Introduction
According to global cancer statistics for 2022,
breast cancer (BC) is the second most frequently diagnosed cancer
type, with ~2.3 million new cases (1). As a natural consequence of the growing
and aging of the world population, the burden of BC is expected to
exceed 3 million new cases and 1 million mortalities in the next
two decades (2). BC comprises a
group of diseases, exhibiting different clinical behaviors
(treatment response/resistance, prognosis and metastatic potential)
between subtypes, which reflects its heterogeneous nature (3). Although there are numerous approaches
such as hormonal, targeted drugs and chemotherapy in BC treatment,
drug resistance is a common phenomenon that limits the treatment
efficacy. However, the mechanisms underlying drug resistance are
complex and not yet fully understood (4).
The phosphatidylinositol 3-kinase (PI3K)/protein
kinase B (AKT) pathway is key for numerous cellular processes,
including cell cycle progression, proliferation and apoptosis
(5). Dysregulation of this pathway
is associated with tumor progression and drug resistance in BC.
Mutations in genes involved in the PI3K/AKT pathway, such as
PI3K, AKT1/2/3 and PTEN, may disturb the regulation
of tumor growth, angiogenesis and cell survival, contributing to
cancer development (6). As a
result, this pathway is a promising target for overcoming drug
resistance in BC (7). AKT1, a
serine/threonine kinase encoded by the AKT1 gene, is an
important signaling protein that serves a key role in the PI3K/AKT
pathway (8). AKT1 phosphorylates
several targets, such as the mammalian target of rapamycin (mTOR),
matrix metalloproteinases, cyclin-dependent kinases and vascular
endothelial growth factor, to promote cell survival (9). During the AKT1 activation process,
PI3K is primarily activated through the binding of ligands,
including growth factors, hormones and cytokines, to receptors.
Activated PI3K leads to the formation of
phosphatidylinositol-(3,4,5)-trisphosphate (PIP3) from
phosphatidylinositol-(4,5)-bisphosphate, facilitating the
recruitment of AKT1 to the plasma membrane. The pleckstrin homology
(PH) domain is key for the activation of AKT1, which binds to PIP3
via its PH domain and is subsequently phosphorylated by
3′-phosphoinositide-dependent protein kinase 1 and mTOR complex 2
(10). Missense mutations in the PH
domain of AKT1, including AKT1-E17K (49G>A), -E49K
(145G>A) and -L52R (155T>G), have been demonstrated to
increase AKT1 activity (11–13).
The AKT1-E17K and -L52R mutations have also been observed in
patients with BC (11,14). While the AKT1-E49K mutation
has been reported in bladder cancer and pulmonary sclerosing
hemangioma (12,15), to the best of our knowledge, its
presence in BC has not been reported.
The canonical wingless-type MMTV integration site
family (WNT) pathway (WNT/β-catenin) is an evolutionarily conserved
signaling cascade that regulates key biological processes,
including cell fate determination, organogenesis, tissue
homeostasis and pathological conditions (such as cancer,
osteoporosis and coronary artery disease). Dysregulation of the
WNT/β-catenin pathway is associated with carcinogenesis through
multiple mechanisms of action (16). In BC, various components of the
WNT/β-catenin pathway undergo alteration due to factors such as
mutations, amplifications and post-transcriptional/-translational
modification. These alterations lead to changes in the localization
of β-catenin within the cell (17).
The phosphorylation status of cytosolic β-catenin is key to the
WNT/β-catenin pathway (18). When
the pathway is inactivated, β-catenin binds to the destruction
complex that includes axis inhibition protein (AXIN), adenomatous
polyposis coli, casein kinase 1 (CK-1) and glycogen synthase kinase
3β (GSK-3β). β-catenin is sequentially phosphorylated by CK-1 on
Ser45 and then by GSK-3β on Thr41, Ser37 and Ser33. This
phosphorylation leads to ubiquitination and proteasomal degradation
of β-catenin (19). In
physiological/pathological conditions such as embryogenesis and
carcinogenesis, the WNT/β-catenin pathway is activated by binding
of WNT family members (WNT1, WNT2, WNT3a, etc.) to the Frizzled
(FZD) receptor and its coreceptor, low-density lipoprotein
receptor-related protein (LRP) 6 or LRP5. The formation of the
WNT-FZD-LRP6/LRP5 complex mediates the assembly of the destruction
complex around the receptor, resulting in inhibition of destruction
complex-mediated β-catenin phosphorylation and hence accumulation
of β-catenin in the cytosol. This accumulation triggers the
translocation of β-catenin to the nucleus, where it initiates the
expression of target genes essential for cell survival,
proliferation, differentiation and migration (20).
Mutations in the catenin β-1 (CTNNB1) gene,
which encodes β-catenin, are observed in numerous types of cancer
and are predominantly all localized in exon 3. The mutations are
primarily found in the phosphorylation sites of β-catenin
(N-terminal region; S33, S37, T41 and S45), leading to resistance
to phosphorylation. As a result, β-catenin accumulates in the cell,
eventually translocating to the nucleus (21). Missense mutations in the
phosphorylation sites of β-catenin include CTNNB1-S33P
(97T>C), -T41A (121A>G) and -S45F (134C>T). The presence
of these mutations is associated with an increase in cellular
proliferation (22–29). CTNNB1-T41A and -S45F have
been reported in BC (30), whereas
the presence of CTNNB1-S33P remains unclear.
Paclitaxel (PTX) is a microtubule-stabilizing agent
from the taxane class of plant alkaloids that is commonly used in
the treatment of BC. PTX induces apoptosis in cancer cells through
various mechanisms of action, including activation of antitumor
immunity, the regulation of mitochondrial function, cell cycle
arrest, downregulation of the PI3K/AKT pathway and production of
reactive oxygen species (31,32).
Despite its potent apoptotic effects via these mechanisms, the
efficacy of PTX is limited, primarily due to the development of
drug resistance (33).
Understanding the molecular mechanisms underlying
PTX resistance and identifying predictive biomarkers for PTX
resistance in BC treatment is key for improving therapeutic
outcomes. To the best of our knowledge, no established biomarker
exists to predict PTX resistance. Therefore, the present study
explored the potential effects of AKT1 (E17K/E49K/L52R) and
CTNNB1 (S33P/T41A/S45F) mutations, which are known to
enhance AKT1 activity (11–13) and β-catenin stability (21), on PTX resistance in in vitro
models of BC. The results of the present study may serve as a guide
for clinical studies associated with PTX resistance.
