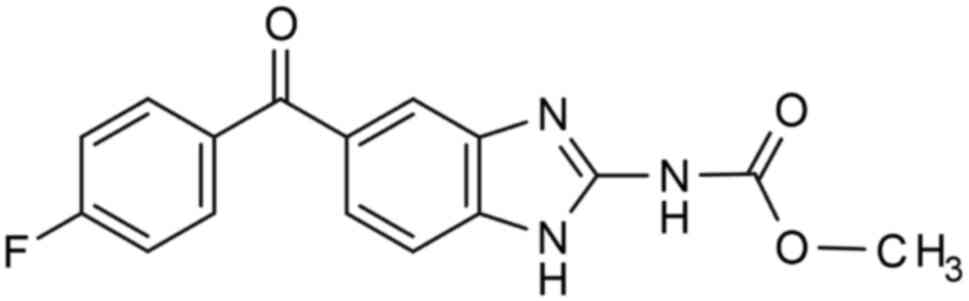
Introduction to flubendazole
Flubendazole, also known as
[5-(4-fluorobenzoyl)-1H-benzimidazole-2-yl]-carbamic acid methyl
ester, is a benzimidazole carbamate anthelmintic drug which was
first reported by Janssen Company (1) in the 1970s (Fig. 1). The fundamental mechanism of
action of benzimidazole drugs against parasites is to bind tubulin
in parasites, thereby disrupting the microtubule structure,
blocking the normal function of the cytoskeleton, interfering with
the normal movement and metabolism of the parasites, and ultimately
leading to the death of the parasites (2–4).
Pharmacological safety and
pharmacokinetics
As an antiparasitic drug, flubendazole has been used
for the treatment of parasites in humans and animals for >40
years and its safety has been well established (1). Previous studies on pigs, hens,
pheasants, dogs and rats have shown that flubendazole is a
well-tolerated nonteratogenic drug (5,6). The
low solubility of flubendazole in aqueous systems results in low
absorption into the blood, which is partially responsible for the
high safety profile of oral flubendazole for the treatment of
intestinal parasitic infections in both animals and humans
(1,7,8).
Notably, a recent study reported that flubendazole affects the
overall developmental processes and causes developmental
neurotoxicity in zebrafish (9),
which suggests that clinicians should be aware of the potential
toxicity of flubendazole. The metabolites of flubendazole are
predominantly reduced flubendazole, followed by hydrolyzed
flubendazole, when it is administered intravenously or enterally
(10,11). The pharmacokinetics of flubendazole
have been previously summarized by Čáňová et al (1) and Chen et al (12).
Repurposing of flubendazole for
anticancer effects
In 2010, Spagnuolo et al (13) first reported that flubendazole has
anticancer effects on leukemia and myeloma. Michaelis et al
(14) reported the effects of
flubendazole on the viability of a panel of cancer cell lines and
showed that 117/321 (36.4%) of cancer cell lines had an
IC90 value <1 µM, and 31/321 (9.7%) cell lines had an
IC90 value between 1–5 µM. It was also reported that
leukemia, multiple myeloma and neuroblastoma cells were the most
sensitive to flubendazole. Currently, flubendazole has been
reported to have anticancer effects on colon cancer, breast cancer,
neuroblastoma, melanoma, glioma, esophageal cancer, lung cancer,
prostate cancer and hepatocellular carcinoma (1,12,15).
This drug repurposing approach can leverage the existing safety,
dosage and pharmacokinetic data of drugs, shorten the time for new
drug development, save substantial costs in the preclinical, phase
I and phase II clinical stages, greatly reduce the cost of drug
development, improve the efficiency of drug utilization and help to
address the issue of drug resistance (16–18).
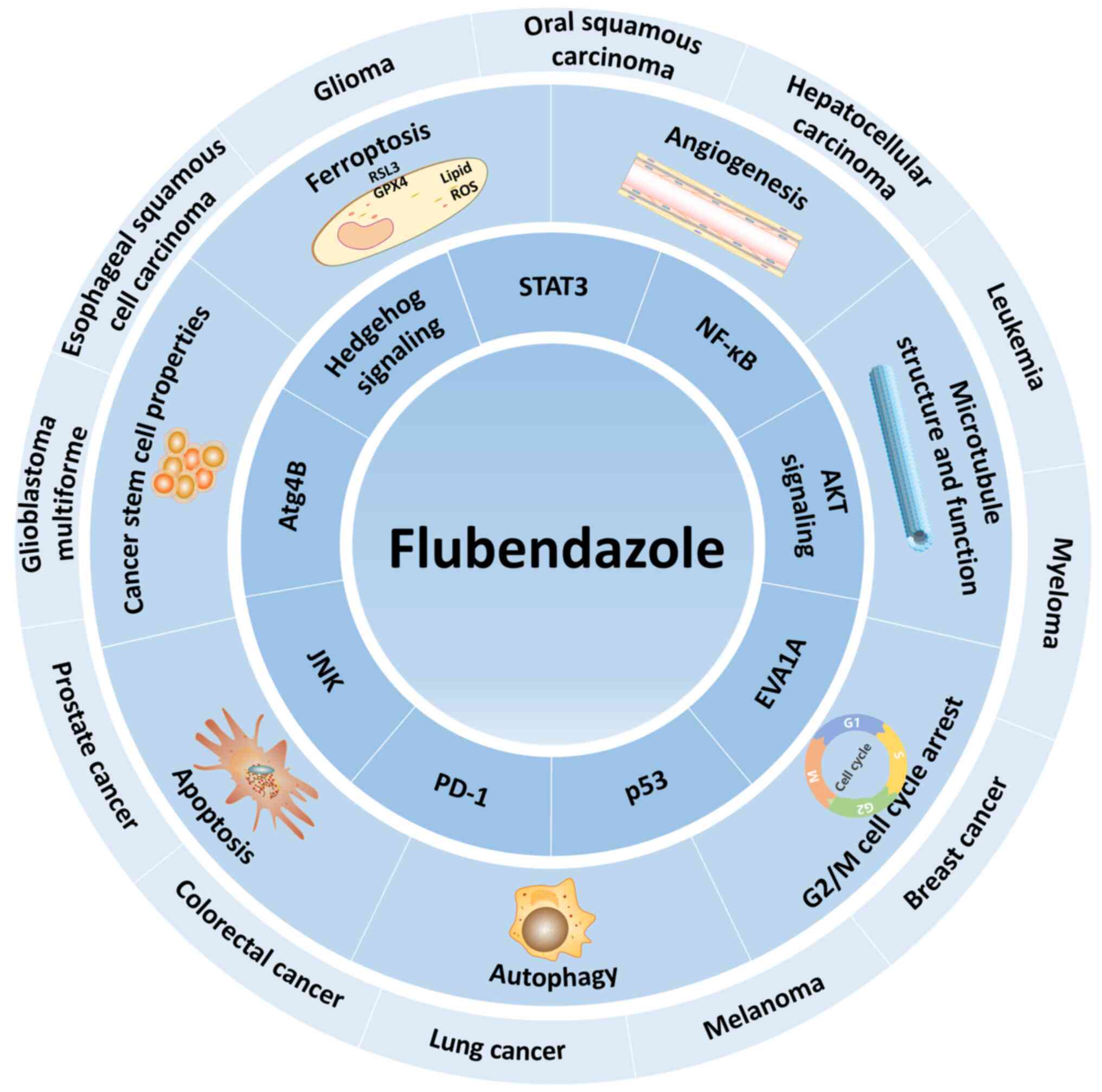
The anticancer effects and mechanisms of flubendazole in various
cancers have been summarized (Fig.
2; Table I).
 | Table I.Summary of anticancer targets and
mechanism molecules of flubendazole in different cancers. |
Table I.
