Introduction
Chronic inflammation has been identified as the key
contributing factor to the malignant state (1). Inflammation occurs when the organism
responds to pathogens or to physical or chemical damage to
eliminate the source of the damage and restore homeostasis
(2). However, the complex crosstalk
between cytokines, chemokines, growth factors and reactive oxygen
species (ROS) produced by inflammatory cells and injured
parenchymal cells during chronic inflammation may lead to
carcinogenesis (3,4), which has been well demonstrated in
inflammatory bowel disease-associated colorectal cancer
(IBD-CRC).
IBD is a group of inflammatory disorders which
manifest in the intestines, including Crohn's disease (CD) and
ulcerative colitis (UC) (5). IBD is
currently thought to be related to chronic inflammation caused by
autoimmune factors and intestinal microbiota. The chronic
inflammatory disease can feature extensive infiltration of
inflammatory cells in a biopsy specimen of the intestinal mucosa
and unconventional dysplasia can frequently be detected during
endoscopy (6). The severity of
colon inflammation is an independent risk factor for UC-associated
CRC and increased inflammatory insults may contribute to higher
rates of genomic instability and malignant transformation in
nonconventional dysplasia (7).
Compared to sporadic CRC (sCRC), IBD-CRC follows a different
clinicopathological, molecular and risk profile and can be
considered a complication of chronic bowel inflammation (8).
The overall incidence of CRC in patients with IBD is
thought to increase almost linearly with disease duration. The
incidence of IBD-CRC is related to the disease course of IBD, so it
is difficult to give an accurate numerical indication of the annual
incidence of IBD-CRC. The cumulative probability of developing CRC
in patients with UC is 2% at 10 years, 8% at 20 years and 18% at 30
years (9). A meta-analysis based on
population-based cohort studies concluded that the pooled
standardized incidence ratio of CRC in all patients with IBD in
population-based studies was 1.7 [95% confidence interval (CI),
1.2–2.2]. Cumulative risks of CRC were 1, 2 and 5% after a disease
duration of 10, 20 and >20 years, respectively (10). In part, this is attributed to the
emergence of novel targeted agents targeting inflammatory pathways
in colitis and a paradigm shift in management aimed at histological
remission. The colonic inflammatory load in patients with IBD has
been effectively reduced, leading to an improvement in their
disease course and prognosis.
In the present review, the key relationships between
chronic inflammation and IBD-CRC pathogenesis were explored.
Specifically, the study delved into the oncogenic mechanisms of the
primary inflammatory signalling pathways that arise during colonic
inflammation and the genomic changes that result from oxidative
stress. In addition, the interplay between the tumour
microenvironment, gut microbiota and host immune factors in the
development of inflammatory carcinogenesis in IBD-CRC was explored.
Furthermore, the significance of anti-inflammatory therapy for
IBD-CRC was discussed to provide ideas for clinical management.
Inflammatory mediators and signalling
pathways in IBD-CRC
The main difference between IBD-CRC and sporadic CRC
in terms of pathogenesis is thought to be in the damaging effects
of oxidative stress resulting from the crosstalk between
inflammatory signalling pathways and epithelial cells and various
inflammatory cells, which in turn leads to the development of an
inflammatory-multifocal dysplasia-cancer sequence. This section
describes the inflammatory mediators and signalling pathways
associated with IBD-CRC.
Nuclear factor κB (NF-κB). The transcription factor
NF-κB is a significant regulator of epithelial cell integrity and
intestinal immune homeostasis. Its upstream activation by various
mechanisms, such as innate and adaptive immune responses and the
release of pro-inflammatory cytokines [e.g. tumour necrosis factor
(TNF)-α and interleukin (IL)-1β], causes NF-κB to translocate to
the nucleus to promote the transcription of cell cycle genes
(11), inhibits apoptosis and
further increases the transcription of pro-inflammatory cytokines.
Activation of NF-κB depends on the induction of NF-κB inhibitors by
IκB kinase (IKK) (12) (Table I) (13–29).
 | Table I.Mechanisms of action of NF-κB in
IBD-CRC. |
Table I.
Mechanisms of action of NF-κB in
IBD-CRC.
| Related
molecule | Main findings | (Refs.) |
|---|
| NF-κB | • Tumour
suppression by deletion of IKKβ in intestinal epithelial cells is
associated with increased apoptosis of epithelial cells during
tumour promotion. | (13,14) |
|
| • Removal of IKKβ
from myeloid cells reduced the expression of pro-inflammatory
cytokines that may act as tumour growth factors, without affecting
apoptosis. |
|
|
| • CAFs produce
pro-inflammatory and tissue remodelling molecules via NF-κB
transcription, regulating the production of pro-inflammatory and
oncogenic mediators, which in turn regulate tumour properties and
the microenvironment, leading to tumour progression. |
|
| β-catenin | Regulation of NF-κB
activity by β-catenin via TNFRSF19 may contribute to colorectal
tumorigenesis. | (15) |
| TNFR1 |
TNF-α/TNFR1-mediated signalling enhances
the expression of chemokines KC/CXCL1 and MCP-1/CCL2, which
regulate the infiltration of neutrophils and macrophages involved
in the development and progression of colitis-associated
cancers. | (16) |
| TNFR2 | • TNFR2 signaling
in intestinal epithelial cells may be directly involved in the
development of IBD-CRC with persistent colitis. | (17,18) |
|
| • The NF-κB pathway
is activated in colonic epithelia from DSS-administered mice in
association with upregulation of TNFR2 rather than TNFR1. |
|
|
| • IL-6- and
TNFα-induced TNFR2 expression in colon cancer cells is mediated
primarily by STAT3, providing evidence that TNFR2 may contribute to
the tumor-promoting roles of STAT3. |
|
| IFN | IFN gene
stimulating factor may inhibit colorectal tumourigenesis by
limiting the activation of NF-κB and STAT3 signalling pathways and
further inhibiting increased levels of the pro-inflammatory
cytokines IL-6 and KC. | (19) |
| RIPK3 | RIPK3 deficiency
can lead to uncontrolled activation of NF-κB, STAT3, AKT and
Wnt-β-catenin signalling pathways, enhancing the ability of
intestinal epithelial cells to proliferate aberrantly in a
persistent inflammatory microenvironment and promoting CRC. | (20) |
| TLR4 | • TLR4 in
intestinal epithelial cells is required for the recruitment and
activation of COX-2 expressing macrophages. | (21,22) |
|
| • TLR4 activation
appears to promote the development of colitis-associated cancer by
mechanisms including enhanced COX-2 expression and increased EGFR
signaling. |
|
| TLR9 | • The protein
expression level of TLR9 is gradually upregulated during the
development of CRC. | (23) |
|
| • The expression
level of TLR9 was found to be positively correlated with that of
NF-κB and Ki67. |
|
|
| • TLR9 may play an
important role in the development of IBD-CRC by regulating NF-κB
signaling. |
|
| IL-17RD | The absence of
IL-17RD had no effect on the proliferation of normal or tumourous
intestinal epithelial cells, but there was elevated expression of
pro-inflammatory tumourigenic cytokines (e.g. IL-17A and IL-6) and
an increase in STAT3 tyrosine phosphorylation. | (24) |
| EGFR | TNF-α, IL-1β and
IFN-γ pro-inflammatory cytokines mediated by EGFR signaling in
macrophages have an important role in IBD-CRC. | (25) |
| Smad7 | Low expression of
IL17A caused by the Smad7 expression in tumor-infiltrating CD4 (+)
T cells enabled the TNF-α-mediated killing of cancer cells both
in vitro and in vivo, thus indicating that the
Smad7-mediated plastic effect on the T-cell phenotype protects
against CRC. | (26) |
| Claudin-1 | • TNF-α is able to
mediate Claudin-1, which has a regulatory role in the tumourigenic
capacity of colon cancer cells. | (27) |
|
| • Dysregulation of
Claudin-1 expression plays a key role in inflammation-induced colon
cancer growth and progression through regulation of ERK and Src
signaling. |
|
| Src kinase | Src kinase
activation enhances the response of epithelial cells to TNF-α,
leading to increased invasion through mechanisms that involve
production of reactive oxygen intermediates. | (28) |
| Mutant P53 CRC | Mutant p53 prolongs
NF-κB activation and promotes chronic inflammation and
inflammation-associated. | (29) |
The NF-κB signalling pathway is involved in
inflammation-associated tumourigenesis and development and
represents a critical molecular link between inflammation and
cancer. Furthermore, the tumour-promoting role of NF-κB in
different cells varies. IKKβ in intestinal epithelial cells
inhibits apoptosis via the mitochondrial pathway, promoting
inflammation-associated tumourigenesis (13). By contrast, IKKβ in myeloid cells
mainly affects tumour multiplication and size by regulating
pro-inflammatory mediators such as cyclooxygenase (COX)-2, matrix
metalloproteinase (MMP)-9 and ROS (13). Cancer-associated fibroblasts (CAFs)
regulate the production of pro-inflammatory and oncogenic mediators
by activating NF-κB, which regulates tumour characteristics and the
microenvironment, leading to tumour progression (14). This activity is similar to the
function of IKKβ in myeloid cells. NF-κB can activate signal
transducer and activator of transcription 3 (STAT3) by increasing
IL-6 transcription, and STAT3 activation is essential for sustained
NF-κB activation in tumour cells. NF-κB activation is also
necessary for sustained NF-κB activation in tumour cells (30).
STAT3. Phosphorylation activation of STAT3, another
essential transcription factor linking IBD and IBD-CRC, can provide
transcriptional nodes for cancer cells to autonomously initiate
transcription of tumour-promoting genes associated with
proliferation and survival (31,32).
Numerous STAT3-induced regulated genes reactivate the STAT3 pathway
and maintain a stable feedback loop between tumour immune and
stromal cells in the tumour microenvironment (Table II) (32–47).
| Table II.Mechanisms of action of STAT3 in
IBD-CRC. |
Table II.
Mechanisms of action of STAT3 in
IBD-CRC.
| Related
molecule | Main findings | (Refs.) |
|---|
| STAT3 | • STAT3 has the
ability to mediate IL-6 and IL-11-dependent IEC survival and
promote proliferation through G1 and G2/M cell cycle
progression. | (32–34) |
|
| • T cells within
the local stroma of tumours are highly activated in myeloid STAT3
knockout IBD-CRC model mice. Myeloid cells may mediate immune
evasion of tumours through STAT3 signaling. |
|
|
| • STAT3 signaling
in the tumour microenvironment induces an increase in IL-23
secretion by tumour-associated macrophages, while decreasing IL-12
secretion by dendritic cells and upregulating IL-10 secretion by
Treg cells, thereby altering the balance of tumour immunity towards
oncogenesis. |
|
| STAT3 mTORC1 | Inflammation
promotes the development of IBD-CRC via the STAT3 and mTORC1
pathways. | (35,36) |
| IL-6 | • IL-6 mediated by
the transcription factor STAT3 produced by intrinsic layer myeloid
cells protects normal and premalignant epithelial cells from
apoptosis. | (37,38) |
|
| The
NF-κB-IL-6-STAT3 cascade is an important regulator of the
proliferation and survival of tumour-initiating IECs. |
|
|
| • IL-6
trans-signalling in epithelial cells induced by
macrophage-derivedIL-6/soluble IL-6 receptor α has a crucial role
in the development of IBD-CRC. |
|
| IL-11 | • The IL-11/STAT3
signalling axis is a more potent driver of gastrointestinal tumour
progression than IL-6. | (39–41) |
|
| Colitis-induced
IL11 activates STAT3 in colon crypt epithelial cells. |
|
| IL-6/IL-11 | IL-6/IL-11
activates STAT3 in tumour-associated fibroblasts and is associated
with poor prognosis. | (42) |
| RORγt | FoxP3+RORγt+ Tregs
drive colitis-associated CRC growth by activating |
|
|
| STAT3 in tumour
cells and promoting uncontrolled expression of IL-6 in
tumour-infiltrating dendritic cells | (43) |
| IL-21 | IL-21 promotes an
inflammatory environment in IBD-CRC through increased | (44) |
|
| T-cell infiltration
and increased expression of IL-6 and IL-17A. |
|
| S1P | • S1P is essential
for the production of the multifunctional NF-κB-regulated cytokine
IL-6, the sustained activation of the transcription factor STAT3
and the upregulation of the S1P receptor S1PR1, linking chronic
inflammation and IBD-CRC. | (45,46) |
|
| • STAT3-induced
S1PR1 expression is crucial for persistent STAT3 activation in
tumours. |
|
| Sphk1 | Intestinal
epithelial deletion of Sphk1 prevents colitis-associated cancer
development by inhibition of epithelial STAT3 activation. | (47) |
In intestinal epithelial cells, STAT3 can inhibit
apoptosis by inducing the expression of B-cell lymphoma extra large
protein, survivin and heat shock protein 70 and can promote cell
proliferation by controlling the expression of G1/S and G2/M cell
cycle regulators (32). In addition
to activation in tumour cells, STAT3 signalling is essential for
the differentiation of pro-inflammatory type 17 T-helper cells
(Th17 cells), inhibition of dendritic cell maturation and
maintenance of the immunosuppressive function in forkhead box P3
(Foxp3)+ T-regulatory (Treg) cells. Constitutive activation of
STAT3 in various tumour-infiltrating immune cells, such as
dendritic cells and macrophages, will lead to sustained secretion
of pro-inflammatory cytokines and shift the local microenvironment
towards an immunosuppressive direction (45,48).
IL-6 and IL-11 can activate STAT3 transcription via
Janus kinase (JAK), affecting downstream inflammatory signalling
(47). The NF-κB/IL-6/JAK/STAT3
cascade is an important regulatory pathway for the proliferation
and survival of tumour-initiating intestinal epithelial cells
(37). IL-6/soluble IL-6 receptor α
produced by macrophages can induce IL-6 transduction signalling in
epithelial cells, which has a vital role in the development of
IBD-CRC (37). The activation of
STAT3 by most signalling molecules is mainly transduced by the
activation of STAT3 by IL-6.
