Introduction
Hemophagocytic lymphohistiocytosis (HLH), also
termed ‘hemophagocytic syndrome’ or ‘macrophage activation
syndrome’, is a life-threatening condition characterized by
pathological inflammatory responses (1). Clinical manifestations of HLH include
recurrent fever, cytopenia, splenomegaly and sepsis-like syndrome,
potentially culminating in multiple organ failure (MOF). HLH is
classified into primary and secondary forms. Primary HLH is a
hereditary condition predominantly observed in pediatric
populations. Secondary HLH stems from malignancies, autoimmune
diseases or idiopathic causes (2).
Although numerous malignancies can accompany HLH,
lymphoma-associated hemophagocytic syndrome (LAHS) is particularly
prevalent. Malignant lymphoma is categorized into low-grade and
high-grade lymphoma. It is worth noting that LAHS can manifest as a
symptom of high-grade lymphoma rather than low-grade lymphoma
(3,4) and is associated with poor prognosis
(5). Although LAHS is usually
treated with corticosteroid therapy and cytotoxic chemotherapy for
lymphoma (6), the significance of
surgical intervention remains unclear, as no surgical cases have
been reported to date.
The present study described a challenging case of
lymphoma-associated splenomegaly, which led to severe HLH following
splenectomy. It was possible to save the patient through timely
splenectomy to reduce the tumor burden, enabling safe and effective
steroid pulse therapy and chemotherapy.
Case report
A 71-year-old man with a medical history of
hypertension and hyperuricemia presented to the primary hospital,
Japan Baptist Hospital's Department of Internal Medicine (Kyoto,
Japan), with fatigue and anorexia in February 2023. Laboratory test
results revealed anemia [hemoglobin (Hb), 10 g/dl; normal range,
12–16 g/dl], thrombocytopenia [platelet count (Plt),
100×103/µl; normal range, 150–450×103/µl],
elevated lactate dehydrogenase levels (LDH, 627 U/l; normal range,
140–271 U/l) and a markedly elevated ferritin level (6,210 ng/ml;
normal range, 15–500 ng/ml). In addition, the patient's soluble
interleukin 2 receptor (sIL-2R) level was significantly increased
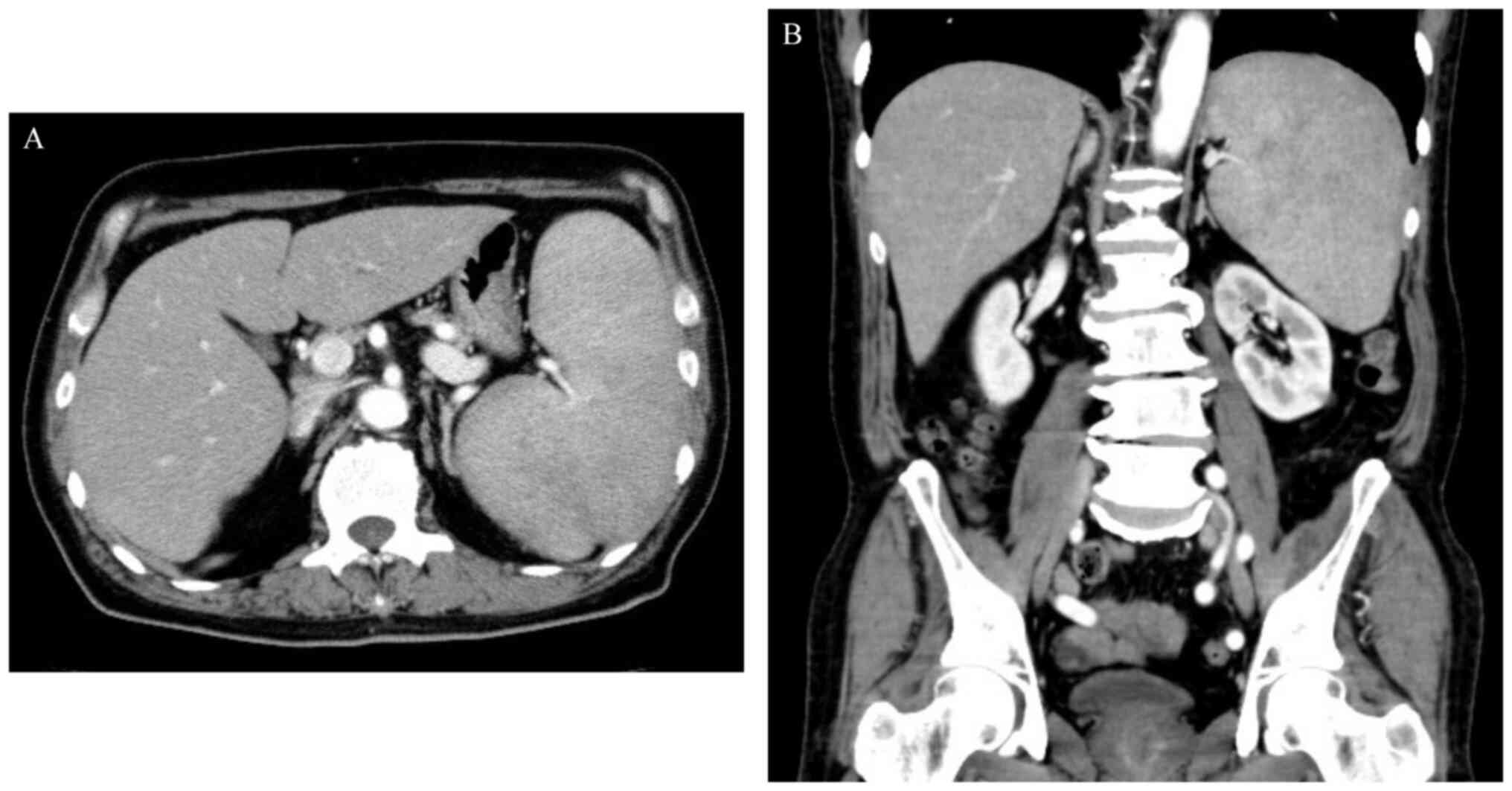
at 11,328 U/ml (normal range, 122–496 U/ml). Abdominal computed
tomography (CT) revealed splenomegaly, without enlarged lymph nodes
(Fig. 1).