Summary of anticancer targets and
mechanism molecules of flubendazole in different cancers.
| Cancer type | Experimental
model | Concentration or
dose | Synergetic
drug |
Effects/targets | Key molecules | (Refs.) |
|---|
| Leukemia and
myeloma | OCI-AML2 and OPM2
cells; xenograft model | 0.5, 1 or 2 µM; 20
or 50 mg/kg (i.p.) | Vinblastine | Microtubule
structure and function, G2 cell cycle arrest and mitotic
catastrophe | Tubulin
polymerization | (13) |
| Breast cancer | MDA-MB-231, BT-549,
SK-BR-3 and MCF-7 cells; xenograft model | 0.125, 0.25 or 0.5
µM; 25 mg/kg (i.p.) | Fluorouracil and
doxorubicin |
CD44high/CD24low
subpopulation, self-renewal related genes, EMT and G2/M cell cycle
arrest | Tubulin
polymerization | (23) |
| Breast cancer | MDA-MB-231, Hs578T,
BT-549 and 4T1 cells; xenograft model | 0.1, 0.25 or 0.5
µM; 10 mg/kg (i.p.) | N/A | Apoptosis, G2/M
cell cycle arrest, cancer stem cell-like properties and
angiogenesis | STAT3 | (26) |
| Breast cancer | BT474, SKBR3,
JIMT-1 and MDA-MB-453 cells; xenograft model | 0.1, 0.25 or 0.5
µM; 20 mg/kg (i.p.) | N/A | G2/M cell cycle
arrest, apoptosis, HER2/HER3 heterodimerization, cancer stem
cell-like properties and trastuzumab-resistance | HER2/AKT
signaling | (29) |
| Breast cancer | DA-MB-231 and
MDA-MB-468 cells; xenograft models | 0.25, 0.5, 1 or 2
µM; 10, 20, or 40 mg/kg (i.p.) | N/A | Autophagy and
apoptosis | EVA1A | (41) |
| Breast cancer | MDA-MB-231 and
MCF-7 cells | 0.25, 0.5 or 1
µM | N/A | Mitochondrial outer
membrane permeability and mitochondrial function | EVA1A | (42) |
| Breast cancer | MCF-7 and
MDA-MB-231 cells; xenograft model | 1.5, 0.5, 1 or 2
µM; 15 mg/kg | Paclitaxel | Aberrant mitosis
and apoptosis | HIF1α/PI3K/AKT
signaling | (66) |
| Breast cancer | MDA-MB-231
cells | 750 nM | N/A | Autophagy and
reactive oxygen species production | Atg4B | (76) |
| Melanoma | A-375, BOWES and
RPMI-7951 cells | 1 µM | N/A | G2/M cell cycle
arrest, microtubular damage, mitotic catastrophe and apoptosis | Tubulin
polymerization and p53 | (25) |
| Melanoma | A-375, BOWES and
RPMI-7951 cells; 3 patients with melanoma | 1 µM | N/A | G2/M cell cycle
arrest, mitotic catastrophe, apoptosis and autophagy | JNK and Noxa | (31) |
| Melanoma | MDA-MB-435 cells;
xenograft models | 1 µM; 200 mg/kg
(i.p.) or 20 mg/kg (i.t.) | N/A | Angiogenesis, PD-1
and myeloid-derived suppressor cells | STAT3 | (51) |
| Melanoma | B16F10 and Jurkat
cells; xenograft models | 10 µM; 200 mg/kg
(i.p.) | N/A | PD-1 expression,
genes in cancer-associated pathways and immunological signature
gene sets | PD-1 | (67) |
| Colon cancer | SW480 and SW620
cells | 1 or 2 µM | N/A | Microtubule
organization, tubulin content, mitotic catastrophe and
senescence | Tubulin
polymerization | (24) |
| Colon cancer | HCT116, RKO and
SW480 cells; 12 patients with colorectal cancer | 0.3, 0.6 or 1.2 µM;
10 or 30 mg/kg (i.p.) | 5-fluorouracil | Autophagy,
apoptosis and nuclear translocation of STAT3 | STAT3 | (39) |
| Colon cancer | SW480 and SW620
cells | 1 µM | N/A | Cell adhesion and
migration | NF-κB | (60) |
| Intestinal
cancer | SW480, SW620, HCT8
and Caco-2 cells | 0.2, 0.25, 0.3,
0.4, 0.5 or 1 µM | Paclitaxel | G2/M cell cycle
arrest | N/A | (32) |
| Prostate
cancer | PC-3, DU145 and
RWPE-1 cells; xenograft model | 0.1, 0.5 or 1 µM;
10 mg/kg (i.p.) | 5-fluorouracil | G2/M cell cycle
arrest and ferroptosis | p53 | (30) |
| Oral squamous
carcinoma | PE/CA-PJ15, DOK,
H376 and GF cells | 0.1 or 0.25 µM | N/A | Proliferation,
migration and cadherin switching | N/A | (22) |
| Glioma | SF-268 and T-98G
cells; xenograft model | 0.25 or 0.5 µM; 25
mg/kg (i.p.) | N/A | G2/M cell cycle
arrest and apoptosis | p53 | (33) |
| Glioblastoma
multiforme | U87-MG and U251-MG
cells; xenograft model | 0.125, 0.25 or 0.5
µM; 12.5, 25 or 50 mg/kg (i.p.) | N/A | DNA synthesis, G2/M
cell cycle arrest, pyroptosis and apoptosis | p53 and NF-κB | (34) |
| Hepatocellular
carcinoma | SNU449, PLC/PRF/5,
Hep3B, HepG2, Huh7, MHCC-97H, MHCC-LM3, HCC-LY10 and HEK-293T
cells; xenograft model | 0.25, 0.5 or 1 µM;
40 mg/kg (i.p.) | Lenvatinib | Apoptosis and G2/M
cell cycle arrest | Hedgehog | (35) |
| Lung cancer | H460, A549, PC-9
BEAS-2b and human umbilical vein endothelial cells; xenograft
model | 0.5, 1 or 2 µM; 10
or 20 mg/kg (i.p.) | N/A | Apoptosis, nuclear
translocation of STAT3 and autophagy | STAT3 | (40) |
| Esophageal squamous
cell carcinoma | EC9706 and TE1
cells | 1, 2 or 4 µM | Doxorubicin | Apoptosis and
cytotoxicity of doxorubicin | NF-κB | (61) |
Anticancer effects of flubendazole
Inhibition of microtubule structure
and function
Microtubules, which are composed of α- and β-tubulin
heterodimers, form slender, tube-like polymers that are crucial
components of the cytoskeleton in all eukaryotic cells. They
participate in processes such as cell division, intracellular
transport, signal transduction and the maintenance of cell shape,
polarity and integrity. During mitosis, the replicated chromosomes
are separated into two identical sets before the cell divides into
two daughter cells (19,20). The primary function of microtubules
is to form a mitotic spindle that ensures the proper segregation
and reassembly of chromosomes (19,20).
The pivotal role of microtubules in mitosis makes them paramount
targets for anticancer drugs (21).
Flubendazole can specifically bind to tubulin and inhibit its
polymerization, thereby exerting its anticancer effects (1,22).
In leukemia and myeloma, flubendazole induces cell
death by binding to the polymerization site of tubulin, preventing
the assembly of tubulin subunits and thus inhibiting the formation
and stability of microtubules, which in turn affects cell cycle
progression and induces mitotic catastrophe (13). Notably, flubendazole acts by binding
to a site on tubulin that is similar to but distinct from the site
targeted by vinblastine, which is why cells resistant to
vinblastine due to overexpression of P-glycoprotein remain
sensitive to flubendazole (13).
Hou et al (23) reported
that in breast cancer, the inhibition of tubulin polymerization by
flubendazole leads to spindle abnormalities and the formation of
monopolar spindles. These monopolar spindles fail to properly
segregate chromosomes into two daughter cells, causing cell
division failure and further inhibiting the proliferation of cancer
cells. As microtubules are essential for spindle formation during
the G2/M phase of the cell cycle, flubendazole inhibits the
proliferation of cancer cells by suppressing tubulin polymerization
and causing cell cycle arrest. By affecting tubulin polymerization,
flubendazole may indirectly impact the self-renewal capacity of
cancer stem cells, as the maintenance and function of these stem
cells depend on normal cell cycle progression and cell division
processes (19,23). The inhibition of tubulin
polymerization may also affect epithelial-mesenchymal transition
(EMT), a critical process through which cancer cells acquire
invasive and metastatic capabilities. By disrupting the normal
function of microtubules, flubendazole may inhibit the EMT process,
thereby reducing the migration and invasion of cancer cells
(22,23). Additionally, flubendazole enhances
the cytotoxic effects of 5-fluorouracil (5-FU) and doxorubicin on
breast cancer cells through cell cycle arrest caused by the
inhibition of tubulin polymerization, which increases the
sensitivity of cells to chemotherapeutic drugs (23). Furthermore, in colon cancer,
melanoma and triple-negative breast cancer (TNBC), flubendazole can
inhibit tubulin polymerization, induce mitotic catastrophe and
cause cell cycle arrest (24–26).
Inducing cell cycle arrest
The cell cycle refers to the series of orderly
processes that a cell undergoes from the end of one division to the
end of the next. The cell cycle is primarily divided into two
phases: Interphase and M phase, also known as mitosis. Interphase
is further subdivided into the G1 phase, S phase and G2 phase, with
the S phase being the period when DNA replication occurs. In the M
phase, genetic material is accurately distributed to two daughter
cells (27). During the G2 phase,
the cell conducts a final check on DNA replication completed in the
S phase, ensuring that no errors occur. At the G2/M transition,
centrosomes, which are microtubule organizing centers present in
animal cells, begin to replicate and migrate to opposite poles of
the cell. Given that microtubules are key structures in the process
of cell division and are responsible for forming the spindle
apparatus and aiding in chromosome separation, factors affecting
microtubule polymerization or function can lead to G2/M arrest
(28).