Materials and methods
Site-directed mutagenesis
The AKT1 and CTNNB1 mutations were
created using a site-directed mutagenesis kit (cat. no. 200519;
Agilent Technologies, Inc.) on the pCMV6 vectors with C-terminal
Myc-DDK tag carrying AKT1 (cat. no. RC220361; OriGene
Technologies, Inc.) and CTNNB1 (cat. no. RC208947; OriGene
Technologies, Inc.) following the manufacturer's protocol.
Mutagenic primers were designed using the specific design program
accessible online at agilent.com/genomics/qcpd and synthesized by
Integrated DNA Technologies, Inc. (Table I).
 | Table I.Mutagenic primers used to generate
AKT1 and CTNNB1 mutations. |
Table I.
Mutagenic primers used to generate
AKT1 and CTNNB1 mutations.
| Mutation | Primer sequence
(5′-3′) |
|---|
| AKT1 |
|
| E17K
(Glu17Lys) | Forward,
GCACAAACGAGGGAAGTACATCAAGAC |
|
49G>A | Reverse,
GTCTTGATGTACTTCCCTCGTTTGTGC |
| E49K
(Glu49Lys) | Forward,
GATGTGGACCAACGTAAGGCTCCCCTCAAC |
|
145G>A | Reverse,
GTTGAGGGGAGCCTTACGTTGGTCCACATC |
| L52R
(Leu52Arg) | Forward,
CGTGAGGCTCCCCGCAACAACTTCTCTG |
|
155T>G | Reverse,
CAGAGAAGTTGTTGCGGGGAGCCTCACG |
| CTNNB1 |
|
| S33P
(Ser33Pro) | Forward,
CAGTCTTACCTGGACCCTGGAATCCATTC |
|
97T>C | Reverse,
GAATGGATTCCAGGGTCCAGGTAAGACTG |
| T41A
(Thr41Ala) | Forward,
CATTCTGGTGCCACTGCCACAGCTCCTTCTC |
|
121A>G | Reverse,
GAGAAGGAGCTGTGGCAGTGGCACCAGAATG |
| S45F
(Ser45Phe) | Forward,
CTACCACAGCTCCTTTTCTGAGTGGTAAAG |
|
134C>T | Reverse,
CTTTACCACTCAGAAAAGGAGCTGTGGTAG |
Transformation and plasmid
isolation
Escherichia coli cells (cat. no. C3030-03;
Invitrogen; Thermo Fisher Scientific, Inc.) were used to transform
mutant and wild-type (WT) AKT1/CTNNB1 pCMV6 vectors,
as well as an empty pCMV6 vector (EV) (cat. no. PS100001; OriGene
Technologies, Inc.), following the manufacturer's protocol based on
the heat-shock method (34). The
transformed cell mixtures were plated onto lysogeny broth (LB) agar
(cat. no. L3147; Sigma-Aldrich; Merck KGaA) containing 25 µg/ml
kanamycin (KAN) (cat. no. 60615; Sigma-Aldrich; Merck KGaA) in
Petri dishes and incubated at 37°C overnight. Since the vectors
contained the KAN-resistance gene, the cells transformed with these
vectors proliferated and formed colonies on LB agar containing KAN.
Then the colonies containing transformed cells were collected by a
loop and cultured in LB at 37°C for 8 h (cat. no. L3522;
Sigma-Aldrich; Merck KGaA). The DNA was purified using an isolation
kit (cat. no. 12123; Qiagen, Inc.), according to the manufacturer's
instructions. The DNA samples were sequenced using next-generation
sequencing by RefGen Gene Research and Biotechnology Company to
verify the efficiency of the transformation.
Cell culture
MCF-7 (cat. no. HTB-22) and MDA-MB-231 (cat. no.
HTB-26) BC cell lines were obtained from the American Type Culture
Collection. MCF-7 cells were maintained in EMEM (cat. no.
320-026-CL; Wisent Biotechnology) supplemented with 10% fetal
bovine serum (FBS; cat. no. FBS-11A; Capricorn Scientific GmbH),
100 U/ml penicillin, 100 µg/ml streptomycin (cat. no. 450-201-EL;
Wisent Biotechnology) and 0.01 mg/ml recombinant human insulin
(cat. no. I9278; Sigma-Aldrich; Merck KGaA). MDA-MB-231 cells were
maintained in RPMI-1640 (cat. no. 350-007-CL; Wisent Biotechnology)
supplemented with 10% FBS and 100 U/ml penicillin/100 µg/ml
streptomycin. The cells were cultured in a humidified incubator
with 5% CO2 at 37°C. The cell lines were passaged four
times prior to use, with transfection performed at passage four.
Following transfection, the cells were passaged a fifth time prior
to drug treatment. To minimize cellular stress and ensure more
reliable results, a cell scraper was used during passaging.
Plasmid DNA transfection
Transient transfection of the plasmid DNA (5 µg)
into the cells was performed using the MegaTran 2.0 polymer-based
plasmid DNA transfection agent (cat. no. TT210003; OriGene
Technologies, Inc.) following the manufacturer's recommendations.
MCF-7 (3×106) and MDA-MB-231 (1.5×106) cells
were seeded in Petri dishes at 37°C for 24 h. Subsequently, the
cells were transfected at 37°C for 17–18 h. Afterward, the medium
containing the transfection complex was removed, fresh medium was
added and the cells were incubated at 37°C for 24 h to allow for
proliferation. At the end of 24 h, the cells were collected to
perform subsequent experiments. The EV served as the transfection
control.
Cell viability assay
The viability of cells was analyzed using MTS (cat.
no. G111; Promega Corporation). MCF-7 (5×104) and
MDA-MB-231 (1×104) cells were seeded into 96-well
plates. PTX (cat. no. T7402; Sigma-Aldrich; Merck KGaA) was
dissolved in DMSO and serially diluted (1:1) to obtain 16 different
PTX doses (100–0.003 µg/ml). The cells were exposed to PTX at 37°C
for 24, 48 and 72 h. DMSO was used as the negative control. A total
of 20 µl MTS/phenazine methosulfate (cat. no. P9625; Sigma-Aldrich;
Merck KGaA) mixture (20:1, volume:volume) was added and the plates
were maintained for 1–4 h. The absorbance values were determined at
490 nm and cell death was determined using the following formula:
Cell death (%)=[1-(absorbance of the sample/absorbance of the
control)] ×100.
