Introduction
An abundance of blood loss during surgery escalates
the likelihood of post-surgical peritoneal metastasis in patients
with gastric cancer (GC) (1,2).
Platelets (PLTs) support malignant behaviors, especially
hematogenous metastases, in various cancer types (3–5). In
our prior study, we established that PLTs interact with GC cells,
forming complexes that amplify their malignant characteristics
through direct contact (6). Surgery
is a very effective treatment, but it does cause some bleeding. The
PLTs present in this bleeding promote peritoneal metastasis. If we
can inhibit PLTs-GC cells interaction with PLTs activation by
intraperitoneal administration during or after surgery, we can
further improve the prognosis of patients undergoing GC surgery. In
this study, we propose that interactions between PLTs and GC cells
during surgery intensify the spread of GC cells in the peritoneum.
Our objective is to investigate the molecular mechanisms driving
this process and evaluate the therapeutic potential of mitigating
peritoneal metastasis by inhibiting interactions between PLTs and
GC cells.
A multitude of molecular entities and biological
pathways are instrumental in the activation of PLTs. In PLTs, the
C-type lectin-like receptor 2 (CLEC-2) plays a crucial role in
activation, which is mediated by Src and Syk (7–9).
Podoplanin (PDPN), a ligand for CLEC-2, is present on renal
podocytes, lymphatic endothelial cells, and cancer cells (10,11).
Glycoprotein (GP)-VI, a primary collagen receptor, is crucial for
PLT activation and function induced by collagen (12,13).
Among the various mediators released during PLT activation
(14), transforming growth factor
(TGF)-β plays crucial roles in the development and progression of
various cancer types (15–17). Our previous study demonstrated that
blocking the interaction between galectin (Gal)-3 and GPVI reduces
the effects of PLTs on GC cells (18).
In this study, we examined the involvement of the
PLT activation pathway and TGF-β released from activated PLTs in
the enhanced malignancy of GC cells via cancer cell-PLT
interactions. Furthermore, we investigated their therapeutic
potential to prevent peritoneal dissemination of GC cells. Our
findings indicate that the Src family plays a pivotal role in
enhancing the metastasis of GC cells facilitated by PLTs, thereby
presenting a potential therapeutic target for mitigating peritoneal
metastasis in individuals with GC.
Materials and methods
Reagents
We assessed the inhibitory impacts of various
substances on the malignant behaviors induced by PLTs in GC cells.
SB-431542, a TGF-β receptor kinase inhibitor (TRKI), was purchased
from Cayman Chemical Company (Ann Arbor, Michigan, USA). SB-431542
(1 µM) was used in the working solution for pre-incubation with GC
cells for 1 h before each assay. PP2, an Src kinase inhibitor, was
purchased from AdipoGen Life Sciences (San Diego, CA, USA), and its
10 µM solution was used in the working solution for pre-incubation
with PLTs for 10 min before each assay. Syk inhibitor R406 was
purchased from InvivoGen (San Diego, CA, USA), and its 1 µM
solution was used in the working solution for pre-incubation with
PLTs for 10 min before each assay.
Cell lines and cell culture
We utilized two GC cell lines, NUGC-3 and MKN74,
procured from the Japanese Collection of Research Bioresources Cell
Bank (Osaka, Japan). These were propagated in Roswell Park Memorial
Institute-1640 medium (supplied by Thermo Fisher Scientific Inc.,
Waltham, MA, USA), enriched with 100 U/ml of penicillin (obtained
from Sigma-Aldrich, Merck KGaA, Darmstadt, Germany), 100 µg/ml of
streptomycin (also from Sigma-Aldrich, Merck KGaA), and 10% fetal
bovine serum (FBS; sourced from Thermo Fisher Scientific Inc.). The
culture conditions were maintained at 37°C in a 5% CO2
environment.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was isolated from NUGC-3 cells utilizing
the miRNeasy Mini Kit (Qiagen, Hilden, Germany) as per the
guidelines provided by the manufacturer. The NanoDrop 2000
spectrophotometer (Thermo Fisher Scientific, Inc.) was employed to
ascertain the total RNA concentration, and 1 µg of total RNA was
converted to cDNA using the HighCapacity cDNA Reverse Transcription
Kit (Thermo Fisher Scientific Inc.), following the manufacturer's
protocol. The levels of transcripts were determined using specific
primer sets and SYBR Green Master Mix (Thermo Fisher Scientific,
Inc.) via RT-qPCR. The RT-qPCR conditions were set as follows: An
initial preheating for 10 min at 95°C, succeeded by 40 cycles of
95°C for 15 sec and 60°C for 60 sec.
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH)
mRNA levels served as internal controls for normalization, and
Gal-3 mRNA levels were calculated using the 2−ΔΔCq
method (19). Primer sequences were
designed using Primer3Plus (https://www.bioinformatics.nl/cgibin/primer3plus/primer3plus.cgi)
with the most conserved region of each sequence obtained from the
National Center for Biotechnology Information database (https://www.ncbi.nlm.nih.gov/nuccore).
Each experiment was conducted in triplicate.
The primers used for the RT-qPCR assay were as
follows: matrix metalloproteinase (MMP)-9 (forward
5′-CCAACTACGACCGGGACAAG-3′ and reverse 5′-AAGTGAA-GGGGAAGACGCAC-3′)
and GAPDH (forward 5′-GTCTCCTCTGACTTCAACAGCG-3′ and reverse
5′-ACCACCCTGTTGCTGTAGCCAA-3′).
PLT preparation
PLTs were sourced from healthy volunteers and
isolated from whole blood, following the method outlined in our
previous study (20). The PLTs were
immediately utilized in a variety of experiments post-extraction.
They were adjusted to a final concentration of 1×105/µl
for use in the subsequent experiments.
