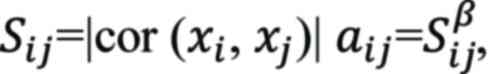Introduction
Pancreatic adenocarcinoma (PAAD) is a highly
aggressive malignancy characterized by a dismal prognosis and
significant economic burden on healthcare systems worldwide
(1,2). The disease is often diagnosed at
advanced stages, leading to limited treatment options and a
five-year survival rate of <10%. Current therapeutic strategies,
including surgical resection, chemotherapy and targeted therapies,
have shown limited efficacy, primarily due to the tumor's complex
microenvironment and the presence of resistance mechanisms
(2). Despite advancements in
understanding the molecular underpinnings of PAAD, there remains a
critical gap in identifying reliable prognostic biomarkers that can
stratify patients based on their disease status and guide
personalized treatment approaches (3). The complex tumor microenvironment and
significant heterogeneity among PAAD subtypes contribute to this
bleak outlook, making it imperative to identify molecular
signatures that can aid in both diagnosis and treatment
decision-making.
Recent advancements in machine learning have opened
new frontiers in cancer biology, allowing for the identification
and characterization of biomarkers that correlate with clinical
outcomes (4,5). One intriguing area of research is the
exploration of damage-associated molecular patterns (DAMPs), which
are endogenous signals released by stressed or damaged cells,
playing critical roles in inflammation and tumor progression
(6,7). DAMPs, including high-mobility group
(HMG) box 1, heat shock proteins and other nucleic acids, can
activate innate immune responses and modulate the tumor
microenvironment (8,9). Their contributions to cancer
pathogenesis and prognosis have sparked interest in establishing a
predictive score, termed DAMPscore, to classify cancer patients
based on their distinct molecular profiles (10).
The present study focuses on the expression of
DAMP-related genes in PAAD and their potential prognostic
implications. Previous research has established a link between
DAMPs and tumor progression (11),
yet the specific role of DAMP-related gene expression in
stratifying patients with PAAD remains underexplored. By employing
Consensus Clustering, three distinct patient clusters were
identified, and it was hypothesized that these clusters reflect
varying DAMP statuses, which may correlate with disease prognosis.
Notably, the present analysis revealed 141 differentially expressed
genes with significant prognostic value across multiple cohorts,
underscoring the relevance of DAMP-related gene expression in PAAD.
The identification of six pivotal genes through Least Absolute
Shrinkage and Selection Operator (LASSO) Cox regression further
highlighted the potential of the DAMPscore as a robust prognostic
tool, demonstrating superior performance compared to existing
models. Additionally, the functional exploration of PLEK2 suggested
its involvement in key signaling pathways associated with immune
response and tumor dynamics, thereby reinforcing the clinical
significance of DAMP-related gene expression in PAAD.
Collectively, these findings illuminate the
intricate relationship between DAMPs and PAAD, presenting a novel
avenue for enhancing prognostic assessments and therapeutic
strategies in this challenging malignancy.
Materials and methods
Public data acquisition and
processing
The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov) was used to retrieve
the TCGA-PAAD cohort. The datasets GSE85916, GSE71729 and GSE62452
were sourced from the Gene Expression Omnibus (GEO) database
(https://ncbi.nlm.nih.gov/gds). The
ICGC_AU cohort was collected from the International Cancer Genomics
Consortium (ICGC) data portal (https://dcc.icgc.org/). The E-MTAB-6134 cohort was
downloaded from the ArrayExpress database (https://www.ebi.ac.uk/arrayexpress/). The mRNA data
and proteomic data of PLEK2 were downloaded from the Clinical
Proteomic Tumor Analysis Consortium (CPTAC)
[https://hupo.org/Clinical-Proteome-Tumor-Analysis-Consortium-(CPTAC)].
Immunohistochemical images of PLEK2 was directly downloaded from
The Human Protein Atlas (HPA; http://www.proteinatlas.org/). All datasets were
processed according to the methods outlined in a previous study by
our group (12). In addition, the
three GEO datasets (GSE85916, GSE71729 and GSE62452) were
integrated into a Meta-cohort using the sva package in R (version
4.1.3) for further analyses. All datasets utilized in this study
were obtained from public repositories, thereby negating the need
for additional ethical approval.
Consensus clustering
Consensus clustering was performed utilizing the
‘ConsensusClusterPlus’ package in R (13). The analysis was executed with the
following parameters: The maximum number of clusters (k) (MaxK)=10;
the number of resampling iterations (reps)=1,000, the proportion of
samples randomly selected in each resampling iteration (pItem)=0.8,
the proportion of features (variables) retained in each resampling
iteration (pFeature)=1, the clustering algorithm applied
(clusterAlg)=‘pam’, the method for handling missing data during
correlation calculations (corUse)=‘complete.obs’ and seed=123,456.
The optimal number of clusters was determined based on the
following criteria: i) Cumulative distribution function (CDF) Plot:
The CDF plot, generated by the ConsensusClusterPlus package, was
analyzed to assess the proportion of variance explained by the
clustering solution for different values of k. The plot was
examined for an ‘elbow point’ or stabilization in the curve, which
indicates the point beyond which increasing the number of clusters
does not significantly improve the clustering solution. ii)
Consensus matrix: The consensus matrix, which measures the
frequency with which pairs of samples are clustered together across
multiple resampling iterations, was also carefully examined. A
clear and stable block-like structure in the consensus matrix was
sought to identify well-defined clusters.
Construction of DAMPscore in PAAD
Differentially expressed genes (DEGs) among the
subgroups identified through consensus clustering were extracted
using the ‘limma’ package in R. Following this, these DEGs were
subjected to univariate Cox regression analysis. Genes that
demonstrated a prognostic P<0.05 in both the TCGA-PAAD and
Meta-cohort were incorporated into a LASSO regression model applied
to the Meta-cohort. The genes selected were then analyzed using a
multivariate Cox regression model. The key genes and their
respective coefficients were utilized to calculate the risk score
for each patient using the following formula: Score=0.12012 × ADM +
0.05077 × HMGA2 + 0.10670 × EREG + 0.16538 × SLC16A3 + 0.06207 ×
LAMC2 + 0.27480 × IGF2BP2. The DAMPscore for patients in each
cohort was computed using the following formula:
DAMPscore=(score-Min)/abs(Max), in accordance with previous
research findings (12,14).
Weighted gene co-expression network
analysis (WGCNA)
WGCNA was performed using R software, as detailed in
previous studies by our group (15,16).
In summary, transcriptional expression data from each dataset were
utilized to construct a gene co-expression network, following the
exclusion of genes and samples with excessive missing values. The
correlation between nodes was assessed by creating an adjacency
matrix using the following formula:
where Sij represents the
co-expression similarity, indicating the Pearson correlation
coefficient between two distinct genes, i and j.
xi and xj denote the
transcriptional expression values for genes i and j, while
aij reflects the correlation strength between
these two genes. A scale-free R2 threshold of 0.9 was
established for selecting the appropriate soft-threshold β.
Subsequently, one-step network construction and module detection
methods were applied, with a minimum module size of 200 and a
mergeCutHeight set at 0.25 for module merging. Finally,
module-trait associations were evaluated to pinpoint modules that
were significantly related to the DAMPscore. In addition, the
definitions and calculations for module eigengenes, gene
significance and module significance followed the methodology
described in a prior study (17).
Enrichment analysis
Gene Set Enrichment Analysis (GSEA) was performed
for patients with pancreatic cancer using the ‘clusterProfiler’
package in R. The results of the GSEA were visualized with the
‘GseaVis’ package in R (18). The
analysis utilized the hallmark gene sets database (https://www.gsea-msigdb.org/gsea/msigdb/human/collections.jsp#H).
Cell culture
AsPC-1 cells were sourced from the Cell Bank of the
Chinese Academy of Sciences, whereas PANC-1 cells were obtained
from the American Type Culture Collection. Prior to
experimentation, both cell lines were verified to be free of
mycoplasma contamination. The cells were cultured in RPMI-1640
medium (Invitrogen; Thermo Fisher Scientific, Inc.) supplemented
with 10% fetal bovine serum (FBS; ExCell), and were incubated in a
humidified atmosphere with 5% CO2 at 37°C.