Recent studies have shown that activating the STAT3
signalling axis by IL-11 production by cancer-associated
fibroblasts and myeloid cells is more potent for gastrointestinal
tumour progression than IL-6 (39).
IL-11+ fibroblasts promote tumour progression by secreting IL-11 to
activate colonic tumour epithelial cells and colonic fibroblasts
(42). IL-11 activation of STAT in
cancer-associated fibroblasts often indicates poor prognosis
(35).
Sphingosine kinase 1 (SphK1)/sphingosine-1-phosphate
(S1P)/S1P receptor 1 (S1PR1) axis. The SphK1/S1P/S1PR1 axis lies
between NF-κB and STAT3, and S1P is essential for the production of
the multifunctional NF-κB-regulated cytokine IL-6 and for the
sustained activation of the transcription factor STAT3 (44). Adenomatosis polyposis Coli protein
(APC) is a common genetic mutation in early-stage colorectal cancer
patients. Mucosal proliferation is reduced in APC Min/+ mice with
Sphk1 deficiency in the intestinal epithelium, STAT3 activation is
inhibited and gene expression of cyclin D1 and cMyc is decreased in
tumour cells (46), suggesting that
its role in colitis-associated CRC (CAC) development should not be
underestimated. Recent studies indicate that S1P may influence CD8+
T-cell proliferation and survival in a cell-intrinsic manner
through S1PR4 by expressing phosphoinositide-3-kinase adaptor
protein 1 and leukotriene A4 hydrolase, two downstream gene
products considered to be associated with T cell proliferation
and/or survival, limiting CD8+ T-cell expansion and thus promoting
tumour growth (49), although this
has not been validated in IBD-CRC models.
Wnt/β-catenin signalling. Wnt/β-catenin signalling
has been involved as a major player in cancer development and
progression with its regulatory role in the inflammatory cascade as
well as oxidative stress, both of which are crucial determinants of
cancer (50). The Wnt/β-catenin
signalling pathway regulates the inflammatory cascade response and
oxidative stress, favouring tumour development and progression.
Hyperactivation of the Wnt/β-catenin signalling pathway has been
associated with the development of CRC. There is growing evidence
that mutations in the critical regulators of the Wnt/β-catenin
signalling pathway are associated with most CRCs (51).
β-catenin is a critical component of Wnt signalling.
It is controlled by a destruction complex composed of axis
inhibition protein, APC, casein kinase-1 and glycogen synthase
kinase-3β (52). Being the core
component of the Wnt/β-catenin signalling pathway, β-catenin has a
paramount role in Wnt signal transduction (50) by translocating into the nucleus to
coactivate, with transcription factor4, the transcription of
downstream genes, including c-Myc and cyclin D1, giving rise to the
pathogenic phenotype of CRC, such as proliferation, metastasis,
chemoresistance and recurrence (53).
In most CRCs, the earliest event is APC gene
deletion, leading to nuclear β-catenin accumulation (54). However, in IBD-CRC, APC gene
mutations occur at a lower incidence of early progression.
β-catenin accumulation in IBD-CRC may occur through an
APC-independent pathway, resulting in cancer development. A
transcriptome-based analysis of tumour subtypes showed that the
typical epithelial tumour subtype associated with Wnt/β-catenin
signalling was completely absent in IBD-CRCs (55).
While another clinical trial has shown that
β-catenin expression is strongly connected with CAC (56), β-catenin levels increase
progressively with the progression from dysplasia to carcinoma.
Dysregulation of the Wnt signalling pathway may have an essential
role in IBD-CRC, with 55% of dysplastic lesions and up to 100% of
cancers expressing nuclear β-catenin (57). However, in contrast to the sCRC
pathway, in which early loss of APC function leads to abnormal Wnt
signalling, loss of APC function in IBD-CRC occurs later in <50%
of cases (58). This may be
attributed to the inflammation-driven upregulation of β-catenin in
the early stages of IBD-CRC, which induces APC mutations
independent of Wnt signalling (59).
Although there are currently conflicting reports on
the role of β-catenin in IBD-associated CRC, when considering the
role of the gut microbiota, it was found that the effect of
specific microbiota in the gut on IBD-CRC stems, at least in part,
from the activation of β-catenin signalling. For instance,
Fusobacterium nucleatum regulates β-catenin signalling by
binding its F. nucleatum adhesion protein A (FadA) adhesion
factor to E-cadherin (60).
Bacteroides fragilis secretes a zinc-dependent
metalloprotease toxin that cleaves E-cadherin, leading to β-catenin
nuclear translocation, increased c-Myc expression and cell
proliferation (61). Further below,
carcinogenesis driven by candidate pathogenic bacteria, this will
be described in detail.
IL-23/Th-17 axis. The IL-23/Th-17 axis has a vital
role in inflammation-associated tumour development. During chronic
intestinal inflammation, the breakdown of the epithelial barrier
leads to the activation of dendritic cells by microbial products,
whose secreted IL-23 activates pro-inflammatory cytokines such as
IL-17A and IL-17F by Th17 cells located in the lamina propria
mediated by the IL-6/STAT3 pathway (62). These pro-inflammatory cytokines
stimulate, through activation of the STAT3/NF-κB-dependent pathway,
the production of additional pro-inflammatory cytokines, including
IL-6 and TNF-α, further amplifying the inflammatory response
(63,64). Observations in IL-17A-deficient
dextran sulfate sodium (DSS)-induced mice revealed that the number
of tumours and mean tumour size were significantly reduced in
IL-17A-deficient mice compared to wild-type mice, suggesting that
it has a vital role in the initiation of CAC development and may
indirectly contribute to tumour progression (65). In addition, the IL-23/Th-17 axis can
also promote excessive proliferation of IBD-CRC through mechanisms
such as increased angiogenesis, upregulation of MMP-9 and reduced
infiltration of CD8+ T cells, thus improving its tumourigenic
capacity (66).
TNF-α. TNF-α participating in chronic inflammatory
diseases is an inflammatory mediator associated with carcinogenesis
that promotes the conversion of noncancer cells into tumour stem
cells (67). TNF-α undergoes
trimerisation upon binding to its receptor, activating downstream
signalling pathways, including the NF-κB inflammatory pathway, the
Fas pro-apoptotic pathway and the cellular inhibitor of apoptosis
protein-1 anti-apoptotic pathway (68). Low-level, sustained TNF-α production
induces a tumour phenotype (69).
The tumour-promoting mechanism of TNF-α is based on the generation
of ROS and reactive nitrogen species (RNS), which induce DNA damage
and thus promote tumourigenesis (28). In the presence of TNF-α and IL-1,
PI3K/AKT activates NF-κB signalling by phosphorylating IKK
(70).
The TNF-α/TNF receptor axis, on the one hand, leads
to massive local infiltration of macrophages and neutrophils
through the release of chemokines, which in turn activates
inflammatory pathways such as NF-κB and controls inducible nitric
oxide synthase (iNOS) production; on the other hand, TNF may also
promote tumour angiogenesis by inducing infiltration of macrophages
and neutrophils expressing COX-2.
Azoxymethane (AOM)/DSS-treated colitis mice exhibit
increased TNF-α expression. In AOM/DSS mice deficient in the
central TNF receptor, neutrophil and macrophage infiltration in the
mucosa is reduced, mucosal damage is attenuated and tumour
formation is inhibited (16). The
increase in TNF-α induced by inflammation was mainly produced by
infiltrating inflammatory cells and further activated the NF-κB
signalling pathway in the inflammatory cells, causing widespread
colonic inflammation, which induced carcinogenesis in this
model.
Oxidative stress in CAC
Oxidative stress induced by chronic inflammation has
a central role in IBD-CRC carcinogenesis, which may also be one of
the critical differences in pathogenesis between IBD-CRC and
sCRC.
During the process of the host immune system
combating intestinal pathogenic bacteria and dealing with
pre-existing inflammation, various kinds of recruited inflammatory
cells produce massive amounts of ROS and RNS, which are
collectively known as reactive oxygen and nitrogen species (RONS).
RONS can cause oxidative stress, leading to DNA damage in
intestinal epithelial cells (71).
Patients with IBD presenting with chronic intestinal inflammation
have been found to have increased RONS production and lipid
peroxidation and decreased antioxidant capacity with increased
oxidative DNA damage, which are likely mechanisms that drive
mutagenesis (72).
The accumulation of molecular events and mutations
in somatic cells, followed by their clonal expansion, have been
studied for their role in IBD-CRC pathogenesis. This process leads
to extensive preneoplastic ‘field changes’ in the colonic mucosa
before the development of histological evidence of dysplasia. In
this section, the recently discovered impact of
inflammation-induced oxidative stress on IBD-CRC and the commonly
found molecular oncogenic pathways of IBD-CRC will be
discussed.
Genomic instability caused by
oxidative stress
During acute inflammation, RONS from activated
immune cells can promote tissue repair and regeneration in addition
to their pathogen-killing effects. However, recurrent histological
damage and RONS repair processes also induce permanent DNA damage
(including single- and double-strand breaks, nucleotide
modifications and abrogation sites) and genomic instability in
proliferating intestinal epithelial cells (4).
Apart from oxygen free radicals produced by
inflammatory cells, extracellular free radicals from intestinal
bacteria significantly increase DNA damage in co-clonal epithelial
cells, possibly associated with CRC-associated chromosomal
instability (CIN) (73).
Furthermore, it has been shown that there is a
positive feedback relationship between inflammation and genomic
instability (74). Inflammation is
attributed to mutations that damage DNA through the production of
RONS, exacerbating the inflammation in turn. Despite being
influenced by complex signalling networks and DNA repair
mechanisms, it usually leads to carcinogenesis under long-term
chronic inflammatory conditions (75,76).
The accumulation of ROS and RNS has been observed in
inflammatory tissues of patients with active UC and CD (75). If RONS disrupt proto-oncogenes in
the intestinal epithelium, it may cause alterations in the genetics
and epigenetics of heritable intestinal epithelial cells.
The primary genetic alterations in IBD-CRC differ in
frequency and timing from those in sCRC, which are thought to be
closely related to genomic damage induced by oxidative stress.
Metagenomic studies have found that proto-oncogene p53 mutations
and loss of heterozygosity occur early in IBD-CRC (77). P53 loss of function appears to be an
early event in initiating IBD-CRC pathogenesis (78). P53 critically impacts cell
proliferation and prevents clonal expansion of mutant cells.
Besides being found in >80% of patients with IBD-CRC (79), p53 mutations have been found in
inflamed mucosa of 50% of patients with UC without cancer (80,81).
In vitro studies have shown that the acquisition of mutant
p53 (mutp53) function is manifested in DSS-induced colitis mice by
the rapid emergence of flattened developmentally abnormal lesions
that can progress to invasive carcinomas, accompanied by the
accumulation of mutant p53 and enhanced activation of NF-κB. This
change is similar to the developmental pattern of human IBD-CRC and
may explain the early appearance of p53 mutations in human IBD-CRC
(30).
Loss of function of the oncogene APC and the
proto-oncogene KRAS also occur as early events in IBD-CRC but with
a much lower prevalence than in sCRC (82). APC encodes an essential member of
the β-catenin disruption complex. This complex critically
attenuates the nuclear localisation of β-catenin and its binding to
the TCF family of transcription factors, promoting Wnt signalling.
Amplification of MYC proto-oncogene occurred in 26% of IBD-CRC
samples compared to 26% of sCRC samples. In addition, isocitrate
dehydrogenase 1 mutations were significantly more common in IBD-CRC
and rare in sCRC.
Carcinogenic pathways of
IBD-associated CRC
Inflammation is instrumental in promoting gene
mutations and genomic instability (74). Inflammation-driven genetic
alterations, along with changes at the epigenetic level, may have
an essential role in the tumourigenesis of CRC, particularly in
IBD-CRC (83). Similar to sCRC, the
major oncogenic molecular pathways in IBD-CRC include CIN,
microsatellite instability (MSI) and CpG island methylator
phenotype.
CIN
CIN is usually triggered by mutations in oncogenes
followed by a series of duplications that ultimately lead to an
altered chromosome number (aneuploidy) and chromosomal structural
abnormalities (somatic copy number alterations, deletions,
insertions and amplifications) (84) and possible loss of oncogene
heterozygosity and chromosomal rearrangements. CIN is considered a
significant feature of the pathogenesis of CAC and is found in 80%
of cases (74,76). CIN is associated with a progressive
accumulation of mutations in pro-oncogenes and tumour suppressor
gene. Aneuploidy is a consequence of CIN and occurs before
dysplasia in colonic mucosal biopsy samples from patients with IBD
(85).
Furthermore, there was a statistically significant
relationship between increased aneuploidy and histological
progression of dysplasia/carcinoma in UC (78). This observation highlights the
progressive genomic instability during IBD-CRC tumour
progression.
MSI and hypermethylation pathway
MSI is thought to be a genomic microsatellite
hypermutation phenotype due to faulty operation of the DNA mismatch
repair (MMR) mechanism. Following the inactivation of MMR genes,
mutations continually accumulate in the cancer genome as there is a
lack of ability to repair post-DNA replicative base substitutions
or slippage mistakes at microsatellite sequences. With defective
MMR, there has been a significant increase in mutated genes that
have not been able to be correctly repaired and this ability to
increase mutation frequency is thought to shorten the typical time
frame for adenoma-to-carcinoma formation from 1–2 decades to 1–2
years (86).
Damage to the DNA MMR system induced by oxidative
stress would lead to the accumulation of MSI phenotypes in the
colonic mucosa of patients with IBD-CRC. The MSI phenotype can be
observed in ~15% of patients with IBD-CRC, a rate similar to that
of sCRC (87). In patients with
IBD-CRC, high MSI is closely associated with hMLH1 hypermethylation
and hMSH2 expression deficiency, two mismatch repair genes
(88). In IBD-CRC, proximal tumours
with high mutation rates correlate with MSI and have a higher
predicted neo-epitope load (89).