Functional assays in vitro
We conducted migration and invasion assays utilizing
Falcon Cell Culture Inserts with 8-µm pore membranes (Corning Inc.,
Corning, NY, USA) and Biocoat Matrigel (BD Bioscience, Franklin
Lakes, NJ, USA), following the methodology outlined in our prior
study (6). In brief, NUGC-3
(1×105) and MKN74 (5×105) cells were placed
in the upper chambers of the FBS-free medium, with or without the
presence of PLTs, while the lower chambers were filled with 10% FBS
medium. For the inhibition experiments, the cells were exposed to a
range of inhibitors (SB-431542, PP2, R406) alongside PLTs. Post a
24-h incubation period, cells that failed to migrate or invade
through the pores were eliminated using cotton swabs. The cells
that had successfully migrated and invaded were stained using the
Diff-Quick staining reagent (Sysmex, Kobe, Japan), and the cell
count in four separate fields was determined using the BZ-X710
All-in-One fluorescence microscope (Keyence Corp., Osaka, Japan) at
100× magnification and BZ-X Analyzer Software (Keyence Corp.). Each
experiment was conducted in triplicate.
Establishment of a peritoneal
dissemination mouse model in vivo
In this investigation, male BALB/c-Slc-nu/nu mice of
six weeks old (procured from Japan SLC Inc., Shizuoka, Japan) were
utilized. These mice were accommodated in a hygienic,
temperature-regulated cage setting with a 12-h light-dark cycle.
They were given unrestricted access to a laboratory-standard diet
and water. The mice were arbitrarily segregated into three
clusters, each containing six mice, by animal care specialists not
directly participating in the research. The investigators were kept
unaware of the group assignments. The exclusion criteria
encompassed mortality during the research duration; however, this
study did not witness any such instances. On the initial day,
NUGC-3 cells (3×106) in 500 µl of Hanks' balanced salt
solution (HBSS(−); sourced from FUJIFILM Wako Pure Chemical
Corporation, Osaka, Japan) were administered intraperitoneally into
the mice of the non-treatment (NT) group. In the PLT group, PLTs
were introduced into NUGC-3 cells. The PLTs were calibrated to a
terminal concentration of 1×105/µl for subsequent
experiments. In the inhibition experiments, the PP2 inhibitor was
administered to the mice along with the NUGC-3 cells and PLTs into
the peritoneal cavity. Post intraperitoneal administration, the
mice were accommodated in individual cages. Weekly monitoring of
physical conditions and body weights was conducted. After a span of
five weeks, the mice were euthanized and peritoneal dissemination
was assessed. For anesthesia induction, a concoction of
medetomidine hydrochloride (0.75 mg/kg), midazolam (4 mg/kg), and
butorphanol tartrate (5 mg/kg) was diluted with saline to achieve a
dosage of 5 µl/g body weight and was administered to the mice via
intraperitoneal injection, as delineated earlier (21–24).
Anesthesia was induced using the aforementioned dosage. However, if
the sedation level with anesthesia was deemed insufficient upon
evaluation (e.g., loss of postural reaction and righting reflex,
eyelid reflex, pedal withdrawal reflex in the fore and hind limbs,
and tail pinch reflex), the dosage was escalated or deescalated to
ensure adequate sedation in compliance with the Guide for the Care
and Use of Laboratory Animals (25). As a measure against hypothermia, we
used a circulating warm water blanket as an anesthesia warming
device, which maintained a surface temperature of approximately
37.5°C. In addition, we made efforts to minimize heat loss by using
insulation and drapes covering the chest and abdomen. Furthermore,
we used atipamezole (1 mg/kg, intraperitoneal injection) to prevent
hypothermia (26). Subsequently,
peritoneal dissemination was assessed post tumor removal from the
host. The count, weight, and maximum diameter of the tumors were
recorded. At the culmination of the experiment, all mice were
euthanized via CO2 inhalation (flow rate was regulated
at 30% of the cage volume per minute), and death was confirmed by
the absence of respiration or cardiac activity. All animal
experiments received approval from the Institutional Animal Care
and Use Committee of the University of Yamanashi, Japan (approval
no. A2-14).
Statistical analysis
Quantitative values are presented as either the mean
± standard error or the median. In cases of multiple comparisons,
we employed one-way analysis of variance, followed by the
Bonferroni post-hoc test under conditions of equal variance, to
draw comparisons between each group and the PLT group. In addition,
under conditions of unequal variance, we used the Kruskal-Wallis
test, followed by the Bonferroni post-hoc test, to compare each
group with the PLT group. The threshold for statistical
significance was set at P<0.05. All statistical computations
were performed using JMP 17 (SAS Institute Inc., Cary, NC, USA),
and EZR version 1.54 (Saitama Medical Center, Jichi Medical
University, Saitama, Japan), which is a graphical user interface
for R (The R Foundation for Statistical Computing, Vienna, Austria)
(27).
Results
Involvement of Src-dependent PLT
activation signaling pathway and TGF-β released from PLTs in the
enhanced malignancy by the GC cells-PLT interaction
Several pathways are involved in PLT activation
(28). Membrane protein- or
extracellular matrix protein-mediated PLT activation, followed by
Src family kinase activation, is mediated by integrin, and
fibrinogen (5), CLEC-2, PDPN
(7,29–31),
GPVI and its ligands, Gal-3 (32).
Conversely, the activation of PLTs through G-protein-coupled
receptors, triggered by soluble PLT agonists, is facilitated by the
agonists of G-protein-coupled receptors, which include thrombin,
ADP, 5-hydroxytryptamine, PLT activating factor, and thromboxane
A2.