Design, synthesis and transfection of
small interfering RNA (siRNA)
The coding DNA sequences of the target genes were
obtained from the National Center for Biotechnology Information
(NCBI) (https://www.ncbi.nlm.nih.gov/) and
siRNAs were designed for each gene using the DSIR design website
(http://biodev.extra.cea.fr/DSIR/DSIR.php). The
synthesis was commissioned from Shanghai Jima Pharmaceutical
Technology Co., Ltd. (detailed information can be found in Table SI). Cells were seeded in 6-well
plates and transfected when they reached ~80% confluency. SiRNAs
and plasmids were transfected into the cells using the Lipo8000™
transfection reagent (cat. no. C0533; Biyuntian) according to the
manufacturer's instructions.
Cell Counting Kit-8 (CCK-8) assay
Cells were seeded in a 96-well plate, transfected
with lentivirus for 72 h and then subjected to continuous selection
with puromycin (5 µg/ml) for one week. After that, the cells were
expanded and subjected to the CCK-8 assay. A working solution was
prepared by mixing cell culture medium with CCK-8 reagent (cat. no.
C0038; Beyotime Institute of Biotechnology) at a 10:1 ratio at 37°C
in the dark for 1 h. After discarding the residual medium, 100 µl
of the working solution was added to each well and the cells were
incubated at 37°C for 1 h prior to measurement with a
multifunctional microplate reader (SynergyHTX; Gene Company
Ltd.).
Transwell and invasion assay
For the migration (Transwell) assay,
1×104 cells were seeded in 100 µl of RPMI-1640 medium in
the upper chamber of Transwell® Permeable Supports with
a polycarbonate membrane (8 mm pore size; Corning, Inc.). In the
lower chamber, 800 µl of RPMI-1640 medium supplemented with 30% FBS
was added. For the invasion assay, 1×105 cells were
seeded in 200 µl RPMI-1640 medium on a Matrigel® layer
in the upper chamber of Corning® BioCoat™
Matrigel® Invasion Chambers (8 mm pore size; Corning,
Inc.), with 800 µl RPMI-1640 medium containing 20% FBS added to the
lower chamber. In both assays, cells were incubated at 37°C with 5%
CO2 for 24 h, followed by fixation in 4%
paraformaldehyde at 25°C for 20 min and staining with Giemsa Stain
solution (product no. 32884; Sigma-Aldrich; Merck KGaA) at 25°C for
20 min. The cells that had transgressed to the bottom of the
membrane were visualized under a light microscope and quantified by
counting the number of cells in seven randomly selected fields at
200-fold magnification.
RNA extraction and reverse
transcription-quantitative (RT-q)PCR
Total RNA was extracted using TRIzol and
subsequently quantified with a Nanodrop 1000 spectrophotometer
(Thermo Fisher Scientific, Inc.). cDNA synthesis was performed
according to the manufacturer's instructions (cat. no. A5003;
GoScript™ Reverse Transcriptase from Promega Corp.). RT-qPCR was
conducted as previously described in earlier studies by our group
(19,20). The primer sequences used in the
experiments are listed in Table
SI. Results were normalized to the housekeeping gene
beta-actin, which remains stable across all samples.
Statistical analysis
Data analysis and visualization were performed using
various R packages (version 4.1.3), including ‘tidyverse’,
‘ggplot2’, ‘ggrepel’, ‘survival’, ‘survminer’, ‘survivalROC’,
‘timeROC’, ‘rms’, ‘msigdbr’, ‘scales’, ‘dplyr’, ‘ggalluvial’,
‘VennDiagram’, ‘patchwork’, ‘org.Hs.eg.db’, ‘CompareC’,
‘enrichplot’, ‘glmnet’, ‘aplot’, ‘corrplot’ and ‘pec’. The Pearson
correlation method was utilized for correlation analyses, while
prognosis assessment was conducted using the Kaplan-Meier method
with the log-rank test. ROC curve analysis was performed by using
the timeROC package in R (version 4.1.3). Comparison between two
groups or among three groups was conducted by R software using the
Wilcox rank-sum test or ANOVA. P<0.05 was considered to indicate
statistical significance.
Results
Unsupervised clustering of
DAMP-related genes in PAAD
Drawing upon the expression of 40 DAMP-related genes
(Table SII) (21,22),
Consensus Clustering was employed to stratify patients with PAAD
within the TCGA-PAAD and Meta-cohort datasets. The optimal
parameter for further analysis was determined to be k=3, based on
the consensus matrix evaluated from k=2 to 10 (Fig. 1A, B, D and E; Fig. S1A and B). In the TCGA-PAAD cohort,
176 patients with PAAD were categorized into three clusters: C1, C2
and C3, with the C3 cluster exhibiting the shortest median overall
survival (OS) (Fig. 1C). A similar
trend was observed in the Meta-cohort, where significant OS
differences were noted among the three clusters, with C3 displaying
the worst prognosis (Fig. 1F).
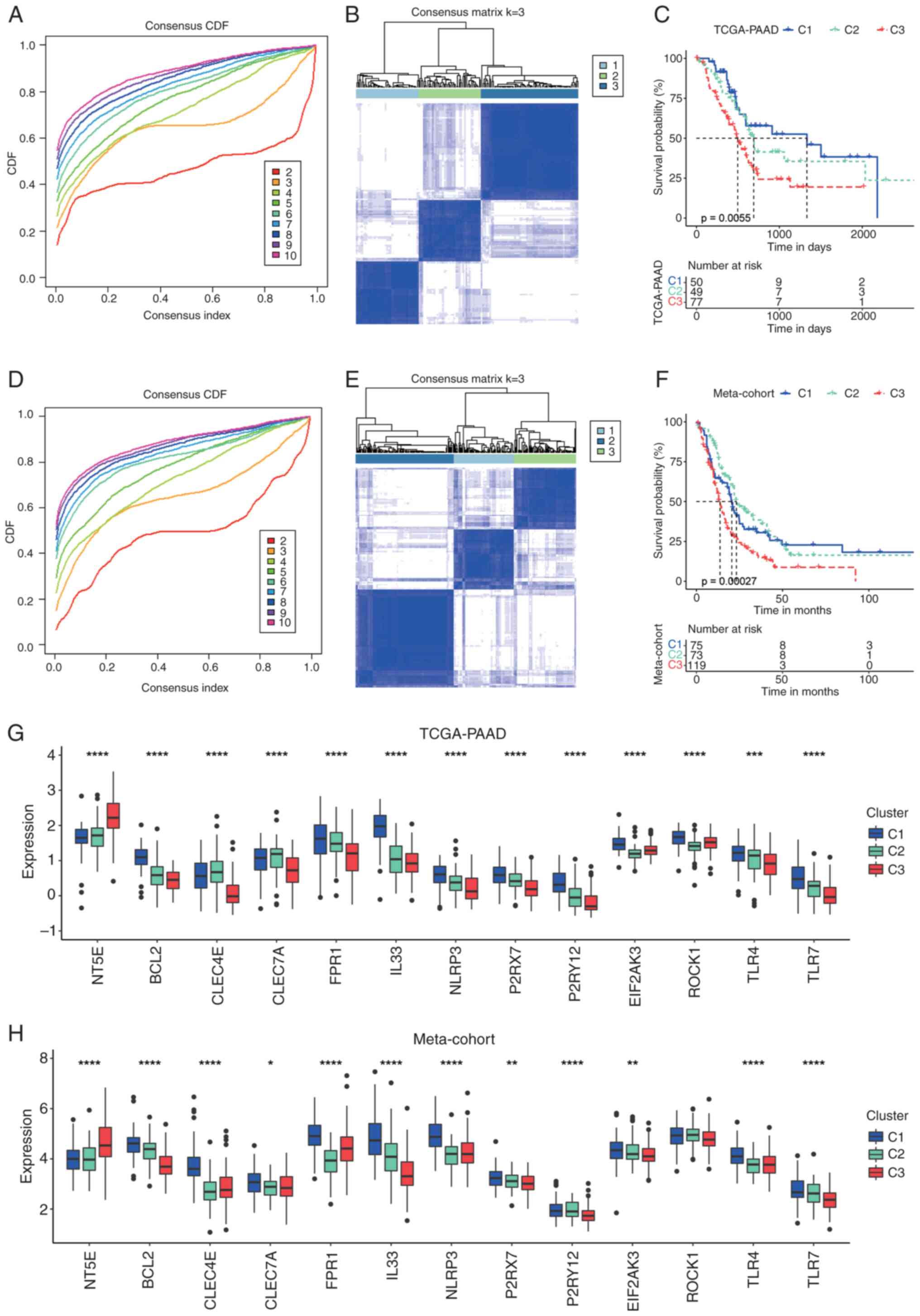 | Figure 1.Consensus clustering of patients with
PAAD based on DAMP-related genes. (A) The CDF curve of consensus
clustering for k=2 to 10 in the TCGA-PAAD cohort. (B) Heatmap
depicting consensus clustering solution (k=3) for DAMP-related
genes in patients with PAAD in the TCGA-PAAD cohort. (C)
Kaplan-Meier curves of OS in the C1, C2 and C3 clusters in the
TCGA-PAAD cohort. (D) CDF curve of consensus clustering for k=2 to
10 in the Meta-cohort. (E) Heatmap depicting consensus clustering
solution (k=3) for DAMP-related genes in patients with PAAD in the
Meta-cohort. (F) Kaplan-Meier curves of OS in the C1, C2 and C3
clusters in the Meta-cohort. (G) Transcriptional expression of
DAMP-related genes in the C1, C2 and C3 clusters in the TCGA-PAAD
cohort. (H) Transcriptional expression of DAMP-related genes in the
C1, C2 and C3 clusters in the Meta-cohort. *P<0.05; **P<0.01;
***P<0.001; ****P<0.0001. ns, no significance; TCGA, The
Cancer Genome Atlas; PAAD, pancreatic adenocarcinoma; CDF,
cumulative distribution function; DAMP, Damage-Associated Molecular
Pattern; OS, overall survival. |
Analysis of the transcriptional expression of the
majority of immunogenic cell death (ICD)-related genes in the
TCGA-PAAD cohort revealed that the C3 cluster was characterized by
notably high expression of NT5E, which is associated with impaired
ICD (21), alongside low expression
of several genes that promote ICD (21,22),
such as TLR4, P2RX7 and FPR1 (Fig.