In leukemia and myeloma cells, flubendazole induces
G2/M cell cycle arrest and mitotic catastrophe by binding to
tubulin and inhibiting microtubule polymerization (13). In breast cancer, flubendazole
directly binds to the colchicine binding site of tubulin,
preventing the polymerization of tubulin subunits, causing spindle
abnormalities and leading to cell cycle arrest in the G2/M phase,
thereby inhibiting the proliferation of cancer cells (23). In TNBC and HER2-positive breast
cancer cells, flubendazole causes G2/M phase arrest of the cell
cycle by binding to tubulin polymerization sites and affecting the
levels of phosphorylated STAT3 (26,29).
Flubendazole can suppress the proliferation of castration-resistant
prostate cancer (CRPC) cells, induce expression of the p53 protein
and increase the expression level of p21, also known as cyclin
dependent kinase inhibitor 1A (CDKN1A), to inhibit the activity of
cell cycle-related proteins such as Cyclin B1 and CDK1, thus
preventing the cell from entering mitosis and causing the cell
cycle to arrest at the G2/M phase (30). Additionally, flubendazole has been
reported to induce G2/M phase cell cycle arrest in melanoma cells
(25,31), intestinal cancer (32), glioma (33), glioblastoma multiforme (GBM)
(34) and hepatocellular carcinoma
(HCC) (35).
Inducing autophagy
Autophagy is a highly regulated intracellular
process in which damaged proteins and organelles are degraded to
maintain cellular homeostasis and provide energy resources. It
serves a pivotal role in cellular stress, aging, the immune
response and the progression of cancer (36). In cancer, autophagy serves a dual
role. It serves as a survival mechanism for tumor cells,
particularly in the context of nutrient scarcity by supporting
uncontrolled cell proliferation. Conversely, if autophagy is
hyperactivated or persists, it may lead to apoptosis (36,37).
LC3 is essential for the formation and maturation of
autophagosomes. There are two forms of LC3: LC3-I and LC3-II.
LC3-II is generated through a process called lipidation, where
LC3-I is conjugated to phosphatidylethanolamine to become LC3-II.
This conversion is a key step in the elongation of the
autophagosome membrane. Therefore, LC3-II is widely used in
research as an indicator of autophagy activity (36,38).
Lin et al (39) and Xie et al (40) reported results on the efficacy of
flubendazole in patients with colorectal cancer (CRC) and non-small
cell lung carcinoma (NSCLC) in 2019 and 2021, respectively. In
vitro experiments showed that flubendazole inhibited the
expression of mTOR and P62, while upregulating the expression of
Beclin-1 and LC3-I/II, thereby activating autophagy in both types
of cancer. The activation of autophagy is associated with the
induction of apoptosis in CRC cells, where activated autophagy can
promote apoptosis. In in vivo tumor xenograft models,
flubendazole reduced tumor volume and weight, and the experimental
results showed that autophagy was activated (39–41).
After treatment with flubendazole, the expression levels of the
autophagy marker protein LC3-II in TNBC cells increased, the p62
protein was degraded and LC3 was converted from LC3-I to LC3-II
(41). Furthermore, the formation
of autophagosomes and autolysosomes induced by flubendazole was
confirmed using the GFP/mRFP-LC3 dual fluorescence reporter system
(41). Flubendazole impaired the
permeability of the mitochondrial outer membrane in breast cancer
cells, which was characterized by a decrease in the mitochondrial
membrane potential, reduced ATP production and increased superoxide
levels, leading to mitochondrial dysfunction. Flubendazole
increased the expression of dynamin-related protein 1 (DRP1),
leading to the aggregation of PTEN-induced putative kinase 1
(PINK1) and the mitochondrial translocation of Parkin, thereby
promoting excessive mitophagy. Excessive mitophagy contributes to
the mitochondrial damage and dysfunction induced by flubendazole,
thus inhibiting the proliferation and migration of breast cancer
cells (42).
Induction of apoptosis
Apoptosis is a form of programmed cell death that
serves a crucial role in the development of an organism, the
maintenance of tissue homeostasis and the elimination of damaged or
aged cells. In the intrinsic pathway of apoptosis, pro-apoptotic
proteins from the BCL-2 family form pores in the outer
mitochondrial membrane, leading to increased mitochondrial membrane
permeability and the subsequent release of cytochrome c.
Cytochrome c then binds to the apoptotic protease activating
factor-1, recruiting and activating procaspase-9. This activation
subsequently triggers the activation of the effector caspases,
caspase-3 and caspase-7, resulting in the process of apoptosis. In
the extrinsic pathway, cell surface death receptors bind to their
ligands, forming the death-inducing signaling complex, which
activates caspase-8 and caspase-10, cleaves the BH3 interacting
domain death agonist protein and promotes the release of cytochrome
c, thereby intersecting with the intrinsic pathway (43,44).
In HER2-positive breast cancer cells treated with
flubendazole, the expression levels of activated caspase-3,
caspase-7 and caspase-8 increase, significantly increasing the
proportion of apoptotic cells, particularly the number of late
apoptotic cells (29). In CRC
cells, flubendazole dose-dependently increases the activity of
caspase-3, a key executor of apoptosis. The aforementioned study
also showed that flubendazole promotes apoptosis by activating
autophagy. The apoptotic effects observed in vitro were
confirmed by in vivo experiments. In a xenograft model,
flubendazole significantly reduced tumor volume and weight and an
increase in the number of apoptotic cells was detected using TUNEL
staining (39). In NSCLC,
flubendazole reduces the expression of BCL-2 by activating the JNK
pathway. Decreased expression levels of BCL-2 leads to the release
of Beclin-1 from the BCL-2-Beclin 1 complex, thereby triggering
autophagy, indicating that flubendazole can induce apoptosis in
NSCLC cells by activating autophagy (40). In TNBC cells, an increase in the
proportion of apoptotic TNBC cells induced by flubendazole was
confirmed using the TUNEL assay and flow cytometry. An increase in
the cleavage of caspase-3, upregulation of Bax and downregulation
of BCL-2, as detected by immunoblotting analysis, suggested that
flubendazole induced apoptosis (26,41).
Inhibition of cancer stem cell (CSC)
properties
CSCs are a unique subset of cells that serve a
crucial role in cancer initiation, progression, metastasis,
therapeutic resistance and recurrence (45,46).
These cells possess characteristics akin to those of stem cells,
such as self-renewal and multilineage differentiation abilities,
distinctive cell division patterns, metabolic phenotypes and robust
resistance to conventional anticancer therapies (45,46).
Studies on the inhibitory effects of flubendazole on the stemness
of CSCs have primarily been conducted in the context of breast
cancer (23,26,29).
Breast CSCs (BCSCs) are a subpopulation of breast cancer cells
characterized by high tumorigenicity and self-renewal. Key BCSC
biomarkers include CD44, CD24, CD49f and aldehyde dehydrogenase
(ALDH) (46,47). CD44 is a transmembrane glycoprotein
which is pivotal for cell survival and pluripotency maintenance in
BCSCs. CD24 is a sialomucin with low or absent expression levels in
BCSCs and is implicated in cell adhesion and metastasis. CD49f, a
laminin-binding glycoprotein, is crucial for cell-extracellular
matrix interactions. ALDH is an enzyme associated with CSC
self-renewal, differentiation and resistance to therapy, which
serves an important role in cancer metabolism and progression.
In 2015, Hou et al (23) reported that flubendazole suppressed
the expression of genes associated with self-renewal in breast
cancer, including c-Myc, Sox2, Oct-4 and Nanog, and reduced the
proportion of the CD44high/CD24low
subpopulation, suggesting that flubendazole decreased the
self-renewal capacity of BCSCs. The ability to form mammospheres is
an indicator of the self-renewal capacity of BCSCs. After treatment
with flubendazole, breast cancer cells exhibited reduced ALDH1
activity, a decrease in the proportion of
CD44high/CD24low phenotype cells and a
decrease in mammosphere formation capacity, indicating the
suppression of stem cell properties in BCSCs (29). Another study focused on TNBC also
reported that flubendazole could significantly affect BCSC-like
properties, including reducing the proportions of
CD24low/CD44high and
CD24high/CD49fhigh subpopulations, decreasing
ALDH1 activity and reducing the capacity for mammosphere formation
(26).
Ferroptosis induction
Ferroptosis, an iron-dependent form of nonapoptotic
cell death, is characterized by uncontrolled lipid peroxidation
within the cell, ultimately leading to the rupture of the cell
membrane. Ferroptosis involves multiple key molecules, including
glutathione peroxidase 4 (GPX4), glutathione (GSH) and solute
carrier family 7 member 11 (SLC7A11). SLC7A11 encodes a
cystine/glutamate transporter responsible for transporting cysteine
into the cell. Cysteine is a key precursor for the synthesis of
GSH, which is one of the primary antioxidants in the cell. GPX4 is
an essential antioxidant enzyme that utilizes GSH as a cofactor to
reduce lipid hydroperoxides, protecting the cell membrane from
oxidative damage. When the function of GPX4 is compromised or the
expression levels of GSH decrease, phospholipids in the cell
membrane become susceptible to lipid peroxidation catalyzed by iron
ions, leading to destruction of the cell membrane and cell death
(48–50).