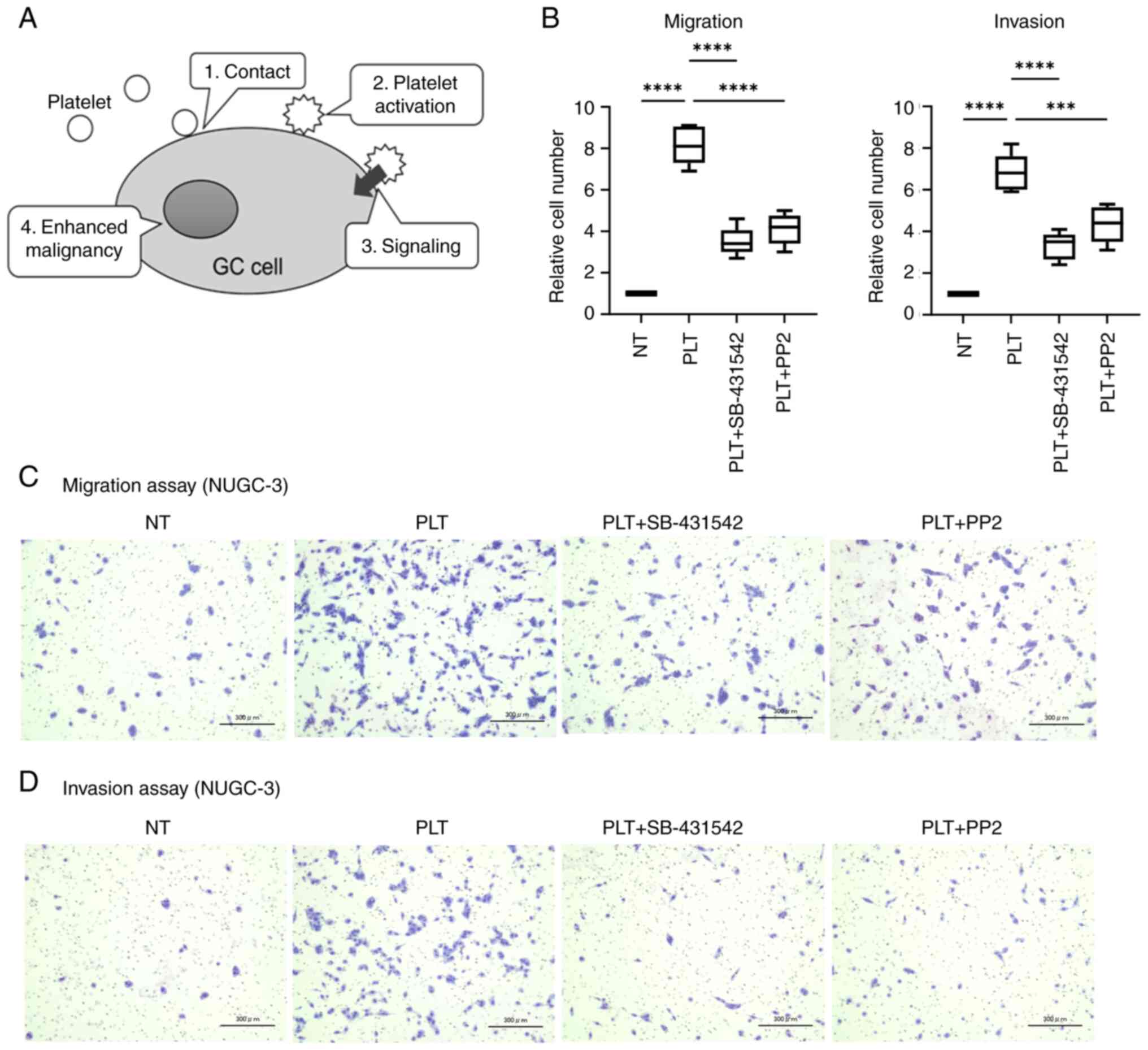
We previously reported that the direct contact
between GC cells and PLTs promotes the malignant behaviors of cells
(6). In our investigation, we
concentrated on the PLT activation pathway, particularly the Src
family kinase activation pathway, which involves direct contact
between GC cells and PLTs (Fig.
1A). To examine this pathway, PP2, a Src kinase inhibitor, was
used to investigate whether Src-dependent PLT activation affects
the behaviors of NUGC-3 cells co-cultured with PLTs. PP2
significantly inhibited the migration of NUGC-3 cells increased via
co-culture with PLTs (50% reduction P<0.001; Fig. 1B). PP2 also inhibited the invasion
NUGC-3 cells increased by PLTs (48% reduction; P=0.009; Fig. 1B). Activated PLTs secrete TGF-β from
their alpha granules (33). Here,
we examined the inhibitory impact of SB-431542 on PLT-enhanced
malignancy of GC cells. SB-431542 effectively suppressed both
PLT-induced migration and invasion of GC cells (55 and 45%
reduction, respectively; P<0.001; Fig. 1B). Images depicting the migration
and invasion assays are presented in Fig. 1C and D. Notably, PP2 and SB-431542
individually did not affect the malignancy of GC cells (Fig. S1). The results obtained using
NUGC-3 cells (Fig. 1) were
consistent with those obtained using MKN74 cells (Fig. S2).
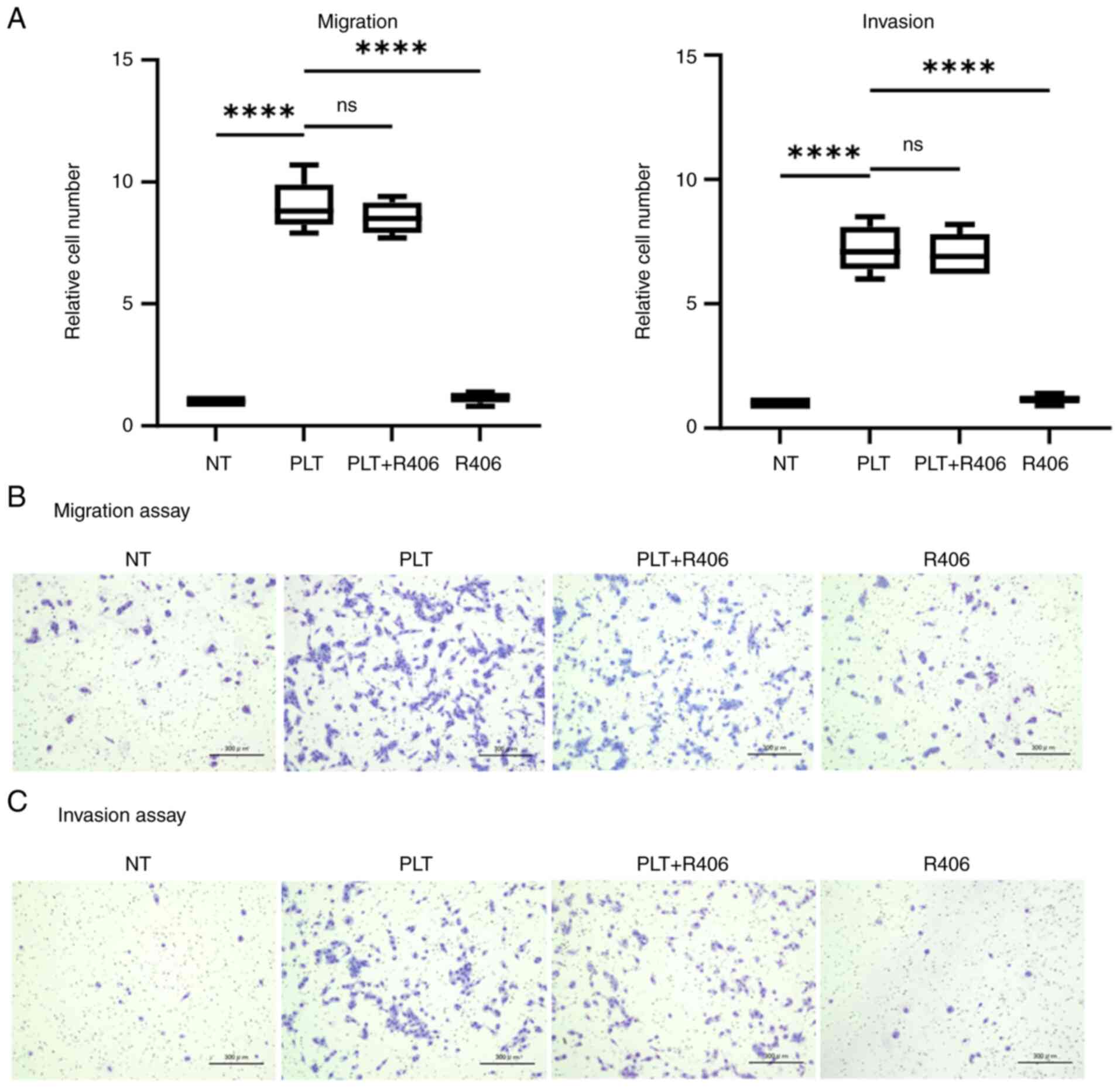
Syk-dependent PLT activation
pathway
PLTs contain two primary types of agonist receptors:
G-protein-coupled receptors and tyrosine kinase pathway receptors,