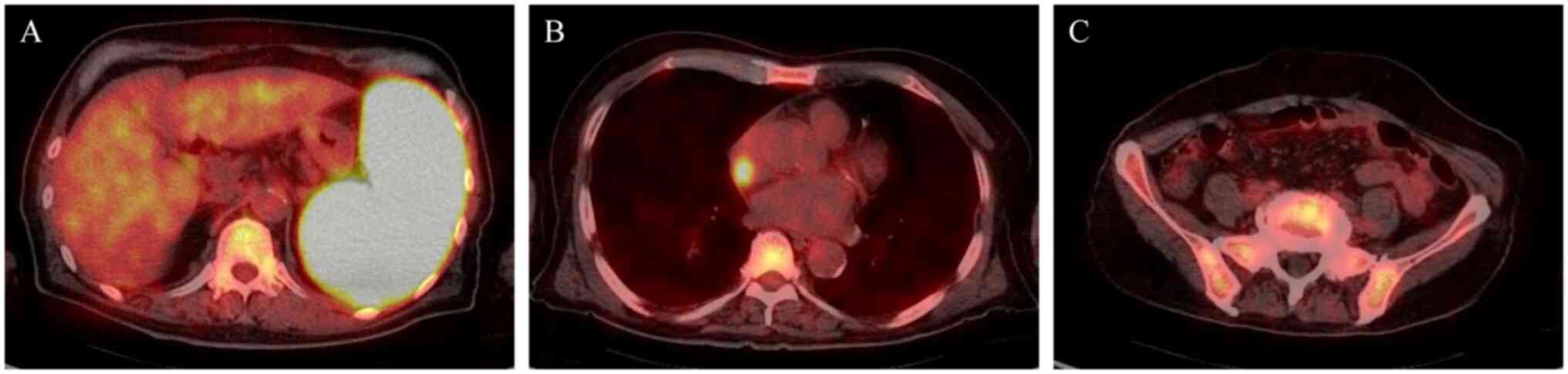
18F-fluorodeoxyglucose-positron emission tomography
(FDG-PET)/CT showed the most pronounced increased uptake of FDG in
the spleen, followed by the right atrium and the bone marrow
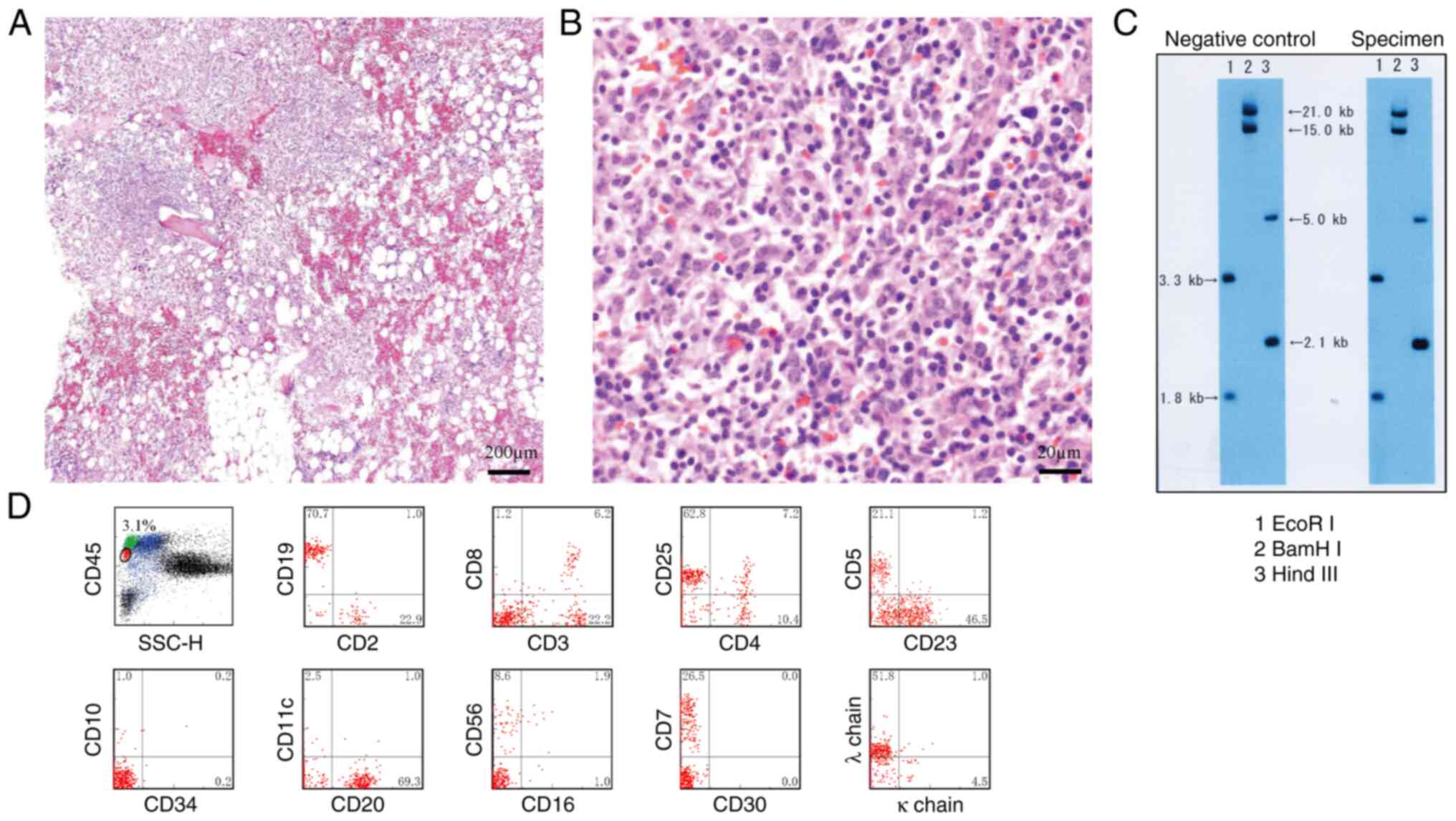
(Fig. 2). The bone marrow biopsy
identified an increase in small lymphocytes with occasional large
cells on Hematoxylin and Eosin (HE) staining (7) (Fig. 3A and
B). Clonality was confirmed through cytogenetic examination of
T cell receptor γ chain Jγ rearrangement (8,9) using
Southern blot analysis (10) with
restriction enzymes EcoRI, BamHI and HindIII (Takara Bio, Inc.)