Such tumours tend to have increased immunogenicity and may respond
better to immunotherapy.
The CpG island methylation phenotype itself overlaps
with MSI pathogenesis. Distinguished from MSI, the hypermethylation
pathway is characterised by the CpG island methylator phenotype,
resulting in the epigenetic silencing of several genes,
particularly tumour suppressor genes. Several studies have explored
the hypermethylation pathways in patients with IBD-CRC. For
instance, promoter hypermethylation of p16INK4a and
p14ARF, which are expected early events in IBD-CRC
carcinogenesis, was found in 100 and 50% of UC-CRC, respectively,
and these two genes have a crucial role in cell cycle inhibition
(90). In addition, CDH1 promoter
methylation was detected in 93% of patients with IBD with colonic
dysplasia, compared with 6% of patients with IBD without dysplastic
lesions (91). This gene is
involved in biological processes that are dysregulated in cancer,
such as the sequence of cell sorting, migration and
differentiation. Death-associated protein kinase (DAPK), a tumour
suppressor gene, also appears to undergo epigenetic modifications
during IBD-CRC carcinogenesis. A study using surgical resection
specimens from patients with UC for DAPK epigenetic analysis
concluded that DAPK silencing induced by promoter hypermethylation
may lead to the accumulation of mucosal DNA damage in UC and,
therefore, may contribute to the development of UC-associated CRC
(92).
Epithelial consensus molecular
subtypes of IBD-CRC
A transcriptome-based analysis of tumour subtypes
showed that the typical epithelial tumour subtype associated with
Wnt signalling was absent in IBD-CRCs; instead, a mesenchymal
stroma-rich subtype predominated (55). IBD-CRCs exhibit a switch from the
epithelial consensus molecular subtype (CMS)2 to the more
mesenchymal CMS4 phenotype [epithelial-mesenchymal transition
(EMT)]. Dysregulation of Wnt signalling activates transforming
growth factor (TGF)-β and results in an ‘immuno-inflammatory’
inhibitory microenvironment enriched with CD4+ cells. EMT involves
loss of tumour cell polarity and cell-cell adhesion to enhance cell
migratory and invasive properties (93).
In IBD-associated CRC, polymeric immunoglobulin
receptor (PIGR) and cytokine oncostatin M (OSMR), which are
involved in mucosal immunity, are dysregulated by epigenetic
modifications. Low expression of PIGR may promote epithelial
barrier dysfunction and persistent inflammation. Increased OSMR
signalling may then favour the establishment of mesenchymal CRC
subtype in patients with IBD (77).
A previous single-cell analysis showed the presence of an activated
mesenchymal cell population in the mesenchyme surrounding the
colonic crypts of patients with IBD. This subpopulation expresses
TNF superfamily member 14, fibroblastic reticulocyte-associated
genes, IL-33 and lysyl oxidase. In addition, it induces factors
that impair epithelial proliferation and maturation and contribute
to oxidative stress and disease severity in vivo (94). Whether this process may contribute
to the cancerous process requires further exploration.
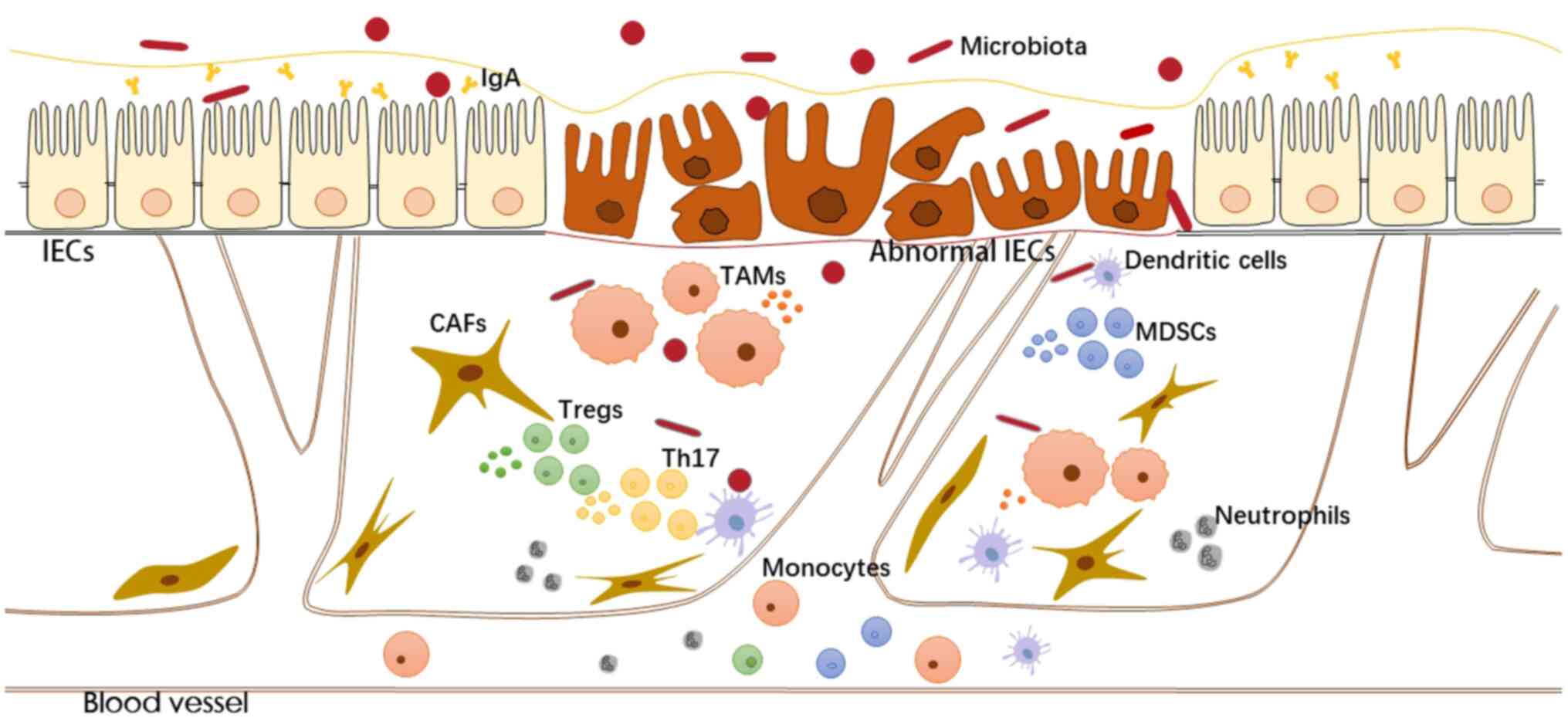
Inflammatory/tumour microenvironment
Various cells in the inflammatory microenvironment
(e.g., epithelial cells, immune cells and stromal cells)
communicate with each other through direct contact or through
secreted cytokines and chemokines, coordinating the ongoing
inflammatory response by autocrine and paracrine ways. The
transformation process of the inflammatory microenvironment to the
immunosuppressive tumour microenvironment has now been documented.
Various immune mediators and the abundance and activation status of
immune cells in the local microenvironment determine the
anti-tumour response and pro-tumour effects of inflammation
(95). The tumour microenvironment
formed by infiltrating immune cells, stromal cells and tumour cells
is essential for maintaining signalling proliferation, resisting
cell death and inducing angiogenesis (Fig. 1).
Myeloid cells promoting immunosuppression. In the
inflammatory microenvironment, myeloid cells are the central cells
that suppress the inflammatory response. However, myeloid cells are
hidden in their original anti-inflammatory function in most
established tumours under the influence of existing tumour cells.
Local infiltration of myeloid cells by the tumour makes it
difficult to contain tumour growth and drives the tumour
microenvironment further toward immunosuppression under the
influence of local immune mediators (96) (Fig.
2).
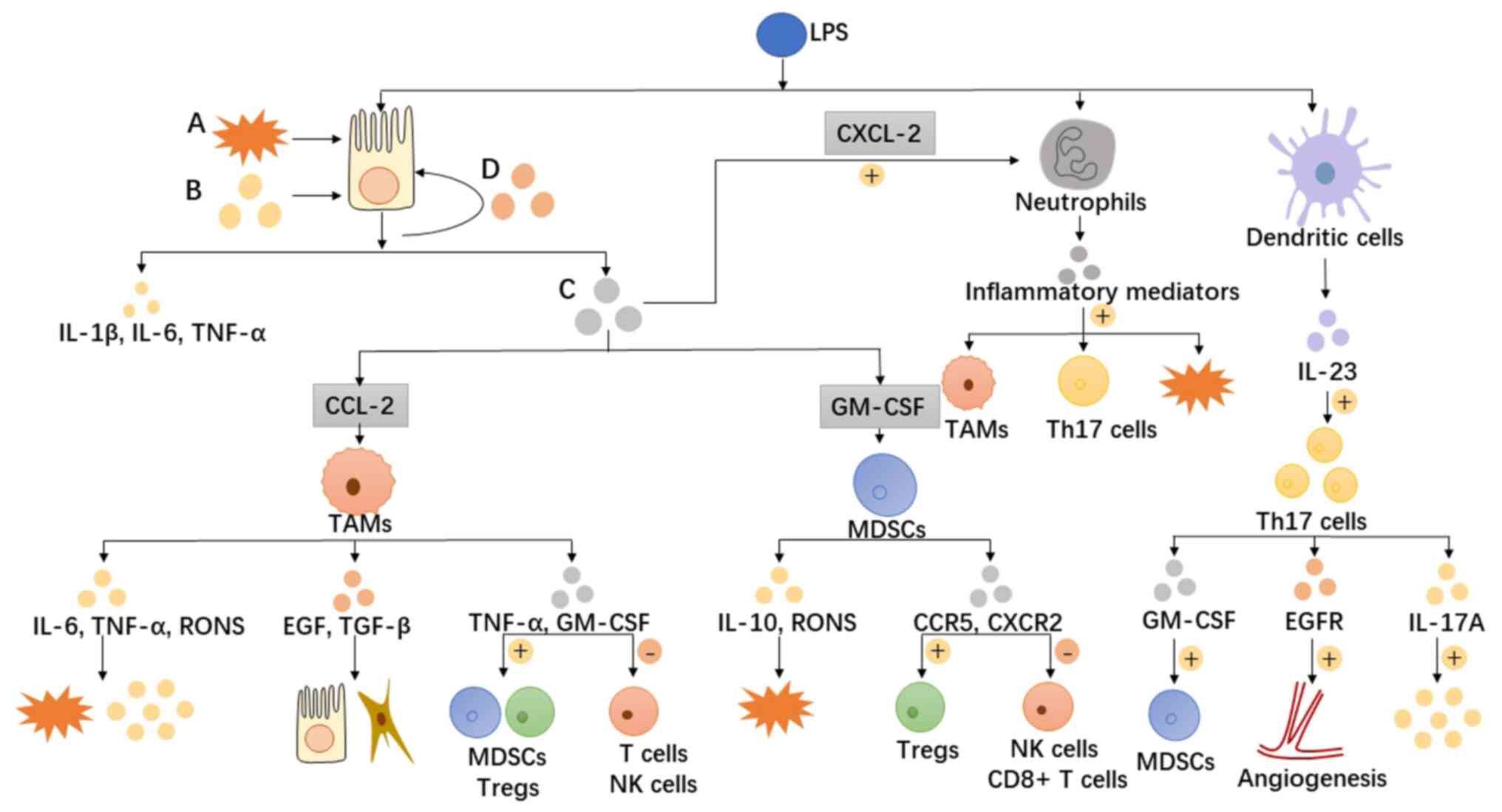 | Figure 2.Disruption of the intestinal mucosal
barrier provides an opportunity for bacterial products (e.g. LPS)
to enter the lamina propria and continuously stimulate recruited
immune cells by chemokines, leading to continuous activation of
inflammatory signalling pathways and the production of large
amounts of inflammatory mediators. Potential mechanisms include the
following: i) Inflammatory factors, such as IL-6, IL-10 and TNF-α,
produced by a large number of infiltrating macrophages, MDSCs and
other inflammatory cells crosstalk via paracrine and autocrine
signals, activating the inflammatory pathways of various cells in
the tumour microenvironment and causing inflammation to induce
uncontrollable development. ii) Cells with immunosuppressive
properties (e.g. MDSCs and Tregs) inhibit cytotoxic CD8+ T-cells
and NK cells either directly or in an indirect manner through the
secretion of chemokines, promoting the establishment of an
immunosuppressive microenvironment. iii) Damage to intestinal
epithelial cells by RONS produced by inflammatory cells further
contributes to the progression of inflammation and malignancy. iv)
The secretion of various growth factors drives angiogenesis,
provides nutrition for tumour cell growth and induces mesenchymal
cell function and differentiation. A, oxidative stress; B,
pro-inflammatory cytokines; C, pro-inflammatory chemokines; and D,
growth factors. LPS, lipopolysaccharide; IL, interleukin; TNF-α,
tumour necrosis factor-α; GM-CSF, granulocyte-macrophage
colony-stimulating factor; TAMs, tumour-associated macrophages;
RONS, reactive oxygen and nitrogen species; TGF-β, transforming
growth factor-β; epidermal growth factor, EGF; Tregs, T regulatory
cells; NK, natural killer; CXCL-2, C-X-C motif ligand 2; CCR5, C-C
motif chemokine receptor 5; EGFR, epidermal growth factor receptor;
MDSC, myeloid-derived suppressor cell. |
Tumour-associated macrophages
(TAMs)
Inflammation-promoting macrophages are a central and
potent component of innate immunity. During colon inflammation,
intestinal epithelial cells stimulated by lipopolysaccharide (LPS)
release chemokines (e.g., C-X-C motif chemokine ligand-2 and C-C
motif chemokine ligand 2) and recruit large numbers of monocytes
and neutrophils (38). Massive
macrophages infiltrate the intestinal lamina propria, triggering
polarisation towards M1 or M2 in response to continuous exposure to
inflammatory microenvironmental signals (97,98).