Western blot analysis
MCF-7 and MDA-MB-231 cells were lysed with RIPA [50
mM Tris-HCl (pH 8), 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium
deoxycholate and 0.1% SDS] and the concentration of the total
protein was assessed by the bicinchoninic acid protein assay kit
(cat. no. BCA1; Sigma-Aldrich; Merck KGaA). A total of 20 µg/lane
total protein was loaded onto the Any kD Mini-PROTEAN tris-glycine
eXtreme (TGX) precast gels (cat. no. 4569034; Bio-Rad Laboratories,
Inc.) and transferred onto a polyvinylidene difluoride membrane
(cat. no. 1704272; Bio-Rad Laboratories, Inc.). The membrane was
blocked in Tris-buffered saline with 0.1% Tween 20 containing 5%
non-fat dry milk at room temperature for 1 h, and was incubated
with anti-Myc-DDK (1:2,000; cat. no. TA50011; OriGene Technologies,
Inc.) and anti-β-actin (1:10,000; cat. no. M01263; Boster
Biological Technology) primary antibodies for 12 h at 4°C. The
anti-Myc-DDK antibody was used due to the C-terminal Myc-DDK tag on
pCMV6 vectors carrying the AKT1 and the CTNNB1 genes.
The membrane was incubated with secondary antibodies (1:10,000;
anti-mouse IgG-HRP; cat. no. sc-2005; Santa Cruz Biotechnology,
Inc.; 1:20,000; anti-rabbit IgG-HRP; cat. no. ab205718; Abcam) for
1 h at room temperature. The membrane was incubated with a
chemiluminescence substrate (cat. no. 1705060; Bio-Rad
Laboratories, Inc.) and imaged using Fusion FX (Vilber Lourmat
Sté). The immunoreactive protein (IRP) bands were quantified using
ImageJ 1.59 software (National Institutes of Health).
Statistical analysis
The experiments were performed in triplicate.
Statistical analysis was performed using SPSS Statistics version 21
(IBM Corp.). The normality of the variables was evaluated using the
Shapiro-Wilk test and all variables were found to be normally
distributed. Descriptive statistics, including the mean and the
standard deviation, were calculated. To compare differences among
>2 groups, one-way ANOVA was performed followed by Tukey's post
hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
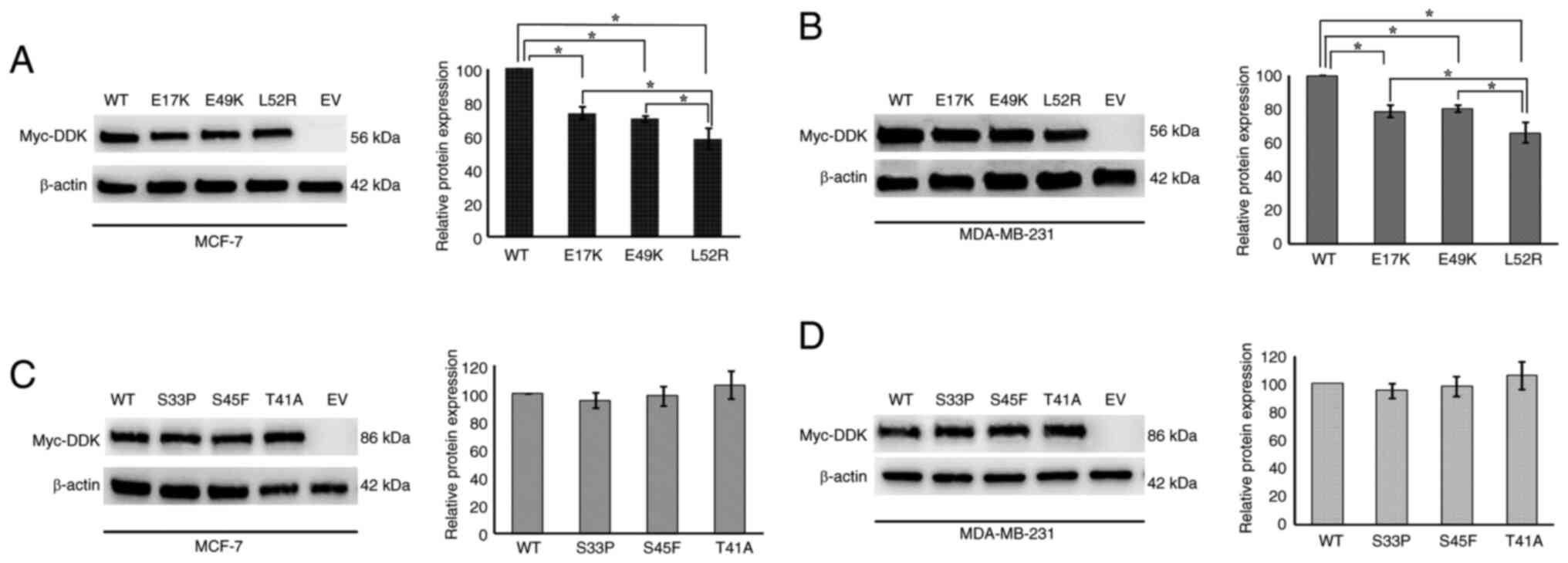
Western blot analysis of
AKT1/CTNNB1-WT and mutant proteins
BC cells were transfected with the EV and vectors
carrying AKT1/CTNNB1-WT and mutations, followed by western
blot analysis. As expected, no IRP band was observed for the EV.
The presence of IRP bands in the samples from AKT1/CTNNB1-WT
and mutant-transfected cells confirmed the success of the
transfection process (Fig. 1). IRP
levels of AKT1-E17K/E49K/L52R were significantly lower
compared with those of AKT1-WT in both cell lines
(P<0.05). AKT1-L52R mutant IRP levels were significantly
lower than those of AKT1-E17K and -E49K (P<0.05; Fig. 1A and B). However, no significant
changes were observed between the IRP levels of
CTNNB1-WT/S33P/T41A/S45F (P>0.05; Fig. 1C and D).