both essential for PLT activation. Tyrosine kinase pathway
receptors, including CLEC-2, FcγRIIA, and GPVI, are linked to Syk
activation (34–38). Here, R406, a Syk inhibitor, was used
to determine whether the effect of PLTs on GC cells is abolished by
suppressing Syk.
R406 failed to suppress the enhanced migration and
invasion of NUGC-3 cells in co-culture with PLTs (Fig. 2A). Images depicting the migration
and invasion assays are presented in Fig. 2B and C. Similar to PP2 and
SB-431542, R406 alone did not affect the malignancy of GC
cells.
Therapeutic effect of PLT activation
inhibition on peritoneal dissemination
In the mouse model, administration of PLTs with
NUGC-3 significantly increased the number, weight, and maximum
diameter of peritoneal tumors (P=0.004, P<0.001, and P<0.001,
respectively). However, co-administration of PP2 markedly decreased
the number, weight, and maximum diameter of peritoneal tumors
compared to those in the PLT group [reduction: 38% (P=0.0013), 37%
(P=0.0017), and 34% (P=0.0031), respectively; Fig. 3A and B].
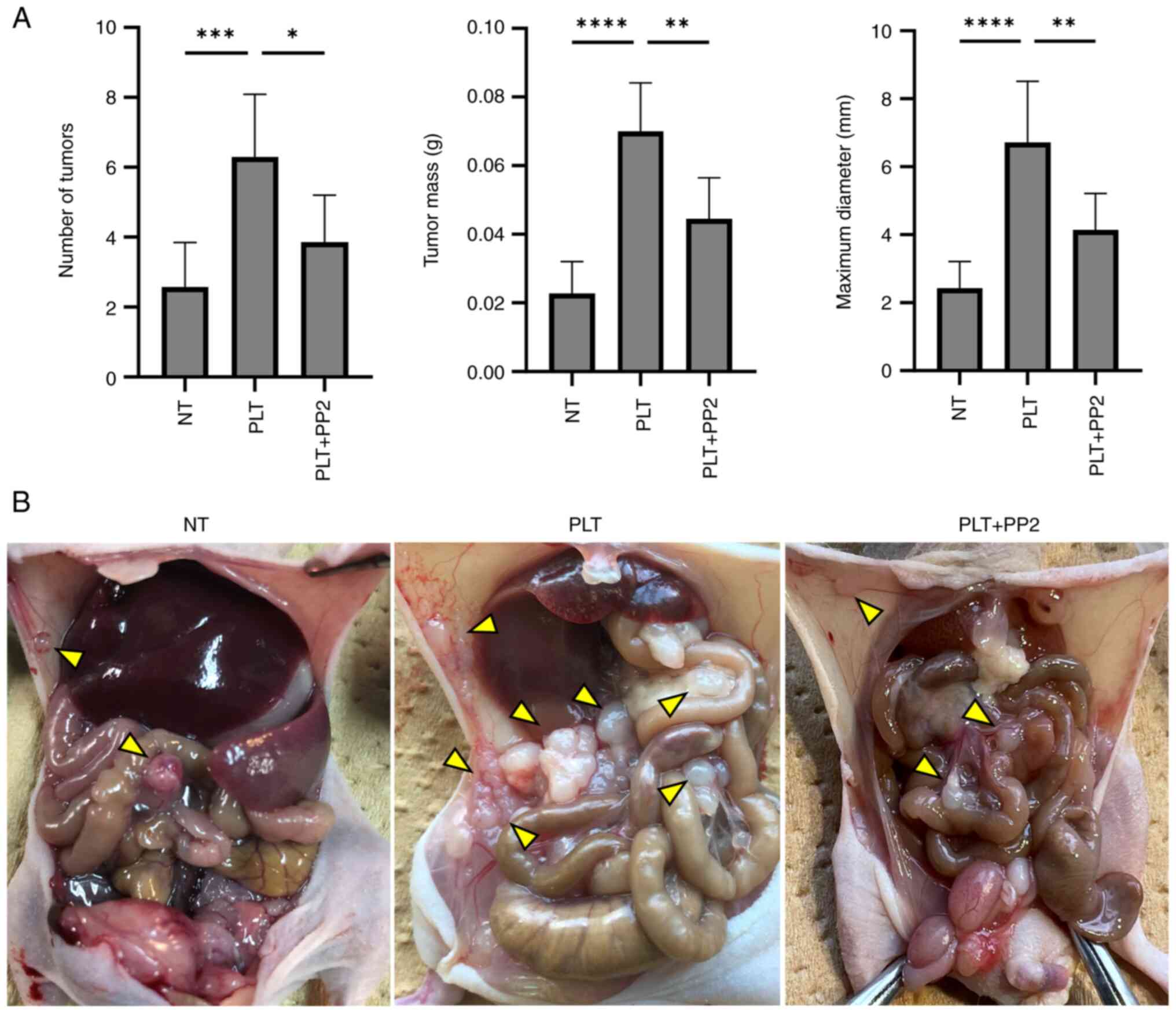 | Figure 3.Therapeutic effect of PP2 (Src family
kinase inhibitor) against peritoneal dissemination. (A) Number,
weight and maximum diameter of peritoneal tumors, and (B) findings
from the abdominal cavity (arrowheads indicate peritoneal
dissemination). The number, weight and maximum diameter of
peritoneal tumors were enhanced by PLT contact (P=0.004, P<0.001
and P<0.001, respectively) and reduced by PP2 [reduction, 38%
(P=0.013), 37% (P=0.0017) and 34% (P=0.0031), respectively].