(Fig. 3C).In addition, clonality
was supported by flow cytometric analysis with CD45 gating
(11) (Fig. 3D). Flow cytometry utilized a panel
of pre-diluted antibodies from BD Biosciences, including CD45
(PerCP) (cat. no. 347464), CD2 (FITC) (cat. no. 347593), CD19
(BV421) (cat. no. 562440), CD3 (PE) (cat. no. 347347), CD8 (BV510)
(cat. no. 563919), CD4 (APC-H7) (cat. no. 641398), CD25 (BV421)
(cat. no. 562442), CD5 (FITC) (cat. no. 347303), CD23 (BV421) (cat.
no. 562707), CD10 (PE) (cat. no. 340921), CD20 (APC-H7) (cat. no.
641396), CD11c (BV510) (cat. no. 563026), CD16 (BV510) (cat. no.
563830), CD56 (APC) (cat. no. 341025), CD30 (FITC) (cat. no.
341644), CD7 (APC) (cat. no. 561604), kappa light chains (PE) (cat.
no. 346601), and lambda light chains (APC-H7) (cat. no. 656648),
all used according to the manufacturer's protocol; however, a
definitive diagnosis of the histological subtype could not be
established.
Despite the abnormal laboratory data, the patient's
activities of daily living were relatively preserved. After
comprehensive assessment under outpatient conditions, elective
surgery was planned to obtain a histological diagnosis two weeks
after the initial presentation. On admission, 2 days
preoperatively, the patient exhibited a spike fever of 39°C and
progressive cytopenia (Hb, 8.6 g/dl; and Plt, 62×103/µl)
was observed. The next day (the day before surgery), 20 units of
platelet concentrate were transfused, but the Plt count had dropped
to 55×103/µl on the operative day. Due to the patient's
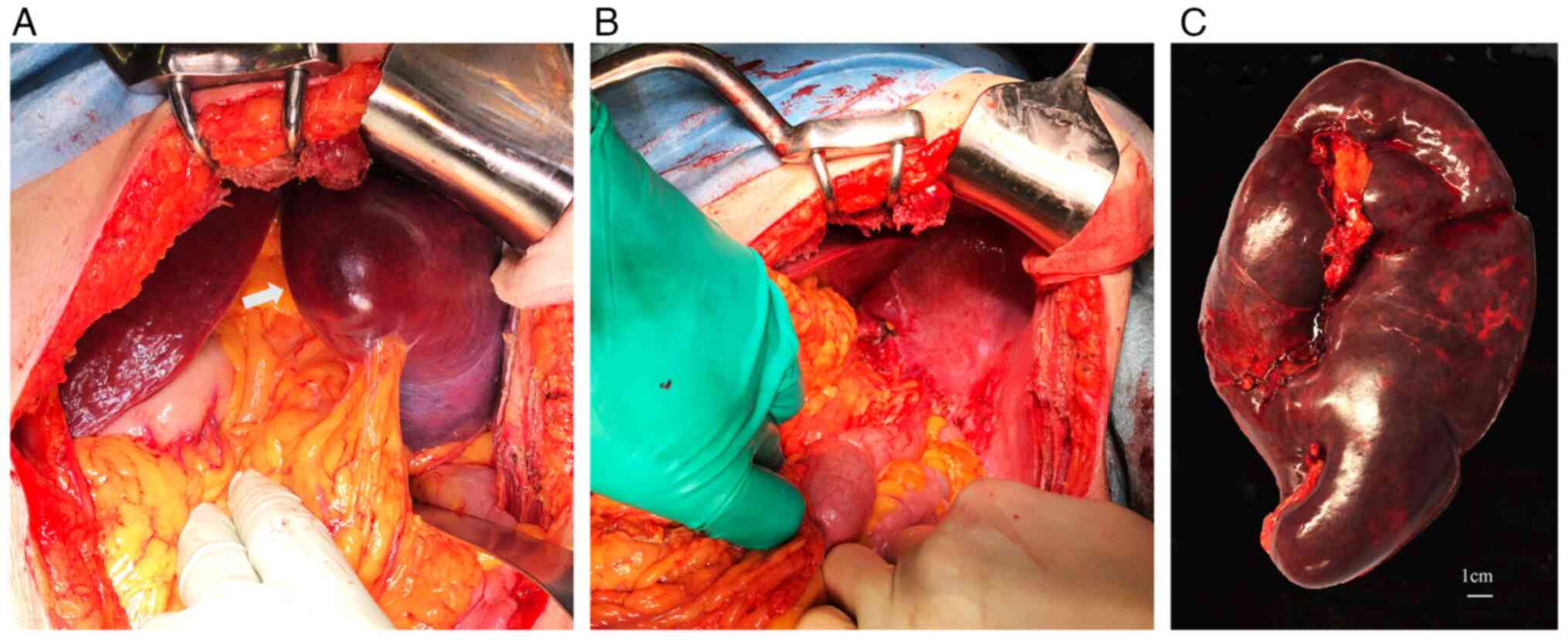
significant splenomegaly, laparotomy was selected instead of
laparoscopy. Splenectomy was eventually performed via a left
subcostal incision (Fig. 4A). The
procedure involved the separate ligation of the splenic artery and
vein, and the spleen was extracted by stapling the tail of the
pancreas at the splenic hilum (Fig.