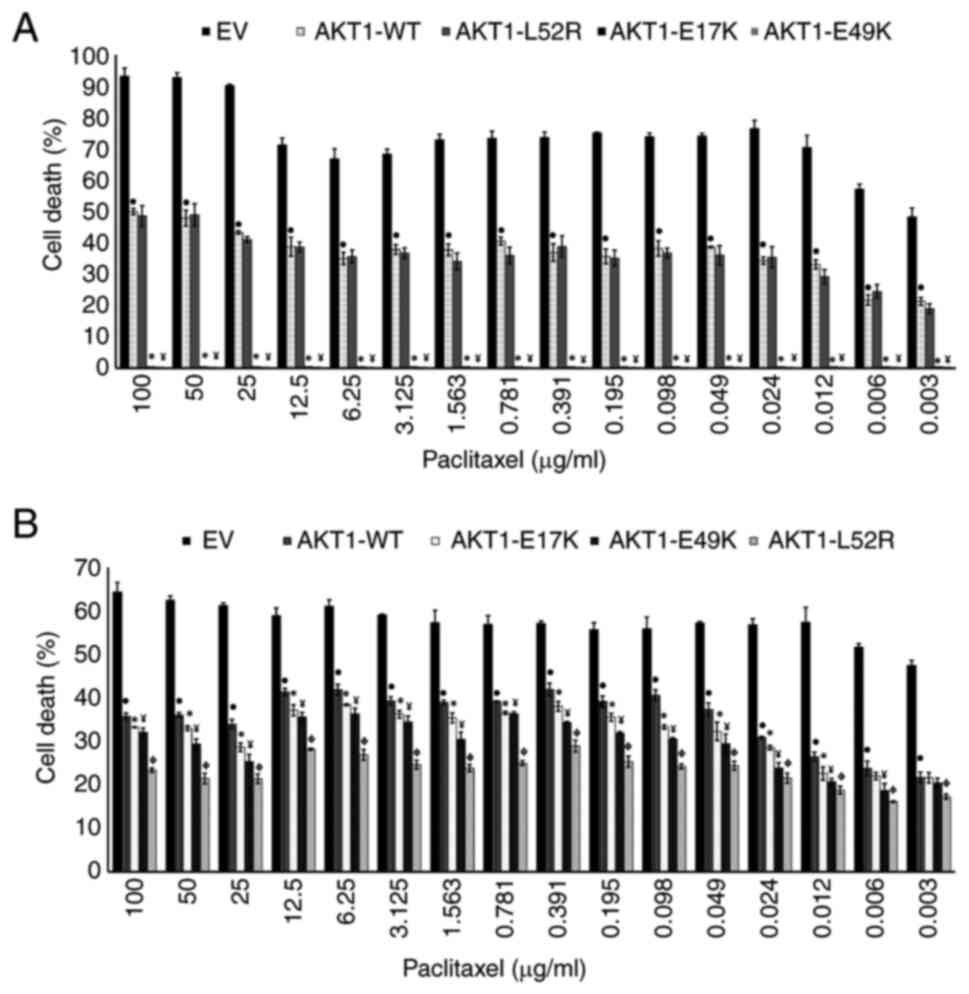
Impact of AKT1 mutations on PTX
resistance in BC cells
BC cells were incubated with PTX for 24, 48 and 72
h. The PTX responses were similar in both EV-transfected and
non-transfected BC cells (P>0.05; Table SI, Table SII, Table SIII, Table SIV, Table SV, Table SVI), indicating that the EV did not
have an effect.
PTX induced cell death in EV-transfected MCF-7 cells
at 24, 48 and 72 h; however, PTX induced cell death in MCF-7 cells
transfected with AKT1-WT only at 72 h. The death of
AKT1-WT-transfected MCF-7 cells was significantly decreased
compared with EV-transfected MCF-7 cells at all exposure times
(P<0.05; Table SI, Table SII, Table SIII). These findings reveal that
AKT1-WT is associated with PTX resistance in MCF-7
cells.
PTX exhibited no cytotoxic effects on the cells
transfected with AKT1 mutations at 24 and 48 h. Following 72
h, death was observed in the cells carrying AKT1-L52R, while
PTX remained ineffective in cells expressing AKT1-E17K and
-E49K mutations. In addition, the death of cells expressing
AKT1-E17K and -E49K was significantly lower compared with
that of cells expressing AKT1-WT (P<0.05); however, no
significant difference was noted between AKT1-L52R and
AKT1-WT (P>0.05; Table
SI, Table SII, Table SIII). These findings indicate that
AKT1-E17K and -E49K conferred enhanced resistance to PTX in
MCF-7 cells compared with AKT1-WT (Fig. 2A).
In both EV-transfected and
AKT1-WT-transfected MDA-MB-231 cells, PTX induced death at
24, 48 and 72 h. In AKT1-WT-transfected cells, cell death
was reduced compared with EV-transfected cells at 48 and 72 h
(P<0.05; Table SIV, Table SV, Table VI). These results indicated that
AKT1-WT was associated with PTX resistance in MDA-MB-231
cells, which was similar to the findings noted in MCF-7 cells.
The effects of AKT1 mutations on the PTX
response in MDA-MB-231 cells were also evaluated. The PTX-induced
death was significantly higher in cells carrying AKT1-WT
compared with those harboring
AKT1-E17K/E49K/AKT1-L52R (P<0.05; Table SIV, Table SV, Table VI). These results indicated that
AKT1-E17K/E49K/L52R induced higher PTX resistance in
MDA-MB-231 cells than AKT1-WT (Fig. 2B).
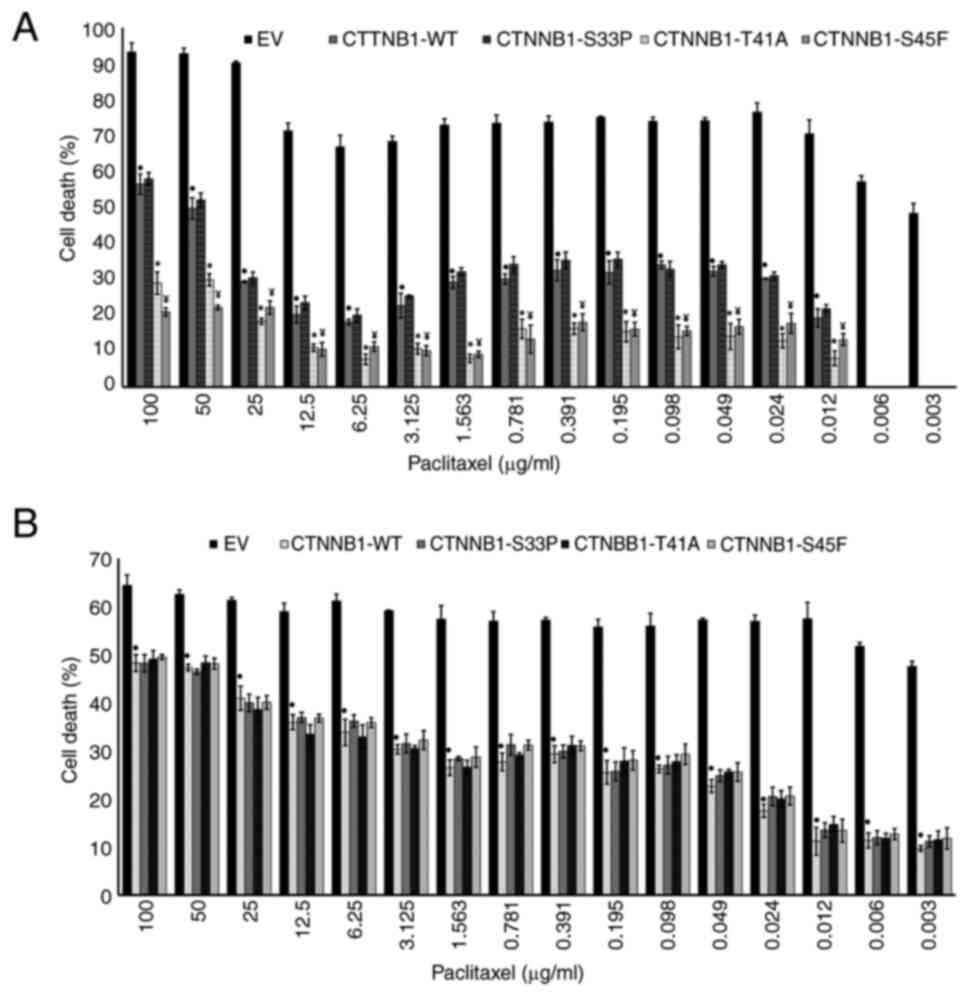
Impact of CTNNB1 mutations on PTX
resistance in BC cells
BC cells were incubated with PTX for 24, 48 and 72
h, and cell death was evaluated. The similar PTX responses in both
EV-transfected and non-transfected BC cells confirmed there was no
effect exerted by EV on cell death (P>0.05; Table SVII, Table SVIII, Table SIX, Table SX, Table SXI, Table XII).