*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs. PLT.
NT, non-treatment; PLT, platelet. |
Discussion
Peritoneal dissemination in GC leads to a poor
prognosis, and many studies are exploring treatment options to
prevent it (39,40). Various risk factors for peritoneal
dissemination in GC have been identified. Arita et al
(1) demonstrated that substantial
intraoperative blood loss elevates the risk of peritoneal
dissemination recurrence in GC patients. Similarly, Kamei et
al (2), through multivariate
analysis, found a significant correlation between the extent of
intraoperative bleeding and peritoneal dissemination recurrence,
establishing excessive blood loss as an independent risk factor for
the recurrence of peritoneal dissemination. Intraoperative blood
loss in patients frequently necessitates clinical blood
transfusions, potentially resulting in immunosuppressive effects.
However, the abovementioned studies clearly demonstrate that
intraoperative bleeding is not associated with other recurrence
patterns, such as nodal or hematogenous metastases. This may be due
to the localized effects of intraoperative blood loss within the
peritoneal cavity. A multitude of research has established the
existence of free-floating cancer cells within the peritoneal
cavity in patients suffering from advanced GC (41,42).
During the perioperative period, cancer cells might show enhanced
interactions with the components of blood present in the peritoneal
cavity. By interacting with circulating tumor cells, PLTs play a
role in promoting hematogenous metastasis (43,44).
Therefore, we hypothesized that intraperitoneally released cancer
cells come in contact with PLTs due to intraoperative bleeding,
thereby exacerbating peritoneal metastasis by enhancing cancer
malignancy via interactions between cancer cells and PLTs. We
previously demonstrated that direct interactions with PLTs
significantly promote the malignant behaviors of GC cells,
especially migration and invasion occur through mechanisms
associated with epithelial-mesenchymal transition (EMT) (6). Furthermore, we revealed that
inhibition of such direct interactions completely suppresses
PLT-induced peritoneal dissemination in GC (18).
In this study, we examined the specific mechanisms
that drive the increased malignant behaviors of GC cells when in
direct contact with PLTs. Notably, we identified the involvement of
the Src-mediated PLT activation pathway and PLT-released TGF-β in
the enhanced malignancy of GC cells. We previously demonstrated
that GC cell-PLT contact enhances cell migration and invasion by
increasing MMP9 expression via EMT. In this examination, we
assessed the alterations in MMP9 expression levels in GC cells
co-cultured with PLTs and treated with PP2 and SB-431542. Our
results confirmed that MMP9 expression was significantly suppressed
in GC cells (Fig. S3). These
results suggest that enhanced migration and invasion of cancer
cells can be inhibited via suppression of EMT by inhibiting TGF-β
release from PLTs. TGF-β has been reported to induce EMT in cancer
cells and promote invasion and metastasis in previous studies
(45,46). In fact, our previous study has
revealed that various EMT-related molecules, in addition to MMP9,
fluctuate in GC cell lines co-cultured with PLTs (6). Also, Wiercinska et al (47) established a 3D model of
TGF-β-induced invasion in breast cancer. Using this model, they
demonstrated that Smad3 and Smad4 are important for TGF-β-induced
invasion by inducing MMP2 and MMP9 (47). Based on these reports,
TGF-β-Smad2/Smad3-MMP2/MMP9 may also play an important role in GC
cells. Therefore, anti-TGF-β strategies targeting the
PLT-activating cascade may aid in cancer treatment.
In contrast to PP2, anti-PLT agent R406 did not
exert considerable inhibitory effect on cancer cell-PLT
interactions in this study. This may be because R406 inhibits the
activation signal derived from CLEC-2 more strongly than that
derived from GPVI (48). Our
findings are consistent with a previous report that GPVI is more
important than CLEC-2 in GC cell-PLT interactions (18) although the contribution of Src
family kinases and Syk in PLT activation mediated by direct
interaction between galectin-3 and GPVI remains still unclear. In
addition, Src family kinase phosphorylates molecules involved in
PLT activation, including those beyond Syk (such as PI3K). It is
possible that the lack of significant effects observed with Syk
inhibitors may be due to the involvement of a Syk-independent
pathway (49).
To validate the results obtained in vitro, we
extended our investigation to study the suppressive effects of PP2
on PLT-induced GC cells, utilizing a mouse model of peritoneal
dissemination. In line with the in vitro findings, our in
vivo analyses indicated a significant augmentation in
peritoneal dissemination when PLTs and GC cells were concurrently
administered to mice. These observations imply that PLTs may play a
role in promoting the peritoneal spread of free cancer cells during
intraoperative bleeding. Therefore, necessary precautions must be
taken to prevent or limit the interactions between PLTs and GC
cells in clinical settings. Additionally, intraoperative bleeding
should be mitigated to prevent hemorrhage during surgeries.