4B). The operation lasted for 100 min, with an estimated blood
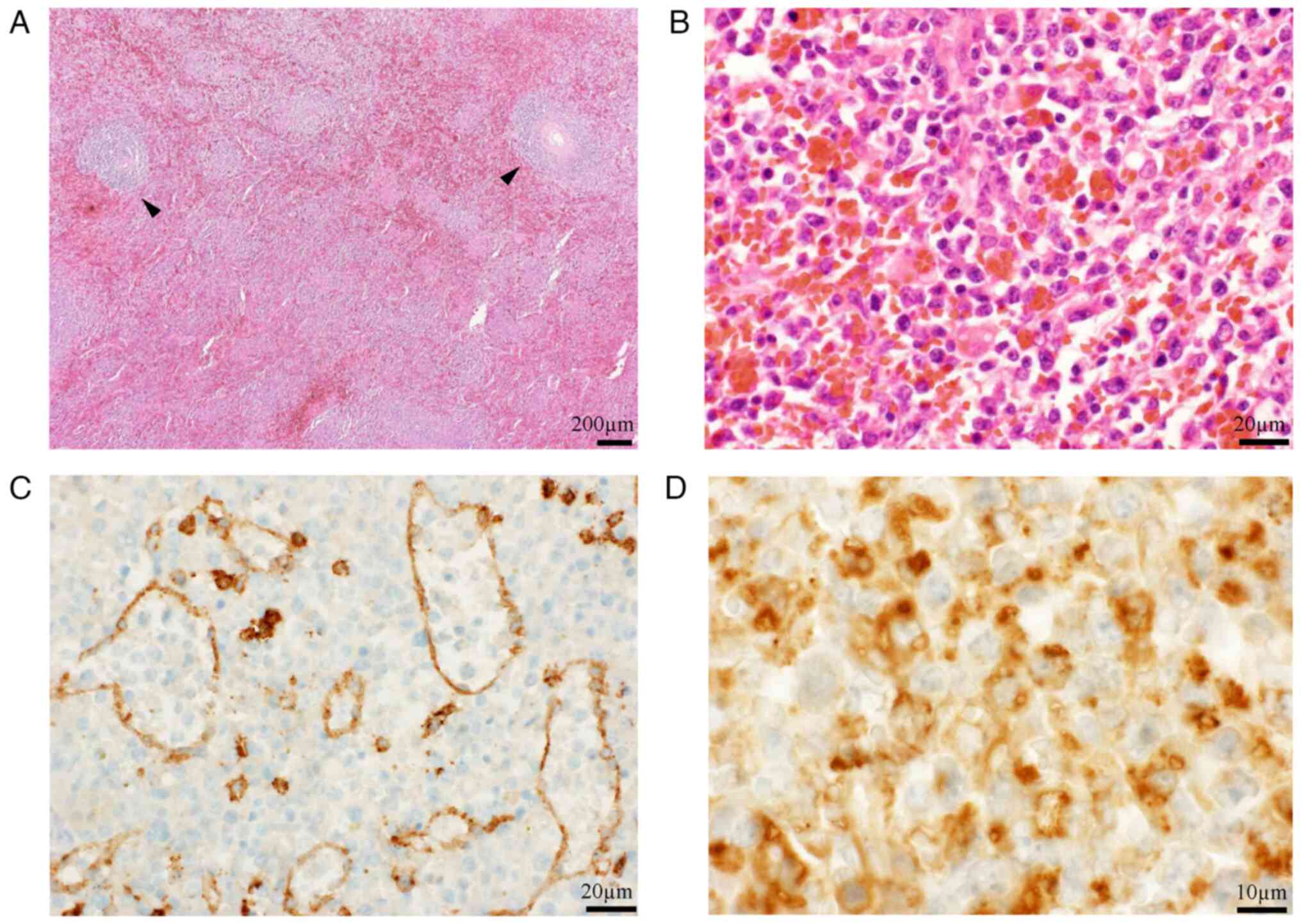
loss of 600 ml. The excised specimen weighed 1,100 g (Fig. 4C). Subsequent histological
examination confirmed the diagnosis of low-grade B-cell lymphoma
with focal transformation to high-grade lymphoma with severe
hemophagocytosis on HE staining (7)
(Fig. 5). To confirm red-pulp, but
not white-pulp, involvement of the lymphoma, immunohistochemistry
was performed using an anti-CD8 antibody (cat. no. M7103; DAKO) at
a dilution of 1:100, and Bentana ultraView DAB universal kit (Roche
Diagnostics) for identification of sinus endothelial cells
according to the manufacturers' standard protocols. The lymphoma
predominantly involved the red pulp with some remaining atrophic
white pulp, and the histological diagnosis was
immunohistochemically confirmed using a CD68 probe (cat. no.