In CRPC, flubendazole can induce p53 protein
expression. p53 binds to the promoter region of SLC7A11, inhibiting
its transcription, which results in decreased expression of the
SLC7A11 protein. The reduced expression level of the SLC7A11
protein results in a decreased ability of the cell to take up
cysteine, which in turn leads to a decrease in GSH levels. A
decrease in GSH levels leads to a reduction in the activity of
GPX4. A decrease in GPX4 activity results in the accumulation of
lipid hydroperoxides, which can react with polyunsaturated fatty
acids in the cell membrane, initiating lipid peroxidation. The
accumulation of lipid peroxidation ultimately leads to damage to
the cell membrane and cell death, resulting in ferroptosis
(30).
Inhibition of angiogenesis
Treatment of TNBC cells with flubendazole inhibits
the activation of STAT3, which consequently leads to a reduction in
cancer angiogenesis. This is associated with a decrease in
microvessel density and the expression level of VEGF. In a
xenograft model of TNBC, flubendazole suppressed tumor growth,
angiogenesis and metastasis to the lungs and liver, which coincided
with reduced levels of MMP-2 and MMP-9 in circulating blood
(26). In primary tumors formed by
subcutaneous inoculation of melanoma MDA-MB-435 cells in xenograft
models, flubendazole almost completely inhibited the expression of
the cancer endothelial cell marker CD31, indicating that
flubendazole is a potent inhibitor of cancer angiogenesis. STAT3
serves a role in regulating angiogenesis and flubendazole achieves
its antiangiogenic effects through the inhibition of STAT3
(51).
Molecular mechanisms of the anticancer
activity of flubendazole
STAT3
STAT3 is a latent cytoplasmic transcription factor
that serves a pivotal role in various cellular processes, including
cell proliferation, differentiation, inflammation, apoptosis,
angiogenesis and immune responses (52). Under normal physiological
conditions, STAT3 activation is tightly regulated. However, STAT3
is constitutively activated in many types of cancers, contributing
to cancer growth and metastasis by regulating the expression of
various target genes involved in cell survival, oncogenesis, cancer
progression and stemness (53,54).
The multifaceted role of STAT3 in cancer includes acting both as an
oncogene and a cancer suppressor factor, depending on the specific
cellular microenvironment and cancer type (55,56).
In breast cancer cells, flubendazole significantly
inhibits the activation of STAT3, promoting TNBC cells to exhibit a
marked increase in apoptosis, reducing the stem-like
characteristics of BCSCs, decreasing the expression of VEGF and
suppressing the process of EMT. In in vivo experiments,
flubendazole significantly reduced the number of lung and liver
metastatic foci by inhibiting STAT3 activation and decreasing the
levels of MMP-2 and MMP-9 in circulating blood (26). In primary tumors formed by
subcutaneous inoculation of melanoma MDA-MB-435 cells in xenograft
models, flubendazole reduced the phosphorylation of STAT3,
particularly at the Tyr705 site, exerting antiangiogenic effects by
inhibiting STAT3 (51). In in
vivo experiments, flubendazole completely inhibited programmed
cell death protein-1 (PD-1) expression in cancer tissue by
inhibiting STAT3 without affecting PD-L1 levels (51). Myeloid-derived suppressor cells
(MDSCs) are a group of myeloid cells in the cancer microenvironment
that inhibit immune responses and promote cancer progression
through various mechanisms, facilitating immune evasion and
metastasis. Flubendazole reduced the number of MDSCs in melanoma
tissue, indicating that flubendazole may weaken the
immunosuppressive effect of cancer by reducing the number of MDSCs,
thereby contributing to the anticancer immune response (51). A previous study showed that
flubendazole also affects the function of MDSCs by inhibiting the
activity of STAT3 (51). In human
CRC, flubendazole can block the activation and nuclear
translocation of STAT3 induced by IL-6, leading to the inhibition
of the transcription of STAT3 target genes, such as MCL1 apoptosis
regulator, BCL-2 family member, VEGF and baculoviral IAP repeat
containing 5. These genes are closely related to the antiapoptotic
characteristics of cancer cells, angiogenesis and metastasis
(39). In NSCLC, flubendazole can
block the phosphorylation of STAT3 in a dose- and time-dependent
manner, thereby regulating the transcription of STAT3 target genes
and exerting anticancer effects through apoptosis and autophagy. As
observed in CRC, flubendazole treatment of NSCLC can also inhibit
the phosphorylation and nuclear localization of STAT3 induced by
IL-6, thus inhibiting the activation of STAT3 and reducing the
expression levels of VEGF and MCL-1 proteins related to STAT3,
thereby affecting cancer angiogenesis and cell survival (40).
NF-κB
NF-κB is a pivotal transcription factor that
is ubiquitously present in various types of cells and comprises
five family members (p65, RelB, c-Rel, NF-κB1 and NF-κB2) (57). NF-κB serves a crucial role in
controlling cell proliferation, inflammation, cellular stress
responses, immune responses and apoptosis (57,58).
NF-κB typically binds to the inhibitory protein IκB in the
cytoplasm. Upon cellular reception of external signals, such as
inflammatory factors, stress or injury, the IKK complex is
activated and phosphorylates IκB, leading to its degradation and
the release of NF-κB into the nucleus, where it activates the
expression of target genes (57,58).
NF-κB is a complex regulatory factor that may serve different roles
at various stages of cancer development. It directly participates
in the development of cancer by promoting the proliferation of
cancer cells, inhibiting apoptosis, promoting angiogenesis and
stimulating invasion and metastasis (57–59).
In particular, it enhances the migratory and invasive capabilities
of cancer cells by inducing EMT, thereby facilitating distant
metastasis (57–59).
In CRC, fenbendazole not only affects cell adhesion
and migration but also inhibits the phosphorylation of NF-κB p65, a
key transcription factor associated with inflammation, survival,
proliferation and metastasis. By employing RNA silencing technology
to knock down NF-κB p65 in SW620 cells, the inhibitory effect on
multiple cancer metastasis markers, such as intercellular adhesion
molecule 1, epithelial cell adhesion molecule, integrin α5, β1 and
α-tubulin, was enhanced. Fenbendazole suppresses the metastasis of
CRC cells by inhibiting the activation of NF-κB p65, thereby
reducing the expression levels of proteins related to cancer
metastasis (60). In esophageal
squamous cell carcinoma (ESCC), fenbendazole can inhibit the
activation of IKK and decrease the phosphorylation of NF-κB p65.
After 24 h of treatment with fenbendazole, it can induce the
cleavage of poly ADP-ribose polymerase (PARP) and reduce the
expression of the antiapoptotic protein BCL-2 while upregulating
the expression of the proapoptotic protein Bim, ultimately leading
to the apoptosis of ESCC cells. When used in combination with the
chemotherapeutic drug doxorubicin, fenbendazole may enhance the
toxicity of doxorubicin in ESCC cells by inhibiting the NF-κB
signaling pathway, demonstrating a synergistic effect (61). NF-κB is hyperactivated in GBM, and
fenbendazole can trigger pyroptosis in GBM cells through the
NF-κB/NLR family pyrin domain containing 3 (NLRP3)/gasdermin D
(GSDMD) pathway (34). Pyroptosis
is a form of programmed cell death closely related to inflammatory
processes and is characterized by cell swelling, cell membrane
rupture and the release of proinflammatory cytokines, such as IL-1β
and IL-18. NF-κB mainly promotes the production of proinflammatory
factors, such as pro-IL-1β, pro-IL-18, NLRP3 and caspase-1, which
serve key roles in pyroptosis. In particular, NF-κB can promote the
activation of the NLRP3 inflammasome, which in turn promotes the
release of IL-1β and IL-18 mediated by caspase-1 and the cleavage
of GSDMD (34). Therefore,
fenbendazole can not only trigger pyroptosis in glioblastoma cells,
but also induce mitochondria-dependent apoptosis (34).
AKT signaling
The PI3K/AKT/mTOR signaling pathway serves a crucial
role in promoting cell survival, growth and cell cycle progression,
with extensive cross-regulation with other cellular signaling
networks. PI3K catalyzes the conversion of phosphatidylinositol
4,5-bisphosphate to phosphatidylinositol 3,4,5-trisphosphate
(PIP3). The generation of PIP3 is a prerequisite for activation of
the AKT protein. AKT is present in the cytoplasmic matrix in an
inactive form. Upon activation of PI3K and production of PIP3, AKT
is recruited to the plasma membrane and activated through
phosphorylation by pyruvate dehydrogenase kinase 1 and mTOR complex
2. Activated AKT can migrate to the cytoplasm and nucleus,
phosphorylating multiple substrate proteins that regulate key
cellular functions such as proliferation, survival and metabolism.
mTOR is a crucial regulator of cell growth and proliferation and
primarily controls protein synthesis, cytoskeletal organization,
cell growth and cell metabolism (62–64).