1G). By contrast, the C1 cluster exhibited the highest
expression levels of most ICD-facilitating genes, including IL33,
FPR1 and TLR7. A comparable expression pattern for these genes was
also observed among the three clusters in the Meta-cohort (Fig. 1H). Given the observed differences in
prognosis and the expression profiles of ICD-related genes across
these three clusters, it may be hypothesized that the C1 cluster
reflects a pro-DAMP status and the C3 cluster an anti-DAMP status,
while the C2 cluster occupies an intermediate position between the
two.
Construction of DAMPscore in PAAD
To further elucidate the key factors influencing the
release of DAMP-related molecules in PAAD, differential gene
analysis was we conducted on the aforementioned C1 and C3 clusters.
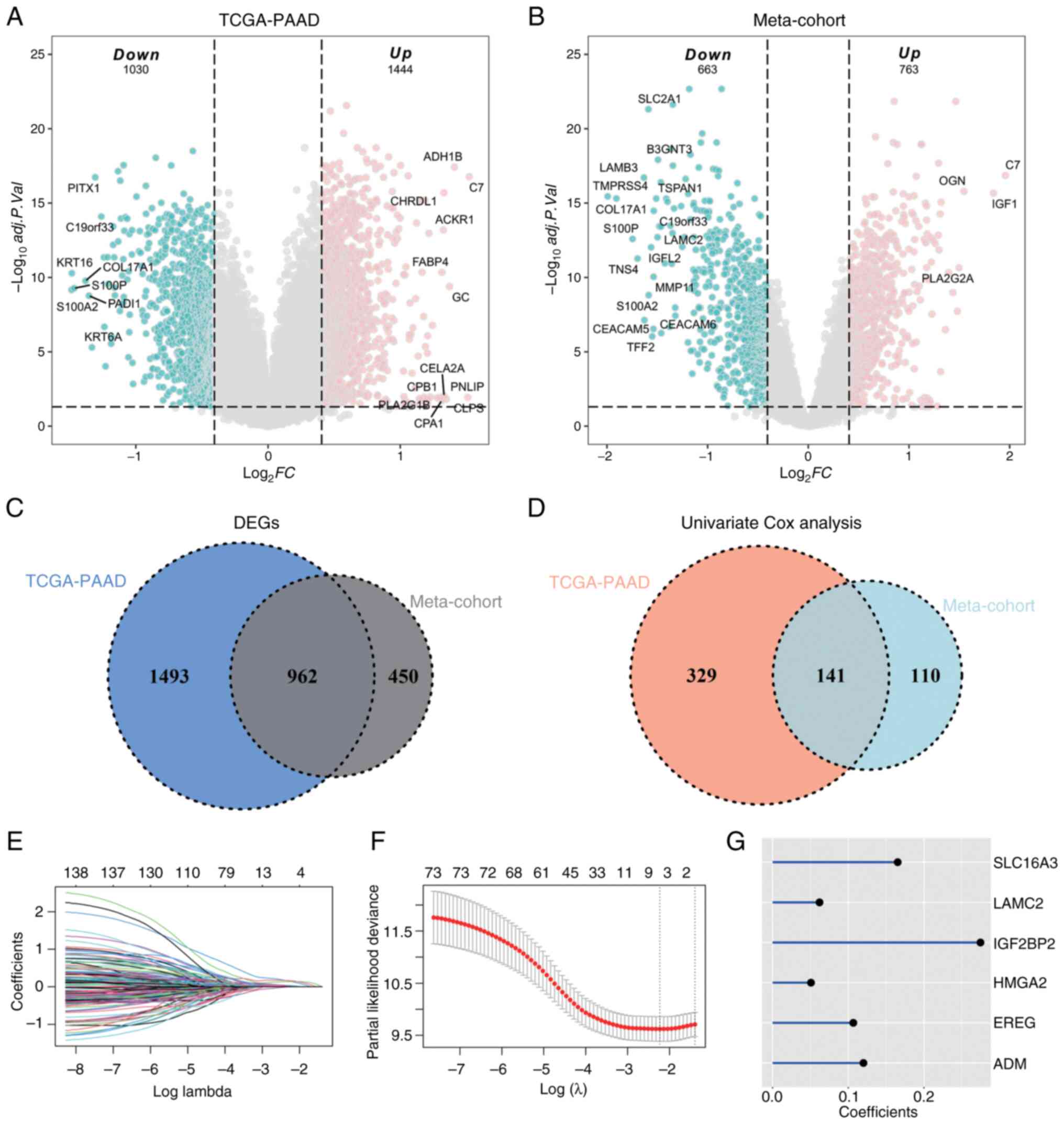
As illustrated in Fig. 2A, a total
of 1,444 upregulated and 1,030 downregulated genes from the
TCGA-PAAD cohort fulfilled the criteria of an absolute fold change
of ≥1.5 and an adjusted P<0.05. Similarly, in the Meta-cohort,
763 upregulated genes and 663 downregulated genes met the same
threshold (Fig. 2B). Among the
differential genes identified from both cohorts, 962 common DEGs
were found (Table SIII; Fig. 2C). These common DEGs were subjected
to univariate Cox analyses within both the TCGA-PAAD dataset and
the Meta-cohort, with 141 genes demonstrating significant
prognostic significance (P<0.05) in both datasets (Fig. 2D). To identify the pivotal genes
that differentiate between the C1 and C3 clusters, LASSO Cox
regression analyses were performed (Fig. 2E and F). Following the determination
of the optimal value of λ, six genes were selected for further
multivariate Cox regression analysis, which allowed for the
calculation of their respective coefficients (Fig. 2G). The six identified genes included
(SLC16A3, LAMC2, IGF2BP2, HMGA2, EREG and ADM). Utilizing the
transcriptional expressions and corresponding coefficients of these
nine genes, the DAMPscore was derived for each sample.
Prognostic value of DAMPscore in
PAAD
As depicted in Fig.
3A, within the training cohort (Meta-cohort), patients with
PAAD presenting a high DAMPscore, based on the median DAMPscore
threshold established in the cohort, exhibited markedly shorter
median OS compared to those with a low DAMPscore (P<0.0001).
Similarly, in the three independent GEO datasets (GSE85916,
GSE71729 and GSE62452) that were not integrated, using the median
DAMPscore as a cutoff, patients with a higher DAMPscore
demonstrated progressively worse prognoses (Fig. 3B-D). Furthermore, the prognostic
value of the DAMPscore was validated across three external datasets
(E-MTAB-6134, ICGC-AU and TCGA-PAAD). As illustrated in Fig. 3E-G, applying the median DAMPscore as
a cutoff across all cohorts revealed that patients with a lower
DAMPscore in these three cohorts experienced significantly
prolonged median OS.