PTX induced death in transfected MCF-7 cells with
CTNNB1-WT at 24, 48 and 72 h. The death of MCF-7 cells
carrying CTNNB1-WT was significantly lower compared with
that of EV-transfected MCF-7 cells (P<0.05; Table SVII, Table SVIII, Table SIX). These findings revealed that
CTNNB1-WT was associated with PTX resistance in MCF-7 cells.
Furthermore, the effects of CTNNB1 mutation on the PTX
resistance in MCF-7 cells were assessed. PTX exhibited cytotoxic
effects on cells expressing CTNNB1-S45F and -T41A only at 72
h. PTX-induced death was observed in cells transfected with
CTNNB1-S33P at 24, 48 and 72 h (Table SVII, Table SVIII, Table SIX). PTX-induced cell death of
CTNNB1-WT was higher compared with that of
CTNNB1-S45F and -T41A (P<0.05); however, no significant
difference (P>0.05) was noted between CTNNB1-WT and -S33P
(Fig. 3A). These results indicated
that CTNNB1-T41A and -S45F display higher PTX resistance
compared with CTNNB1-WT in MCF-7 cells.
In MDA-MB-231 cells, PTX induced different death in
cells transfected with CTNNB1-WT. The death of cells
expressing CTNNB1-WT was significantly decreased compared
with that of EV-transfected cells (P<0.05; Table SX, Table SXI, Table SXII). These findings suggest that
CTNNB1-WT was associated with PTX resistance in MDA-MB-231
cells.
The impact of CTNNB1 mutations on PTX
resistance in MDA-MB-231 cells was also evaluated (Table SX, Table SXI, Table SXII). The death levels of the cells
expressing CTNNB1-S33P/S45F/T41A were comparable with those
of cells expressing CTNNB1-WT (P>0.05; Fig. 3B). These findings demonstrated that
CTNNB1-S33P/S45F/T41A mutations did not confer additional
PTX resistance in MDA-MB-231 cells compared with
CTNNB1-WT.
Discussion
BC is a heterogeneous group of diseases with
different morphology and molecular structures, exhibiting varying
biological behaviors and prognoses. Given the heterogeneous nature
of BC, the primary challenge in its treatment is drug resistance.
The identification of the molecular mechanisms underlying drug
resistance and potential predictive biomarkers are key for
overcoming this (35,36). The majority of the commonly used
predictive biomarkers in BC treatment are estrogen receptor (ER)α,
progesterone receptor (PR) and epidermal growth factor 2 (HER2).
ERα and PR are used to guide endocrine therapy, while HER2 is used
to determine the potential efficacy of anti-HER2 targeted therapies
(37).
In the present study, cell lines (MCF-7 and
MDA-MB-231) with different genetic profiles were used. The MCF-7
cell line represents a typical luminal subtype of BC characterized
by ER(+)/PR(+)/HER2(−) (38). By
contrast, the MDA-MB-231 cell line is a typical example of triple
negative (TN)BC [ER(−)/PR(−)/HER2(−)] (39). The molecular pathophysiology of TNBC
remains poorly understood and the primary strategy of the treatment
is taxane-based chemotherapy. By contrast, endocrine therapy is the
primary treatment strategy for ER(+)/HER2(−) BC; however,
resistance is a common issue in patients with advanced BC, leading
to recurrence. To decrease the risk of recurrence, taxane-based
chemotherapy is often recommended. Nevertheless, the response rates
are generally lower in ER(+)/HER2(−) patients with BC compared with
those with HER2(+) or TNBC (40,41).
PTX is a taxane-class chemotherapeutic agent
commonly used in BC treatment; however, its effectiveness is
limited by drug resistance. The underlying mechanisms of PTX
resistance remain unclear. To date, mutations in genes such as
multidrug resistance 1, β-tubulin and p53 have been implicated in
PTX resistance (33). In addition,
elevated PI3K/AKT signaling in BC contributes to PTX resistance via
specific mechanisms, such as promoting cell survival, enhancing
anaerobic metabolism and inhibiting apoptosis (42). Genetic alterations in the PI3K/AKT
pathway are frequently observed in BC, including gain-of-function
mutations in PIK3CA and AKT1, as well as mutations in
PIK3R1, AKT2, AKT3 and PTEN. These alterations
highlight the key role of this pathway in breast carcinogenesis
(11,43,44).
AKT1-E17K/E49K/L52R are the PH domain
mutations resulting in AKT1 activation in a PI3K-independent manner
(11–13). The elevated levels of PI3K/AKT
signaling due to these mutations lead to enhanced cell survival and
oncogenic transformation (12,13,45–48).
In the present study, western blot analysis indicated that
AKT1-E17K/E49K/L52R protein levels were lower compared with
those of AKT1-WT in both cell lines. Also, AKT1-L52R
protein levels were lower than those of AKT1-E17K and -E49K.
The decreased levels of mutant proteins may be due to several
factors, including altered protein stability, improper folding or
differential interactions with cellular components (such as
chaperones and downstream signaling partners). These mutations may
affect the overall structural integrity or function of the protein,
resulting in a decreased expression level. Additionally, the L52R
mutation may disrupt critical interactions within the AKT1 protein,
affecting its stability or trafficking within the cell.
The potential effects of AKT1-E17K/E49K/L52R
mutations on PTX resistance in BC cells were also investigated
in vitro, as well as their impact on protein activity.