Here, in vivo analyses demonstrated that
co-administration of PP2 significantly attenuated PLT-enhanced
peritoneal dissemination compared to single administrations alone.
The outcomes of our study underscore the pivotal role of Src family
kinases in peritoneal dissemination, thereby spotlighting them as
potential therapeutic targets for preventing recurrence in GC
patients (Fig. S4). In this study,
inhibition of downstream signaling by PP2 and TRKI suppressed
PLT-enhanced malignancy in vitro. TGF-β signaling has
connections to a multitude of diseases such as malignancies and
inflammatory and fibrotic conditions (50). Therefore, suppressing TGF-β may
attenuate the biological functions of diseased cells. To verify
this, we investigated the effects of PP2 on GC cells. Inhibition of
PLT activation leads to excessive bleeding. However, Src knockout
mice did not exhibit excessive bleeding, similar to a previous
report (51), suggesting that PP2
does not cause bleeding. Here, no adverse events associated with
PP2 were observed in in vivo experiments.
We demonstrated that PP2 inhibited PLT-enhanced
malignancy of NUGC-3, in vitro and in vivo, however,
there remains some issues that needs further investigations about
the mechanism of the effect of PP2 in the future. First, we will
conduct a quantification experiment to determine the extent to
which PLTs are activated and the amount of TGF-β released when
NUGC-3 is co-cultured with PLTs. Second, we will analyze in detail
the signaling profiles in the NUGC-3 cells when NUGC-3 and PLTs
come into direct contact with each other since our previous report
has reported that direct contact between NUGC-3 and PLTs is also
important for NUGC-3 malignancy (6,18).
Finally, we will confirm the synergistic effect of combining the
direct contact inhibitor shown to be effective in our previous
study with PP2. By verifying these in future experiments, it may be
possible to further elucidate the mechanism of enhanced malignant
behavior of GC cells induced with interaction between GC cells and
PLTs.
In our study, we demonstrated the use of PP2, and we
are considering intraperitoneal administration during or after
surgery. Suppressing the activation of PLTs, which cannot be
completely removed by postoperative washing alone, using PP2, could
potentially inhibit the enhancement of malignancy in GC cells and
consequently suppress peritoneal dissemination. These new treatment
options can be a very attractive therapy that can improve the
prognosis of GC patients undergoing surgery, and could potentially
be a blessing for patients suffering from refractory cancer.
This study has some limitations. First, the mouse
model was intraperitoneally administered with the Src family kinase
inhibitor PP2. However, to fully inhibit the PLT activation
resulting from PLT-GC cell interactions, administration via oral or
intravenous routes would be more appropriate. Second, side effects
of PP2, especially its effect on hemostasis, could not be
determined in this study, warranting further investigation.
Although similar mechanisms may be involved in different cancer
types, further exploration of specific molecules and pathways is
warranted for each type of cancer.
In conclusion, this study revealed that Src family
kinase inhibitors suppressed the PLT-enhanced malignancy of GC
cells in vitro and that PLT interaction with GC cells
markedly increased their peritoneal dissemination. However,
inhibition of Src-mediated PLT activation significantly suppressed
the PLT-enhanced peritoneal dissemination of GC cells in
vivo. Overall, our findings suggest that targeting the
Src-mediated PLT activation pathway holds promise as a therapeutic
strategy to prevent peritoneal dissemination in GC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study received partial funding from the Japan
Society for the Promotion of Science, specifically through the JSPS
KAKENHI grants numbered 20K17642 and 20K09031.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
TN, RS, SF, YH, KM, KaT, SM, KaS, KoT, KeS, YK, HA,
HK, NT, TS, KSI and DI conceived the study idea and designed the
study. TN and RS confirm the authenticity of all raw data. KM, SF,
KaT, SM, KaS and KeS collected and assembled the data. YH, KoT, YK,
HA and HK analyzed and interpreted the data. All authors have made
contributions to the composition of this manuscript, and read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethic Committee of the
University of Yamanashi (approval no. 2159; Chuo, Japan) for
patient experiments. The study was approved by the Animal Care and
Use Committee at the University of Yamanashi (approval no. A2-14;
Chuo, Japan) for animal experiments. The study adhered to the
ethical standards outlined in The Declaration of Helsinki and its
subsequent amendments (52). All
volunteers provided written informed consent for the use of their
samples. All animal procedures were conducted in accordance with
the ARRIVE 2.0 guidelines (53) and
the National Institutes of Health's Guide for the Care and Use of
Laboratory Animals (25).
Patient consent for publication
Written informed consent for publication was
obtained from all healthy volunteers.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CLEC-2
|
C-type lectin-like receptor 2
|
|
EMT
|
epithelial-mesenchymal transition
|
|
Gal-3
|
galectin-3
|
|
GC
|
gastric cancer
|
|
GPVI
|
glycoprotein VI
|
|
NT
|
non-treatment
|
|
PDPN
|
podoplanin
|
|
PLT
|
platelet
|
|
TRKI
|
transforming growth factor-β receptor
kinase inhibitor
|
References
|
1
|
Arita T, Ichikawa D, Konishi H, Komatsu S,
Shiozaki A, Hiramoto H, Hamada J, Shoda K, Kawaguchi T, Hirajima S,
et al: Increase in peritoneal recurrence induced by intraoperative
hemorrhage in gastrectomy. Ann Surg Oncol. 22:758–764. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kamei T, Kitayama J, Yamashita H and
Nagawa H: Intraoperative blood loss is a critical risk factor for
peritoneal recurrence after curative resection of advanced gastric
cancer. World J Surg. 33:1240–1246. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Labelle M, Begum S and Hynes RO: Direct
signaling between platelets and cancer cells induces an
epithelial-mesenchymal-like transition and promotes metastasis.