Bacterial products and Th1 cytokines induce macrophage polarisation
towards pro-inflammatory, cytotoxic and antigen-presenting M1,
whereas Th2 cytokines polarise macrophages towards
immunomodulatory, pro-angiogenic M2 (99).
TAMs are the most abundant myeloid cell population
infiltrating solid tumours, predominantly displaying an M2
phenotype as crucial for promoting tumour inflammation and
mediating immunosuppression (100). TAMs have been found to have poor
antigen-presenting capacity and limited anti-tumour responses.
Reduced killing capacity of TAMs has been associated with reduced
iNOS expression (101).
The tumour-promoting effects of TAMs are
multifaceted. TAMs infiltrated in the TME, on the one hand, promote
the formation of the inflammatory microenvironment through a large
amount of released inflammatory mediators (e.g., IL-6, TNF-α and
RONS) (102); on the other hand,
they further provide proliferation signals through the supply of
mitogenic growth mediators [e.g., epidermal growth factor (EGF) and
TGF-β] to maintain the uncontrolled proliferation of epithelial and
mesenchymal cells (103). In
addition, TAMs have recruitment effects on immunosuppressive cells
such as myeloid-derived suppressor cells (MDSCs) and Tregs and
inhibitory effects on T cells and natural killer (NK) cells,
resulting in the suppression of immune activity in the local
microenvironment (104,105). In the early stages of IBD-CRC,
monocyte-like macrophages are recruited into the local
microenvironment and promote the expansion of Th17 cells by
producing IL-1β, which further exacerbates dysbiosis and
inflammation, leading to a vicious cycle and contributing to
tumourigenesis (102).
MDSCs
Similar to TAMs, MDSCs [including monocytic MDSCs
and polymorphonuclear MDSCs (PMN-MDSCs)] are also premature myeloid
cells. Although they overlap with monocytes and granulocytic
myeloid cells, they represent two cell populations with distinct
immune phenotypes. MDSCs can suppress both adaptive and innate
immunity. Infiltration of MDSCs may promote tumourigenesis on the
one hand through activation of the STAT3 and NF-κB pathways
(106), and on the other hand, may
influence the function of other cells through chemokines. MDSCs,
which express C-X-C motif chemokine receptor 2, can directly
inhibit cytocidal cytotoxicity of NK cells and CD8+ T cells
(107,108), accelerating tumour growth
(109). Furthermore, C-C motif
chemokine receptor 5-expressing MDSCs can also induce local
aggregation of Tregs in tumours (110), promoting immunosuppression. Mouse
models with breast cancer have additionally shown that MDSCs can
affect tumour immunity by increasing IL-10 production and
inhibiting macrophage secretion of IL-12 (111).
T cells that promote immunosuppression.
Tumour-infiltrating T cells have been associated with improved
clinical prognosis and survival of patients with CRC (112,113). Tumour-killing type 1 T-helper
cells, CD4+ T cells and CD8+ T cells have anti-tumour effects.
However, these types of cells are regulated by other
immunosuppressive cells in the tumour microenvironment, which, to a
certain extent, maintains the local inflammatory state and leads to
an immunosuppressive state in the local microenvironment.
Th17 cells
The IL-23/Th17 signalling pathway has an important
role in inflammation-associated tumours. IL-23 produced by
inflammatory dendritic cells infiltrated in the tumour
microenvironment induces the polarisation of Th17 cells.
Tumour-infiltrating γδT17 cells further activate downstream
inflammatory signals by secreting inflammatory factors such as
IL-17A. In addition, IL-8, TNF-α and granulocyte-macrophage
colony-stimulating factor secreted by Th17 cells chemotactically
attract immunosuppressive PMN-MDSC and maintain immunosuppressive
activity (114–116).
Treg cells
Treg cells modulate adaptive immunity, suppressing
inflammation in chronic inflammatory diseases. Treg cells promote
the clearance of pathogenic bacteria by establishing a protective
immune response during intestinal infections (117,118). However, the cytotoxic effects of
Treg cells on antitumourigenic CD8+ T cells have suggested that
these cells are immunosuppressive in the tumour environment
(119), and different
subpopulations of Treg cells exhibit other immune properties in an
environmentally dependent manner.
Massive infiltration of RAR-related orphan receptor
(ROR)γt+Foxp3+ cells has been observed in the intestinal lamina
propria of UC and colon cancer (120), a subpopulation of Foxp3+ Treg
cells induced in a chronic inflammatory environment. This
subpopulation of Tregs can exhibit certain features of Th17 cells,
such as secretion of IL-17A, which has been shown to have a
pathogenic role in the pathogenesis of infiltrative CRC (121). In addition, this group of cells
promotes the production of inflammatory cytokines, such as IL-17A,
IL-1, IFN-γ and TNF-α, by colitis cells (120), which supports local inflammatory
responses and oxidative stress insults.
Treg cells can perform an immunosuppressive function
by inhibiting the anti-tumour response of CD4+ T cells and CD8+ T
cells, contributing to the formation of an immunosuppressive tumour
microenvironment (122). The
co-expressed transcription factors Foxp3 and RORγt on Treg cells
can drive IBD-CRC growth by activating STAT3 in tumour cells and
promoting uncontrolled expression of IL-6 in tumour-infiltrating
dendritic cells (38).
Mesenchymal cells that promote immunosuppression.
Mesenchymal stromal cells in tumours, also known as CAFs, are a
significant component of the tumour stroma, which secrete growth
factors and inflammatory ligands. In the tumour microenvironment,
CAFs with distinct phenotypes are activated by bacterial products
(e.g., LPS) and cytokines (e.g., TNF-α) (123) to promote tumour progression
through the expression of a wide range of growth and inflammatory
factors [e.g., IL-8, vascular endothelial growth factor (VEGF) and
fibroblast growth factor 2] (124–127), resulting in a sustained
inflammatory environment. Factors from diverse cellular and tumour
sources shape the phenotype of heterogeneous CAFs (128). Similar to the polarisation of
TAMs, CAFs show a high degree of plasticity. The function of CAFs
has been shown to impact the development of the tumour
microenvironment substantially and has been found to have partial
anti-tumour activity. Studies on the role of fibroblasts in the
IBD-CRC model are summarised in Table
III (14,39,42,123–130).
| Table III.Functions of fibroblasts in the
IBD-CRC model in response to different signals. |
Table III.
Functions of fibroblasts in the
IBD-CRC model in response to different signals.
| Related
molecule | Main findings | (Refs.) |
|---|
| NF-κB | CAFs produce
pro-inflammatory and tissue remodelling molecules via NF-κB
transcription, regulating the production of pro-inflammatory and
oncogenic mediators, which in turn regulate tumour properties and
the microenvironment leading to tumour progression. | (14) |
| IL-11 | Tumour cells induce
IL-11+ fibroblasts; a feed-forward loop between colon tumour
epithelial cells and IL-11+ fibroblasts via secretion of IL-11 may
contribute to tumour development. | (39) |
| IL-6/IL-11 | IL-6/IL-11
activates STAT3 in tumour-associated fibroblasts and is associated
with poor prognosis. | (42) |
| FGF-1 and | CAFs promote tumour
growth and angiogenesis by secreting FGF-1 and | (128) |
| FGF-3 | FGF-3, which
promote the production of MMP-7 and mitogen-activated protein
kinase kinase/extracellular signal-regulated kinase in tumour
cells. |
|
| Tpl2 | Tpl2 expressed by
IMF suppresses epithelial tumorigenesis by inhibiting the HGF/c-Met
pathway. | (123) |
| EREG | CAFs can stimulate
the activation of ERK in tumour cells through the production of
EREG, which directly promotes the proliferation of tumour
cells. | (124) |
| CCL3-CCR5 | CCL3-CCR5 in the
tumour microenvironment mediates CAF accumulation and promotes CAF
proliferation and heparin-binding epidermal growth factor-like
growth factor expression. | (125) |
| Tenascin-C | • In the early
stages of IBD-CRC, tenascin-C produced by IMFs affects tumour
development via integrin αvβ3-mediated angiogenesis.of IBD-CRC
via | (126,127) |
|
| •
Tenascin-C-derived peptide TNIIIA2 may contribute to the formation
activation of stromal fibroblasts based on β1-integrin
activation. |
|
| IMP1 | The tumour
suppressor role of stromal IMP1 and its ability to modulate
protumorigenic factors suggest that the status of IMP1 is important
for the initiation and growth of epithelial tumours. | (129) |
| PGE | Locally
infiltrating CAF and neutrophils express EP2, which synergises with
TNF-α to amplify activation of the NF-κB inflammatory cascade
response and amplify recruitment to neutrophils through autocrine
amplification of CXCL1, resulting in the formation of the tumour
microenvironment. | (130) |
Gut microbiota and host immune factors
Human gut microbiota is a complex community of 100
trillion microorganisms, including various types of bacteria,
fungi, protists and viruses, which is considered a ‘microbial
organ’. The intestinal immune system of the host interacts with the
microbiota and produces a variety of metabolites or components.
These metabolites maintain the normal metabolism, proliferation and
differentiation of epithelial cells and have essential
physiological functions such as protective activities against
pathogens and immune system regulation (131,132). It has also been shown that gut
microbiota is involved in the brain-gut axis of brain-gut
communication, thereby affecting the mental and neurological
functions of the host (133).
However, dysbiosis disrupts the mucosal barrier,
leading to prolonged inflammation and cancer. Current research on
the impact of the gut microbiota on the progression of IBD towards
CRC has advanced. This section describes the intricate
physiological role of the gut microbiota in maintaining host health
and the latest information on the function of gut dysbiosis in
colorectal carcinogenesis and progression (134).
Physiological role of the gut
microbiota
The gut microbiota and its metabolites have a
multifaceted role in inhibiting inflammation, maintaining the
mucosal barrier and modulating immunity during interactions with
the host. The gut microbiota can produce a wide range of
metabolites using exogenous undigested dietary substrates (e.g.,
dietary fibre) and endogenous compounds [e.g., bile acid (BA)
metabolites] to provide energy and nutrients to the host. Certain
bacterial species, such as Bifidobacteria, are involved in the
biosynthesis of various components such as vitamin K and B
(135).
Several bacteria, such as Firmicutes, Bacteroidetes
and certain anaerobic gut microorganisms, can provide short-chain
fatty acids (SCFAs) with biologically active components through the
fermentation of dietary fibre (136). SCFAs produced in the gut are
rapidly absorbed by host epithelial cells. They are a significant
source of energy for colonic epithelial cells and have systemic
immunomodulatory and anti-inflammatory effects (136). The most abundant SCFAs in the
colon are acetate, propionate and butyrate (13,15,16),
which promote the proliferation of beneficial bacteria, stimulate
Treg cells and macrophages to reduce the production of inflammatory
mediators and increase the oxygen consumption of colonic epithelial
cells, resulting in the enhancement of immunomodulation and
intestinal barrier function (137). Butyrate has been found to enhance
intestinal barrier function by activating AMPK, Akt and other
signalling pathways to promote tight junction assembly in a
dose-dependent manner (138).
Furthermore, during colitis, butyrate induces T cell-independent
immunoglobulin A (IgA) secretion in the colon through activation of
free fatty acid receptor 3 [also known as G protein-coupled
receptor (GPR)41] and carboxylic acid receptor 2 (also known as
GPR109A), as well as inhibition of histone deacetylase, which
restores the epithelial barrier function under inflammatory
conditions (139).
In addition to producing substances that regulate
energy metabolism in colonic epithelial cells, the gut microbiota
can synthesise several neurochemicals that can affect both the
central nervous and peripheral gut systems. For instance,
γ-aminobutyric acid (GABA) is an important inhibitory
neurotransmitter in the brain and numerous neuropsychiatric
disorders have been linked to GABA dysfunction, which is also
thought to be connected to the brain-gut axis connection.
The gut microbiota has also reportedly been involved
in synthesising substances such as BA, cholesterol and conjugated
fatty acids and branched-chain amino acids (140). Bile salts escaping during
enterohepatic circulation become substrates for gut microbial
metabolism. Bacteria, including Clostridium, Bifidobacterium,
Bacteroides, Listeria and Lactobacillus, are involved in
deconjugating bile acids, which is vital for the gut microbiota to
mediate immune regulation. On the one hand, bile acids can produce
direct antimicrobial effects by disrupting bacterial biomembrane.
On the other hand, the bile acid receptors farnesoid X receptor
(FXR) and Takeda G protein-coupled receptor 5 can be activated by
bile acid derivatives, which are essential for the regulation of
intestinal inflammation and tumourigenesis. For example, activated
FXR signalling suppresses CRC progression by antagonising the
Wnt/β-catenin pathway and inhibiting cytokine signalling 3 gene
transcription.
Another significance of the gut microbiota for the
host is its fundamental role in the differentiation of the host's
immune system. Symbiotic bacteria in the gut are able to not only
competitively inhibit pathogenic pathogens in the gut, but also
indirectly defend against pathogens by activating the immune
response. For instance, LPS and flagellin derived from the gut
microbiota enhance the expression of antimicrobial peptide and
RegIIIγ from epithelial cells by stimulating Toll-like receptor
(TLR)4+ stromal cells and TLR5+ CD103+ dendritic cells (141). The gut microbial metabolite
butyrate modulates T-cell differentiation and proliferation. It
enhances Treg cell function and induces their differentiation
(142), inhibits IL-17 levels as
well as Th17 cells in peripheral blood and colon tissues of rats
with 2,4,6-trinitrobenzenesulfonic acid-induced colitis (143), and suppresses the proliferation of
CD4+ and CD8+ T cells in a dose-dependent manner (144).
Host recognition of microbial metabolites relies on
pattern recognition receptors (PRRs), essential for initiating the
innate immune response (145). One
of the most well-studied PRRs is the TLR, a transmembrane protein
expressed in intestinal epithelial cells and innate immune cells.