AKT1-E17K and -E49K mutations conferred increased PTX
resistance in MCF-7 cells compared with AKT1-WT. In
MDA-MB-231 cells, all three AKT1 mutations (E17K/E49K/L52R)
were associated with increased PTX resistance. In our previous
study, the association between AKT1-E17K/E49K/L52R mutations
and cetuximab, 5-fluorouracil, 7-ethyl-10-hydroxycamptothecin
(SN-38), oxaliplatin and irinotecan resistance in colorectal cancer
cells was identified (49). The
present study indicated similarities to those observed in
colorectal cancer regarding drug resistance. Studies performed on
patients with BC harboring an AKT1-E17K mutation have
reported that the use of AKT inhibitors alone (50,51) or
in combination with PTX (52) or
fulvestrant (53) increases the
response to treatment. The findings of the present study regarding
the AKT1-E17K mutation and PTX resistance in BC are
consistent with those reported in the literature (49–53).
To the best of our knowledge, the association between
AKT1-E49K and -L52R mutations and drug resistance has not
been previously reported in BC.
In addition to the PI3K/AKT pathway, the
WNT/β-catenin pathway is associated with BC. Elevated levels of
nuclear and/or cytoplasmic β-catenin are commonly observed in BC
tissue and are associated with poor clinical outcomes and
chemoresistance (54–57). Genetic and epigenetic alterations in
the WNT/β-catenin pathway lead to enhanced transcription of
WNT-targeted genes (such as AXIN2, cyclin D1, epidermal growth
factor receptor and myelocytomatosis viral oncogene homolog) in BC
(58).
CTNNB1-S33P/T41A/S45F mutations (in exon 3)
decrease ubiquitination-mediated proteolysis of β-catenin, causing
its nuclear accumulation and activation (22–24,27,28).
Therefore, in the present study, the association between PTX
resistance and the WNT/β-catenin pathway was explored. The results
indicate that CTNNB1-S45F and -T41A induce PTX resistance,
whereas CTNNB1-S33P did not affect the PTX response in MCF-7
cells. By contrast, CTNNB1-S33P/T41A/S45F mutations did not
cause changes in PTX response in MDA-MB-231 cells. The varying
effects of CTNNB1-T41A and -S45F mutations on PTX resistance
may be attributed to the different genetic profiles of these cell
lines. To the best of our knowledge, the relationship between
CTNNB1/S33P/T41A/S45F mutations and drug resistance has
rarely been investigated (49). A
comprehensive analysis of patients with BC performed by Ozcan
(59) identified CTNNB1 as a
reliable biomarker for predicting the response to taxane-based
chemotherapy in ER(+)/HER2(−) BC. The findings of the present study
regarding MCF-7 are consistent with previous studies (49,59).
Yoo et al (60) reported that while high mutation
rates in TP53, PIK3CA, BRCA2 and ATM serine/threonine
kinase genes are observed in patients with TNBC, CTNNB1
mutations are rare. Furthermore, Geyer et al (61) reported that activation of the
WNT/β-catenin pathway is notably associated with the TNBC phenotype
with poor clinical outcomes but is not triggered by the
CTNNB1 exon 3 mutations. The aforementioned studies support
the findings of the present study on the association between
CTNNB1 mutation and PTX response in MDA-MB-231 cells and
suggest that CTNNB1 mutations do not drive PTX resistance in
TNBC. Therefore, the activation of the WNT/β-catenin pathway in
TNBC may not be linked to CTNNB1 exon 3 mutations, or this
activation could be driven by mutations in other exons of
CTNNB1 or genes associated with the WNT/β-catenin
pathway.
In conclusion, to the best of our knowledge, the
present study is the first to demonstrate the association of
AKT1 and CTNNB1 mutations with PTX resistance in BC
cells. AKT1-E17K/E49K and CTNNB1-S45F/T41A mutations
may serve as predictive biomarkers for PTX resistance in patients
with BC and ER(+)/PR(+)/HER2(−), while AKT1-E17K/E49K/L52R
mutations may be used as indicators of PTX resistance in patients
with TNBC. The emergence of resistance to cancer therapies
highlights the necessity for personalized treatment approaches.
Although the association between AKT1/CTNNB1
mutations and PTX resistance has been established in the present
study, the underlying molecular mechanisms, particularly the roles
of these mutations in PTX resistance via the PI3K/AKT and
WNT/β-catenin pathways, remain unclear. To the best of our
knowledge, the phosphorylation status of AKT1 and β-catenin, as
well as their downstream effectors such as mTOR and GSK-3β, have
not been investigated. Furthermore, considering the highly
heterogeneous nature of BC, the use of only MCF-7 and MDA-MB-231
cell lines limits the generalizability of the present findings.
Finally, the absence of combination therapy involving PTX and other
widely used chemotherapeutics, and the lack of in vivo
experiments are key limitations of the present study. Future
studies should incorporate additional HER2(+) BC cell lines, such
as BT474 and SK-BR-3, to represent the various BC subtypes. Future
studies should also include gene silencing/overexpression
experiments, protein-protein interaction assays and
phosphorylation-specific analyses to explore the roles of these
mutations in PTX resistance via the PI3K/AKT and WNT/β-catenin
pathways. To validate the clinical implications of the in
vitro results, future studies should also incorporate in
vivo xenograft tumor models.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Scientific Research
Projects Coordination Unit of Istanbul University (grant no.
TDK-2018-30153).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
GAU and PAS designed the study, analyzed and
interpreted data and wrote the manuscript. GAU and GHC performed
the experiments. GAU and PAS confirm the authenticity of all the
raw data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
BC
|
breast cancer
|
|
PTX
|
paclitaxel
|
|
PI3K
|
phosphatidylinositol 3-kinase
|
|
AKT
|
protein kinase B
|
|
WT
|
wild-type
|
References
|
1
|
Bray F, Laversanne M, Sung H, Ferlay J,
Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 74:229–263. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Arnold M, Morgan E, Rumgay H, Mafra A,
Singh D, Laversanne M, Vignat J, Gralow JR, Cardoso F, Siesling S
and Soerjomataram I: Current and future burden of breast cancer:
Global statistics for 2020 and 2040. Breast. 66:15–23. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Turashvili G and Brogi E: Tumor
heterogeneity in breast cancer. Front Med (Lausanne). 4:2272017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tang Y, Wang Y, Kiani MF and Wang B:
Classification, treatment strategy, and associated drug resistance
in breast cancer. Clin Breast Cancer. 16:335–343. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shi X, Wang J, Lei Y, Cong C, Tan D and
Zhou X: Research progress on the PI3K/AKT signaling pathway in
gynecological cancer. Mol Med Rep. 19:4529–4535. 2019.PubMed/NCBI
|
|
6
|
Noorolyai S, Shajari N, Baghbani E,
Sadreddini S and Baradaran B: The relation between PI3K/AKT
signaling pathway and cancer. Gene. 698:120–128. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dong C, Wu J, Chen Y, Nie J and Chen C:
Activation of PI3K/AKT/mTOR pathway causes drug resistance in
breast cancer. Front Pharmacol. 12:6286902021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cidado J and Park BH: Targeting the
PI3K/Akt/mTOR pathway for breast cancer therapy. J Mammary Gland
Biol Neoplasia. 17:205–216. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nicholson KM and Anderson NG: The protein
kinase B/Akt signaling pathway in human malignancy. Cell Signal.
