDS |
| del (7q) | 14.6 | N | Not treated | Dead | 3.2 |
|
|
|
|
| IA (6 cycles) | recurrence |
|
|
|
|
|
|
|
|
|
|
|
|
| VIP (2 cycles) |
|
|
|
|
|
|
|
|
|
| 3 | 68 | M | Osteosarcoma | Not treated | NED | MDS | 46, XY, del
(20) (q11,2) |
| 112.4 | N | Not treated | Alive | 29.7+ |
|
|
|
|
|
|
|
| [13]/46, idem, der
(Y) t |
|
|
|
|
|
|
|
|
|
|
|
|
|
| (Y;1) (q12: q12),
del (11) |
|
|
|
|
|
|
|
|
|
|
|
|
|
| (q14) [7] |
|
|
|
|
|
|
| 4 | 59 | F | UPS | MMCA (6
cycles) | NED | AML | −7[13] |
| 21.0 | N | Not treated | Dead | 6.2 |
| 5 | 47 | M | Chondrosarcoma | MAID (2
cycles) | NED | FL | Not tested |
| 11.2 | Y | IFRT | Alive | 38.7+ |
|
|
|
|
| ICE (2 cycles) |
|
|
|
|
|
|
|
|
|
|
|
|
|
| CYVADIC (4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| cycles) |
|
|
|
|
|
|
|
|
|
| 6 | 18 | F | Ewing sarcoma | IA (6 cycles) | NED | AML | Not tested |
| 31.2 | Y | HDAC | Alive | 154.9+ |
|
|
|
|
|
|
|
|
|
|
|
| induction- |
|
|
|
|
|
|
|
|
|
|
|
|
|
| HSCT |
|
|
| 7 | 16 | F | Osteosarcoma | (MM)CA | Distant | AML | inv(11)(p15q22) |
| 25.7 | Y | AD | Dead | 13.9 |
|
|
|
|
| (2 cycles) | recurrence |
| [17]/46,ide |
|
|
| induction- |
|
|
|
|
|
|
| (M)ICE (4
cycles) |
|
|
m,del(13)(q12q14)[3] |
|
|
| f/u loss |
|
|
|
|
|
|
| CA (6 cycles) |
|
|
|
|
|
|
|
|
|
| 8 | 44 | M | UPS | MAID (2
cycles) | Distant | ALL |
t(4;11)(q21;q23)[20] |
| 49.5 | Y | CTX+PD | Dead | 0.9 |
|
|
|
|
| MMCA (2
cycles) | recurrence |
|
|
|
|
|
|
|
|
|
|
|
|
| IP (2 cycles) |
|
|
|
|
|
|
|
|
|
|
|
|
|
| ICE (8 cycles) |
|
|
|
|
|
|
|
|
|
| 9 | 21 | M | Osteosarcoma | MMCA (6
cycles) | NED | AML | inv(16)(p13.1q22) |
| 18.5 | Y | AD | Alive | 88.3+ |
|
|
|
|
|
|
|
| [9]/46,X Y[6] |
|
|
| induction + |
|
|
|
|
|
|
|
|
|
|
|
|
|
| HDAC |
|
|
|
|
|
|
|
|
|
|
|
|
|
| consolidation |
|
|
| 10 | 50 | M | UPS | ICE (5 cycles) | Distant | AML |
t(8;16)(p11.2;p13.3) |
| 24.9 | Y | AD | Alive | 27.8+ |
|
|
|
|
| MAID (6
cycles) | recurrence |
| [16]/4 6,XY[4] |
|
|
| induction + |
|
|
|
|
|
|
|
|
|
|
|
|
|
| HDAC |