Flubendazole can significantly reduce the
phosphorylation levels of HER2 and human epidermal growth factor
receptor 3 (HER3) in HER2-positive breast cancer cells, thereby
decreasing the levels of phosphorylated AKT, indicating that
flubendazole may inhibit the PI3K/AKT signaling pathway by
suppressing the formation of HER2/HER3 heterodimers and the
activation of AKT. p95HER2 is a C-terminal truncated form of the
HER2 receptor with tyrosine kinase activity that is associated with
resistance to trastuzumab, also known as Herceptin. Flubendazole
can significantly reduce the expression level of p95HER2,
contributing to the inhibition of the AKT signaling pathway
(29). Hypoxia-inducible factor 1α
(HIF1α) is a key transcription factor in the cellular response to
hypoxic environments that activates multiple genes associated with
angiogenesis, glycolysis and cell survival. In cancer, the abnormal
expression of HIF1α is closely related to the aggressiveness,
metastasis and therapeutic resistance of cancers (65). RNA sequencing (RNA-seq) showed
changes in a number of pathways controlling cellular metabolic
processes in MDA-MB-231 breast cancer cells treated with
flubendazole combined with paclitaxel, particularly a significant
reduction in the expression levels of HIF1α, phosphorylated
(p)-PI3K, p-AKT and p-mTOR. This suggests that flubendazole may
produce a synergistic anticancer effect by reducing the expression
level of HIF1α and inhibiting the PI3K/AKT signaling pathway.
Furthermore, in paclitaxel-resistant breast cancer cells (MCF-7/PTX
cells), treatment with flubendazole inhibited the PI3K/AKT pathway
and reduced the level expression of HIF1α, demonstrating the
potential to reverse paclitaxel resistance in breast cancer
treatment (66). In addition, the
expression of a number of genes related to immune responses,
cellular metabolism and cell signaling was altered in human Jurkat
cells treated with flubendazole. In particular, flubendazole
upregulated genes associated with the PI3K/AKT signaling pathway,
which is closely related to the effector function of T cells
(67).
Eva-1 homolog A (EVA1A)
EVA1A is a transmembrane protein associated with
lysosomes and the endoplasmic reticulum that serves a crucial role
in regulating autophagy and apoptosis. In cancer cells, EVA1A can
interact with autophagy-related proteins, such as autophagy related
16 like 1, to promote the formation of autophagosomes, thereby
inhibiting the proliferation of cancer cells. Furthermore, EVA1A
can also induce cancer cell apoptosis by affecting the activation
of caspase cascades (68).
Zhen et al (41) reported that flubendazole induced
apoptosis and autophagy in TNBC cells. RNA-seq analysis showed that
the expression of EVA1A was upregulated in TNBC cells after
treatment with flubendazole. Silencing EVA1A with small interfering
RNA significantly restored the reduced proliferation ability of
TNBC cells and decreased the autophagy and apoptosis induced by
flubendazole. Molecular docking was used to predict the binding
mode of flubendazole with EVA1A, and the results suggested that
flubendazole may interact with the Trp135, Thr113 and Asn110
residues of EVA1A. Subsequently, three site mutants of EVA1A
(EVA1AW135A, EVA1AT113A and
EVA1AN110A) were constructed and it was reported that
only EVA1AT113A significantly weakened the inhibitory
effect of flubendazole on MDA-MB-231 cells. This indicates that
Thr113 may be a key amino acid residue involved in the binding of
flubendazole to EVA1A and could serve an important role in
regulating the proliferation and autophagy of TNBC cells. After
treatment of breast cancer cells with flubendazole, the expression
levels of DRP1 and PINK1 increased, promoting the mitochondrial
translocation of Parkin to induce mitophagy by targeting EVA1A.
Silencing EVA1A partially blocked DRP1 expression and Ser616
phosphorylation induced by flubendazole, reduced the colocalization
of mitochondria and autophagosomes and decreased the expression of
PINK1, Parkin and p-ParkinSer65. Conversely, overexpression of
EVA1A in breast cancer cells can trigger DRP1-mediated mitophagy
and significantly inhibit cell growth and proliferation (42).
p53
The p53 protein, encoded by the TP53 gene, is a
crucial cancer suppressor. The functionality of p53 is complex, as
it directly regulates the transcription of >300 target genes and
indirectly affects the expression of thousands of genes. Its
functions include cell cycle regulation, promotion of apoptosis,
induction of cellular senescence, facilitation of DNA damage
repair, maintenance of genomic stability, regulation of stem cell
fate, suppression of metastasis, modulation of the balance between
cell death and survival and regulation of glucose, lipid, amino
acid, nucleotide, iron and redox metabolism (69).
Different melanoma cell lines exhibit distinct
responses of p53 to flubendazole treatment. In A-375 cells, the p53
protein expression level significantly increased following
treatment with flubendazole, while in BOWES cells, the p53 protein
level remained stable. In RPMI-7951 cells, no p53 protein
expression was detected in either the control group or the
flubendazole-treated group, which is consistent with their known
p53 null status (25). In CRPC,
flubendazole can induce p53 protein expression. p53 increases
p21/CDKN1A to inhibit the activity of Cyclin B1 and CDK1, thereby
preventing cells from entering mitosis; it also transcriptionally
represses SLC7A11 to reduce the uptake of cysteine within the cell,
which in turn decreases the activity of GPX4 and triggers
ferroptosis. Moreover, the activation of p53 may enhance the
cytotoxicity of 5-FU, as the anticancer effect of 5-FU is partly
dependent on the p53 signaling pathway (30). In GBM, flubendazole affects the p53
signaling pathway in a dose-dependent manner, upregulating the
expression of the cancer suppressor proteins p53 and p21 and
downregulating the expression of cyclin B1. The p53/p21/cyclin B1
signaling pathway induces cell cycle arrest in GBM cells, thereby
promoting apoptosis (30).
PD-1
PD-1 is an immune checkpoint receptor expressed on
the surface of various immune cells, including activated T cells, B
cells, dendritic cells, monocytes, myeloid cells and natural killer
cells. Within the cancer microenvironment, the interaction between
PD-1 and its ligand, programmed death ligand 1 (PD-L1), is one of
the critical mechanisms by which cancer cells undergo immune
escape. The activation of PD-1 signaling can inhibit T cell
receptor-mediated cytotoxicity and the proliferation of
CD8+ T cells, thereby preventing the clearance of cancer
cells by the immune system. In cancer therapy, blockade of the
PD-1/PD-L1 pathway can restore the ability of T cells to recognize
and kill cancer cells, thus preventing immune escape (70,71).
In 2019, Li et al (51) reported that flubendazole could
suppress the levels of PD-1 in human melanoma but had no effect on
PD-L1 levels. Flubendazole inhibits the expression of PD-1 by
suppressing STAT3, which may help enhance the immune system's
response to cancer cells, thereby inhibiting cancer growth and
metastasis. In 2023, Li et al (67) reported that flubendazole could
reduce PD-1 protein expression levels in B16F10 melanoma cells from
C57BL/6J mice without affecting PD-L1 levels. A reduction in PD-1
was accompanied by an increase in CD3+ T cell
infiltration, indicating that flubendazole may enhance the
anticancer immune response by modulating PD-1 in the immune
microenvironment. RNA-seq analysis of human Jurkat cells treated
with flubendazole showed that flubendazole downregulated the mRNA
expression level of PD-1 and upregulated the expression level of
the transcription factor AP-1 family member Jun. When Jurkat T
cells were pretreated with the AP-1 inhibitor T5224, the
suppressive effect of flubendazole on PD-1 protein expression was
blocked by T5224, indicating that the ability of flubendazole to
inhibit PD-1 is, at least, partially dependent on the activation of
AP-1.
JNK
JNK is a member of the MAPK family and is involved
in a variety of cellular activities, including cell proliferation,
differentiation, survival and death. The role of JNK in cancer is
complex and multifaceted, and its specific function depends on a
multitude of factors, such as the type of cancer cell and the
microenvironment in which it resides. JNK can positively regulate
autophagy, which can promote the survival of cancer cells under
conditions of nutrient deficiency or stress, counteracting
apoptosis. On the other hand, under conditions of sustained
activation, JNK can activate proapoptotic proteins such as Bax and
Bak, leading to cell apoptosis (72,73).
In melanoma cells treated with flubendazole, the
activity of JNK gradually increased during the 24 to 48 h treatment
period, peaking at 48 h in most cell types. The activation of JNK
can lead to the phosphorylation of antiapoptotic proteins such as
BCL-2, BCL-xL and MCL1 apoptosis regulator, which, once
phosphorylated, undergo degradation or loss of function, ultimately
promoting apoptosis. When melanoma cells were pretreated with the
JNK-specific inhibitor SP600125 followed by the addition of
flubendazole, the rate of apoptosis significantly decreased,
confirming the important role of JNK in the apoptosis induced by
flubendazole (31).