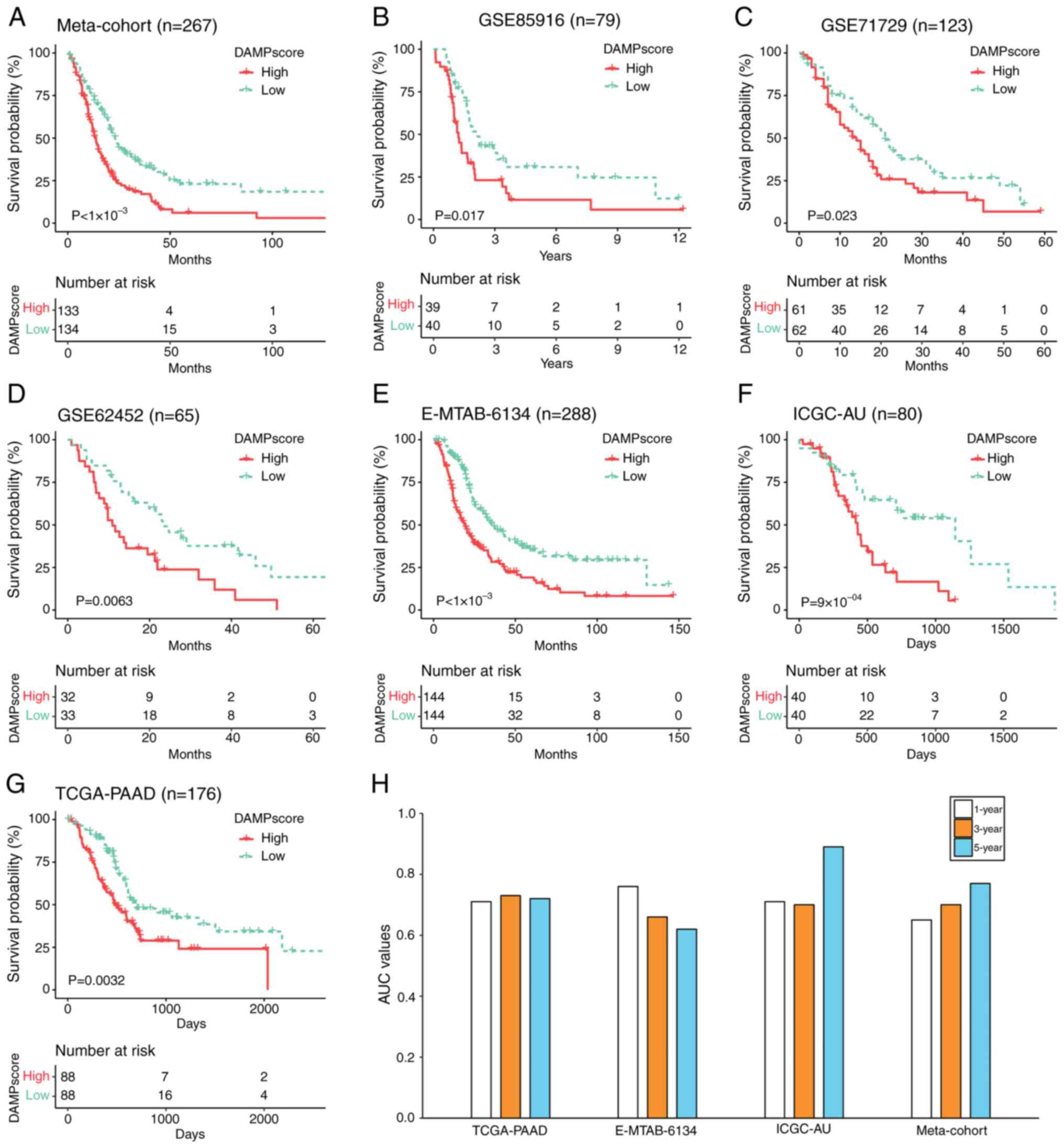 | Figure 3.Prognostic evaluation of the
DAMPscore in PAAD. (A-G) Kaplan-Meier curves of overall survival in
patients with PAAD with high and low DAMPscore in datasets (A) from
the Meta-cohort, (B) GSE85916, (C) GSE71729, (D) GSE62452, (E)
E-MTAB-6134, (F) ICGC-AU and (G) TCGA-PAAD. (H) The 1-, 3- and
5-year AUCs of DAMPscore in the TCGA-PAAD, E-MTAB-6134, ICGC-AU and
Meta-cohort. DAMP, Damage-Associated Molecular Pattern; TCGA, The
Cancer Genome Atlas; PAAD, pancreatic adenocarcinoma; AUC, area
under the receiver operating characteristic curve. |
To further evaluate the prognostic predictive
capability of the DAMPscore in patients with PAAD at the baseline,
a ROC curve analysis was performed and the corresponding area under
the curve (AUC) values for the 1-, 3- and 5-year survival rates
were calculated. In the Meta-cohort, the AUC values for the 1-, 3-
and 5-year DAMPscore were 0.65, 0.70 and 0.77, respectively. In the
TCGA-PAAD cohort, the AUC values for the same time intervals were
0.71, 0.73 and 0.72. For the E-MTAB-6134 cohort, the AUC values
were 0.76, 0.66 and 0.62, while in the ICGC-AU cohort, they were
0.71, 0.70 and 0.89 (Fig. 3H).
These findings collectively suggest that DAMPscore demonstrates a
substantial prognostic performance in PAAD.
Comparison of DAMPscore and other
prognostic models
PAAD exhibits subtype-specific prognostic
heterogeneity. Bailey et al (23) established four molecular subtypes:
i) Squamous (TP53/KDM6A mutations, TP63ΔN activation, endodermal
gene hypermethylation); ii) progenitor (FOXA2/3-PDX1-MNX1
signatures); iii) immunogenic (immunosuppressive pathway
enrichment); and iv) ADEX (KRAS-driven exocrine/endocrine
differentiation via NR5A2/RBPJL and NEUROD1/NKX2-2). Crucially, the
squamous subtype independently predicts poor survival (23). While the DAMPscore distribution
showed non-significant inter-subtype variation (P=0.274; Fig. S1C), its gradient pattern suggests a
complementary value to molecular subtyping for precision oncology
applications. To further assess the prognostic utility of the
DAMPscore in patients with PAAD, the concordance index (C-index)
for the DAMPscore in comparison to other clinical characteristics,
including T stage, N stage and age, was calculated. As depicted in
Fig. 4A, the DAMPscore achieved the
highest C-index among the evaluated metrics. In addition, 25
previously published mRNA-related prognostic signatures were
included for comparative analysis (Table SIV). These signatures are linked to
various factors such as gemcitabine resistance, cancer-associated
fibroblasts, DNA and RNA methylation, pyroptosis, immune cell
infiltration, mitophagy, invasion and other biological processes
(24–29). Beyond its moderate C-index in the
TCGA-PAAD cohort (Fig. 4B), the
DAMPscore exhibited superior performance in the E-MTAB-6134
(Fig. 4C), GSE62452 (Fig. 4D), GSE85916 (Fig. 4E) and Meta-cohort (Fig. 4F). Furthermore, within the ICGC-AU
and GSE71729 cohorts, the DAMPscore's C-index ranked third among
all prognostic models assessed (Fig. 4G
and H).