TLRs have an essential role in activating the innate immune
mechanism and maintaining the integrity of the intestinal
epithelial barrier by recognising highly conserved structural
molecules expressed by microbial pathogens (146). TLR4 pre-expression is upregulated
in intestinal epithelial cells during UC development and is further
increased in CAC, suggesting an essential role in the transition
from inflammation to cancer (147,148).
The gut microbiota controls the levels of
antimicrobial peptides and immunoglobulin IgA by regulating the
differentiation of intestinal lymphoid tissues and the number of
lymphocytes, which then handles the development and maturation of
the immune system.
Carcinogenesis driven by candidate
pathogenic bacteria
Dysregulation of the interaction between the host
and its microbiota leads to a loss of host immune tolerance
(149), which induces immune
responses. This aberrant immune response is thought to link
inflammation to cancer (136). For
instance, an increased abundance of pathogenic bacteria that can
cling to the intestinal epithelium correlates with increased
intestinal permeability. Alterations in the diversity and
composition of the gut microbiota would lead to the induction of
intestinal inflammation by regulating the expression of
inflammatory genes (150).
In patients with IBD with an impaired mucus layer of
the gastrointestinal tract, the luminal microbiota can penetrate
the epithelial cells, causing proliferative and inflammatory
processes (151). The microbiota
activates the host's innate and adaptive immune response by
producing metabolites that act as antigens, which cause
inflammation and promote tumour growth. Mucous membranes under the
influence of inflammatory damage are more susceptible to bacterial,
particularly pathogenic, stimulation, which causes the submucosa to
be exposed to more antigens, which leads to a vicious, positive
feedback loop of mucosal damage (152).
The onset and progression of CRC do not depend on
the species prevalent but on the entire metabolic functional
pathway of the microbiota. Metagenomic studies have observed
altered diversity and abundance of the gut microbiota in patients
with IBD and CRC. In a recent survey of patients with CAC, Richard
et al (153) reported that
the CAC group had a significantly altered gut microbiota and
reduced α-diversity compared to a healthy population and patients
with sporadic tumours. Fusobacterium and Ruminococcus genera
appeared to be significantly reduced in patients with IBD-CRC
compared to those with sCRC (154). Indeed, evidence suggests that
patients with CAC may experience changes in the composition of
their gut microbiota at different stages. Specifically, CAC can
show a significant increase in Akkermansia, Fusobacterium,
Peptostreptococcus, Streptococcus and Ruminococcus at advanced
stages, while Granulicatella and Lactobacillus were reduced
(155).
Some of these genera have been shown to contribute
to colorectal carcinogenesis through direct oncogenic effects or by
participating in or promoting chronic inflammation-mediated
epithelial cell damage (136). Gut
microbes differentially affect DNA damage, DNA methylation,
chromatin structure and non-coding RNA expression in CRCs. Several
genes and pathways altered by gut microbes have been implicated in
the development of CRC, particularly those involved in cell
proliferation and Wnt signalling.
Fusarium nucleatum is associated with
developing cancers of the oral cavity and gastrointestinal tract.
FadA inhibits the tumour suppressor activity of E-cadherin by
binding to E-cadherin in epithelial and malignant cells, leading to
increased levels of β-catenin-regulated transcription. This process
leads to the expression of inflammatory molecules, such as NF-κB,
IL-6, IL-8 and IL-18 and activation of TLR2/TLR4 (156). It has also been shown that F.
nucleatum accelerates DNA methylation of cancer-specific genes
in UC patients and inhibits cytotoxicity of NK cells through the
expression of Fap2 protein, a lectin expressed by F.
nucleatum (157).
Enterotoxigenic Bacteroides fragilis (ETBF)
is positively associated with active IBD and CRC. Activation of
Wnt/β-linker protein and NF-κB signalling by secretion of B.
fragilis toxin cleaves E-calmodulin. This cleavage increases
the permeability of the intestinal barrier and the signal
transduction of E-cadherin and β-catenin in intestinal epithelial
cells, giving the organism the potential for co-clonal oncogenic
transformation (61). Furthermore,
ETBF induces the STAT3 pathway and the Th17 cancer pathway by
driving the local infiltration of Tregs, which promotes the
pre-expression of inflammatory factors (e.g., IL-17, IL-8) and
establishes an immunosuppressive tumour microenvironment,
contributing to the development of IBD-CRC (158–161).
The polyketide genotoxin colistin, produced by E.
coli via polyketide synthase (Pks), can cause DNA damage upon
contact with epithelial cells and acts synergistically with host
inflammation to create a tumour-promoting microenvironment
(162). Colonic inflammation has
been further shown to promote the genotoxic effects of Pks+ E.
coli and facilitate E. coli colonisation of the mucosa,
leading to an increase in colistin-induced DNA damage to colonic
epithelial cells, which allows this bacterial strain to exert its
oncogenic activity (162).
In the stool flora of patients with CRC,
Enterococcus faecalis is significantly more abundant than in
healthy individuals. The possible pathogenic role of E.
faecalis in CRC is multifaceted. On the one hand, E.
faecalis activates macrophage MMP-9, which disrupts the
integrity of the intestinal monolayer and induces an inflammatory
response in the intestine. On the other hand, ROS and extracellular
superoxide produced by E. faecalis lead to genomic
instability, causing damage to colonic DNA and inducing mutations
that can lead to cancer (163).
It has been suggested that genotoxic indoleamine
derived from Morganella morganii may affect multiple aspects
of host biology, including direct DNA damage activity, triggering
or exacerbating inflammatory processes, and cancer in abiotic mice
(164). Bacteria produce
genotoxicity that can increase species' competitive ability to grow
in an inflammatory microenvironment, affecting microbiota
composition. Dysbiotic microbiota will further contribute to the
vicious cycle of inflammation and DNA damage in patients with IBD
and ultimately lead to tumourigenesis.
The current consensus is that dysbiosis is present
in both IBD and CRC and that dysbiosis may lead to disruption of
the mucosal barrier and persistence of inflammation and cancer. It
is thought that dysbiosis leads to an increase in bacteria such as
E. coli and ETBF, which leads to a breakdown of the intestinal
mucosal barrier, allowing more bacteria to move from the lumen of
the bowel to the inside of the tissue. This condition leads to
chronic tissue inflammation and the release of inflammatory and
pro-cancer mediators increases the risk of developing CRC. This
positive feedback loop of dysbiosis may underlie the
inflammation-abnormal proliferation-cancer sequence. Further
studies are required to elucidate the impact of host factors and
pathogenic microbial interactions in patients with IBD-CRC.
Implications of anti-inflammatory therapy
for IBD-CRC
The primary treatment for CRC includes surgical
resection and chemotherapy. Furthermore, radiotherapy is used
before or after surgery. In the case of metastatic CRC, targeted
therapies such as anti-EGF receptor antibodies and anti-VEGF
receptor antibodies are employed (165). For CRC, which evolves from IBD,
anti-inflammatory therapy may be an effective treatment modality to
delay tumour progression.
Chronic inflammation and oxidative stress are common
pathological processes that accompany and contribute to cancer.
Numerous epidemiological studies have shown that anti-inflammatory
therapies can be used to target the chronic inflammatory
microenvironment of tumours, contributing to therapeutic recovery
from established cancers. It has been demonstrated that
non-steroidal anti-inflammatory drugs (NSAIDs) can slow tumour
growth by acting on chronic inflammation and oxidative stress.
NSAIDs are peroxisome proliferator-activated receptor γ (PPARγ)
agonists and, therefore, downregulate the aberrant Wnt/β-catenin
pathway in cancer, which may have a positive effect on both cancer
prevention and tumour treatment (166).
Acetyl salicylic acid (aspirin) is the most widely
consumed NSAID, which also exhibits anticancer properties (167). Studies on four CRC cell lines
(i.e., HCT116, SW948, LoVo and SW480) have shown that aspirin
inhibits the transcription of genes regulated by the β-catenin-TCF
complex, including Cyclin D1 (168). Aspirin-mediated phosphorylation of
GSK-3β and β-catenin resulted in attenuating its downstream
inflammatory signalling. In addition, aspirin regulates both
β-catenin and COX-2 in colon cancer cells (169), exerting anti-cancer effects.
The mechanism of action of mesalazine can also be
used to treat and prevent CRC. This is because mesalazine has a
comprehensive effect on the pathways that lead to cancer
development and progression (i.e., the Wnt/β-catenin pathway and
PPARγ), as well as affecting the cell cycle or inhibiting the
proliferation of cancer stem cells, which are the primary cause of
CRC recurrence. Mesalazine also inhibits the COX and lipoxygenase
pathways, thereby inhibiting the release of prostaglandin E2 and
leukotrienes, which are closely related to inflammation (170). The ability of 5-aminosalicylic
acid (5-ASA) to inhibit peroxynitrite-mediated DNA strand breaks,
scavenge peroxynitrite and affect peroxynitrite-mediated formation
of free radicals is partly responsible for the anti-inflammatory
and anti-cancer effects of 5-ASA (171).
Conclusions
The pathogenesis of IBD-CRC has not yet been clearly
described. To the best of our knowledge, this is the first review
of the pathogenesis of IBD-CRC that includes molecular mechanisms,
oxidative stress, immune mechanisms and gut microbiota studies. The
review is novel in that it provides a comprehensive summary of
recent advances in the field. Anti-inflammatory therapies may be an
effective treatment as the number of treatments available
increases, and further research is needed before the efficacy of
these therapies for IBD-CRC can be demonstrated. For instance, in
terms of disease pathogenesis, further exploration of the tumour
microenvironment and histological studies on patients with IBD-CRC
may be worthwhile; in terms of treatment, more targeted therapies
and development of novel drugs for IBD-CRC and metastatic tumours
may improve the prognosis for these patients.
Acknowledgements
Not applicable.
Funding
The present study was supported by The Science and Technology
Agency Jilin Province (grant nos. 20210402013GH and
20200201343JC).
Availability of data and materials
Not applicable.
Authors' contributions
ZW, YC, HBS, YQL and TYT drafted the manuscript.
ZW, YC and HBS organized the structure of the manuscript. Funding
acquisition for this study was performed by TYT and YQL. All
authors read and approved the final version of the manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
IBD
|
inflammatory bowel disease
|
|
CRC
|
colorectal cancer
|
|
IBD-CRC
|
inflammatory bowel disease-associated
colorectal cancer
|
|
ROS
|
reactive oxygen species
|
|
UC
|
ulcerative colitis
|
|
sCRC
|
sporadic CRC
|
|
CD
|
Crohn's disease
|
|
NF-κB
|
nuclear factor κB
|
|
TNF-α
|
tumour necrosis factor-α
|
|
IKK
|
IκB kinase
|
|
CAF
|
cancer-associated fibroblast
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
JAK
|
Janus kinase
|
|
S1P
|
sphingosine-1-phosphate
|
|
APC
|
adenomatosis polyposis coli
protein
|
|
DSS
|
dextran sulfate sodium
|
|
MMP
|
matrix metalloproteinase
|
|
RNS
|
reactive nitrogen species
|
|
IL
|
interleukin
|
|
AOM
|
azoxymethane
|
|
RONS
|
reactive oxygen and nitrogen
species
|
|
CIN
|
chromosomal instability
|
|
MSI
|
microsatellite stability
|
|
MMR
|
mismatch repair
|
|
DAPK
|
death-associated protein kinase
|
|
CMS
|
consensus molecular subtype
|
|
PIGR
|
polymeric immunoglobulin receptor
|
|
OSMR
|
cytokine oncostatin M
|
|
TAM
|
tumour-associated macrophage
|
|
LPS
|
lipopolysaccharide
|
|
TGF-β
|
transforming growth factor-β
|
|
Treg
|
T regulatory
|
|
MDSC
|
myeloid-derived suppressor cell
|
|
PMN-MDSC
|
polymorphonuclear MDSC
|
|
SCFA
|
short-chain fatty acid
|
|
GABA
|
γ-aminobutyric acid
|
|
FXR
|
farnesoid X receptor
|
|
PRR
|
pattern recognition receptor
|
|
TLR
|
Toll-like receptor
|
|
Ig
|
immunoglobulin
|
|
CAC
|
colitis-associated CRC
|
|
ETBF
|
enterotoxigenic Bacteroides
fragilis
|
|
NSAID
|
non-steroidal anti-inflammatory
drug
|
|
GPR
|
G protein-coupled receptor
|
|
COX
|
cyclooxygenase
|
References
|
1
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Medzhitov R: Origin and physiological
roles of inflammation. Nature. 454:428–435. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Elinav E, Nowarski R, Thaiss CA, Hu B, Jin
C and Flavell RA: Inflammation-induced cancer: Crosstalk between
tumours, immune cells and microorganisms. Nat Rev Cancer.
13:759–771. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Singh N, Baby D, Rajguru JP, Patil PB,
Thakkannavar SS and Pujari VB: Inflammation and cancer. Ann Afr
Med. 18:121–126. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang YZ and Li YY: Inflammatory bowel
disease: Pathogenesis. World J Gastroenterol. 20:91–99. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Choi WT, Yozu M, Miller GC, Shih AR,
Kumarasinghe P, Misdraji J, Harpaz N and Lauwers GY:
Nonconventional dysplasia in patients with inflammatory bowel
disease and colorectal carcinoma: A multicenter clinicopathologic
study. Mod Pathol. 33:933–943. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nguyen ED, Wang D, Lauwers GY and Choi WT:
Increased histologic inflammation is an independent risk factor for
nonconventional dysplasia in ulcerative colitis. Histopathology.
81:644–652. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lukas M: Inflammatory bowel disease as a
risk factor for colorectal cancer. Dig Dis. 28:619–624. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Eaden JA, Abrams KR and Mayberry JF: The
risk of colorectal cancer in ulcerative colitis: A meta-analysis.