|
|
|
|
|
|
|
|
|
|
|
|
|
| consolidation |
|
|
| 11 | 68 | M | UPS |
| Distant | MM |
| Trisomy 1q | 18.6 | Y | Rd | Alive | 17.6+ |
|
|
|
|
|
| recurrence |
|
|
|
|
|
|
|
|
| 12 | 21 | M | Osteosarcoma | (MM)CA | Distant | AML | der(2)t(2;11) |
| 45 | N | Not treated | Dead | 0.4 |
|
|
|
|
| (6 cycles) | recurrence |
|
(q31;p15),der(11) |
|
|
|
|
|
|
|
|
|
|
| ICE (10
cycles) |
|
| add(11)(p15)t(2;11) |
|
|
|
|
|
|
|
|
|
|
| Gemcitabine + |
|
| [19]/46,XY[1] |
|
|
|
|
|
|
|
|
|
|
| docetaxel (4
cycles) |
|
|
|
|
|
|
|
|
|
|
|
|
|
| ICE (1 cycle) |
|
|
|
|
|
|
|
|
|
| 13 | 45 | M | Osteosarcoma | MMCA (4
cycles) | NED | CML | t(9,22)(q34;q11.2) | BCR/A BL1 | 110.6 | Y | Imatinib | Alive | 131.1+ |
|
|
|
|
|
|
|
| [19]/4 6,
XY[1] | rearrangement |
|
|
|
|
|
| 14 | 9 | F | Osteosarcoma | CA (3 cycles) | NED | ALL |
68~72,XXY,+1,-2,- |
| 48.3 | Y | VPDL-6MP- | Dead | 11.2 |
|
|
|
|
| IB (1 cycle) |
|
| 3,der(4)t(4;5)(q25;q13), |
|
|
| MTX |
|
|
|
|
|
|
| CA (2 cycles) |
|
| −5,-7,+8,- |
|
|
|
|
|
|
|
|
|
|
| IB (1 cycle) |
|
|
9,+10,+11,add(13)(p11. |
|
|
|
|
|
|
|
|
|
|
| CA (2 cycles) |
|
|
2),-15,-16,-17,- |
|
|
|
|
|
|
|
|
|
|
| IB (1 cycle) |
|
|
18,+19,+20,+21,+5~7m |
|
|
|
|
|
|
|
|
|
|
|
|
|
| ar/46,XY[17] |
|
|
|
|
|
|
| 15 | 35 | M | Osteosarcoma | CA (5 cycles) | NED | MDS | Not tested |
| 65.8 | Y | NA | Dead | 12.6 |
| 16 | 20 | M | Osteosarcoma | CA (7 cycles) | Distant | MDS |
| del(5q31) | 123.7 | Y | Azacitidine | Alive | 9.4+ |
|
|
|
|
| ICE (6 cycles) | recurrence |
|
| del(7q) |
|
|
|
|
|
|
|
|
|
|
|
|
|
| del(20q32) |
|
|
|
|
|
| 17 | 9 | M | Ewing sarcoma | IA (6 cycles) | NED | ALL | Not tested |
| 12.8 | Y | HSCT | Dead | 36.3 |
| 18 | 72 | M | Liposarcoma | ICE (3 cycles) | Local | AA | 46, XY |
| 30.9 | Y | CsA + | Dead | 0.8 |
|
|
|
|
|
| recurrence |
|
|
|
|
| Eltrombopag |
|
|
SHMs
Among the 2,953 patients with sarcoma included in
the present study, 18 developed SHMs. The most prevalent SHMs were
AML (n=6, 33.3%) and myelodysplastic syndrome (n=5, 27.7%).