NCL-CD68-KP1; Novocastra) at a dilution of 1:400 (Fig. 5). Thus, splenic marginal-zone
lymphoma (commonly seen in this organ) could not be included in the
histologic differential diagnosis; the lymphoma was finally
characterized as unclassifiable. The patient was diagnosed with
stage IVB disease according to the Ann Arbor Classification System
(12).
Despite having undergone surgery, the pre-existing
symptoms of intermittent fever and fatigue did not improve
postoperatively. The patient exhibited low blood pressure and
required temporary dopamine administration. On postoperative day
(POD) 3, a rapid decrease in the Plt count (62×103/µl)
and elevation in LDH (1,948 U/l), aspartate aminotransferase (AST)
(234 U/l; normal range, 8–48 U/l), total bilirubin (T-Bil) (6.6
mg/dl; normal range, 0.3–1.2 mg/dl), direct bilirubin (D-Bil) (4.5
mg/dl; normal range, 0.0–0.3 mg/dl) and C-reactive protein (CRP)
levels (18.56 mg/dl; normal range, <0.5 mg/dl) were observed
(Fig. 6). CT scanning did not
detect any postoperative complications, such as pancreatic leakage.
To exclude the possibility of drug-induced liver damage or
potential embolism, medication adjustments were made and heparin
was administered. However, the patient's condition further
deteriorated.
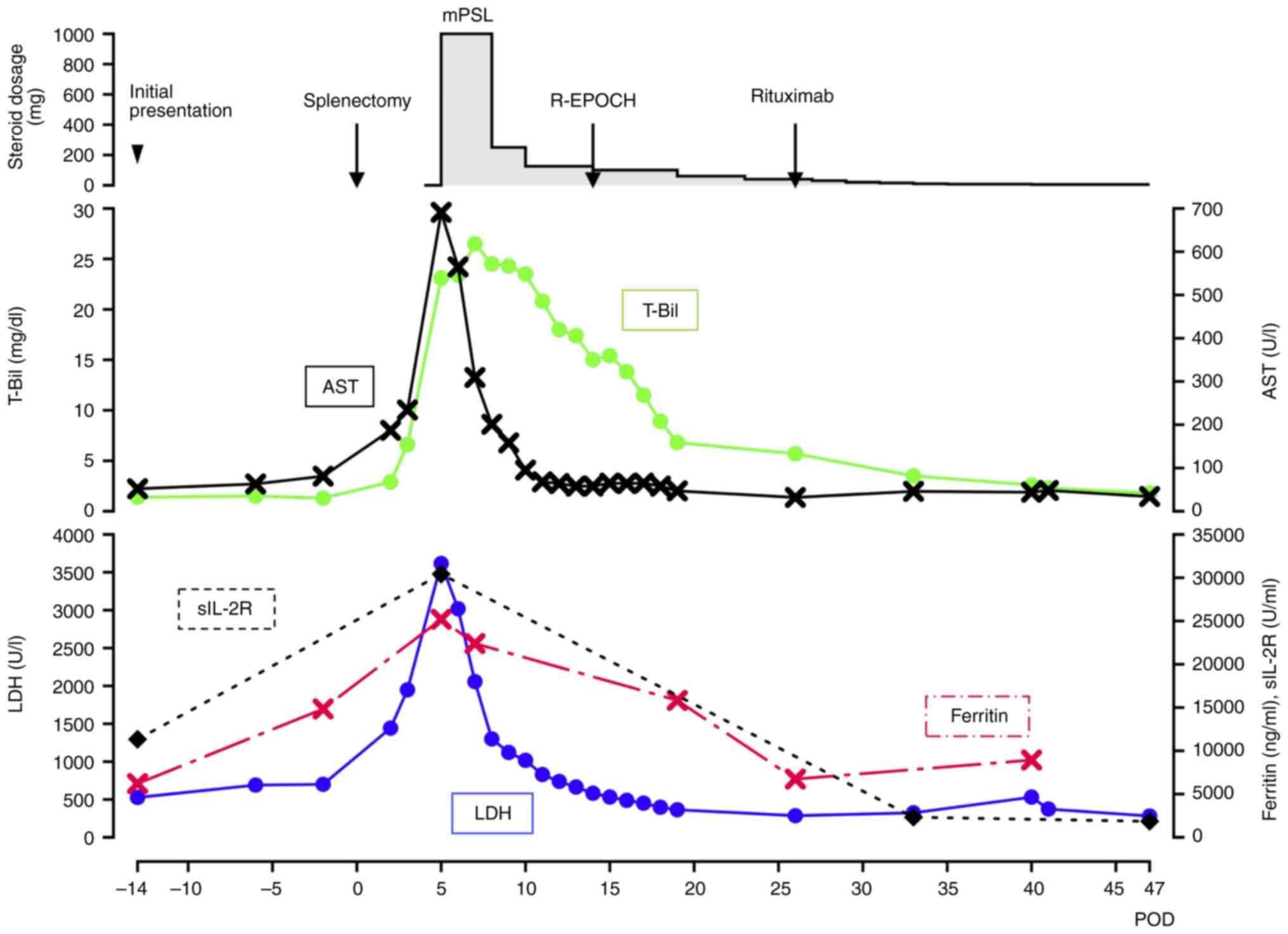 | Figure 6.Detailed treatment course of the
patient. Line graph illustrating changes in key biochemical markers
(T-Bil, AST, LDH, Ferritin and sIL-2R) over time, starting from the
preoperative period. Note that a significant improvement is
observed following steroid pulse therapy and subsequent
chemotherapy for the lymphoma. The steroid administration was
switched from intravenous injection of mPSL to oral intake of
prednisolone on POD 14. T-Bil, total bilirubin (normal range,
0.3–1.2 mg/dl); AST, aspartate transaminase (normal range, 8–48
U/l); LDH, lactate dehydrogenase (normal range, 140–271 U/l);
Ferritin (normal range, 15–500 ng/ml); sIL-2R, soluble interleukin
2 receptor (normal range, 122–496 U/ml); mPSL, methylprednisolone;
POD, postoperative day; R-EPOCH, Rituximab, Etoposide, Prednisone,
Oncovin, Cyclophosphamide and Hydroxydaunorubicin. |
Consequently, the patient was transferred to a
tertiary care institution on POD 5, where he was admitted to the
intensive care unit (ICU). Laboratory reassessment revealed the
following abnormalities: Decrease in Hb (8.3 g/dl) and Plt
(47×103/µl); and significant elevations in LDH (3,616
U/l), AST (691 U/l), alanine aminotransferase (112 U/l; normal
range, 7–55 U/l), T-Bil (21 mg/dl), D-Bil (17 mg/dl), creatinine
(1.77 mg/dl; normal range, 0.5–1.2 mg/dl), CRP (23.44 mg/dl),
ferritin (25,197 ng/ml) and sIL-2R (30,420 U/ml). The patient also
exhibited hypoglycemia, with a blood glucose level of 25 mg/dl
(normal range: 70–99 mg/dl), indicative of hepatic failure.