Cancer Cell. 20:576–590. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rothwell PM, Wilson M, Price JF, Belch JF,
Meade TW and Mehta Z: Effect of daily aspirin on risk of cancer
metastasis: A study of incident cancers during randomised
controlled trials. Lancet. 379:1591–1601. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shirai T, Inoue O, Tamura S, Tsukiji N,
Sasaki T, Endo H, Satoh K, Osada M, Sato-Uchida H, Fujii H, et al:
C-type lectin-like receptor 2 promotes hematogenous tumor
metastasis and prothrombotic state in tumor-bearing mice. J Thromb
Haemost. 15:513–525. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Saito R, Shoda K, Maruyama S, Yamamoto A,
Takiguchi K, Furuya S, Hosomura N, Akaike H, Kawaguchi Y, Amemiya
H, et al: Platelets enhance malignant behaviours of gastric cancer
cells via direct contacts. Br J Cancer. 124:570–573. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Suzuki-Inoue K: Roles of the
CLEC-2-podoplanin interaction in tumor progression. Platelets. 1–7.
2018.(Epub ahead of print). PubMed/NCBI
|
|
8
|
Suzuki-Inoue K, Fuller GL, García A, Eble
JA, Pöhlmann S, Inoue O, Gartner TK, Hughan SC, Pearce AC, Laing
GD, et al: A novel Syk-dependent mechanism of platelet activation
by the C-type lectin receptor CLEC-2. Blood. 107:542–549. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Suzuki-Inoue K, Inoue O and Ozaki Y: Novel
platelet activation receptor CLEC-2: From discovery to prospects. J
Thromb Haemost. 9 (Suppl 1):S44–S55. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Breiteneder-Geleff S, Soleiman A, Kowalski
H, Horvat R, Amann G, Kriehuber E, Diem K, Weninger W, Tschachler
E, Alitalo K and Kerjaschki D: Angiosarcomas express mixed
endothelial phenotypes of blood and lymphatic capillaries:
Podoplanin as a specific marker for lymphatic endothelium. Am J
Pathol. 154:385–394. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fujita N and Takagi S: The impact of
Aggrus/podoplanin on platelet aggregation and tumour metastasis. J
Biochem. 152:407–413. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Moroi M, Jung SM, Okuma M and Shinmyozu K:
A patient with platelets deficient in glycoprotein VI that lack
both collagen-induced aggregation and adhesion. J Clin Invest.
84:1440–1445. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sugiyama T, Okuma M, Ushikubi F, Sensaki
S, Kanaji K and Uchino H: A novel platelet aggregating factor found
in a patient with defective collagen-induced platelet aggregation
and autoimmune thrombocytopenia. Blood. 69:1712–1720. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mammadova-Bach E, Mangin P, Lanza F and
Gachet C: Platelets in cancer. From basic research to therapeutic
implications. Hamostaseologie. 35:325–336. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Derynck R, Turley SJ and Akhurst RJ: TGFβ
biology in cancer progression and immunotherapy. Nat Rev Clin
Oncol. 18:9–34. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ishimoto T, Miyake K, Nandi T, Yashiro M,
Onishi N, Huang KK, Lin SJ, Kalpana R, Tay ST, Suzuki Y, et al:
Activation of transforming growth factor beta 1 signaling in
gastric cancer-associated fibroblasts increases their motility, via
expression of rhomboid 5 homolog 2, and ability to induce
invasiveness of gastric cancer cells. Gastroenterology.
153:191–204.e16. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tauriello DVF, Palomo-Ponce S, Stork D,
Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, Sevillano M,
Ibiza S, Cañellas A, Hernando-Momblona X, et al: TGFβ drives immune
evasion in genetically reconstituted colon cancer metastasis.
Nature. 554:538–543. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nakayama T, Saito R, Furuya S, Shoda K,
Maruyma S, Takiguchi K, Shiraishi K, Akaike H, Kawaguchi Y, Amemiya
H, et al: Inhibition of cancer cell-platelet adhesion as a
promising therapeutic target for preventing peritoneal
dissemination of gastric cancer. Oncol Lett. 26:5382023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Satoh K, Fukasawa I, Kanemaru K, Yoda S,
Kimura Y, Inoue O, Ohta M, Kinouchi H and Ozaki Y: Platelet
aggregometry in the presence of PGE(1) provides a reliable method
for cilostazol monitoring. Thromb Res. 130:616–621. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kawai S, Takagi Y, Kaneko S and Kurosawa
T: Effect of three types of mixed anesthetic agents alternate to
ketamine in mice. Exp Anim. 60:481–487. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Scala M, Nishikawa M, Ito H, Tabata H,
Khan T, Accogli A, Davids L, Ruiz A, Chiurazzi P, Cericola G, et
al: Variant-specific changes in RAC3 function disrupt
corticogenesis in neurodevelopmental phenotypes. Brain.
145:3308–3327. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Narikiyo K, Mizuguchi R, Ajima A, Shiozaki
M, Hamanaka H, Johansen JP, Mori K and Yoshihara Y: The claustrum
coordinates cortical slow-wave activity. Nat Neurosci. 23:741–753.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Olajide OJ, Gbadamosi IT, Yawson EO,
Arogundade T, Lewu FS, Ogunrinola KY, Adigun OO, Bamisi O, Lambe E,
Arietarhire LO, et al: Hippocampal degeneration and behavioral
impairment during alzheimer-like pathogenesis involves glutamate
excitotoxicity. J Mol Neurosci. 71:1205–1220. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals, . Guide for the care and use of laboratory animals. 8th
edition. Washington (DC): National Academies Press (US); 2011
|
|
26
|
Tashiro M and Tohei A: Recommended doses
of medetomidine-midazolam-butorphanol with atipamezole for
preventing hypothermia in mice. J Vet Med Sci. 84:445–453. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kanda Y: Investigation of the freely
available easy-to-use software ‘EZR’ for medical statistics. Bone
Marrow Transplant. 48:452–458. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Estevez B and Du X: New concepts and
mechanisms of platelet activation signaling. Physiology (Bethesda).