14:381–395. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Basu A and Lambring CB: Akt isoforms: A
family affair in breast cancer. Cancers. 13:34452021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Carpten JD, Faber AL, Horn C, Donoho GP,
Briggs SL, Robbins CM, Hostetter G, Bpguslawski S, Moses TY, Savage
S, et al: A transforming mutation in the plekstrin homology domain
of AKT1 in cancer. Nature. 448:439–444. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Askham JM, Platt F, Chambers PA, Snowden
H, Taylor CF and Knowles MA: AKT1 mutations in bladder cancer:
Identification of a novel oncogenic mutation that can co-operate
with E17K. Oncogene. 29:150–155. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Parikh C, Janakiraman V, Wu WI, Foo CK,
Kljavin NM, Chaudhuri S, Stawiski E, Lee B, Lin J, Li H, et al:
Disruption of PH-kinase domain interactions leads to oncogenic
activation of AKT in human cancers. Proc Natl Acad Sci USA.
109:19368–19373. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hinz N and Jücker M: Distinct functions of
AKT isoforms in breast cancer: A comprehensive review. Cell Commun
Signal. 17:1–29. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jung SH, Kim MS, Lee SH, Park HC, Choi HJ,
Maeng L, Min KO, Kim J, Park TI, Shin OR, et al: Whole-exome
sequencing identifies recurrent AKT1 mutations in sclerosing
hemangioma of lung. Proc Natl Acad Sci USA. 113:10672–10677. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
MacDonald BT, Tamai K and He X:
Wnt/β-catenin signaling: Components, mechanisms, and diseases. Dev
Cell. 17:9–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu X, Zhang M, Xu F and Jiang S: Wnt
signaling in breast cancer: Biological mechanisms, challenges and
opportunities. Mol Cancer. 19:1652020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Clevers H: Wnt/β-catenin signaling in
development and disease. Cell. 127:469–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kimelman D and Xu W: β-Catenin destruction
complex: Insights and questions from a structural perspective.
Oncogene. 25:7482–7491. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu J, Xiao Q, Xiao J, Niu C, Li Y, Zhang
X, Zhou Z, Shu G and Yin G: Wnt/β-catenin signaling: Function,
biological mechanisms, and therapeutic opportunities. Signal
Transduc Target Ther. 7:32022. View Article : Google Scholar
|
|
21
|
Gao C, Wang Y, Broaddus R, Sun L, Xue F
and Zhang W: Exon 3 mutations of CTNNB1 drive tumorigenesis: A
review. Oncotarget. 9:5492–5508. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Van Nhieu JT, Renard CA, Wei Y, Cherqui D,
Zafrani ES and Buendia MA: Nuclear accumulation of mutated
β-catenin in hepatocellular carcinoma is associated with increased
cell proliferation. Am J Pathol. 155:703–710. 1999. View Article : Google Scholar
|
|
23
|
Tanaka Y, Kato K, Notohara K, Hojo H,
Ijiri R, Miyake T, Nagahara N, Sasaki F, Kitagawa N, Nakatani Y and
Kobayashi Y: Frequent β-catenin mutation and cytoplasmic/nuclear
accumulation in pancreatic solid-pseudo papillary neoplasm. Cancer
Res. 61:8401–8404. 2001.PubMed/NCBI
|
|
24
|
Machin P, Catasus L, Pons C, Muñoz J,
Matias-Guiu X and Prat J: CTNNB1 mutations and β-catenin expression
in endometrial carcinomas. Hum Pathol. 33:206–212. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lévy L, Neuveut C, Renard CA, Charneau P,
Branchereau S, Gauthier F, Nhieu JTV, Cherqui D, Petit-Bertron AF,
Mathieu D and Buendia MA: Transcriptional activation of
interleukin-8 by β-catenin-Tcf4. J Biol Chem. 277:42386–42393.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Austinat M, Dunsch R, Wittekind C,
Tannapfel A, Gebhardt R and Gaunitz F: Correlation between
β-catenin mutations and expression of Wnt-signaling target genes in
hepatocellular carcinoma. Mol Cancer. 7:21–30. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hamada S, Futamura N, Ikuta K, Urakawa H,
Kozawa E, Ishiguro N and Nishida Y: CTNNB1 S45F mutation predicts
poor efficacy of meloxicam treatment for desmoid tumors: A pilot
study. PLoS One. 9:e963912014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
An J, Woo HY, Lee Y, Kim HS, Jeong J and
Kim SK: Clinicopathological features of 70 desmoid-type
fibromatoses confirmed by β-catenin immunohistochemical staining
and CTNNB1 mutation analysis. PLoS One. 16:e02506192021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Oulès B, Mourah S, Baroudjian B, Jouenne
F, Delyon J, Louveau B, Gruber A, Lebbe C and Battistella M:
Clinicopathologic and molecular characterization of melanomas
mutated for CTNNB1 and MAPK. Virchows Arch. 480:475–480. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Norkowski E, Masliah-Planchon J, Le
Guellec S, Trassard M, Courrèges JB, Charron-Barra C, Terrier P,
Bonvalot S, Coindre JM and Laé M: Lower rate of CTNNB1 mutations
and higher rate of APC mutations in desmoid fibromatosis of the
breast: A series of 134 tumors. Am J Surg Pathol. 44:1266–1273.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li G, Xu D, Sun J, Zhao S and Zheng D:
Paclitaxel inhibits proliferation and invasion and promotes
apoptosis of breast cancer cells by blocking activation of the
PI3K/AKT signaling pathway. Adv Clin Exp Med. 29:1337–1345. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhao S, Tang Y, Wang R and Najafi M:
Mechanisms of cancer cell death induction by paclitaxel: An updated
review. Apoptosis. 27:647–667. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Alalawy AI: Key genes and molecular
mechanisms related to Paclitaxel Resistance. Cancer Cell Int.
24:2442024. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Froger A and Hall JE: Transformation of
plasmid DNA into E. coli using the heat shock method. J Vis Exp.