However, since coagulation markers, such as prothrombin time and
activated partial thromboplastin clotting time, were normal, plasma
exchange therapy was deemed unnecessary (13).
The diagnosis of concurrent HLH was made and
treatment with immunosuppressive therapy, steroid pulse therapy at
1,000 mg/day for 3 days, was immediately initiated under mechanical
ventilation and continuous hemodiafiltration. Following this
intervention, liver dysfunction was gradually ameliorated; the
steroid dosage was tapered down (Fig.
6). The patient was extubated on POD 12 and subsequent
chemotherapy for lymphoma with the R-EPOCH regimen (Rituximab,
Etoposide, Prednisone, Oncovin, Cyclophosphamide and
Hydroxydaunorubicin) was initiated on POD 14. Considering the
hepatic dysfunction and tumor load, the initial dose of the regimen
was reduced to 50%, and the administration of rituximab was delayed
until POD 26. Bradycardia with a heart rate of 40 beats/min was
transiently observed, presumably due to the infiltration of
lymphoma into the sinoatrial node (Fig.
2B). However, there was no need for the placement of a
pacemaker. After chemotherapy initiation, this bradycardia
gradually improved, and further improvements in liver function were
observed. The patient was discharged from the ICU on POD 23 and
hemodialysis was discontinued on POD 30.
After experiencing gastric ulcer bleeding on POD 19
and acute pancreatitis on POD 27 as complications of steroid
treatment, which were successfully managed with hemostasis and
endoscopic drainage, a second course of R-EPOCH therapy was
initiated on POD 77. After the patient's condition stabilized, he
was transferred back to our primary hospital on POD 93 and was
discharged home on POD 168. At eight months after the onset, the
patient remains alive with improved performance status and his
treatment is ongoing, specifically continuing with consolidation
therapy for lymphoma. He is undergoing outpatient consultations
with a frequency of approximately once every two weeks.
Discussion
HLH comprises a spectrum of conditions driven by
uncontrolled inflammatory reactions. Among secondary HLH,
malignancy-related HLH is predominantly associated with non-Hodgkin
lymphomas. Of note, LAHS demonstrates a dismal prognosis with an
average survival of only 37 days after treatment initiation
(5), and this condition has been
extensively studied (14,15). LAHS is itself a fatal condition,
which often results in death despite intensive treatment with
corticosteroid pulse therapy and cytotoxic chemotherapy (4,16–20).
Splenomegaly, or an increased standardized uptake
value on FDG-PET, is a frequent manifestation of numerous
hematological disorders, including malignant lymphomas (21). An enlarged spleen may act as a
reservoir for malignant cells, potentially accelerating disease
progression. Splenectomy can alleviate related symptoms, such as
abdominal discomfort and cytopenia, while allowing for diagnosis
based on histopathological examination. This procedure may also
offer therapeutic benefits, particularly in cases of B-cell splenic
lymphoma (22–24). There is a distinct subset of HLH
cases of unknown etiology that poses a clinical challenge. In such
cases, splenectomy has been shown to serve as an effective
diagnostic and therapeutic strategy (25–27),
while a clinical trial is currently underway to validate its
efficacy (NCI02862054). It can be inferred from these observations
that the role of splenectomy in the context of splenic lymphoma and
secondary HLH of idiopathic etiology has been established.
Malignant HLH is associated with MOF, leading to
high rates of morbidity and mortality. In the present case, the
preoperative progressive decrease in Hb and Plt indicates that
signs of MOF were already present. If only chemotherapy had been
administered without surgical intervention, there would have been a
potential risk of triggering a severe tumor lysis syndrome and
exacerbating pre-existing HLH due to the high tumor burden
(28). Therefore, without
splenectomy, the patient may have experienced more disastrous
outcomes, such as the progression of MOF, splenic rupture (29), and even death. Retrospectively, the
splenectomy did have a role in reducing the tumor burden, which in
turn facilitated safer and more effective chemotherapy.
However, a critical question remains in the present
case: Despite the removal of a significant tumor mass, why did the
patient still develop severe HLH? Based on the postoperative course
of the case, it is evident that surgery alone failed to halt the
development of HLH, with hypotension observed postoperatively,
suggesting a concurrent cytokine storm. Initially, it was assumed
that his splenic lymphoma was primarily indolent. However, highly
elevated ferritin levels at the presentation suggested the presence
of HLH preoperatively. Pathological findings of the spleen
supported this, further revealing high-grade lymphoma
transformation. There is a high likelihood that the acute
transformation occurred immediately before surgery. On the other
hand, the widespread progression of HLH may have resulted from
several factors during the surgical procedure: The surgical
stimulation could have activated the lymphoma cells to secrete
large amounts of soluble factors, such as TNF-α (30), or the operative procedure may have
dispersed cellular components into the systemic circulation
(31).