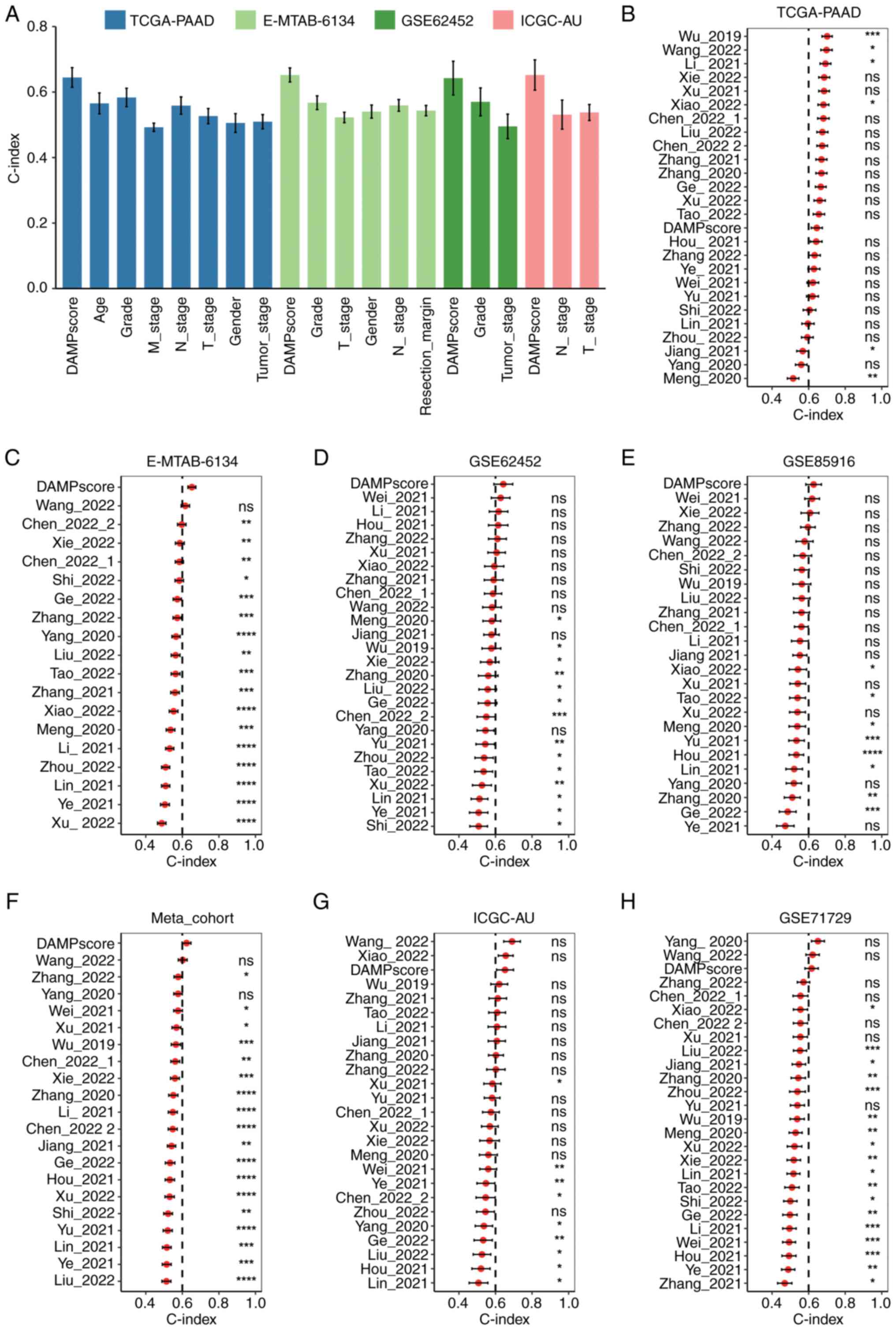 | Figure 4.Comparison of DAMPscore and other
factors in prognostic prediction. (A) The C-index (95% confidence
interval) of DAMPscore and clinical features in the TCGA-PAAD,
E-MTAB-6134, ICGC-AU and GSE62452 cohorts. (B-H) The C-index (95%
confidence interval) of DAMPscore and 25 previously published mRNA
signatures in the (B) TCGA-PAAD, (C) E-MTAB-6134, (D) GSE62452, (E)
GSE85916, (F) Meta-cohort, (G) ICGC-AU and (H) GSE71729.
*P<0.05; **P<0.01; ***P<0.001; ****P<0.0001. ns, no
significance; DAMP, Damage-Associated Molecular Pattern; TCGA, The
Cancer Genome Atlas; PAAD, pancreatic adenocarcinoma. |
Identification of DAMPscore-related
hub genes in PAAD
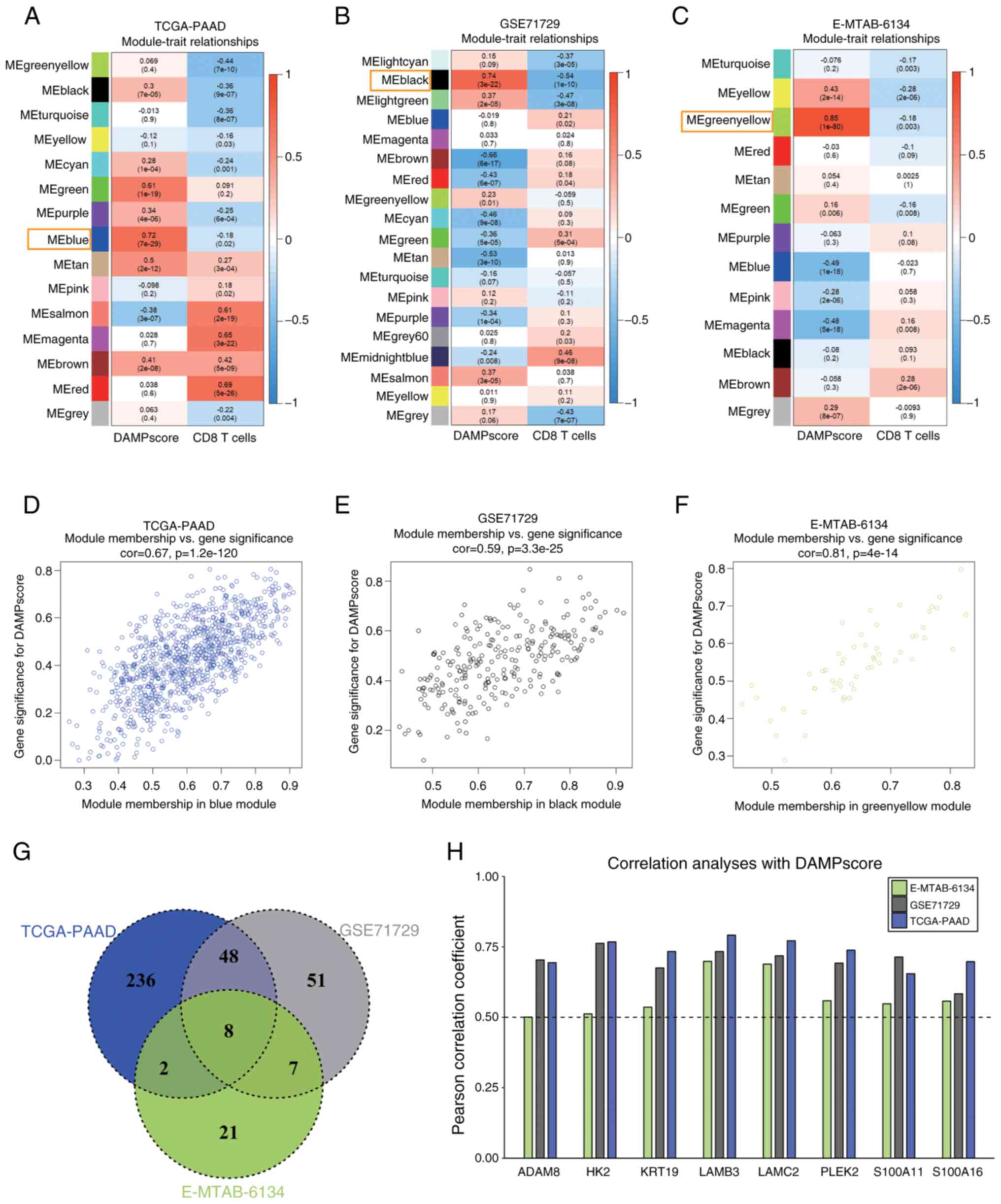
To identify hub genes potentially involved in DAMPs
release and immune response induction, WGCNA was employed to select
gene modules that exhibit a strong correlation with both the
DAMPscore and CD8 T-cell infiltration across the TCGA-PAAD
(Fig. 5A), GSE71729 (Fig. 5B) and E-MTAB-6134 (Fig. 5C) cohorts. The analyses revealed
that the blue module in TCGA-PAAD (r=0.72, P=7×10−29),
the black module in GSE71729 (r=0.74, P=3×10−22) and the
green-yellow module in E-MTAB-6134 (r=0.85, P=1×10−80)
were significantly positively correlated with DAMPscore. In
addition, these modules displayed a notable negative correlation
with CD8 T-cell infiltration (Fig.