Autophagy-related protein 4B
(Atg4B)
Atg4B is a cysteine protease that is responsible for
cleaving the C-terminal amino acids of Atg8 family proteins,
including LC3 and gamma-aminobutyric acid A receptor-associated
protein, thereby regulating the elongation and closure of
autophagosomes. Additionally, Atg4B removes lipids from the Atg8
proteins that have fulfilled their function, thus promoting the
maturation of autophagosomes, which is crucial for the proper
formation and function of autophagosomes (74,75).
In TNBC, molecular docking and dynamics simulations
suggested that flubendazole could form a stable complex with Atg4B.
The binding of flubendazole to Atg4B leads to alterations in the
activity of Atg4B, subsequently affecting the autophagic process.
Furthermore, treatment with flubendazole significantly increased
the production of reactive oxygen species (ROS) in MDA-MB-231
cells. ROS have been reported to regulate the substrate preference
and activity of Atg4B, and flubendazole may facilitate autophagy by
modulating ROS levels (76).
Hedgehog (Hh) signaling
The Hh signaling pathway is a conserved signaling
pathway that serves a crucial role in embryonic development, the
regulation of cell proliferation, differentiation, tissue
regeneration and stem cell maintenance. This pathway is composed of
core components, such as secreted proteins of the Hh family,
including Sonic hedgehog (Shh), its receptors [Patched (Ptch) and
Smoothened (Smo)] and the Gli family of transcription factors
(Gli1, Gli2 and Gli3). In the absence of Hh signaling, Ptch
inhibits the activity of Smo, thereby suppressing downstream
signaling. When the Hh ligand binds to Ptch, the inhibition of Smo
is lifted, activating downstream signals that ultimately regulate
the expression of target genes through Gli transcription factors.
Overactivation of the Hh signaling pathway can promote the
proliferation, survival and dedifferentiation of cancer cells while
also inhibiting apoptosis. Moreover, Hh signaling is associated
with interactions with the cancer microenvironment, affecting
cancer angiogenesis, invasion and metastasis (77,78).
Proprotein convertase subtilisin/kexin type 9
(PCSK9) is an enzyme involved in cholesterol metabolism that is
primarily responsible for regulating the levels of low-density
lipoprotein cholesterol in the blood. High expression levels of
PCSK9 in HCC tissues is correlated with a poor prognosis. PCSK9 can
promote the carcinogenicity and metastasis of HCC by activating the
Hh signaling pathway, particularly through the upregulation of Smo
and Gli1 (35). Jin et al
(35) revealed that treatment with
flubendazole reduces the expression of the PCSK9 protein in HCC,
increasing the uptake of cholesterol by HCC cells. This leads to
the accumulation of cholesterol in the cell membrane, thereby
inhibiting the activation of Smo and the activation of the Hh
signaling pathway, resulting in the downregulation of Smo and Gli1
proteins. Furthermore, the combination of flubendazole with another
multikinase inhibitor, lenvatinib, was more effective in treating
HCC compared with lenvatinib alone, suggesting that flubendazole
may affect the Hh signaling pathway by inhibiting PCSK9 and
producing a synergistic effect with existing anticancer drugs
(35).
Conclusions and future perspectives
The present review summarized that flubendazole can
exert anticancer effects by inhibiting microtubule structure and
function, inducing G2/M cell cycle arrest, inducing autophagy and
apoptosis, suppressing cancer stem cell properties, inducing
ferroptosis and inhibiting angiogenesis. Its anticancer mechanisms
involve molecules and pathways such as the STAT3, NF-κB, AKT,
EVA1A, p53, PD-1, JNK, Atg4B and Hh signaling pathways.
Further investigation is needed into the anticancer
properties of flubendazole. First, the identification of additional
cancer types sensitive to flubendazole is essential. Michaelis
et al (14) screened a
number of cancer cell lines with flubendazole and reported that the
IC90 of flubendazole for 26 types of cancers was ≤5 µM.
Future studies should consider the impact of flubendazole on a
range of understudied cancers, including but not limited to Ewing
sarcoma, head and neck cancer and medulloblastoma, cervical,
ovarian, gastric, urothelial, renal cell and thyroid cancers
(14). This research will enrich
the current understanding of the anticancer effects of flubendazole
and may reveal novel mechanisms of action. Second, enhancing the
sophistication of experimental models is crucial for bolstering the
reliability of findings. Notably, the majority of studies reported
to date are based on cell line research, with a subset utilizing
xenograft models. A limited number, specifically three studies,
have employed primary patient cells for validation in patients with
neuroblastoma (14), melanoma
(31) and colorectal cancer
(39). Therefore, future studies
could benefit from employing patient-derived xenograft models
(79), organoids (80,81)
and conditionally reprogrammed cells (82,83).
Third, the discovery of synergistic drug combinations with
established chemotherapeutics, such as vinblastine, doxorubicin,
paclitaxel, 5-FU and lenvatinib, in addition to the pleiotropic
effects of flubendazole, is a compelling avenue for maximizing
therapeutic efficacy and overcoming drug resistance (13,23,30,32,35,39,61,66).
Fourth, due to its poor aqueous solubility and low oral absorption
of flubendazole (1,5,84),
addressing bioavailability challenges is essential. Innovative
strategies such as high-oil-content nanoemulsions (85), novel nanocrystal formulation via
microfluidization (86) and
biphasic dissolution combined with the cylinder method have
previously shown promise (87).
Finally, the development of novel administration modalities, such
as pulmonary delivery and intravesical chemotherapy, presents
opportunities for the use of flubendazole in a targeted and
effective manner. Compared with intravenous administration, the
pulmonary route offers unique advantages for drug delivery due to
the large surface area of the alveoli, thin epithelial barriers and
rich blood supply, allowing for higher drug concentrations in the
lungs (88). The exploration of
inhalable formulations of flubendazole is an area of active
research (89). Intravesical
chemotherapy is a well-established treatment for superficial
bladder urothelial carcinoma (90),
and the application of flubendazole in this context could
facilitate direct interaction with urothelial cancer cells. These
avenues of research hold promise for advancing the therapeutic
utility of flubendazole in cancer treatment.
Acknowledgements
Not applicable.
Funding
This work was financially supported by the Jingzhou Science and
Technology Bureau (grant no. 2023HC19).
Availability of data and materials
Not applicable.
Authors' contributions
XX and ZZ were responsible for searching the
literature and writing the original manuscript draft. HP and SC
reviewed and edited the manuscript. SC and HP conceptualized the
manuscript and approved the final version to be published. Data
authentication is not applicable. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Čáňová K, Rozkydalová L and Rudolf E:
Anthelmintic flubendazole and its potential use in anticancer
therapy. Acta Medica (Hradec Kralove). 60:5–11. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Friedman PA and Platzer EG: Interaction of
anthelmintic benzimidazoles with Ascaris suum embryonic tubulin.
Biochim Biophys Acta. 630:271–278. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lacey E: Mode of action of benzimidazoles.
Parasitol Today. 6:112–115. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Venugopal S, Kaur B, Verma A, Wadhwa P,
Magan M, Hudda S and Kakoty V: Recent advances of benzimidazole as
anticancer agents. Chem Biol Drug Des. 102:357–376. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Michiels M, Hendriks R, Heykants J and van
den Bossche H: The pharmacokinetics of mebendazole and flubendazole
in animals and man. Arch Int Pharmacodyn Ther. 256:180–191.
1982.PubMed/NCBI
|
|
6
|
Bossche HV, Thienpont D and Janssens PG:
Chemotherapy of Gastrointestinal Helminths. Springer; Berlin: pp.
p7191985
|
|
7
|
Nath J, Paul R, Ghosh SK, Paul J, Singha B
and Debnath N: Drug repurposing and relabeling for cancer therapy:
Emerging benzimidazole antihelminthics with potent anticancer
effects. Life Sci. 258:1181892020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tweats DJ, Johnson GE, Scandale I,
Whitwell J and Evans DB: Genotoxicity of flubendazole and its
metabolites in vitro and the impact of a new formulation on in vivo
aneugenicity. Mutagenesis. 31:309–321. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim J, Bang J, Ryu B, Kim CY and Park JH:
Flubendazole exposure disrupts neural development and function of
zebrafish embryos (Danio rerio). Sci Total Environ. 898:1653762023.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Moreno L, Alvarez L, Mottier L, Virkel G,
Bruni SS and Lanusse C: Integrated pharmacological assessment of
flubendazole potential for use in sheep: Disposition kinetics,
liver metabolism and parasite diffusion ability. J Vet Pharmacol
Ther. 27:299–308. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Krízová V, Nobilis M, Prusková L, Chládek
J, Szotáková B, Cvilink V, Skálová L and Lamka J: Pharmacokinetics
of flubendazole and its metabolites in lambs and adult sheep (Ovis
aries). J Vet Pharmacol Ther. 32:606–612. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen C, Ding Y, Liu H, Sun M, Wang H and
Wu D: Flubendazole plays an important anti-tumor role in different
types of cancers. Int J Mol Sci. 23:5192022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Spagnuolo PA, Hu J, Hurren R, Wang X,
Gronda M, Sukhai MA, Di Meo A, Boss J, Ashali I, Beheshti Zavareh
R, et al: The antihelmintic flubendazole inhibits microtubule
function through a mechanism distinct from Vinca alkaloids and
displays preclinical activity in leukemia and myeloma. Blood.