Additionally, three patients (n=3, 16.6%) developed acute
lymphoblastic leukemia, whereas one each (n=1, 5.5%) developed
aplastic anemia (AA), chronic myeloid leukemia, follicular lymphoma
and multiple myeloma. Regarding the SHMs incidence according to the
type of treatments, two of 1,295 patients (0.15%) who had a history
of surgery alone developed SHMs, while 16 of 1,658 patients (0.96%)
who underwent both surgery and chemotherapy developed SHMs. The
incidence of SHMs differed significantly between the two groups
(P<0.001) (Table SII). The SHMs
occurred at a median patient age of 44 (range, 10–77) years. The
latency from sarcoma diagnosis to SHM occurrence was 30 (range,
11–121) months. A total of 10 out of 18 patients (55.5%) succumbed
to SHM. The median OS period after SHM diagnosis was 15.7 (range,
0.4–154.9) months. A summary of the clinical history of the 18
patients who were diagnosed with SHMs following sarcoma is provided
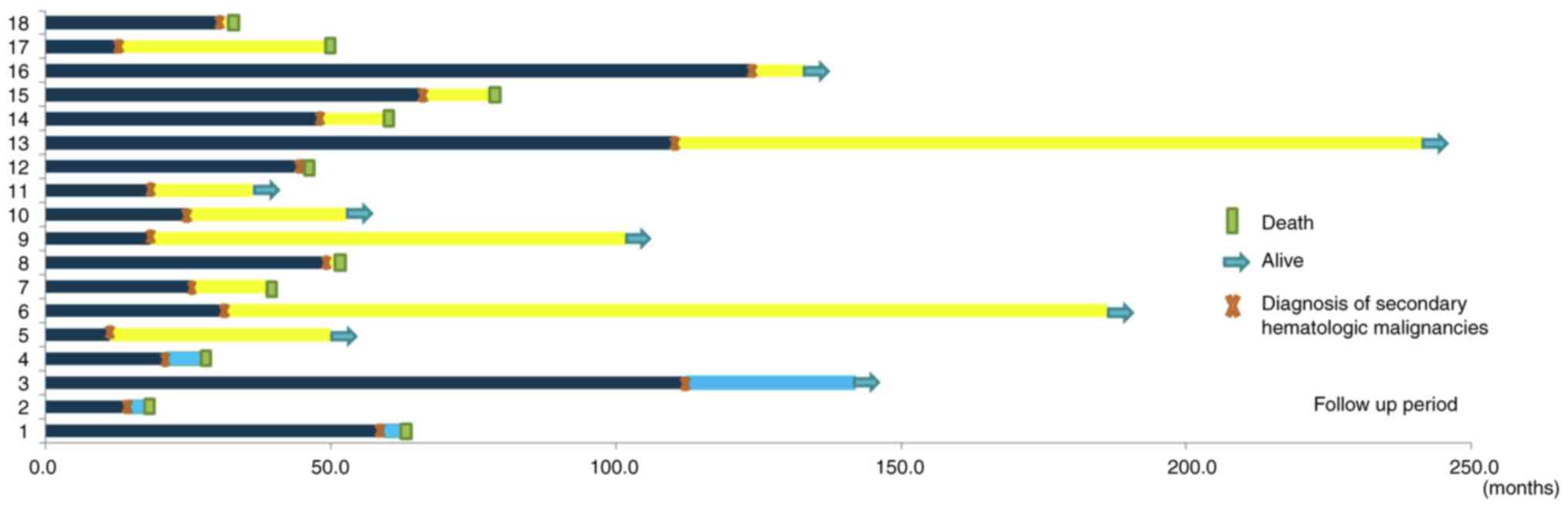
in Fig. 2.
Of these 18 patients, 14 had available data from
cytogenetic studies. Among these 14, four patients (22.2%) had
monosomy 7 or 7q/5q deletions. Additional information regarding the
cytogenetic study data for each of these patients is provided in
Table II.
A total of five patients (n=5, 27.8%) did not
receive treatment for SHMs. Among them, three patients did not
receive treatment due to metastatic sarcoma, poor performance
status and the risk of cytopenia-related infection. The other
patients had no evidence of sarcoma disease (NED) at the time of
SHMs diagnosis but had pancytopenia with pneumonia; thus, the
patient refused further treatment. The last patient who already had
NED for sarcoma did not need further treatment for SHMs because of
low-risk myelodysplastic syndrome. All patients succumbed except
for one patient who did not require chemotherapy (Table SIII).