2532007.doi: 10.3791/253. PubMed/NCBI
|
|
35
|
Ke X and Shen L: Molecular targeted
therapy of cancer: The progress and future prospect. Front Med.
1:69–75. 2017.
|
|
36
|
Tarighati E, Keivan H and Mahani H: A
review of prognostic and predictive biomarkers in breast cancer.
Clin Exp Med. 23:1–16. 2023.PubMed/NCBI
|
|
37
|
Duffy MJ and Crown J: A personalized
approach to cancer treatment: How biomarkers can help. Clin Chem.
54:1770–1779. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Beaver JA, Gustin JP, Yi KH, Rajpurohit A,
Thomas M, Gilbert SF, Rosen DM, Park BH and Lauring J: PIK3CA and
AKT1 mutations have distinct effects on sensitivity to targeted
pathway inhibitors in an isogenic luminal breast cancer model
system. Clin Cancer Res. 19:5413–5422. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Welsh J: Animal models for studying
prevention and treatment of breast cancer. Animal Models for the
Study of Human Disease. Conn PM: Academic Press; Cambridge: pp.
997–1018. 2013, View Article : Google Scholar
|
|
40
|
Waks AG and Winer EP: Breast cancer
treatment: A review. JAMA. 321:288–300. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Burguin A, Diorio C and Durocher F: Breast
cancer treatments: Updates and new challenges. J Pers Med.
11:8082021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kaboli PJ, Imani S, Jomhori M and Ling KH:
Chemoresistance in breast cancer: PI3K/Akt pathway inhibitors vs
the current chemotherapy. Am J Cancer Res. 11:51552021.PubMed/NCBI
|
|
43
|
Kim MS, Jeong EG, Yoo NJ and Lee SH:
Mutational analysis of oncogenic AKT E17K mutation in common solid
cancers and acute leukaemias. Br J Cancer. 98:1533–1535. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lauring J, Park BH and Wolff AC: The
phosphoinositide-3-kinase-Akt mTOR pathway as a therapeutic target
in breast cancer. J Natl Compre Cancer Netw. 11:670–678. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Guo G, Qiu X, Wang S, Chen Y, Rothman PB,
Wang Z and Chen JL: Oncogenic E17K mutation in the plekstrin
homology domain of AKT1 promotes v-Abl-mediated pre-B-cell
transformation and survival of Pim-deficient cells. Oncogene.
29:3845–3853. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yang WL, Zhang X and Lin HK: Emerging role
of Lys-63 ubiquitination in protein kinase and phosphatase
activation and cancer development. Oncogene. 29:4493–4503. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yi KH, Axtmayer J, Gustin JP, Rajpurohit A
and Lauring J: Functional analysis of non-hotspot AKT1 mutants
found in human breast cancers identifies novel driver mutations:
Implications for personalized medicine. Oncotarget. 4:29–34. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
De Marco C, Malanga D, Rinaldo N, De Vita
F, Scrima M, Lovisa S, Fabris L, Carriero MV, Franco R, Rizzuto A,
et al: Mutant AKT1-E17K is oncogenic in lung epithelial cells.
Oncotarget. 6:39634–39650. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hasbal-Celikok G, Aksoy-Sagirli P,
Altiparmak-Ulbegi G and Can A: Identification of AKT1/β-catenin
mutations conferring cetuximab and chemotherapeutic drug resistance
in colorectal cancer treatment. Oncol Lett. 21:1–10. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Davies BR, Guan N, Logie A, Crafter C,
Hanson L, Jacobs V, James N, Dudley P, Jacques K, Ladd B, et al:
Tumors with AKT1 E17K mutations are rational targets for single
agent or combination therapy with AKT inhibitors. Mol Can Ther.
14:2441–2451. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hyman DM, Smyth LM, Donoghue MTA, Westin
SN, Bedard PL, Dean EJ, Bando H, El–Khoueiry AB, Pérez–Fidalgo JA,
Mita A, et al: AKT inhibition in solid tumors with AKT1 mutations.
J Clin Oncol. 35:2251–2259. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Schmid P, Abraham J, Chan S, Wheatley D,
Brunt AM, Nemsadze G, Baird RD, Park YH, Hall PS, Perren T, et al:
Capivasertib plus paclitaxel versus placebo plus paclitaxel as
first-line therapy for metastatic triple-negative breast cancer:
The PAKT trial. J Clin Oncol. 38:423–433. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Smyth LM, Tamura K, Oliveira M, Ciruelos
EM, Mayer IA, Sablin MP, Biganzoli L, Ambrose HJ, Ashton J,
Barnicle A, et al: Capivasertib, an AKT kinase inhibitor, as
monotherapy or in combination with fulvestrant in patients with
AKT1 E17K-Mutant, ER-positive metastatic breast cancer. Clin Cancer
Res. 26:3947–3957. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lin SY, Xia W, Wang JC, Kwong KY, Spohn B,
Wen Y, Pestell RG and Hung MC: Beta-catenin, a novel prognostic
marker for breast cancer: Its roles in cyclin D1 expression and
cancer progression. Proc Natl Acad Sci USA. 97:4262–4266. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ryo A, Nakamura M, Wulf G, Liou YC and Lu
KP: Pin1 regulates turnover and subcellular localization of
beta-catenin by inhibiting its interaction with APC. Nat Cell Biol.
3:793–801. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Li S, Li S, Sun Y and Li L: The expression
of β-catenin in different subtypes of breast cancer and its
clinical significance. Tumor Biol. 35:7693–7698. 2014. View Article : Google Scholar
|
|
57
|
Samant C, Kale R, Pai KSR, Nandakumar K
and Bhonde M: Role of Wnt/β-catenin pathway in cancer drug
resistance: Insights into molecular aspects of major solid tumors.
Biochem Biophys Res Commun. 729:1503482024. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Mukherjee N and Panda CK: Wnt/β-catenin
signaling pathway as chemotherapeutic target in breast cancer: An
update on pros and cons. Clin Breast Cancer. 20:361–370. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ozcan G: PTCH1 and CTNNB1 emerge as
pivotal predictors of resistance to neoadjuvant chemotherapy in
ER+/HER2-breast cancer. Front Oncol. 13:12164382023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yoo TK, Lee WS, Kim J, Kim MK, Park IA,
Kim JH and Han W: Mutational analysis of triple-negative breast
cancer using targeted kinome sequencing. J Breast Cancer.
25:1642022. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Geyer FC, Lacroix-Triki M, Savage K,
Arnedos M, Lambros MB, MacKay A, Natrajan R and Reis-Filho JS:
β-catenin pathway activation in breast cancer is associated with
triple-negative phenotype but not with CTNNB1 mutation. Mod Pathol.
24:209–231. 2011. View Article : Google Scholar : PubMed/NCBI
|