115:4824–4833. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Michaelis M, Agha B, Rothweiler F,
Löschmann N, Voges Y, Mittelbronn M, Starzetz T, Harter PN, Abhari
BA, Fulda S, et al: Identification of flubendazole as potential
anti-neuroblastoma compound in a large cell line screen. Sci Rep.
5:82022015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Son DS, Lee ES and Adunyah SE: The
antitumor potentials of benzimidazole anthelmintics as repurposing
drugs. Immune Netw. 20:e292020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schipper LJ, Zeverijn LJ, Garnett MJ and
Voest EE: Can Drug repurposing accelerate precision oncology?
Cancer Discov. 12:1634–1641. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hijazi MA, Gessner A and El-Najjar N:
Repurposing of chronically used drugs in cancer therapy: A chance
to grasp. Cancers (Basel). 15:31992023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kawczak P, Feszak I, Brzeziński P and
Bączek T: Structure-activity relationships and therapeutic
applications of retinoids in view of potential benefits from drug
repurposing process. Biomedicines. 12:10592024. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fletcher DA and Mullins RD: Cell mechanics
and the cytoskeleton. Nature. 463:485–492. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gudimchuk NB and McIntosh JR: Regulation
of microtubule dynamics, mechanics and function through the growing
tip. Nat Rev Mol Cell Biol. 22:777–795. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jordan MA and Wilson L: Microtubules as a
target for anticancer drugs. Nat Rev Cancer. 4:253–265. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kralova V, Hanušová V, Caltová K, Špaček
P, Hochmalová M, Skálová L and Rudolf E: Flubendazole and
mebendazole impair migration and epithelial to mesenchymal
transition in oral cell lines. Chem Biol Interact. 293:124–132.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hou ZJ, Luo X, Zhang W, Peng F, Cui B, Wu
SJ, Zheng FM, Xu J, Xu LZ, Long ZJ, et al: Flubendazole,
FDA-approved anthelmintic, targets breast cancer stem-like cells.
Oncotarget. 6:6326–6340. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Králová V, Hanušová V, Rudolf E, Čáňová K
and Skálová L: Flubendazole induces mitotic catastrophe and
senescence in colon cancer cells in vitro. J Pharm Pharmacol.
68:208–218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Čáňová K, Rozkydalová L, Vokurková D and
Rudolf E: Flubendazole induces mitotic catastrophe and apoptosis in
melanoma cells. Toxicol In Vitro. 46:313–322. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Oh E, Kim YJ, An H, Sung D, Cho TM,
Farrand L, Jang S, Seo JH and Kim JY: Flubendazole elicits
anti-metastatic effects in triple-negative breast cancer via STAT3
inhibition. Int J Cancer. 143:1978–1993. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jamasbi E, Hamelian M, Hossain MA and
Varmira K: The cell cycle, cancer development and therapy. Mol Biol
Rep. 49:10875–10883. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rieder CL and Cole R: Microtubule
disassembly delays the G2-M transition in vertebrates. Curr Biol.
10:1067–1070. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim YJ, Sung D, Oh E, Cho Y, Cho TM,
Farrand L, Seo JH and Kim JY: Flubendazole overcomes trastuzumab
resistance by targeting cancer stem-like properties and HER2
signaling in HER2-positive breast cancer. Cancer Lett. 412:118–130.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhou X, Zou L, Chen W, Yang T, Luo J, Wu
K, Shu F, Tan X, Yang Y, Cen S, et al: Flubendazole, FDA-approved
anthelmintic, elicits valid antitumor effects by targeting P53 and
promoting ferroptosis in castration-resistant prostate cancer.
Pharmacol Res. 164:1053052021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rudolf K and Rudolf E: An analysis of
mitotic catastrophe induced cell responses in melanoma cells
exposed to flubendazole. Toxicol In Vitro. 68:1049302020.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Králová V, Hanušová V, Staňková P,
Knoppová K, Čáňová K and Skálová L: Antiproliferative effect of
benzimidazole anthelmintics albendazole, ricobendazole, and
flubendazole in intestinal cancer cell lines. Anticancer Drugs.
24:911–919. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou X, Liu J, Zhang J, Wei Y and Li H:
Flubendazole inhibits glioma proliferation by G2/M cell cycle
arrest and pro-apoptosis. Cell Death Discov. 4:182018. View Article : Google Scholar
|
|
34
|
Ren LW, Li W, Zheng XJ, Liu JY, Yang YH,
Li S, Zhang S, Fu WQ, Xiao B, Wang JH and Du GH: Benzimidazoles
induce concurrent apoptosis and pyroptosis of human glioblastoma
cells via arresting cell cycle. Acta Pharmacol Sin. 43:194–208.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jin W, Yu J, Su Y, Lin H, Liu T, Chen J,
Ge C, Zhao F, Geng Q, Mao L, et al: Drug repurposing flubendazole
to suppress tumorigenicity via PCSK9-dependent inhibition and
potentiate lenvatinib therapy for hepatocellular carcinoma. Int J
Biol Sci. 19:2270–2288. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Glick D, Barth S and Macleod KF:
Autophagy: Cellular and molecular mechanisms. J Pathol. 221:3–12.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Debnath J, Gammoh N and Ryan KM: Autophagy
and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol.
24:560–575. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mizushima N, Noda T, Yoshimori T, Tanaka
Y, Ishii T, George MD, Klionsky DJ, Ohsumi M and Ohsumi Y: A
protein conjugation system essential for autophagy. Nature.
395:395–398. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lin S, Yang L, Yao Y, Xu L, Xiang Y, Zhao
H, Wang L, Zuo Z, Huang X and Zhao C: Flubendazole demonstrates
valid antitumor effects by inhibiting STAT3 and activating
autophagy. J Exp Clin Cancer Res. 38:2932019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xie X, Cai X, Tang Y, Jiang C, Zhou F,
Yang L, Liu Z, Wang L, Zhao H, Zhao C and Huang X: Flubendazole
elicits antitumor effects by inhibiting STAT3 and activating
autophagy in non-small cell lung cancer. Front Cell Dev Biol.
9:6806002021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhen Y, Zhao R, Wang M, Jiang X, Gao F, Fu
L, Zhang L and Zhou XL: Flubendazole elicits anti-cancer effects
via targeting EVA1A-modulated autophagy and apoptosis in
triple-negative breast cancer. Theranostics. 10:8080–8097. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhen Y, Yuan Z, Zhang J, Chen Y, Fu Y, Liu
Y, Fu L, Zhang L and Zhou XL: Flubendazole induces mitochondrial
dysfunction and DRP1-mediated mitophagy by targeting EVA1A in
breast cancer. Cell Death Dis. 13:3752022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Carneiro BA and El-Deiry WS: Targeting
apoptosis in cancer therapy. Nat Rev Clin Oncol. 17:395–417. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen C, Liu J, Lin X, Xiang A, Ye Q, Guo
J, Rui T, Xu J and Hu S: Crosstalk between cancer-associated
fibroblasts and regulated cell death in tumors: Insights into
apoptosis, autophagy, ferroptosis, and pyroptosis. Cell Death
Discov. 10:1892024. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Huang B, Yan X and Li Y: Cancer stem cell
for tumor therapy. Cancers (Basel). 13:48142021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Borlongan MC, Saha D and Wang H: Tumor
microenvironment: A niche for cancer stem cell immunotherapy. Stem
Cell Rev Rep. 20:3–24. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Conde I, Ribeiro AS and Paredes J: Breast
cancer stem cell membrane biomarkers: Therapy targeting and
clinical implications. Cells. 11:9342022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chen X, Kang R, Kroemer G and Tang D:
Broadening horizons: The role of ferroptosis in cancer. Nat Rev
Clin Oncol. 18:280–296. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wang S, Guo Q, Zhou L and Xia X:
Ferroptosis: A double-edged sword. Cell Death Discov. 10:2652024.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Li Y, Acharya G, Elahy M, Xin H and
Khachigian LM: The anthelmintic flubendazole blocks human melanoma
growth and metastasis and suppresses programmed cell death
protein-1 and myeloid-derived suppressor cell accumulation. Cancer
Lett. 459:268–276. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lee H, Jeong AJ and Ye SK: Highlighted
STAT3 as a potential drug target for cancer therapy. BMB Rep.