Discussion
In the present single-center retrospective study,
the analysis of SHM occurrence in patients treated for sarcoma
revealed that SHMs occurred rarely. Among the patients included in
the present study, osteosarcoma was the most common sarcoma, while
therapy-related myeloid neoplasms (t-MNs) were the most common
among the SHMs. The prognosis of patients who developed SHMs was
poor. Although they rarely occur, the risk of SHMs in patients who
have received treatment for sarcoma, especially chemotherapy, and
who have shown long-term survival, should be considered.
The incidence rate of SHM after sarcoma treatment
was 0.6%, similar to the incidence rate of 0.79% reported
previously (23). There is a
complex mechanism involving multiple factors regarding the
occurrence of SHM. According to a previous study, DNA damage and
mutations caused by chemotherapy and radiation therapy were
reported to be the causes of SHMs (15). Chemotherapy, or RT, does not just
focus on tumor cells; it also impacts normal cells. Critically,
prolonged exposure can interfere with the genes that regulate the
growth and specialization of hematopoietic stem and precursor
cells, potentially leading to the emergence of a neoplastic myeloid
clone (16). Since the degree of
occurrence of DNA damage and mutation varies depending on multiple
factors that each patient has, SHMs may have occurred only in some
patients who received chemotherapy. Previous studies have also
identified factors associated with SHMs, such as patient age, the
sensitivity of the organs, and RT treatment dose (12).
The analysis of the present study revealed a
significant difference of the incidence of SHMs according to
previous treatment history, consistent with those of other studies.
Considering this result, chemotherapy may be associated with the
occurrence of SHMs though there is a potential bias in the present
results. Multivariate analysis would have been useful in the
present study to identify other risk factors, but due to the nature
of the data, the analysis was unable to be performed. A
larger-scale study in the future is necessary to identify factors
associated with SHM occurrence.
Among the patients in the present study, 16 (88.8%)
received chemotherapy. Among these patients, doxorubicin/adriamycin
was the most frequently administered chemotherapy agent (93%),
followed by ifosfamide (81%) and cisplatin (68%). Additionally,
methotrexate, carboplatin and etoposide, which correspond to
similar real-world practices, were used. A study conducted across
four European countries, including patients with soft tissue
sarcoma, reported that 68.4 and 40.2% of patients were administered
doxorubicin/adriamycin and ifosfamide, respectively (24). Furthermore, an investigation of
patients with bone sarcoma in Korea revealed that high-dose of
methotrexate, doxorubicin/adriamycin, and cisplatin and also
vincristine, doxorubicin/adriamycin, cyclophosphamide, etoposide,
and ifosfamide were used as first-line chemotherapy in most
patients, similar to the findings of the present study (25).
Alkylating agents and topoisomerase II inhibitors
are chemotherapeutic agents commonly known to cause t-MNs.
Alkylating agents modify DNA, resulting in DNA cross-linking,
double-stranded breaks, mutations and cytotoxicity. In particular,
t-MN is commonly characterized by chromosomal abnormalities
involving chromosome 5 and/or 7, complex karyotypes and TP53
aberrations (26). Conversely,
topoisomerase II inhibitors hinder the rejoining of DNA strands
cleaved during replication, thereby inducing double-stranded DNA
breaks. The two prevalent mutations are t(11q23.3) and t(21q22.1)
(27). In the present cohort study,
ten patients were diagnosed with t-MN after chemotherapy. A total
of eight out of ten underwent cytogenetic testing, five of whom
exhibited cytogenetic abnormalities commonly associated with t-MN.
The latency period for developing t-MN is typically 5–7 years for
alkylating agents and 2–3 years for topoisomerase II inhibitors
(28). In the patient group of the
present study, the median latency period for developing t-MN
following chemotherapy was relatively short, at 28.5 (range,
14.6–123.7) months. Recent studies have reported varying latency
periods for t-MN appearance, ranging from 1 to 10 years; therefore,
the results of the present study do not differ considerably from
previously reported results (26).
Considering the well-known risk factors of chemotherapy or
radiation therapy for AA (29), it
is reasonable to suspect that the patients in the present study who
underwent chemotherapy had a high risk of developing
therapy-related AA. The results of the present study were
consistent with those of previous studies on SHMs, suggesting that
prior treatment for the primary tumor is a considerable risk
factor.