Gut. 48:526–535. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lutgens MWMD, van Oijen MGH, van der
Heijden GJMG, Vleggaar FP, Siersema PD and Oldenburg B: Declining
risk of colorectal cancer in inflammatory bowel disease: An updated
meta-analysis of population-based cohort studies. Inflamm Bowel
Dis. 19:789–799. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu F, Xia Y, Parker AS and Verma IM: IKK
biology. Immunol Rev. 246:239–253. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Karin M and Ben-Neriah Y: Phosphorylation
meets ubiquitination: The control of NF-[kappa]B activity. Annu Rev
Immunol. 18:621–663. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Greten FR, Eckmann L, Greten TF, Park JM,
Li ZW, Egan LJ, Kagnoff MF and Karin M: IKKbeta links inflammation
and tumorigenesis in a mouse model of colitis-associated cancer.
Cell. 118:285–296. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Koliaraki V, Pasparakis M and Kollias G:
IKKβ in intestinal mesenchymal cells promotes initiation of
colitis-associated cancer. J Exp Med. 212:2235–2251. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schön S, Flierman I, Ofner A, Stahringer
A, Holdt LM, Kolligs FT and Herbst A: β-catenin regulates NF-κB
activity via TNFRSF19 in colorectal cancer cells. Int J Cancer.
135:1800–1811. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Popivanova BK, Kitamura K, Wu Y, Kondo T,
Kagaya T, Kaneko S, Oshima M, Fujii C and Mukaida N: Blocking
TNF-alpha in mice reduces colorectal carcinogenesis associated with
chronic colitis. J Clin Invest. 118:560–570. 2008.PubMed/NCBI
|
|
17
|
Hamilton KE, Simmons JG, Ding S, Van
Landeghem L and Lund PK: Cytokine induction of tumor necrosis
factor receptor 2 is mediated by STAT3 in colon cancer cells. Mol
Cancer Res. 9:1718–1731. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Onizawa M, Nagaishi T, Kanai T, Nagano K,
Oshima S, Nemoto Y, Yoshioka A, Totsuka T, Okamoto R, Nakamura T,
et al: Signaling pathway via TNF-alpha/NF-kappaB in intestinal
epithelial cells may be directly involved in colitis-associated
carcinogenesis. Am J Physiol Gastrointest Liver Physiol.
296:G850–G859. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhu Q, Man SM, Gurung P, Liu Z, Vogel P,
Lamkanfi M and Kanneganti TD: Cutting edge: STING mediates
protection against colorectal tumorigenesis by governing the
magnitude of intestinal inflammation. J Immunol. 193:4779–4782.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bozec D, Iuga AC, Roda G, Dahan S and
Yeretssian G: Critical function of the necroptosis adaptor RIPK3 in
protecting from intestinal tumorigenesis. Oncotarget.
7:46384–46400. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fukata M, Hernandez Y, Conduah D, Cohen J,
Chen A, Breglio K, Goo T, Hsu D, Xu R and Abreu MT: Innate immune
signaling by Toll-like receptor-4 (TLR4) shapes the inflammatory
microenvironment in colitis-associated tumors. Inflamm Bowel Dis.
15:997–1006. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fukata M, Chen A, Vamadevan AS, Cohen J,
Breglio K, Krishnareddy S, Hsu D, Xu R, Harpaz N, Dannenberg AJ, et
al: Toll-like receptor-4 promotes the development of
colitis-associated colorectal tumors. Gastroenterology.
133:1869–1881. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Luo Q, Zeng L, Tang C, Zhang Z, Chen Y and
Zeng C: TLR9 induces colitis-associated colorectal carcinogenesis
by regulating NF-κB expression levels. Oncol Lett. 20:1102020.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Girondel C, Lévesque K, Langlois MJ,
Pasquin S, Saba-El-Leil MK, Rivard N, Friesel R, Servant MJ,
Gauchat JF, Lesage S and Meloche S: Loss of interleukin-17 receptor
D promotes chronic inflammation-associated tumorigenesis. Oncogene.
40:452–464. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hardbower DM, Coburn LA, Asim M, Singh K,
Sierra JC, Barry DP, Gobert AP, Piazuelo MB, Washington MK and
Wilson KT: EGFR-mediated macrophage activation promotes
colitis-associated tumorigenesis. Oncogene. 36:3807–3819. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rizzo A, De Mare V, Rocchi C, Stolfi C,
Colantoni A, Neurath MF, Macdonald TT, Pallone F, Monteleone G and
Fantini MC: Smad7 induces plasticity in tumor-infiltrating Th17
cells and enables TNF-alpha-mediated killing of colorectal cancer
cells. Carcinogenesis. 35:1536–1546. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bhat AA, Ahmad R, Uppada SB, Singh AB and
Dhawan P: Claudin-1 promotes TNF-α-induced epithelial-mesenchymal
transition and migration in colorectal adenocarcinoma cells. Exp
Cell Res. 349:119–127. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kawai N, Tsuji S, Tsujii M, Ito T,
Yasumaru M, Kakiuchi Y, Kimura A, Komori M, Sasaki Y, Hayashi N, et
al: Tumor necrosis factor alpha stimulates invasion of
Src-activated intestinal cells. Gastroenterology. 122:331–339.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cooks T, Pateras IS, Tarcic O, Solomon H,
Schetter AJ, Wilder S, Lozano G, Pikarsky E, Forshew T, Rosenfeld
N, et al: Mutant p53 prolongs NF-κB activation and promotes chronic
inflammation and inflammation-associated colorectal cancer. Cancer
Cell. 23:634–646. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee H, Herrmann A, Deng JH, Kujawski M,
Niu G, Li Z, Forman S, Jove R, Pardoll DM and Yu H: Persistently
activated Stat3 maintains constitutive NF-kappaB activity in
tumors. Cancer Cell. 15:283–293. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Atreya R and Neurath MF: Signaling
molecules: The pathogenic role of the IL-6/STAT-3 trans signaling
pathway in intestinal inflammation and in colonic cancer. Curr Drug
Targets. 9:369–374. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bollrath J, Phesse TJ, von Burstin VA,
Putoczki T, Bennecke M, Bateman T, Nebelsiek T, Lundgren-May T,
Canli O, Schwitalla S, et al: gp130-mediated Stat3 activation in
enterocytes regulates cell survival and cell-cycle progression
during colitis-associated tumorigenesis. Cancer Cell. 15:91–102.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pathria P, Gotthardt D, Prchal-Murphy M,
Putz EM, Holcmann M, Schlederer M, Grabner B, Crncec I, Svinka J,
Musteanu M, et al: Myeloid STAT3 promotes formation of
colitis-associated colorectal cancer in mice. OncoImmunology.
4:e9985292015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang H, Wang DH, Yang X, Sun Y and Yang
CS: Colitis-induced IL11 promotes colon carcinogenesis.
Carcinogenesis. 42:557–569. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Heichler C, Scheibe K, Schmied A, Geppert
CI, Schmid B, Wirtz S, Thoma OM, Kramer V, Waldner MJ, Büttner C,
et al: STAT3 activation through IL-6/IL-11 in cancer-associated
fibroblasts promotes colorectal tumour development and correlates
with poor prognosis. Gut. 69:1269–1282. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
He Z, He X, Chen Z, Ke J, He X, Yuan R,
Cai Z, Chen X, Wu X and Lan P: Activation of the mTORC1 and STAT3
pathways promotes the malignant transformation of colitis in mice.
Oncol Rep. 32:1873–1880. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Matsumoto S, Hara T, Mitsuyama K, Yamamoto
M, Tsuruta O, Sata M, Scheller J, Rose-John S, Kado S and Takada T:
Essential roles of IL-6 trans-signaling in colonic epithelial
cells, induced by the IL-6/soluble-IL-6 receptor derived from
lamina propria macrophages, on the development of
colitis-associated premalignant cancer in a murine model. J
Immunol. 184:1543–1551. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rizzo A, Di Giovangiulio M, Stolfi C,
Franzè E, Fehling HJ, Carsetti R, Giorda E, Colantoni A, Ortenzi A,
Rugge M, et al: RORγt-expressing Tregs drive the growth of
colitis-associated colorectal cancer by controlling IL6 in
dendritic cells. Cancer Immunol Res. 6:1082–1092. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Putoczki TL, Thiem S, Loving A, Busuttil
RA, Wilson NJ, Ziegler PK, Nguyen PM, Preaudet A, Farid R, Edwards
KM, et al: Interleukin-11 is the dominant IL-6 family cytokine
during gastrointestinal tumorigenesis and can be targeted
therapeutically. Cancer Cell. 24:257–271. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Grivennikov S, Karin E, Terzic J, Mucida
D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H,
Eckmann L and Karin M: IL-6 and Stat3 are required for survival of
intestinal epithelial cells and development of colitis-associated
cancer. Cancer Cell. 15:103–113. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Stolfi C, Rizzo A, Franzè E, Rotondi A,
Fantini MC, Sarra M, Caruso R, Monteleone I, Sileri P,
Franceschilli L, et al: Involvement of interleukin-21 in the
regulation of colitis-associated colon cancer. J Exp Med.
208:2279–2290. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nishina T, Deguchi Y, Ohshima D, Takeda W,
Ohtsuka M, Shichino S, Ueha S, Yamazaki S, Kawauchi M, Nakamura E,
et al: Interleukin-11-expressing fibroblasts have a unique gene
signature correlated with poor prognosis of colorectal cancer. Nat
Commun. 12:22812021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liang J, Nagahashi M, Kim EY, Harikumar
KB, Yamada A, Huang WC, Hait NC, Allegood JC, Price MM, Avni D, et
al: Sphingosine-1-phosphate links persistent STAT3 activation,
chronic intestinal inflammation, and development of
colitis-associated cancer. Cancer Cell. 23:107–120. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lee H, Deng J, Kujawski M, Yang C, Liu Y,
Herrmann A, Kortylewski M, Horne D, Somlo G, Forman S, et al:
STAT3-induced S1PR1 expression is crucial for persistent STAT3
activation in tumors. Nat Med. 16:1421–1428. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kortylewski M, Kujawski M, Wang T, Wei S,
Zhang S, Pillon-Thomas S, Niu G, Kay H, Mulé J, Kerr WG, et al:
Inhibiting Stat3 signaling in the hematopoietic system elicits
multicomponent antitumor immunity. Nat Med. 11:1314–1321. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Park SB, Choi B, Lee BJ, Kim NJ, Jeong YA,
Joo MK, Kim HJ, Park JJ, Kim JS, Noh YS and Lee HJ: Intestinal
epithelial deletion of sphk1 prevents colitis-associated cancer
development by inhibition of epithelial STAT3 activation. Dig Dis
Sci. 65:2284–2293. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Becker C, Fantini MC, Schramm C, Lehr HA,
Wirtz S, Nikolaev A, Burg J, Strand S, Kiesslich R, Huber S, et al:
TGF-beta suppresses tumor progression in colon cancer by inhibition
of IL-6 trans-signaling. Immunity. 21:491–501. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kortylewski M, Xin H, Kujawski M, Lee H,
Liu Y, Harris T, Drake C, Pardoll D and Yu H: Regulation of the
IL-23 and IL-12 balance by Stat3 signaling in the tumor
microenvironment. Cancer Cell. 15:114–123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Olesch C, Sirait-Fischer E, Berkefeld M,
Fink AF, Susen RM, Ritter B, Michels BE, Steinhilber D, Greten FR,
Savai R, et al: S1PR4 ablation reduces tumor growth and improves
chemotherapy via CD8+ T cell expansion. J Clin Invest.
130:5461–5476. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tewari D, Bawari S, Sharma S, DeLiberto LK
and Bishayee A: Targeting the crosstalk between canonical
Wnt/β-catenin and inflammatory signaling cascades: A novel strategy
for cancer prevention and therapy. Pharmacol Ther. 227:1078762021.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
White BD, Chien AJ and Dawson DW:
Dysregulation of Wnt/β-catenin signaling in gastrointestinal
cancers. Gastroenterology. 142:219–232. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Clevers H and Nusse R: Wnt/β-catenin
signaling and disease. Cell. 149:1192–1205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Bian J, Dannappel M, Wan C and Firestein
R: Transcriptional regulation of Wnt/β-catenin pathway in
colorectal cancer. Cells. 9:21252020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Takayama T, Ohi M, Hayashi T, Miyanishi K,
Nobuoka A, Nakajima T, Satoh T, Takimoto R, Kato J, Sakamaki S and
Niitsu Y: Analysis of K-ras, APC, and beta-catenin in aberrant
crypt foci in sporadic adenoma, cancer, and familial adenomatous
polyposis. Gastroenterology. 121:599–611. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Guinney J, Dienstmann R, Wang X, de
Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda
G, Angelino P, et al: The consensus molecular subtypes of
colorectal cancer. Nat Med. 21:1350–1356. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Brown JB, Lee G, Managlia E, Grimm GR,
Dirisina R, Goretsky T, Cheresh P, Blatner NR, Khazaie K, Yang GY,
et al: Mesalamine inhibits epithelial beta-catenin activation in
chronic ulcerative colitis. Gastroenterology. 138:595–605,
605.e1-e3. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Claessen MMH, Schipper MEI, Oldenburg B,
Siersema PD, Offerhaus GJA and Vleggaar FP: WNT-pathway activation
in IBD-associated colorectal carcinogenesis: Potential biomarkers
for colonic surveillance. Cell Oncol. 32:303–310. 2010.PubMed/NCBI
|
|
58
|
Chakrabarty S, Varghese VK, Sahu P,
Jayaram P, Shivakumar BM, Pai CG and Satyamoorthy K: Targeted
sequencing-based analyses of candidate gene variants in ulcerative
colitis-associated colorectal neoplasia. Br J Cancer. 117:136–143.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ma B and Hottiger MO: Crosstalk between
Wnt/β-catenin and NF-κB signaling pathway during inflammation.