52:415–423. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Parri E, Kuusanmaki H, van Adrichem AJ,
Kaustio M and Wennerberg K: Identification of novel regulators of
STAT3 activity. PLoS One. 15:e02308192020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Carpenter RL and Lo HW: STAT3 target genes
relevant to human cancers. Cancers (Basel). 6:897–925. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tolomeo M and Cascio A: The multifaced
Role of STAT3 in cancer and its implication for anticancer therapy.
Int J Mol Sci. 22:6032021. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhang HF and Lai R: STAT3 in cancer-friend
or foe? Cancers (Basel). 6:1408–1440. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yu H, Lin L, Zhang Z, Zhang H and Hu H:
Targeting NF-κB pathway for the therapy of diseases: Mechanism and
clinical study. Signal Transduct Target Ther. 5:2092020. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Taniguchi K and Karin M: NF-κB,
inflammation, immunity and cancer: Coming of age. Nat Rev Immunol.
18:309–324. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mirzaei S, Saghari S, Bassiri F, Raesi R,
Zarrabi A, Hushmandi K, Sethi G and Tergaonkar V: NF-κB as a
regulator of cancer metastasis and therapy response: A focus on
epithelial-mesenchymal transition. J Cell Physiol. 237:2770–2795.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Hanušová V, Skálová L, Králová V and
Matoušková P: The effect of flubendazole on adhesion and migration
in SW480 and SW620 colon cancer cells. Anticancer Agents Med Chem.
18:837–846. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Tao J, Zhao H, Xie X, Luo M, Gao Z, Sun H
and Huang Z: The anthelmintic drug flubendazole induces cell
apoptosis and inhibits NF-κB signaling in esophageal squamous cell
carcinoma. Onco Targets Ther. 12:471–478. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Glaviano A, Foo ASC, Lam HY, Yap KCH,
Jacot W, Jones RH, Eng H, Nair MG, Makvandi P, Geoerger B, et al:
PI3K/AKT/mTOR signaling transduction pathway and targeted therapies
in cancer. Mol Cancer. 22:1382023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Sabbah DA, Hajjo R, Bardaweel SK and Zhong
HA: Targeting the PI3K/AKT signaling pathway in anticancer
research: A recent update on inhibitor design and clinical trials
(2020–2023). Expert Opin Ther Pat. 34:141–158. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Cocco S, Leone A, Roca MS, Lombardi R,
Piezzo M, Caputo R, Ciardiello C, Costantini S, Bruzzese F, Sisalli
MJ, et al: Inhibition of autophagy by chloroquine prevents
resistance to PI3K/AKT inhibitors and potentiates their antitumor
effect in combination with paclitaxel in triple negative breast
cancer models. J Transl Med. 20:2902022. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kierans SJ and Taylor CT: Regulation of
glycolysis by the hypoxia-inducible factor (HIF): Implications for
cellular physiology. J Physiol. 599:23–37. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhou Y, Liao M, Li Z, Ye J, Wu L, Mou Y,
Fu L and Zhen Y: Flubendazole enhances the inhibitory effect of
paclitaxel via HIF1α/PI3K/AKT signaling pathways in breast cancer.
Int J Mol Sci. 24:151212023. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Li Y, Wu B, Hossain MJ, Quagliata L,
O'Meara C, Wilkins MR, Corley S and Khachigian LM: Flubendazole
inhibits PD-1 and suppresses melanoma growth in immunocompetent
mice. J Transl Med. 21:4672023. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zhao H, Liu H, Yang Y and Wang H: The
emerging role of EVA1A in different types of cancers. Int J Mol
Sci. 23:66652022. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Liu Y, Su Z, Tavana O and Gu W:
Understanding the complexity of p53 in a new era of tumor
suppression. Cancer Cell. 42:946–967. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Tang Q, Chen Y, Li X, Long S, Shi Y, Yu Y,
Wu W, Han L and Wang S: The role of PD-1/PD-L1 and application of
immune-checkpoint inhibitors in human cancers. Front Immunol.
13:9644422022. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Yi M, Zheng X, Niu M, Zhu S, Ge H and Wu
K: Combination strategies with PD-1/PD-L1 blockade: Current
advances and future directions. Mol Cancer. 21:282022. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Wu Q, Wu W, Fu B, Shi L, Wang X and Kuca
K: JNK signaling in cancer cell survival. Med Res Rev.
39:2082–2104. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Abdelrahman KS, Hassan HA, Abdel-Aziz SA,
Marzouk AA, Narumi A, Konno H and Abdel-Aziz M: JNK signaling as a
target for anticancer therapy. Pharmacol Rep. 73:405–434. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Yang G, Li Y, Zhao Y, Ouyang L, Chen Y,
Liu B and Liu J: Targeting Atg4B for cancer therapy: Chemical
mediators. Eur J Med Chem. 209:1129172021. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Park NY, Jo DS and Cho DH:
Post-translational modifications of ATG4B in the regulation of
autophagy. Cells. 11:13302022. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhang L, Guo M, Li J, Zheng Y, Zhang S,
Xie T and Liu B: Systems biology-based discovery of a potential
Atg4B agonist (Flubendazole) that induces autophagy in breast
cancer. Mol Biosyst. 11:2860–2866. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Jing J, Wu Z, Wang J, Luo G, Lin H, Fan Y
and Zhou C: Hedgehog signaling in tissue homeostasis, cancers, and
targeted therapies. Signal Transduct Target Ther. 8:3152023.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Jiang J: Hedgehog signaling mechanism and
role in cancer. Semin Cancer Biol. 85:107–122. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Aslani S and Saad MI: Patient-derived
xenograft models in cancer research: Methodology, applications, and
future prospects. Methods Mol Biol. 2806:9–18. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Yan HHN, Chan AS, Lai FPL and Leung SY:
Organoid cultures for cancer modeling. Cell Stem Cell. 30:917–937.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Huang W, Xu Z, Li S, Zhou J and Zhao B:
Living biobanks of organoids: Valuable resource for translational
research. Biopreserv Biobank. Jul 3–2024.(Epub ahead of print).
doi: 10.1089/bio.2023.0142. View Article : Google Scholar
|
|
82
|
Cao J, Chan WC and Chow MSS: Use of
conditional reprogramming cell, patient derived xenograft and
organoid for drug screening for individualized prostate cancer
therapy: Current and future perspectives (review). Int J Oncol.
60:522022. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Liu W, Ju L, Cheng S, Wang G, Qian K, Liu
X, Xiao Y and Wang X: Conditional reprogramming: Modeling
urological cancer and translation to clinics. Clin Transl Med.
10:e952020. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
McKellar QA, Galbraith EA and Baxter P:
Oral absorption and bioavailability of fenbendazole in the dog and
the effect of concurrent ingestion of food. J Vet Pharmacol Ther.
16:189–198. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Yukuyama MN, Guimaraes LMF, Segovia RS,
Lameu C, de Araujo GLB, Löbenberg R, de Souza A, Henostroza MAB,
Folchini BR, Peroni CM, et al: Malignant wound-the influence of oil
components in flubendazole-loaded nanoemulsions in A549 lung cancer
xenograft-bearing mice. J Drug Deliv Sci Technol. 78:1039632022.
View Article : Google Scholar
|
|
86
|
de Souza Gonçalves D, Yukuyama MN, Saito
Miyagi MY, Vieira Silva TJ, Lameu C, Bou-Chacra NA and de Araujo
GLB: Revisiting flubendazole through nanocrystal technology:
Statistical design, characterization and its potential inhibitory
effect on xenografted lung tumor progression in mice. J Clust Sci.
34:261–272. 2023. View Article : Google Scholar
|
|
87
|
Yukuyama MN, Zuo J, Park C, Yousef M,
Henostroza MAB, de Araujo GLB, Bou-Chacra NA and Löbenberg R:
Biphasic dissolution combined with modified cylinder method-a new
promising method for dissolution test in drug-loaded nanoemulsions.
Int J Pharm. 632:1225542023. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Kosmidis C, Sapalidis K, Zarogoulidis P,
Sardeli C, Koulouris C, Giannakidis D, Pavlidis E, Katsaounis A,
Michalopoulos N, Mantalobas S, et al: Inhaled cisplatin for NSCLC:
Facts and results. Int J Mol Sci. 20:20052019. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Miyagi MYS, de Oliveira Faria R, de Souza
GB, Lameu C, Tagami T, Ozeki T, Bezzon VDN, Yukuyama MN, Bou-Chacra
NA and de Araujo GLB: Optimizing adjuvant inhaled chemotherapy:
Synergistic enhancement in paclitaxel cytotoxicity by flubendazole
nanocrystals in a cycle model approach. Int J Pharm.
644:1233242023. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Holzbeierlein JM, Bixler BR, Buckley DI,
Chang SS, Holmes R, James AC, Kirkby E, McKiernan JM and Schuckman
AK: Diagnosis and treatment of non-muscle invasive bladder cancer:
AUA/SUO guideline: 2024 Amendment. J Urol. 211:533–538. 2024.
View Article : Google Scholar : PubMed/NCBI
|