5A-C).
Furthermore, the gene module membership correlation
(cor.geneModuleMembership) and gene trait significance correlation
(cor.geneTraitSignificance) were computed for the blue module in
TCGA-PAAD (Fig. 5D), the black
module in GSE71729 (Fig. 5E) and
the green-yellow module in E-MTAB-6134 (Fig. 5F) in order to screen for hub genes.
The thresholds were set to |cor.geneTraitSignificance| >0.5 and
|cor.geneModuleMembership| >0.5. This approach identified a
total of 394 genes in the blue module of TCGA-PAAD, 114 genes in
the black module of GSE71729 and 38 genes in the green-yellow
module of E-MTAB-6134 that met the established criteria (Fig. 5G).
A total of eight hub genes were subsequently
identified, including (ADAM6, HK2, KRT19, LAMB3, LAMC2, PLEK2,
S100A11 and S100A16), all of which exhibited a strong positive
correlation with the DAMPscore across the three datasets (Fig. 5G and H).
Exploration of the function of PLEK2
in PAAD
Among the eight core genes, LAMB3, LAMC2 and PLEK2
showed the highest correlations with the DAMP score (Fig. 5H). While the roles of LAMB3 and
LAMC2 in pancreatic cancer have been extensively studied (30–32),
research regarding the functional involvement of PLEK2 in this
context remains limited.
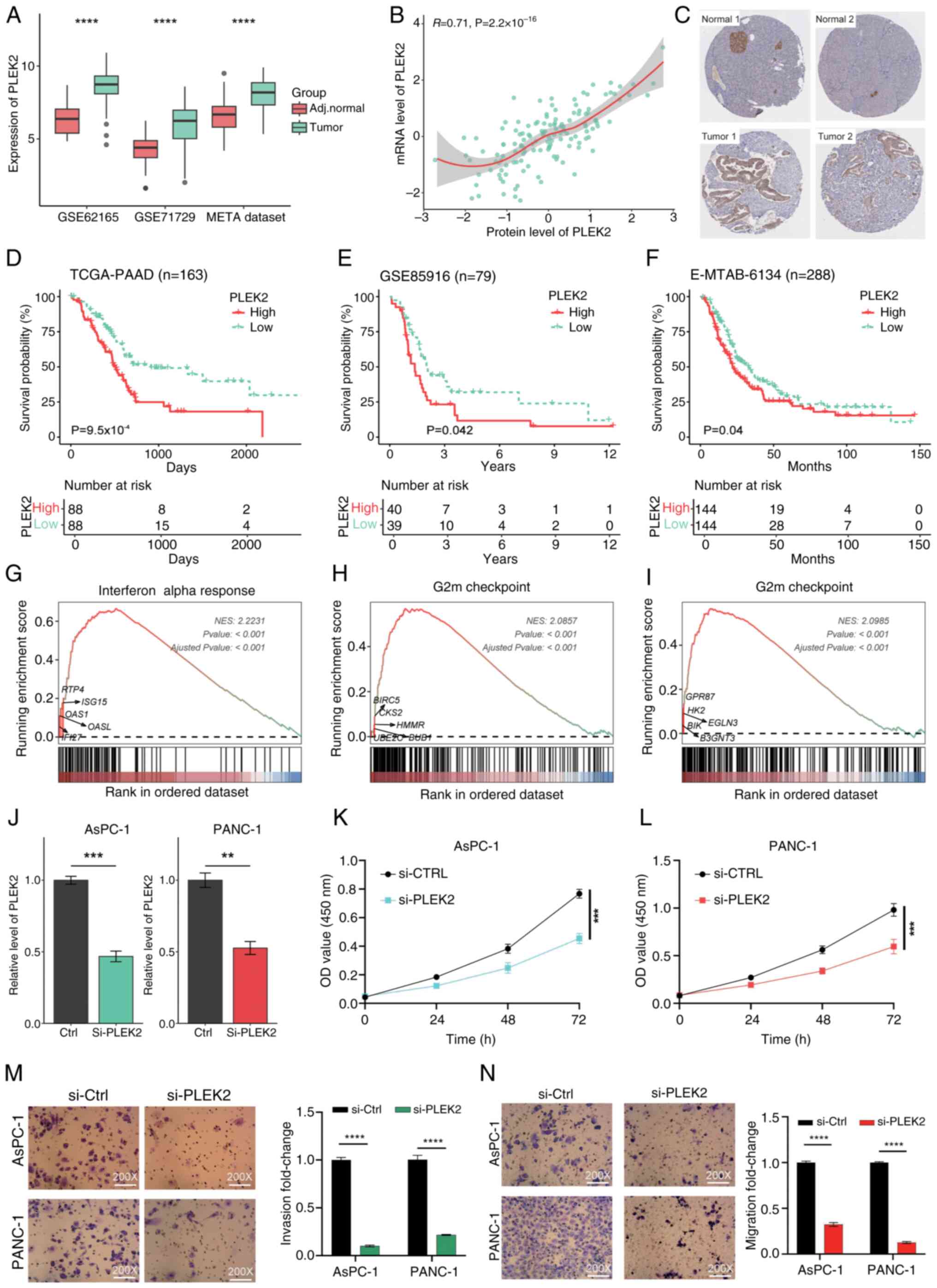
Initially, the expression levels of PLEK2 in PAAD
were assessed. Transcriptional analyses across three independent
datasets indicated that PLEK2 expression was significantly higher
in tumor tissues compared to adjacent normal tissues (Fig. 6A). Furthermore, by integrating the
mRNA data and proteomic data from the CPTAC, a strong positive
correlation between PLEK2 mRNA levels and protein expression in
pancreatic cancer was found (R=0.71, P<2.2×10−16;
Fig. 6B).
Immunohistochemical analysis from the HPA revealed
that PLEK2 is predominantly expressed in islet tissues of normal
pancreatic tissue, while it is nearly undetectable in exocrine
gland tissues (Fig. 6C). By
contrast, PLEK2 expression was significantly increased in
pancreatic cancer tissues (Fig.
6C). Using the median expression level of PLEK2 as a cutoff, it
was observed that higher levels of PLEK2 expression were associated
with poorer prognosis across three independent PAAD datasets
(Fig. 6D-F).
Enrichment analysis indicated that tissues with
elevated PLEK2 expression were primarily enriched in hallmark
signaling pathways, including interferon alpha response (Fig. 6G), G2M checkpoint (Fig. 6H) and glycolysis (Fig. 6I). The interferon alpha response is
closely related to immune cell infiltration (33), suggesting that PLEK2 may play a role
in DAMP release. The G2M checkpoint is associated with cell
proliferation, while glycolysis significantly impacts tumor growth,
invasion and treatment resistance (34).
Subsequently, PLEK2 expression was knocked down in
AsPC-1 and PANC-1 cells, and RT-qPCR confirmed the success of the
interference of PLEK2 expression (Fig.
6J). CCK-8 assays indicated that silencing PLEK2 in PAAD cells
promoted cell proliferation (Fig.
6L). In addition, Transwell assays demonstrated that knockdown
of PLEK2 significantly impaired the migration and invasion
capabilities of PAAD cells (Fig. 6M and
N).
Discussion
The role of DAMPs in the context of PAAD has
garnered increasing attention in recent years due to their
potential to influence tumor behavior and patient outcomes
(7,9). The present study aimed to evaluate the
prognostic value of DAMP-related scores derived from a cohort of 40
DAMP-associated genes (21,22). Through meticulous bioinformatics
analyses, patients with PAAD were stratified into three distinct
clusters using Consensus Clustering: C1, C2 and C3. This
classification underscores the dichotomy within the tumor
microenvironment, wherein C1 is hypothesized to represent a
pro-DAMP status, C3 an anti-DAMP status and C2 embodying an
intermediate state. Notably, the C3 cluster was defined by markedly
elevated expression of NT5E - a gene linked to impaired ICD -
coupled with suppressed expression of key ICD-promoting genes,
including TLR4, P2RX7, and FPR1 (Fig.
1G). Conversely, the C1 cluster demonstrated the highest
transcriptional activity of genes associated with ICD facilitation,
such as IL-33, FPR1 and TLR7. Mechanistically, NT5E (in concert
with CD39) catalyzes the conversion of extracellular ATP into
adenosine, a metabolite known to suppress antitumor immunity
through immunosuppressive pathways (35,36).