The HLH-2004 diagnostic criteria are well-known
(2). However, adult patients may
exhibit symptoms that are difficult to distinguish from sepsis or
MOF of other causes. Delayed diagnosis of LAHS may have fatal
consequences (17,18). Currently, no standardized treatment
strategy exists, and whether an HLH-directed or malignancy-directed
approach should be adopted (32,33)
remains unclear. In the case of the present study, upon his
transfer to a tertiary hospital on POD 5, the patient met 6 of the
8 HLH-2004 diagnostic criteria, leading to the identification of
his condition. Finally, immunosuppressive therapy preceded,
stabilizing the patient's condition and allowing us to commence
chemotherapy. At the time of conclusion of the present study, eight
months postoperatively, the patient has achieved a longer survival
than typical reported cases.
In conclusion, while splenic lymphoma typically
manifests with low-grade lymphoma, it can transform into a
high-grade lymphoma, which can accompany severe complications such
as HLH and MOF. In the present case, splenectomy served not only in
the diagnosis but also in tumor cytoreduction before chemotherapy.
Furthermore, through interdisciplinary collaboration between the
surgical and hematology teams, the patient's condition was
successfully treated by performing a timely splenectomy to reduce
the tumor burden, followed by steroid pulse therapy and
chemotherapy. Of note, surgery and subsequent chemotherapy may be
life-saving in certain cases of LAHS, as this strategy may assist
in avoiding chemotherapy-related fatal complications.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
HM, KO and KK participated in the surgical
procedures. MSh, YI, AU, MSu, TS and KK were involved in clinical
management. HM drafted the manuscript. MSh, YI, AU, MSu, TS, KO and
KK revised the manuscript for important intellectual content. All
the authors have read and approved the final manuscript. HM and KK
have independently reviewed and confirm all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Written informed consent was obtained from the
patient for the publication of this case report and the
accompanying images.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HLH
|
hemophagocytic lymphohistiocytosis
|
|
MOF
|
multiple organ failure
|
|
LAHS
|
lymphoma-associated hemophagocytic
syndrome
|
|
Hb
|
hemoglobin
|
|
Plt
|
platelet count
|
|
LDH
|
lactate dehydrogenase
|
|
sIL-2R
|
soluble interleukin 2 receptor
|
|
CT
|
computed tomography
|
|
FDG-PET
|
18F-fluorodeoxyglucose-positron emission tomography
|
|
HE
|
Hematoxylin and Eosin
|
|
POD
|
postoperative day
|
|
AST
|
aspartate aminotransferase
|
|
T-Bil
|
total bilirubin
|
|
D-Bil
|
direct bilirubin
|
|
CRP
|
C-reactive protein
|
|
ICU
|
intensive care unit
|
|
R-EPOCH
|
Rituximab, Etoposide, Prednisone,
Oncovin, Cyclophosphamide and Hydroxydaunorubicin
|
|
SUV
|
standardized uptake value
|
|
mPSL
|
methylprednisolone
|
References
|
1
|
La Rosée P, Horne A, Hines M, von Bahr
Greenwood T, Machowicz R, Berliner N, Birndt S, Gil-Herrera J,
Girschikofsky M, Jordan MB, et al: Recommendations for the
management of hemophagocytic lymphohistiocytosis in adults. Blood.
133:2465–2477. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Henter JI, Horne A, Aricó M, Egeler RM,
Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski
J and Janka G: HLH-2004: Diagnostic and therapeutic guidelines for
hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer.
48:124–131. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lee JC and Logan AC: Diagnosis and
management of adult malignancy-associated hemophagocytic
lymphohistiocytosis. Cancers (Basel). 15:18392023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pasvolsky O, Zoref-Lorenz A, Abadi U,
Geiger KR, Hayman L, Vaxman I, Raanani P and Leader A:
Hemophagocytic lymphohistiocytosis as a harbinger of aggressive
lymphoma: A case series. Int J Hematol. 109:553–562. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li F, Li P, Zhang R, Yang G, Ji D, Huang
X, Xu Q, Wei Y, Rao J, Huang R and Chen G: Identification of
clinical features of lymphoma-associated hemophagocytic syndrome
(LAHS): An analysis of 69 patients with hemophagocytic syndrome
from a single-center in central region of China. Med Oncol.
31:9022014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang H, Xiong L, Tang W, Zhou Y and Li F:
A systematic review of malignancy-associated hemophagocytic
lymphohistiocytosis that needs more attentions. Oncotarget.
8:59977–59985. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fischer AH, Jacobson KA, Rose J and Zeller
R: Hematoxylin and eosin staining of tissue and cell sections. CSH
Protoc. 2008.pdb.prot4986. 2008.
|
|
8
|
Moreau EJ, Langerak AW, van Gastel-Mol EJ,
Wolvers-Tettero IL, Zhan M, Zhou Q, Koop BF and van Dongen JJ: Easy
detection of all T cell receptor gamma (TCRG) gene rearrangements
by Southern blot analysis: Recommendations for optimal results.
Leukemia. 13:1620–1626. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
van Dongen JJ and Wolvers-Tettero IL:
Analysis of immunoglobulin and T cell receptor genes. Part I: Basic
and technical aspects. Clin Chim Acta. 198:1–91. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Southern E: Southern blotting. Nat Protoc.
1:518–525. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim B, Lee ST, Kim HJ and Kim SH: Bone
marrow flow cytometry in staging of patients with B-cell
non-Hodgkin lymphoma. Ann Lab Med. 35:187–193. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Carbone PP, Kaplan HS, Musshoff K,
Smithers DW and Tubiana M: Report of the committee on Hodgkin's
disease staging classification. Cancer Res. 31:1860–1861.