After the SHM diagnosis, 10 patients succumbed.
Among these patients, at the time of SHM diagnosis, four, one, and
five had no evidence of sarcoma, local recurrence, or distant
recurrence, respectively. Regarding the five patients who did not
receive treatment for SHMs, four succumbed. Among the patients,
three had refractory metastatic sarcoma. A previous study suggested
that metastatic soft tissue sarcoma has a poor prognosis (30). Their attending physicians determined
that the risks associated with SHM treatment, such as the risk of
infection, outweighed the potential benefits of chemotherapy for
these patients. On the other hand, the other two patients who did
not received chemotherapy showed no evidence of sarcoma disease.
Although one of them was considered for SHM treatment, it was not
pursued due to complications of pancytopenia with pneumonia caused
by SHMs. The last one did not require chemotherapy because of the
presence of low-risk myelodysplastic syndrome. These findings may
suggest that physicians managing SHM patients consider the status
of sarcoma during their decision-making process about the treatment
of SHMs in real-world practice.
The present study had several limitations. First,
this was a retrospective study, and because of insufficient data,
multivariate analysis was not possible to be performed due to
factors contributing to the occurrence of SHMs. Secondly, soft
tissue and bone sarcomas analyses were not within the capacity of
the present study to be conducted separately. Thirdly, most of the
patients who were diagnosed with SHMs had been treated for sarcoma
before the implementation of NGS at Korea Cancer Center Hospital.
Consequently, no NGS data were available for any of patients
diagnosed with SHMs. Finally, the prevalence of SHMs may have
diminished owing to the loss of follow-up after a sarcoma
diagnosis. Despite these limitations, to the best of the authors'
knowledge, this is the first study focusing on the incidence of
SHMs in all patients with bone and soft tissue sarcoma across all
age groups. Future nationwide studies should identify the risk
factors associated with the development of SHMs.
In conclusion, this is the first study on the
overall incidence and characteristics of SHMs in patients with all
types of sarcoma. As in the case of previous studies, SHMs occurred
infrequently among the patients in the present study diagnosed with
sarcoma. However, patients who received chemotherapy for sarcoma
were more prone to develop SHMs than those who did not.
Additionally, the presence of SHMs was associated with a poor
prognosis. Therefore, caution is needed regarding SHM occurrence in
patients with long-term survival after a diagnosis of sarcoma,
especially in pediatric patients who have received
chemotherapy.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Korea Institute of
Radiological and Medical Sciences, funded by the Ministry of
Science, ICT (MSIT), Seoul, Republic of Korea (grant no.
50574-2024).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
YJJ and HKJ conceived and designed the analysis.
YJJ, HKJ, CBK, WSS, WHC, DGJ, HK, SHY, IIN, HRL and HJK contributed
data and analysis tools. YJJ and HKJ confirm the authenticity of
all the raw data. YJJ and HJK wrote the first draft of the
manuscript. YJJ, HKJ, CBK, WSS, WHC, DGJ, HK, SHY, IIN, HRL and HJK
performed data interpretation, and reviewed and commented on the
manuscript. All authors have read and approved the final version of
the manuscript.
Ethics approval and consent to
participate
The present study was approved (approval no.
2023-05-004) by the Institutional Review Board of Korea Cancer
Center Hospital (Seoul, South Korea) and was conducted in
accordance with the tenets of the Declaration of Helsinki. Due to
the retrospective nature of the current analysis, informed consent
was not required for the present study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
SHM
|
secondary hematological malignancy
|
|
IRB
|
Institutional Review Board
|
|
RT
|
radiotherapy
|
|
AML
|
acute myeloid leukemia
|
|
AA
|
aplastic anemia
|
|
OS
|
overall survival
|
|
NED
|
no evidence of disease
|
|
t-MNs
|
therapy-related myeloid neoplasms
|
References
|
1
|
Burningham Z, Hashibe M, Spector L and
Schiffman JD: The epidemiology of sarcoma. Clin Sarcoma Res.