Front Immunol. 7:3782016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Rubinstein MR, Wang X, Liu W, Hao Y, Cai G
and Han YW: Fusobacterium nucleatum promotes colorectal
carcinogenesis by modulating E-cadherin/β-catenin signaling via its
FadA adhesin. Cell Host Microbe. 14:195–206. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wu S, Morin PJ, Maouyo D and Sears CL:
Bacteroides fragilis enterotoxin induces c-Myc expression and
cellular proliferation. Gastroenterology. 124:392–400. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Iwakura Y and Ishigame H: The IL-23/IL-17
axis in inflammation. J Clin Invest. 116:1218–1222. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wang L, Yi T, Kortylewski M, Pardoll DM,
Zeng D and Yu H: IL-17 can promote tumor growth through an
IL-6-Stat3 signaling pathway. J Exp Med. 206:1457–1464. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Chen XW and Zhou SF: Inflammation,
cytokines, the IL-17/IL-6/STAT3/NF-κB axis, and tumorigenesis. Drug
Des Devel Ther. 9:2941–2946. 2015.PubMed/NCBI
|
|
65
|
Hyun YS, Han DS, Lee AR, Eun CS, Youn J
and Kim HY: Role of IL-17A in the development of colitis-associated
cancer. Carcinogenesis. 33:931–936. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Langowski JL, Zhang X, Wu L, Mattson JD,
Chen T, Smith K, Basham B, McClanahan T, Kastelein RA and Oft M:
IL-23 promotes tumour incidence and growth. Nature. 442:461–465.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Popa C, Netea MG, van Riel PLCM, van der
Meer JWM and Stalenhoef AFH: The role of TNF-alpha in chronic
inflammatory conditions, intermediary metabolism, and
cardiovascular risk. J Lipid Res. 48:751–762. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Chen G and Goeddel DV: TNF-R1 signaling: A
beautiful pathway. Science. 296:1634–1635. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Balkwill F: TNF-alpha in promotion and
progression of cancer. Cancer Metastasis Rev. 25:409–416. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Pikarsky E, Porat RM, Stein I, Abramovitch
R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E and
Ben-Neriah Y: NF-kappaB functions as a tumour promoter in
inflammation-associated cancer. Nature. 431:461–466. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Seidelin JB and Nielsen OH: Continuous
cytokine exposure of colonic epithelial cells induces DNA damage.
Eur J Gastroenterol Hepatol. 17:363–369. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Porter RJ, Arends MJ, Churchhouse AMD and
Din S: Inflammatory bowel disease-associated colorectal cancer:
Translational risks from mechanisms to medicines. J Crohns Colitis.
15:2131–2141. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Huycke MM, Abrams V and Moore DR:
Enterococcus faecalis produces extracellular superoxide and
hydrogen peroxide that damages colonic epithelial cell DNA.
Carcinogenesis. 23:529–536. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Kay J, Thadhani E, Samson L and Engelward
B: Inflammation-induced DNA damage, mutations and cancer. DNA
Repair (Amst). 83:1026732019. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Rachmilewitz D, Stamler JS, Bachwich D,
Karmeli F, Ackerman Z and Podolsky DK: Enhanced colonic nitric
oxide generation and nitric oxide synthase activity in ulcerative
colitis and Crohn's disease. Gut. 36:718–723. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Meira LB, Bugni JM, Green SL, Lee CW, Pang
B, Borenshtein D, Rickman BH, Rogers AB, Moroski-Erkul CA, McFaline
JL, et al: DNA damage induced by chronic inflammation contributes
to colon carcinogenesis in mice. J Clin Invest. 118:2516–2525.
2008.PubMed/NCBI
|
|
77
|
Rajamäki K, Taira A, Katainen R, Välimäki
N, Kuosmanen A, Plaketti RM, Seppälä TT, Ahtiainen M, Wirta EV,
Vartiainen E, et al: Genetic and epigenetic characteristics of
inflammatory bowel disease-associated colorectal cancer.
Gastroenterology. 161:592–607. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Burmer GC, Rabinovitch PS, Haggitt RC,
Crispin DA, Brentnall TA, Kolli VR, Stevens AC and Rubin CE:
Neoplastic progression in ulcerative colitis: Histology, DNA
content, and loss of a p53 allele. Gastroenterology. 103:1602–1610.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Yaeger R, Shah MA, Miller VA, Kelsen JR,
Wang K, Heins ZJ, Ross JS, He Y, Sanford E, Yantiss RK, et al:
Genomic alterations observed in colitis-associated cancers are
distinct from those found in sporadic colorectal cancers and vary
by type of inflammatory bowel disease. Gastroenterology.
151:278–287.e6. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Hussain SP, Amstad P, Raja K, Ambs S,
Nagashima M, Bennett WP, Shields PG, Ham AJ, Swenberg JA, Marrogi
AJ and Harris CC: Increased p53 mutation load in noncancerous colon
tissue from ulcerative colitis: A cancer-prone chronic inflammatory
disease. Cancer Res. 60:3333–3337. 2000.PubMed/NCBI
|
|
81
|
Rabinovitch PS, Dziadon S, Brentnall TA,
Emond MJ, Crispin DA, Haggitt RC and Bronner MP: Pancolonic
chromosomal instability precedes dysplasia and cancer in ulcerative
colitis. Cancer Res. 59:5148–5153. 1999.PubMed/NCBI
|
|
82
|
Redston MS, Papadopoulos N, Caldas C,
Kinzler KW and Kern SE: Common occurrence of APC and K-ras gene
mutations in the spectrum of colitis-associated neoplasias.
Gastroenterology. 108:383–392. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Kraus S and Arber N: Inflammation and
colorectal cancer. Curr Opin Pharmacol. 9:405–410. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Mármol I, Sánchez-de-Diego C, Pradilla
Dieste A, Cerrada E and Rodriguez Yoldi M: Colorectal carcinoma: A
general overview and future perspectives in colorectal cancer. Int
J Mol Sci. 18:1972017. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Rubin CE, Haggitt RC, Burmer GC, Brentnall
TA, Stevens AC, Levine DS, Dean PJ, Kimmey M, Perera DR and
Rabinovitch PS: DNA aneuploidy in colonic biopsies predicts future
development of dysplasia in ulcerative colitis. Gastroenterology.
103:1611–1620. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Carethers JM: Screening for colorectal
cancer in African Americans: Determinants and rationale for an
earlier age to commence screening. Dig Dis Sci. 60:711–721. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Colotta F, Allavena P, Sica A, Garlanda C
and Mantovani A: Cancer-related inflammation, the seventh hallmark
of cancer: Links to genetic instability. Carcinogenesis.
30:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Fujiwara I, Yashiro M, Kubo N, Maeda K and
Hirakawa K: Ulcerative colitis-associated colorectal cancer is
frequently associated with the microsatellite instability pathway.
Dis Colon Rectum. 51:1387–1394. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Din S, Wong K, Mueller MF, Oniscu A,
Hewinson J, Black CJ, Miller ML, Jiménez-Sánchez A, Rabbie R,
Rashid M, et al: Mutational analysis identifies therapeutic
biomarkers in inflammatory bowel disease-associated colorectal
cancers. Clin Cancer Res. 24:5133–5142. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Sato F, Harpaz N, Shibata D, Xu Y, Yin J,
Mori Y, Zou TT, Wang S, Desai K, Leytin A, et al: Hypermethylation
of the p14(ARF) gene in ulcerative colitis-associated colorectal
carcinogenesis. Cancer Res. 62:1148–1151. 2002.PubMed/NCBI
|
|
91
|
Azarschab P, Porschen R, Gregor M, Blin N
and Holzmann K: Epigenetic control of the E-cadherin gene (CDH1) by
CpG methylation in colectomy samples of patients with ulcerative
colitis. Genes Chromosomes Cancer. 35:121–126. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Kuester D, Guenther T, Biesold S, Hartmann
A, Bataille F, Ruemmele P, Peters B, Meyer F, Schubert D, Bohr UR,
et al: Aberrant methylation of DAPK in long-standing ulcerative
colitis and ulcerative colitis-associated carcinoma. Pathol Res
Pract. 206:616–624. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Vincan E and Barker N: The upstream
components of the Wnt signalling pathway in the dynamic EMT and MET
associated with colorectal cancer progression. Clin Exp Metastasis.
25:657–663. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Kinchen J, Chen HH, Parikh K,
Antanaviciute A, Jagielowicz M, Fawkner-Corbett D, Ashley N, Cubitt
L, Mellado-Gomez E, Attar M, et al: Structural remodeling of the
human colonic mesenchyme in inflammatory bowel disease. Cell.
175:372–386.e17. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Lin WW and Karin M: A cytokine-mediated
link between innate immunity, inflammation, and cancer. J Clin
Invest. 117:1175–1183. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Grivennikov SI, Greten FR and Karin M:
Immunity, inflammation, and cancer. Cell. 140:883–899. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Thiesen S, Janciauskiene S, Uronen-Hansson
H, Agace W, Högerkorp CM, Spee P, Håkansson K and Grip O:
CD14(hi)HLA-DR(dim) macrophages, with a resemblance to classical
blood monocytes, dominate inflamed mucosa in Crohn's disease. J
Leukoc Biol. 95:531–541. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Ruffell B, Affara NI and Coussens LM:
Differential macrophage programming in the tumor microenvironment.
Trends Immunol. 33:119–126. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Gordon S: Alternative activation of
macrophages. Nat Rev Immunol. 3:23–35. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Biswas SK, Gangi L, Paul S, Schioppa T,
Saccani A, Sironi M, Bottazzi B, Doni A, Vincenzo B, Pasqualini F,
et al: A distinct and unique transcriptional program expressed by
tumor-associated macrophages (defective NF-kappaB and enhanced
IRF-3/STAT1 activation). Blood. 107:2112–2122. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Dinapoli MR, Calderon CL and Lopez DM: The
altered tumoricidal capacity of macrophages isolated from
tumor-bearing mice is related to reduce expression of the inducible
nitric oxide synthase gene. J Exp Med. 183:1323–1329. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Yang Y, Li L, Xu C, Wang Y, Wang Z, Chen
M, Jiang Z, Pan J, Yang C, Li X, et al: Cross-talk between the gut
microbiota and monocyte-like macrophages mediates an inflammatory
response to promote colitis-associated tumourigenesis. Gut.
70:1495–1506. 2020.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Zhang Y, Sime W, Juhas M and Sjölander A:
Crosstalk between colon cancer cells and macrophages via
inflammatory mediators and CD47 promotes tumour cell migration. Eur
J Cancer. 49:3320–3334. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Martinez FO, Sica A, Mantovani A and
Locati M: Macrophage activation and polarization. Front Biosci.
13:453–461. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
105
|
Maisonneuve C, Tsang DKL, Foerster EG,
Robert LM, Mukherjee T, Prescott D, Tattoli I, Lemire P, Winer DA,
Winer S, et al: Nod1 promotes colorectal carcinogenesis by
regulating the immunosuppressive functions of tumor-infiltrating
myeloid cells. Cell Rep. 34:1086772021. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Ibrahim ML, Klement JD, Lu C, Redd PS,
Xiao W, Yang D, Browning DD, Savage NM, Buckhaults PJ, Morse HC III
and Liu K: Myeloid-derived suppressor cells produce IL-10 to elicit
DNMT3b-dependent IRF8 silencing to promote colitis-associated colon
tumorigenesis. Cell Rep. 25:3036–3046.e6. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Suzuki E, Kapoor V, Jassar AS, Kaiser LR
and Albelda SM: Gemcitabine selectively eliminates splenic
Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and
enhances antitumor immune activity. Clin Cancer Res. 11:6713–6721.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Bronte V, Serafini P, Apolloni E and
Zanovello P: Tumor-induced immune dysfunctions caused by myeloid
suppressor cells. J Immunother. 24:431–446. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Katoh H, Wang D, Daikoku T, Sun H, Dey SK
and Dubois RN: CXCR2-expressing myeloid-derived suppressor cells
are essential to promote colitis-associated tumorigenesis. Cancer
Cell. 24:631–644. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Huang B, Pan PY, Li Q, Sato AI, Levy DE,
Bromberg J, Divino CM and Chen SH: Gr-1+CD115+ immature myeloid
suppressor cells mediate the development of tumor-induced T
regulatory cells and T-cell anergy in tumor-bearing host. Cancer
Res. 66:1123–1131. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Sinha P, Clements VK, Bunt SK, Albelda SM
and Ostrand-Rosenberg S: Cross-talk between myeloid-derived
suppressor cells and macrophages subverts tumor immunity toward a
type 2 response. J Immunol. 179:977–983. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Galon J, Costes A, Sanchez-Cabo F,
Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M,
Berger A, Wind P, et al: Type, density, and location of immune
cells within human colorectal tumors predict clinical outcome.
Science. 313:1960–1964. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Tosolini M, Kirilovsky A, Mlecnik B,
Fredriksen T, Mauger S, Bindea G, Berger A, Bruneval P, Fridman WH,
Pagès F and Galon J: Clinical impact of different classes of
infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in
patients with colorectal cancer. Cancer Res. 71:1263–1271. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Wu P, Wu D, Ni C, Ye J, Chen W, Hu G, Wang
Z, Wang C, Zhang Z, Xia W, et al: γδT17 cells promote the
accumulation and expansion of myeloid-derived suppressor cells in
human colorectal cancer. Immunity. 40:785–800. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Sade-Feldman M, Kanterman J, Ish-Shalom E,
Elnekave M, Horwitz E and Baniyash M: Tumor necrosis factor-α
blocks differentiation and enhances suppressive activity of
immature myeloid cells during chronic inflammation. Immunity.
38:541–554. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
He D, Li H, Yusuf N, Elmets CA, Li J,
Mountz JD and Xu H: IL-17 promotes tumor development through the
induction of tumor promoting microenvironments at tumor sites and
myeloid-derived suppressor cells. J Immunol. 184:2281–2288. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Ohkura N, Kitagawa Y and Sakaguchi S:
Development and maintenance of regulatory T cells. Immunity.