1971.PubMed/NCBI
|
|
13
|
Schwartz J, Padmanabhan A, Aqui N, Balogun
RA, Connelly-Smith L, Delaney M, Dunbar NM, Witt V, Wu Y and Shaz
BH: Guidelines on the use of therapeutic apheresis in clinical
practice-evidence-based approach from the writing committee of the
American society for apheresis: The seventh special issue. J Clin
Apher. 31:149–162. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li B, Guo J, Li T, Gu J, Zeng C, Xiao M,
Zhang W, Li Q, Zhou J and Zhou X: Clinical characteristics of
hemophagocytic lymphohistiocytosis associated with non-hodgkin
B-Cell lymphoma: A multicenter retrospective study. Clin Lymphoma
Myeloma Leuk. 21:e198–e205. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yao S, Jin Z, He L, Zhang R, Liu M, Hua Z,
Wang Z and Wang Y: Clinical features and prognostic risk prediction
of non-hodgkin lymphoma-associated hemophagocytic syndrome. Front
Oncol. 11:7880562021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Che Y, Xu L, Zhang X, Qiu X, Song J, Ding
X and Sun X: Secondary hemophagocytic lymphohistiocytosis triggered
by peripheral T-cell lymphoma: An unusual case report. Clin Case
Rep. 10:e65282022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Paluszkiewicz P, Martuszewski A, Majcherek
M, Kucharska M, Bogucka-Fedorczuk A, Wróbel T and Czyż A:
Hemophagocytic lymphohistiocytosis secondary to peripheral T cell
lymphoma with rapid onset and fatal progression in a young patient:
A case report and review of the literature. Am J Case Rep.
22:e9327652021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Iizuka H, Mori Y, Iwao N, Koike M and
Noguchi M: Hemophagocytic lymphohistiocytosis associated with
Epstein-Barr virus-positive diffuse large B-cell lymphoma, NOS of
bone marrow-liver-spleen type: An autopsy case report. J Clin Exp
Hematop. 61:102–108. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tang S, Zhao C and Chen W: Aggressive
diffuse large B-cell lymphoma with hemophagocytic
lymphohistiocytosis: report of one case. Int J Clin Exp Pathol.
13:2392–2396. 2020.PubMed/NCBI
|
|
20
|
Patel R, Patel H, Mulvoy W and Kapoor S:
Diffuse large B-cell lymphoma with secondary hemophagocytic
lymphohistiocytosis presenting as acute liver failure. ACG Case Rep
J. 4:e682017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hu Y, Zhou W, Sun S, Guan Y, Ma J and Xie
Y: 18F-fluorodeoxyglucose positron emission tomography-based
prediction for splenectomy in patients with suspected splenic
lymphoma. Ann Transl Med. 9:10092021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pata G, Bartoli M, Damiani E, Solari S,
Anastasia A, Pagani C and Tucci A: Still a role for surgery as
first-line therapy of splenic marginal zone lymphoma? Results of a
prospective observational study. Int J Surg. 41:143–149. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kennedy ND, Lê GN, Kelly ME, Harding T,
Fadalla K and Winter DC: Surgical management of splenic marginal
zone lymphoma. Ir J Med Sci. 187:343–347. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Archibald WJ, Baran AM, Williams AM,
Salloum RM, Richard Burack W, Evans AG, Syposs CR and Zent CS: The
role of splenectomy in management of splenic B-cell lymphomas. Leuk
Res. 128:1070532023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ma J, Jiang Z, Ding T, Xu H, Song J, Zhang
J, Xie Y and Wang W: Splenectomy as a diagnostic method in
lymphoma-associated hemophagocytic lymphohistiocytosis of unknown
origin. Blood Cancer J. 7:e5342017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jing-Shi W, Yi-Ni W, Lin W and Zhao W:
Splenectomy as a treatment for adults with relapsed hemophagocytic
lymphohistiocytosis of unknown cause. Ann Hematol. 94:753–760.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fouquet G, Larroche C, Carpentier B,
Terriou L, Urbanski G, Lacout C, Lazaro E, Salmon Gandonnière C,
Perlat A, Lifermann F, et al: Splenectomy for haemophagocytic
lymphohistiocytosis of unknown origin: risks and benefits in 21
patients. Br J Haematol. 194:638–642. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Howard SC, Jones DP and Pui CH: The tumor
lysis syndrome. N Engl J Med. 364:1844–1854. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Han SM, Teng CL, Hwang GY, Chou G and Tsai
CA: Primary splenic lymphoma associated with hemophagocytic
lymphohistiocytosis complicated with splenic rupture. J Chin Med
Assoc. 71:210–213. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Griffin G, Shenoi S and Hughes GC:
Hemophagocytic lymphohistiocytosis: An update on pathogenesis,
diagnosis, and therapy. Best Pract Res Clin Rheumatol.
34:1015152020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hunt DJ, Royer AM and Maxwell RA: Post
splenectomy tumor lysis syndrome. Am Surg. 78:E299–E300. 2012.
View Article : Google Scholar
|
|
32
|
Daver N, McClain K, Allen CE, Parikh SA,
Otrock Z, Rojas-Hernandez C, Blechacz B, Wang S, Minkov M, Jordan
MB, et al: A consensus review on malignancy-associated
hemophagocytic lymphohistiocytosis in adults. Cancer.
123:3229–3240. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Song Y, Wang J, Wang Y, Wu L and Wang Z:
Requirement for containing etoposide in the initial treatment of
lymphoma associated hemophagocytic lymphohistiocytosis. Cancer Biol
Ther. 22:598–606. 2021. View Article : Google Scholar : PubMed/NCBI
|