2:142012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD, Fuchs HE and Jemal
AM: Cancer statistics, 2021. CA Cancer J Clin. 71:7–33. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
WHO Classification of Tumours Editorial
Board, . Soft tissue and bone tumors: WHO Classification of Tumors
Series. 5th edition. Vol 3. International Agency for Research on
Cancer; Lyon: 2020
|
|
4
|
Sung JJ, Kyeong SK, Kyo WL, Jae BP,
Yoon-La C, Jeong IY, Su JL, Dong IC and Sung JK: Distribution and
survival of primary sarcoma in Korea: A single center analysis of
2017 cases. Korean J Clin Oncol. 14:30–36. 2018. View Article : Google Scholar
|
|
5
|
Kim HS, Nam CM, Jang SY, Choi SK, Han M,
Kim S, Moneta MV, Lee SY, Cho JM, Novick D and Rha SY:
Characteristics and treatment patterns of patients with advanced
soft tissue sarcoma in Korea. Cancer Res Treat. 51:1380–1391. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wood ME, Vogel V, Ng A, Foxhall L, Goodwin
P and Travis LB: Second malignant neoplasms: Assessment and
strategies for risk reduction. J Clin Oncol. 30:3734–3745. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kang MJ, Won YJ, Lee JJ, Jung KW, Kim HJ,
Kong HJ, Im JS and Seo HG; Community of Population-Based Regional
Cancer Registries, : Community of population-based regional cancer
registries: Cancer statistics in Korea: Incidence, mortality,
survival, and prevalence in 2019. Cancer Res Treat. 54:330–344.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vogt A, Schmid S, Heinimann K, Frick H,
Herrmann C, Cerny T and Omlin A: Multiple primary tumors:
Challenges and approaches, a review. ESMO Open. 2:e0001722017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Choi DK, Helenowski I and Hijiya N:
Secondary malignancies in pediatric cancer survivors: Perspectives
and review of the literature. Int J Cancer. 135:1764–1773. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bright CJ, Reulen RC, Winter DL, Stark DP,
McCabe MG, Edgar AB, Frobisher C and Hawkins MM: Risk of subsequent
primary neoplasms in survivors of adolescent and young adult cancer
(Teenage and Young Adult Cancer Survivor Study): A
population-based, cohort study. Lancet Oncol. 20:531–545. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chao C, Bhatia S, Xu L, Cannavale KL, Wong
FL, Huang PS, Cooper R and Armenian SH: Incidence, risk factors,
and mortality associated with second malignant neoplasms among
survivors of adolescent and young adult cancer. JAMA Netw Open.
2:e1955362019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Demoor-Goldschmidt C and de Vathaire F:
Review of risk factors of secondary cancers among cancer survivors.
Br J Radiol. 92:201803902019.PubMed/NCBI
|
|
13
|
Mertens AC, Liu Q, Neglia JP, Wasilewski
K, Leisenring W, Armstrong GT, Robison LL and Yasui Y:
Cause-specific late mortality among 5-year survivors of childhood
cancer: The childhood cancer survivor study. J Natl Cancer Inst.