38:414–423. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Wang Z, Friedrich C, Hagemann SC, Korte
WH, Goharani N, Cording S, Eberl G, Sparwasser T and Lochner M:
Regulatory T cells promote a protective Th17-associated immune
response to intestinal bacterial infection with C. rodentium.
Mucosal Immunol. 7:1290–1301. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Chang LY, Lin YC, Mahalingam J, Huang CT,
Chen TW, Kang CW, Peng HM, Chu YY, Chiang JM, Dutta A, et al:
Tumor-derived chemokine CCL5 enhances TGF-β-mediated killing of
CD8(+) T cells in colon cancer by T-regulatory cells. Cancer Res.
72:1092–1102. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Kryczek I, Wu K, Zhao E, Wei S, Vatan L,
Szeliga W, Huang E, Greenson J, Chang A, Roliński J, et al: IL-17+
regulatory T cells in the microenvironments of chronic inflammation
and cancer. J Immunol. 186:4388–4395. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Blatner NR, Mulcahy MF, Dennis KL,
Scholtens D, Bentrem DJ, Phillips JD, Ham S, Sandall BP, Khan MW,
Mahvi DM, et al: Expression of RORγt marks a pathogenic regulatory
T cell subset in human colon cancer. Sci Transl Med.
4:164ra1592012. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Olguín JE, Medina-Andrade I, Molina E,
Vázquez A, Pacheco-Fernández T, Saavedra R, Pérez-Plasencia C,
Chirino YI, Vaca-Paniagua F, Arias-Romero LE, et al: Early and
partial reduction in CD4+Foxp3+ regulatory T
cells during colitis-associated colon cancer induces
CD4+ and CD8+ T cell activation inhibiting
tumorigenesis. J Cancer. 9:239–249. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Koliaraki V, Roulis M and Kollias G: Tpl2
regulates intestinal myofibroblast HGF release to suppress
colitis-associated tumorigenesis. J Clin Invest. 122:4231–4242.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Neufert C, Becker C, Türeci Ö, Waldner MJ,
Backert I, Floh K, Atreya I, Leppkes M, Jefremow A, Vieth M, et al:
Tumor fibroblast-derived epiregulin promotes growth of
colitis-associated neoplasms through ERK. J Clin Invest.
123:1428–1443. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Sasaki S, Baba T, Shinagawa K, Matsushima
K and Mukaida N: Crucial involvement of the CCL3-CCR5 axis-mediated
fibroblast accumulation in colitis-associated carcinogenesis in
mice. Int J Cancer. 135:1297–1306. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Kawamura T, Yamamoto M, Suzuki K, Suzuki
Y, Kamishima M, Sakata M, Kurachi K, Setoh M, Konno H and Takeuchi
H: Tenascin-C produced by intestinal myofibroblasts promotes
colitis-associated cancer development through angiogenesis. Inflamm
Bowel Dis. 25:732–741. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Fujita M, Ito-Fujita Y, Iyoda T, Sasada M,
Okada Y, Ishibashi K, Osawa T, Kodama H, Fukai F and Suzuki H:
Peptide TNIIIA2 Derived from tenascin-C contributes to malignant
progression in colitis-associated colorectal cancer via β1-integrin
activation in fibroblasts. Int J Mol Sci. 20:27522019. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Bai YP, Shang K, Chen H, Ding F, Wang Z,
Liang C, Xu Y, Sun MH and Li YY: FGF-1/-3/FGFR4 signaling in
cancer-associated fibroblasts promotes tumor progression in colon
cancer through Erk and MMP-7. Cancer Sci. 106:1278–1287. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Hamilton KE, Chatterji P, Lundsmith ET,
Andres SF, Giroux V, Hicks PD, Noubissi FK, Spiegelman VS and
Rustgi AK: Loss of stromal IMP1 promotes a tumorigenic
microenvironment in the colon. Mol Cancer Res. 13:1478–1486. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Ma X, Aoki T, Tsuruyama T and Narumiya S:
Definition of prostaglandin E2-EP2 signals in the colon tumor
microenvironment that amplify inflammation and tumor growth. Cancer
Res. 75:2822–2832. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Natividad JMM and Verdu EF: Modulation of
intestinal barrier by intestinal microbiota: Pathological and
therapeutic implications. Pharmacol Res. 69:42–51. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Slack E, Hapfelmeier S, Stecher B,
Velykoredko Y, Stoel M, Lawson MA, Geuking MB, Beutler B, Tedder
TF, Hardt WD, et al: Innate and adaptive immunity cooperate
flexibly to maintain host-microbiota mutualism. Science.
325:617–620. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Nagao-Kitamoto H, Kitamoto S and Kamada N:
Inflammatory bowel disease and carcinogenesis. Cancer Metastasis
Rev. 41:301–316. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Fantini MC and Guadagni I: From
inflammation to colitis-associated colorectal cancer in
inflammatory bowel disease: Pathogenesis and impact of current
therapies. Dig Liver Dis. 53:558–565. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
LeBlanc JG, Laiño JE, del Valle MJ,
Vannini V, van Sinderen D, Taranto MP, de Valdez GF, de Giori GS
and Sesma F: B-group vitamin production by lactic acid
bacteria-current knowledge and potential applications. J Appl
Microbiol. 111:1297–1309. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Quaglio AEV, Grillo TG, De Oliveira ECS,
Stasi LCD and Sassaki LY: Gut microbiota, inflammatory bowel
disease and colorectal cancer. World J Gastroenterol. 28:4053–4060.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Thursby E and Juge N: Introduction to the
human gut microbiota. Biochem J. 474:1823–1836. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Yan H and Ajuwon KM: Butyrate modifies
intestinal barrier function in IPEC-J2 cells through a selective
upregulation of tight junction proteins and activation of the Akt
signaling pathway. PLoS One. 12:e01795862017. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Isobe J, Maeda S, Obata Y, Iizuka K,
Nakamura Y, Fujimura Y, Kimizuka T, Hattori K, Kim YG, Morita T, et
al: Commensal-bacteria-derived butyrate promotes the
T-cell-independent IgA response in the colon. Int Immunol.
32:243–258. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Gomaa EZ: Human gut microbiota/microbiome
in health and diseases: A review. Antonie Van Leeuwenhoek.
113:2019–2040. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Kinnebrew MA, Ubeda C, Zenewicz LA, Smith
N, Flavell RA and Pamer EG: Bacterial flagellin stimulates
Toll-like receptor 5-dependent defense against vancomycin-resistant
Enterococcus infection. J Infect Dis. 201:534–543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Furusawa Y, Obata Y, Fukuda S, Endo TA,
Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et
al: Commensal microbe-derived butyrate induces the differentiation
of colonic regulatory T cells. Nature. 504:446–450. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Zhang M, Zhou Q, Dorfman RG, Huang X, Fan
T, Zhang H, Zhang J and Yu C: Butyrate inhibits interleukin-17 and
generates Tregs to ameliorate colorectal colitis in rats. BMC
Gastroenterol. 16:842016. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Zimmerman MA, Singh N, Martin PM,
Thangaraju M, Ganapathy V, Waller JL, Shi H, Robertson KD, Munn DH
and Liu K: Butyrate suppresses colonic inflammation through
HDAC1-dependent Fas upregulation and Fas-mediated apoptosis of T
cells. Am J Physiol Gastrointest Liver Physiol. 302:G1405–G1415.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Janeway CA Jr: Approaching the asymptote?
Evolution and revolution in immunology. Cold Spring Harb Symp Quant
Biol. 54:1–13. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Kawai T and Akira S: Toll-like receptors
and their crosstalk with other innate receptors in infection and
immunity. Immunity. 34:637–650. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Cario E and Podolsky DK: Differential
alteration in intestinal epithelial cell expression of toll-like
receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect
Immun. 68:7010–7017. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Fukata M, Shang L, Santaolalla R,
Sotolongo J, Pastorini C, España C, Ungaro R, Harpaz N, Cooper HS,
Elson G, et al: Constitutive activation of epithelial TLR4 augments
inflammatory responses to mucosal injury and drives
colitis-associated tumorigenesis. Inflamm Bowel Dis. 17:1464–1473.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Frantz AL, Rogier EW, Weber CR, Shen L,
Cohen DA, Fenton LA, Bruno ME and Kaetzel CS: Targeted deletion of
MyD88 in intestinal epithelial cells results in compromised
antibacterial immunity associated with downregulation of polymeric
immunoglobulin receptor, mucin-2, and antibacterial peptides.
Mucosal Immunol. 5:501–512. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Ahmed I, Roy BC, Khan SA, Septer S and
Umar S: Microbiome, metabolome and inflammatory bowel disease.
Microorganisms. 4:202016. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Parekh PJ, Balart LA and Johnson DA: The
influence of the gut microbiome on obesity, metabolic syndrome and
gastrointestinal disease. Clin Transl Gastroenterol. 6:e912015.
View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Ghouri YA, Tahan V and Shen B: Secondary
causes of inflammatory bowel diseases. World J Gastroenterol.
26:3998–4017. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Richard ML, Liguori G, Lamas B, Brandi G,
da Costa G, Hoffmann TW, Pierluigi Di Simone M, Calabrese C,
Poggioli G, Langella P, et al: Mucosa-associated microbiota
dysbiosis in colitis associated cancer. Gut Microbes. 9:131–142.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Muller M, Hansmannel F, Arnone D, Choukour
M, Ndiaye NC, Kokten T, Houlgatte R and Peyrin-Biroulet L: Genomic
and molecular alterations in human inflammatory bowel
disease-associated colorectal cancer. United European Gastroenterol
J. 8:675–684. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Pan HW, Du LT, Li W, Yang YM, Zhang Y and
Wang CX: Biodiversity and richness shifts of mucosa-associated gut
microbiota with progression of colorectal cancer. Res Microbiol.
171:107–114. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Proença MA, Biselli JM, Succi M, Severino
FE, Berardinelli GN, Caetano A, Reis RM, Hughes DJ and Silva AE:
Relationship between Fusobacterium nucleatum, inflammatory
mediators and microRNAs in colorectal carcinogenesis. World J
Gastroenterol. 24:5351–5365. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Tahara T, Hirata I, Nakano N, Tahara S,
Horiguchi N, Kawamura T, Okubo M, Ishizuka T, Yamada H, Yoshida D,
et al: Potential link between Fusobacterium enrichment and DNA
methylation accumulation in the inflammatory colonic mucosa in
ulcerative colitis. Oncotarget. 8:61917–61926. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Geis AL, Fan H, Wu X, Wu S, Huso DL, Wolfe
JL, Sears CL, Pardoll DM and Housseau F: Regulatory T-cell response
to enterotoxigenic Bacteroides fragilis colonization triggers
IL17-dependent colon carcinogenesis. Cancer Discov. 5:1098–1109.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Wick EC, Rabizadeh S, Albesiano E, Wu X,
Wu S, Chan J, Rhee KJ, Ortega G, Huso DL, Pardoll D, et al: Stat3
activation in murine colitis induced by enterotoxigenic Bacteroides
fragilis. Inflamm Bowel Dis. 20:821–834. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Purcell RV, Permain J and Keenan JI:
Enterotoxigenic Bacteroides fragilis activates IL-8 expression
through Stat3 in colorectal cancer cells. Gut Pathog. 14:162022.
View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Wu S, Rhee KJ, Albesiano E, Rabizadeh S,
Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, et al: A
human colonic commensal promotes colon tumorigenesis via activation
of T helper type 17 T cell responses. Nat Med. 15:1016–1022. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Arthur JC, Perez-Chanona E, Mühlbauer M,
Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B,
Rogers AB, et al: Intestinal inflammation targets cancer-inducing
activity of the microbiota. Science. 338:120–123. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
de Almeida CV, Taddei A and Amedei A: The
controversial role of Enterococcus faecalis in colorectal cancer.
Therap Adv Gastroenterol. 11:17562848187836062018. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Cao Y, Oh J, Xue M, Huh WJ, Wang J,
Gonzalez-Hernandez JA, Rice TA, Martin AL, Song D, Crawford JM, et
al: Commensal microbiota from patients with inflammatory bowel
disease produce genotoxic metabolites. Science. 378:eabm32332022.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Kuipers EJ, Grady WM, Lieberman D,
Seufferlein T, Sung JJ, Boelens PG, van de Velde CJ and Watanabe T:
Colorectal cancer. Nat Rev Dis Primers. 1:150652015. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Vallée A, Lecarpentier Y and Vallée JN:
Targeting the canonical WNT/β-catenin pathway in cancer treatment
using non-steroidal anti-inflammatory drugs. Cells. 8:7262019.
View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Dihlmann S and von Knebel Doeberitz M:
Wnt/beta-catenin-pathway as a molecular target for future
anti-cancer therapeutics. Int J Cancer. 113:515–524. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Dihlmann S, Siermann A and von Knebel
Doeberitz M: The nonsteroidal anti-inflammatory drugs aspirin and
indomethacin attenuate beta-catenin/TCF-4 signaling. Oncogene.
20:645–653. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Tuynman JB, Vermeulen L, Boon EM, Kemper
K, Zwinderman AH, Peppelenbosch MP and Richel DJ: Cyclooxygenase-2
inhibition inhibits c-Met kinase activity and Wnt activity in colon
cancer. Cancer Res. 68:1213–1220. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Sonu I, Lin MV, Blonski W and Lichtenstein
GR: Clinical pharmacology of 5-ASA compounds in inflammatory bowel
disease. Gastroenterol Clin North Am. 39:559–599. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Graham PM, Li JZ, Dou X, Zhu H, Misra HP,
Jia Z and Li Y: Protection against peroxynitrite-induced DNA damage
by mesalamine: Implications for anti-inflammation and anti-cancer
activity. Mol Cell Biochem. 378:291–298. 2013. View Article : Google Scholar : PubMed/NCBI
|