Furthermore, the release of HMG box 1 (HMGB1) - a hallmark of ICD -
requires sequential permeabilization of nuclear and plasma
membranes, representing a terminal post-mortem event. Extracellular
HMGB1 acts as a potent pro-inflammatory mediator by engaging immune
cell surface receptors, including TLR4 (37). Similarly, P2RX7 contributes to ICD
induction via the purinergic receptor-inflammasome-IL-1 signaling
axis (21,38). These findings collectively
underscore the functional divergence of TME clusters in modulating
immunogenic pathways and antitumor responses. This stratification
not only reflects the complexity of DAMP biology in PAAD but also
raises important implications for personalized therapeutic
strategies directed towards modulating the immune landscape.
To further dissect the biological underpinnings of
these clusters, differential gene expression analyses between the
C1 and C3 groups were conducted. The results revealed that 141 DEGs
had significant prognostic implications across both TCGA-PAAD and
Meta-cohort datasets. This extensive overlap in prognostic genes
indicates the potential of using DAMP-related scores to predict
clinical outcomes in patients with PAAD. Employing LASSO Cox
regression helped hone in on 6 pivotal genes, SLC16A3, LAMC2,
IGF2BP2, HMGA2, EREG and ADM, which were able to differentiate
between clusters with contrasting DAMP statuses. The subsequent
computation of DAMPscores, informed by these genes, paves the way
for a refined prognostic tool with clinical applicability.
The ability of the DAMPscore to predict OS was
corroborated through robust ROC curve analyses. Furthermore, its
superior performance in multiple datasets reinforces the necessity
for larger, multi-center studies to validate its clinical utility.
In a comparative analysis of the DAMPscore with 25 existing
prognostic models, it was observed that the DAMPscore exhibited
improved performance across most cohorts. This positions it as a
potentially valuable addition to the existing arsenal of prognostic
assessments in PAAD. Other studies have also found that prognosis
models constructed based on DAMP or ICD-related genes can
effectively guide the prognosis and personalized treatment of
malignant tumors such as melanoma (29,39–41).
The present study further expands the application scope of
prognosis models developed based on DAMP-related genes.
Delving deeper into the biological relevance of the
identified hub genes through WGCNA, it was noted that several of
these genes, such as ADAM6, HK2 and S100, showed a positive
correlation with the DAMPscore across datasets, suggesting their
integral role in tumor biology and DAMP modulation. In particular,
the role of PLEK2 warrants further attention. PLEK2 is a member of
the pleckstrin family of proteins that primarily plays roles in
cell signaling and cytoskeletal dynamics. Several studies have
found that PLEK2 is emerging as an important player in
tumorigenesis, with roles in promoting cell proliferation (42), enhancing metastatic potential
(43) and facilitating immune
escape (44) within cancers.
Enrichment analyses revealed that PAAD tissues overexpressing PLEK2
were closely linked to hallmark signaling pathways, including the
interferon alpha response, G2M checkpoint and glycolysis. The
Interferon alpha response is particularly intriguing, as it serves
as an indicator of immune cell infiltration and participates in the
occurrence of ICD (45,46), hinting that PLEK2 may be a
significant player in DAMP release and the subsequent inflammatory
response within the tumor microenvironment. In advanced
investigations, PLEK2′s functional relevance was confirmed through
knockdown experiments in AsPC-1 and PANC-1 PAAD cell lines. The
findings revealed that silencing PLEK2 expression promoted cell
proliferation, impairing migration and invasion capabilities. This
dual role in cell proliferation and invasive behavior reinforces
its potential as a therapeutic target within the DAMP
framework.
This study presents several noteworthy findings;
however, it is essential to also acknowledge its limitations.
First, the reliance on retrospective data from the TCGA-PAAD and
Meta-cohort datasets may introduce biases inherent to such
datasets, including selection bias and confounding factors that
could affect the generalizability of the present results. In
addition, while the DAMPscore demonstrated promising prognostic
capabilities, its performance varied across different cohorts,
indicating that external validation in larger, independent
populations is necessary to confirm its robustness.
Methodologically, this can be achieved by quantifying expression
levels of DAMPscore-associated genes in patient tumor specimens via
RNA sequencing or RT-qPCR, followed by correlation analyses with
longitudinal prognostic data. Of note, the clinical implementation
of multigene assays such as the 21-gene recurrence score (Oncotype
DX®) in breast cancer (47) - which informs prognosis and guides
therapeutic decision-making-provides a validated paradigm. This
precedent offers critical methodological and translational insights
for further investigating the clinical utility of the DAMPscore in
PAAD. Furthermore, the functional exploration of PLEK2, although
revealing significant insights into its role in PAAD, was limited
to in vitro experiments, which may not fully resemble the
complex tumor microenvironment in vivo. Future studies
should aim to address these limitations by incorporating
prospective data collection, validating the DAMPscore across
diverse populations and exploring the functional implications of
PLEK2 in more comprehensive in vivo models.
In conclusion, the present study validated the
prognostic value of the DAMPscore in PAAD and elucidated its
relationship with underlying molecular mechanisms driving tumor
behavior. The framework established not only advances the
understanding of DAMP biology in cancer but also unlocks
possibilities for novel therapeutic strategies aimed at modulating
the tumor immune response. The interplay between DAMPs, immune
status and tumor metabolism represents a fertile ground for future
investigations that may ultimately lead to enhanced therapeutic
approaches in the challenging landscape of pancreatic cancer.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
The study was designed by WD and JZ. In vitro
experiments were conducted by HW, GL and JZ. Data analysis was
carried out by WD, JL and ZS. Bioinformatics analysis was conducted
by JL and ZS. WD and JZ confirm the authenticity of all raw data.
The manuscript was drafted by WD, JL and HW, and was revised by all
authors All authors have read and approved the final version of the
study.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Klein AP: Pancreatic cancer epidemiology:
Understanding the role of lifestyle and inherited risk factors. Nat
Rev Gastroenterol Hepatol. 18:493–502. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stoffel EM, Brand RE and Goggins M:
Pancreatic cancer: Changing epidemiology and new approaches to risk
assessment, early detection, and prevention. Gastroenterology.
164:752–765. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cai J, Chen H, Lu M, Zhang Y, Lu B, You L,
Zhang T, Dai M and Zhao Y: Advances in the epidemiology of
pancreatic cancer: Trends, risk factors, screening, and prognosis.
Cancer Lett. 520:1–11. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu Z, Liu L, Weng S, Guo C, Dang Q, Xu H,
Wang L, Lu T, Zhang Y, Sun Z and Han X: Machine learning-based
integration develops an immune-derived lncRNA signature for
improving outcomes in colorectal cancer. Nat Commun. 13:8162022.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen S, Jiang L, Gao F, Zhang E, Wang T,
Zhang N, Wang X and Zheng J: Machine learning-based pathomics
signature could act as a novel prognostic marker for patients with
clear cell renal cell carcinoma. Br J Cancer. 126:771–777. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Roh JS and Sohn DH: Damage-associated
molecular patterns in inflammatory diseases. Immune Netw.
18:e272018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ashrafizadeh M, Farhood B, Eleojo Musa A,
Taeb S and Najafi M: Damage-associated molecular patterns in tumor
radiotherapy. Int Immunopharmacol. 86:1067612020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang H, Tohme S, Al-Khafaji AB, Tai S,
Loughran P, Chen L, Wang S, Kim J, Billiar T, Wang Y and Tsung A:
Damage-associated molecular pattern-activated neutrophil
extracellular trap exacerbates sterile inflammatory liver injury.
Hepatology. 62:600–614. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ahmed A and Tait SWG: Targeting
immunogenic cell death in cancer. Mol Oncol. 14:2994–3006. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li X, Kang J, Yue J, Xu D, Liao C, Zhang
H, Zhao J, Liu Q, Jiao J, Wang L and Li G: Identification and
validation of immunogenic cell death-related score in uveal
melanoma to improve prediction of prognosis and response to
immunotherapy. Aging (Albany NY). 15:3442–3464. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yanai H, Hangai S and Taniguchi T:
Damage-associated molecular patterns and Toll-like receptors in the
tumor immune microenvironment. Int Immunol. 33:841–846. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang X, Liu Y, Zhang Z, Tan J, Zhang J,
Ou H, Li J and Song Z: Multi-omics analysis of anlotinib in
pancreatic cancer and development of an anlotinib-related
prognostic signature. Front Cell Dev Biol. 9:6492652021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wilkerson MD and Hayes DN:
ConsensusClusterPlus: A class discovery tool with confidence
assessments and item tracking. Bioinformatics. 26:1572–1573. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qu H, Zhao H, Zhang X, Liu Y, Li F, Sun L
and Song Z: Integrated analysis of the ETS family in melanoma
reveals a regulatory role of ETV7 in the immune microenvironment.