32:162–177. 2017.PubMed/NCBI
|
|
29
|
Hwang BO, Park SY, Cho ES, Zhang X, Lee
SK, Ahn HJ, Chun KS, Chung WY and Song NY: Platelet
CLEC2-podoplanin axis as a promising target for oral cancer
treatment. Front Immunol. 12:8076002021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sasaki T, Shirai T, Tsukiji N, Otake S,
Tamura S, Ichikawa J, Osada M, Satoh K, Ozaki Y and Suzuki-Inoue K:
Functional characterization of recombinant snake venom rhodocytin:
Rhodocytin mutant blocks CLEC-2/podoplanin-dependent platelet
aggregation and lung metastasis. J Thromb Haemost. 16:960–972.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Suzuki-Inoue K: Platelets and
cancer-associated thrombosis: Focusing on the platelet activation
receptor CLEC-2 and podoplanin. Blood. 134:1912–1918. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mammadova-Bach E, Gil-Pulido J,
Sarukhanyan E, Burkard P, Shityakov S, Schonhart C, Stegner D,
Remer K, Nurden P, Nurden AT, et al: Platelet glycoprotein VI
promotes metastasis through interaction with cancer cell-derived
galectin-3. Blood. 135:1146–1160. 2020.PubMed/NCBI
|
|
33
|
Saito M, Ichikawa J, Ando T, Schoenecker
JG, Ohba T, Koyama K, Suzuki-Inoue K and Haro H: Platelet-derived
TGF-β induces tissue factor expression via the Smad3 pathway in
osteosarcoma cells. J Bone Miner Res. 33:2048–2058. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Asazuma N, Ozaki Y, Satoh K, Yatomi Y,
Handa M, Fujimura Y, Miura S and Kume S: Glycoprotein Ib-von
Willebrand factor interactions activate tyrosine kinases in human
platelets. Blood. 90:4789–4798. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Poole A, Gibbins JM, Turner M, van Vugt
MJ, van de Winkel JG, Saito T, Tybulewicz VL and Watson SP: The Fc
receptor gamma-chain and the tyrosine kinase Syk are essential for
activation of mouse platelets by collagen. EMBO J. 16:2333–2341.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Suzuki-Inoue K, Wilde JI, Andrews RK,
Auger JM, Siraganian RP, Sekiya F, Rhee SG and Watson SP:
Glycoproteins VI and Ib-IX–V stimulate tyrosine phosphorylation of
tyrosine kinase Syk and phospholipase Cgamma2 at distinct sites.
Biochem J. 378:1023–1029. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Turner M, Schweighoffer E, Colucci F, Di
Santo JP and Tybulewicz VL: Tyrosine kinase SYK: Essential
functions for immunoreceptor signalling. Immunol Today. 21:148–154.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yanaga F, Poole A, Asselin J, Blake R,
Schieven GL, Clark EA, Law CL and Watson SP: Syk interacts with
tyrosine-phosphorylated proteins in human platelets activated by
collagen and cross-linking of the Fc gamma-IIA receptor. Biochem J.
311:471–478. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huang B, Rouvelas I and Nilsson M: Gastric
and gastroesophageal junction cancer: Risk factors and prophylactic
treatments for prevention of peritoneal recurrence after curative
intent surgery. Ann Gastroenterol Surg. 6:474–485. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kitayama J, Ishigami H, Yamaguchi H,
Sakuma Y, Horie H, Hosoya Y, Lefor AK and Sata N: Treatment of
patients with peritoneal metastases from gastric cancer. Ann
Gastroenterol Surg. 2:116–123. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cieśla S, Lisiecki R, Ławnicka A,
Kudliński B, Ostrowska P, Davì A, Veroux M and Murawa D: Clinical
significance of peritoneal fluid examination for free cancer cells
in patients qualified for surgery for gastric cancer. Front Surg.
8:6858682021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kołomańska MM and Głuszek S: Free cancer
cells in gastric cancer-methods of detection, clinical and
prognostic importance (meta-analysis). Contemp Oncol (Pozn).
24:67–74. 2020.PubMed/NCBI
|
|
43
|
Amo L, Tamayo-Orbegozo E, Maruri N,
Eguizabal C, Zenarruzabeitia O, Riñón M, Arrieta A, Santos S, Monge
J, Vesga MA, et al: Involvement of platelet-tumor cell interaction
in immune evasion. Potential role of podocalyxin-like protein 1.
Front Oncol. 4:2452014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Labelle M and Hynes RO: The initial hours
of metastasis: The importance of cooperative host-tumor cell
interactions during hematogenous dissemination. Cancer Discov.
2:1091–1099. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Takemoto A, Okitaka M, Takagi S, Takami M,
Sato S, Nishio M, Okumura S and Fujita N: A critical role of
platelet TGF-β release in podoplanin-mediated tumour invasion and
metastasis. Sci Rep. 7:421862017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Schlesinger M: Role of platelets and
platelet receptors in cancer metastasis. J Hematol Oncol.
11:1252018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wiercinska E, Naber HPH, Pardali E, van
der Pluijm G, van Dam H and ten Dijke P: The TGF-β/Smad pathway
induces breast cancer cell invasion through the up-regulation of
matrix metalloproteinase 2 and 9 in a spheroid invasion model
system. Breast Cancer Res Treat. 128:657–666. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Spalton JC, Mori J, Pollitt AY, Hughes CE,
Eble JA and Watson SP: The novel Syk inhibitor R406 reveals
mechanistic differences in the initiation of GPVI and CLEC-2
signaling in platelets. J Thromb Haemost. 7:1192–1199. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Manne BK, Badolia R, Dangelmaier C, Eble
JA, Ellmeier W, Kahn M and Kunapuli SP: Distinct pathways regulate
Syk protein activation downstream of immune tyrosine activation
motif (ITAM) and hemITAM receptors in platelets. J Biol Chem.
290:11557–11568. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
David CJ and Massagué J: Contextual
determinants of TGFβ action in development, immunity and cancer.
Nat Rev Mol Cell Biol. 19:419–435. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
De Kock L and Freson K: The (patho)biology
of SRC kinase in platelets and megakaryocytes. Medicina (Kaunas).
56:6332020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
World Medical Association, . World medical
association declaration of Helsinki: Ethical principles for medical
research involving human subjects. JAMA. 310:2191–2194. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Percie du Sert N, Ahluwalia A, Alam S,
Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U,
Emerson M, et al: Reporting animal research: Explanation and
elaboration for the ARRIVE guidelines 2.0. PLoS Biol.
18:e30004112020. View Article : Google Scholar : PubMed/NCBI
|