100:1368–1379. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Friedman DL, Whitton J, Leisenring W,
Mertens AC, Hammond S, Stovall M, Donaldson SS, Meadows AT, Robison
LL and Neglia JP: Subsequent neoplasms in 5-year survivors of
childhood cancer: The childhood cancer survivor study. J Natl
Cancer Inst. 102:1083–1095. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
The Lancet Haematology, . Secondary
haematological malignancies. Lancet Haematol. 10:e792023.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sill H, Olipitz W, Zebisch A, Schulz E and
Wölfler A: Therapy-related myeloid neoplasms: Pathobiology and
clinical characteristics. Br J Pharmacol. 162:792–805. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Strauss SJ, Frezza AM, Abecassis N, Bajpai
J, Bauer S, Biagini R, Bielack S, Blay JY, Bolle S, Bonvalot S, et
al: Bone sarcomas: ESMO-EURACAN-GENTURIS-ERN PaedCan Clinical
Practice Guideline for diagnosis, treatment and follow-up. Ann
Oncol. 32:1520–1536. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
National Comprehensive Cancer Network
(NCCN), . Soft Tissue Sarcoma (Version 2.2023). NCCN; Plymouth
Meeting, PA: 2023, https://www.nccn.org/professionals/physician_gls/pdf/sarcoma.pdfMay
12–2023
|
|
19
|
National Comprehensive Cancer Network
(NCCN), . Bone Cancer (Version 3.2023). NCCN; Plymouth Meeting, PA:
2023, https://www.nccn.org/professionals/physician_gls/pdf/bone.pdfMay
12–2023
|
|
20
|
Bhatia S, Krailo MD, Chen Z, Burden L,
Askin FB, Dickman PS, Grier HE, Link MP, Meyers PA, Perlman EJ, et
al: Therapy-related myelodysplasia and acute myeloid leukemia after
Ewing sarcoma and primitive neuroectodermal tumor of bone: A report
from the Children's Oncology Group. Blood. 109:46–51. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hong KT, Choi JY, Hong CR, Kang HJ, Park
KD and Shin HY: Therapy-related acute myeloid leukemia after the
treatment of primary solid cancer in children: A single-center
experience. J Pediatr Hematol Oncol. 40:e23–e28. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sanford NN, Martin AM, Brunner AM, Cote
GM, Choy E, DeLaney TF, Aizer AA and Chen YL: Secondary acute
leukemia in sarcoma patients: A population-based study. Int J
Radiat Oncol Biol Phys. 100:687–694. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kube SJ, Blattmann C, Bielack SS, Kager L,
Kaatsch P, Kühne T, Sorg B, Kevric M, Jabar S, Hallmen E, et al:
Secondary malignant neoplasms after bone and soft tissue sarcomas
in children, adolescents, and young adults. Cancer. 128:1787–1800.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nagar SP, Mytelka DS, Candrilli SD,
D'yachkova Y, Lorenzo M, Kasper B, Lopez-Martin JA and Kaye JA:
Treatment patterns and survival among adult patients with advanced
soft tissue sarcoma: A retrospective medical record review in the
United Kingdom, Spain, Germany, and France. Sarcoma.
2018:54670572018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jeong H, Im HS, Kim W, Lee JS, Song SY,
Song JS, Cho KJ, Chung HW, Lee MH, Kim JE and Ahn JH: Demographics,
changes in treatment patterns, and outcomes of bone and soft tissue
sarcomas in Korea-a sarcoma-specific, institutional registry-based
analysis. Cancer Manag Res. 13:8795–8802. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu YC, Illar GM, Al Amri R, Canady BC,
Rea B, Yatsenko SA and Geyer JT: Therapy-related myeloid neoplasms
with different latencies: A detailed clinicopathologic analysis.
Mod Pathol. 35:625–631. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cancer Genome Atlas Research Network, .
Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A,
Hoadley K, Triche TJ Jr, Laird PW, et al: Genomic and epigenomic
landscapes of adult de novo acute myeloid leukemia. N Engl J Med.
368:2059–2074. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
McNerney ME, Godley LA and Le Beau MM:
Therapy-related myeloid neoplasms: When genetics and environment
collide. Nat Rev Cancer. 17:513–527. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ruan J and Han B: Effective treatment of
aplastic anemia secondary to chemoradiotherapy using cyclosporine
A. Chin Med J (Engl). 134:2356–2358. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Italiano A, Mathoulin-Pelissier S, Cesne
AL, Terrier P, Bonvalot S, Collin F, Michels JJ, Blay JY, Coindre
JM and Bui B: Trends in survival for patients with metastatic
soft-tissue sarcoma. Cancer. 117:1049–1054. 2011. View Article : Google Scholar : PubMed/NCBI
|