Front Immunol. 11:6127842020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhu Z, Li G, Li Z, Wu Y, Yang Y, Wang M,
Zhang H, Qu H, Song Z and He Y: Core immune cell infiltration
signatures identify molecular subtypes and promote precise
checkpoint immunotherapy in cutaneous melanoma. Front Immunol.
13:9146122022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tang J, Kong D, Cui Q, Wang K, Zhang D,
Gong Y and Wu G: Prognostic genes of breast cancer identified by
gene co-expression network analysis. Front Oncol. 8:3742018.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jun Z: GseaVis: An Implement R Package to
Visualize GSEA Results. 2022.
|
|
19
|
Li G, Song Z, Wu C, Li X, Zhao L, Tong B,
Guo Z, Sun M, Zhao J, Zhang H, et al: Downregulation of NEDD4L by
EGFR signaling promotes the development of lung adenocarcinoma. J
Transl Med. 20:472022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Song Z, Yu Z, Chen L, Zhou Z, Zou Q and
Liu Y: MicroRNA-1181 supports the growth of hepatocellular
carcinoma by repressing AXIN1. Biomed Pharmacother. 119:1093972019.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Garg AD, De Ruysscher D and Agostinis P:
Immunological metagene signatures derived from immunogenic cancer
cell death associate with improved survival of patients with lung,
breast or ovarian malignancies: A large-scale meta-analysis.
Oncoimmunology. 5:e10699382015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu M, Lu JH, Zhong YZ, Jiang J, Shen YZ,
Su JY and Lin SY: Immunogenic cell death-relevant damage-associated
molecular patterns and sensing receptors in triple-negative breast
cancer molecular subtypes and implications for immunotherapy. Front
Oncol. 12:8709142022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bailey P, Chang DK, Nones K, Johns AL,
Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC,
et al: Genomic analyses identify molecular subtypes of pancreatic
cancer. Nature. 531:47–52. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang J, Chen M, Fang C and Luo P: A
cancer-associated fibroblast gene signature predicts prognosis and
therapy response in patients with pancreatic cancer. Front Oncol.
12:10521322022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wei X, Zhou X, Zhao Y, He Y, Weng Z and Xu
C: A 14-gene gemcitabine resistance gene signature is significantly
associated with the prognosis of pancreatic cancer patients. Sci
Rep. 11:60872021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xiao M, Liang X, Yan Z, Chen J, Zhu Y, Xie
Y, Li Y, Li X, Gao Q, Feng F, et al: A DNA-methylation-driven genes
based prognostic signature reveals immune microenvironment in
pancreatic cancer. Front Immunol. 13:8039622022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tao S, Tian L, Wang X and Shou Y: A
pyroptosis-related gene signature for prognosis and immune
microenvironment of pancreatic cancer. Front Genet. 13:8179192022.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhuo Z, Lin H, Liang J, Ma P, Li J, Huang
L, Chen L, Yang H, Bai Y and Sha W: Mitophagy-related gene
signature for prediction prognosis, immune scenery, mutation, and
chemotherapy response in pancreatic cancer. Front Cell Dev Biol.
9:8025282022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang C, Chen Y, Xinpeng Y, Xu R, Song J,
Ruze R, Xu Q and Zhao Y: Construction of immune-related signature
and identification of S100A14 determining immune-suppressive
microenvironment in pancreatic cancer. BMC Cancer. 22:8792022.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang H, Pan YZ, Cheung M, Cao M, Yu C,
Chen L, Zhan L, He ZW and Sun CY: LAMB3 mediates apoptotic,
proliferative, invasive, and metastatic behaviors in pancreatic
cancer by regulating the PI3K/Akt signaling pathway. Cell Death
Dis. 10:2302019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lin H, Yang P, Li B, Chang Y, Chen Y, Li
Y, Liu K, Liang X, Chen T, Dai Y, et al: S100A10 promotes
pancreatic ductal adenocarcinoma cells proliferation, migration and
adhesion through JNK/LAMB3-LAMC2 axis. Cancers (Basel). 15:2022022.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Erice O, Narayanan S, Feliu I,
Entrialgo-Cadierno R, Malinova A, Vicentini C, Guruceaga E, Delfino
P, Trajkovic-Arsic M, Moreno H, et al: LAMC2 regulates key
transcriptional and targetable effectors to support pancreatic
cancer growth. Clin Cancer Res. 29:1137–1154. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Borden EC: Interferons α and β in cancer:
Therapeutic opportunities from new insights. Nat Rev Drug Discov.
18:219–234. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ganapathy-Kanniappan S and Geschwind JF:
Tumor glycolysis as a target for cancer therapy: Progress and
prospects. Mol Cancer. 12:1522013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kepp O, Senovilla L, Vitale I, Vacchelli
E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N,
et al: Consensus guidelines for the detection of immunogenic cell
death. Oncoimmunology. 3:e9556912014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Antonioli L, Pacher P, Vizi ES and Haskó
G: CD39 and CD73 in immunity and inflammation. Trends Mol Med.
19:355–367. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yu M, Wang H, Ding A, Golenbock DT, Latz
E, Czura CJ, Fenton MJ, Tracey KJ and Yang H: HMGB1 signals through
toll-like receptor (TLR) 4 and TLR2. Shock. 26:174–179. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ghiringhelli F, Apetoh L, Tesniere A,
Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G,
Ullrich E, et al: Activation of the NLRP3 inflammasome in dendritic
cells induces IL-1beta-dependent adaptive immunity against tumors.
Nat Med. 15:1170–1178. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li G, Zhang H, Zhao J, Liu Q, Jiao J, Yang
M and Wu C: Machine learning-based construction of immunogenic cell
death-related score for improving prognosis and response to
immunotherapy in melanoma. Aging (Albany NY). 15:2667–2688. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu J, Shi Y and Zhang Y: Multi-omics
identification of an immunogenic cell death-related signature for
clear cell renal cell carcinoma in the context of 3P medicine and
based on a 101-combination machine learning computational
framework. EPMA J. 14:275–305. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu X, Yao S, Feng Y, Li P, Li Y and Xia
S: Construction of a novel damage-associated
molecular-pattern-related signature to assess lung adenocarcinoma's
prognosis and immune landscape. Biomolecules. 14:1082024.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cai T, Yao W, Qiu L, Zhu AR, Shi Z and Du
Y: PLEK2 promotes the proliferation and migration of non-small cell
lung cancer cells in a BRD4-dependent manner. Mol Biol Rep.
49:3693–3704. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shen H, He M, Lin R, Zhan M, Xu S, Huang
X, Xu C, Chen W, Yao Y, Mohan M and Wang J: PLEK2 promotes
gallbladder cancer invasion and metastasis through EGFR/CCL2
pathway. J Exp Clin Cancer Res. 38:2472019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mao D, Zhou Z, Chen H, Liu X, Li D, Chen
X, He Y, Liu M and Zhang C: Pleckstrin-2 promotes tumour immune
escape from NK cells by activating the MT1-MMP-MICA signalling axis
in gastric cancer. Cancer Lett. 572:2163512023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Du B and Waxman DJ: Medium dose
intermittent cyclophosphamide induces immunogenic cell death and
cancer cell autonomous type I interferon production in glioma
models. Cancer Lett. 470:170–180. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Omori R, Eguchi J, Hiroishi K, Ishii S,
Hiraide A, Sakaki M, Doi H, Kajiwara A, Ito T, Kogo M and Imawari
M: Effects of interferon-alpha-transduced tumor cell vaccines and
blockade of programmed cell death-1 on the growth of established
tumors. Cancer Gene Ther. 19:637–643. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Paik S, Tang G, Shak S, Kim C, Baker J,
Kim W, Cronin M, Baehner FL, Watson D, Bryant J, et al: Gene
expression and benefit of chemotherapy in women with node-negative,
estrogen receptor-positive breast cancer. J Clin Oncol.
24:3726–3734. 2006. View Article : Google Scholar : PubMed/NCBI
|