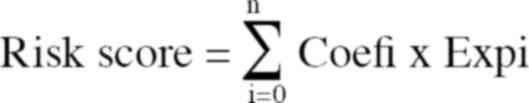Introduction
Cervical squamous cell carcinoma (CSCC) is one of
the most common gynecological cancer types affecting women
worldwide. The International Agency for Research on Cancer (IARC)
reported that there were 60,412 new cases of cervical cancer in
2020, which resulted in 341,831 deaths (1). The high-risk human papillomavirus
continues to be the primary pathogen responsible for CSCC (2). Epigenetic modifications, including DNA
methylation, histone modification, non-coding RNA regulation and
chromatin remodeling, are closely associated with the development
of CSCC (3). However, these
mechanisms do not fully elucidate the pathogenesis of CSCC,
necessitating further exploration of treatment strategies based on
epigenetic modifications.
The most prevalent and reversible RNA alteration
found in mammals is the N6-methyladenosine
(m6A) modification (4).
Other types of RNA modification, such as the
N7-methylguanosine (m7G) modification, which
is mainly defined by the creation of a ‘cap’ structure at the
5′-untranslated region of mRNA, transfer RNA (tRNA), ribosomal RNA
(rRNA) and microRNA (miRNA/miR), have also received attention
recently (5,6). Methyltransferase 1 (METTL1) and its
cofactor, WD repeat domain 4 (WDR4), together form the METTL1/WDR4
complex, which is the most extensively researched regulatory
component of m7G modification. The METTL1/WDR4 complex
is essential for the biological roles of m7G
modification across tRNA, rRNA, miRNA and mRNA (7). For example, METTL1 modifies the
m7G modification of rRNA in bladder cancer, regulating
the ribosome during tRNA-mRNA codon recognition (8). Furthermore, METTL1 knockdown markedly
increases the sensitivity of HeLa cells to 5-fluorouracil,
suggesting that m7G alteration is a viable target to
overcome tumor cell resistance (9).
By controlling the m7G alteration of tRNA, the
METTL1/WDR4 complex increases the production of EGFR protein in
hepatocellular carcinoma, reducing the susceptibility of liver
cancer cells to Lenvatinib (10).
Aberrant m7G alteration is frequently linked to a
variety of tumor outcomes, including the promotion of bladder,
liver and head and neck cancer progression and the possible
inhibition of teratoma progression (11,12).
However, it is currently unclear how the m7G mutation
affects the development of cervical cancer.
Numerous non-coding RNA functions have been
identified as high-throughput sequencing technologies have advanced
(13). Transcripts >200
nucleotides, known as long non-coding RNAs (lncRNAs) (14), are essential for vital biological
processes at the transcriptional, translational and
post-translational stages (15). In
the field of oncology, lncRNAs modulate the expression of target
genes in tumors, thereby altering the biological behaviors of
cancer cells (16). Previous
research has demonstrated the key roles that lncRNAs serve in
cervical cancer growth, metastasis, drug resistance,
immuno-environmental changes and metabolic reprogramming (17). Compared with proteins, lncRNAs are
highly specialized. Novel techniques for targeted therapy can be
derived from clustering tumor subtypes based on the differential
expression patterns of lncRNAs (18–20).
According to an analysis of The Cancer Genome Atlas (TCGA), the
expression levels of lncRNAs are frequently dysregulated in cancer
and has the highest cancer type-specificity, followed by
pseudogenes and then protein-coding genes, which were least subtype
specific and ~18.27% of lncRNAs showed subtype specificity, while
only 10.55% of protein-coding genes were subtype-specific (21–24).
Therefore, from this perspective, dysregulated lncRNAs hold greater
specificity for tumor diagnosis and classification compared with
protein-coding genes, which adds further importance to the
identification of specific lncRNAs as tumor biomarkers.
To the best of our knowledge, research on lncRNAs
linked to the m7G modification in CSCC has not yet been
conducted. Therefore, the aim of the present study was to identify
m7G-related lncRNAs and build prognostic, immune
infiltration and drug-sensitivity models around them, which may be
valuable for CSCC genotyping, diagnosis and prognostic evaluation
in the future.
Materials and methods
Datasets
Transcriptome data and clinical features of patients
with CSCC and normal individuals were retrieved from TCGA
(https://portal.gdc.cancer.gov/) and
Genotype-Tissue Expression (GTEx; https://www.genome.gov/Funded–Programs–Projects/Genotype–Tissue–Expression–Project;
GTEx_ Analysis_2017-06-05_v8_RNASeQCv1.1.9_gene_reads.gct.gz)
databases and a dataset of 260 samples, which included 248
cancerous tissues and 12 normal tissues or adjacent non-cancerous
tissues. Furthermore, 35 m7G-related genes were gathered
from the Gene Set Enrichment Analysis (GSEA; http://www.gsea-msigdb.org/gsea/index.jsp) website and
relevant published literature (25).
The present research was not subject to ethical
committee review as all data was obtained from publicly accessible
databases.
Identification of
m7G-related lncRNAs
Gene annotation probes for the expression matrix
were downloaded from GENCODE (https://www.gencodegenes.org/). Differential analysis
was performed using the ‘limma’ package (RStudio; Posit Software,
PBC) to obtain differentially expressed m7G-related
genes and lncRNAs, with the criteria of |log2[fold
change (FC)]|>2 and false discovery rate (FDR) <0.05. The
differentially expressed genes (DEGs) underwent Pearson's
correlation coefficient analysis (r2) and lncRNAs
meeting the criteria of |coefficients|>0.4 and P<0.05 were
defined as m7G-related lncRNAs.
Development of a prediction model
based on m7G-related lncRNAs
Integration of m7G-related lncRNAs along
with survival durations and statuses, among other clinical data was
conducted. Univariate Cox analysis was conducted using the
‘Survival’ package (RStudio; Posit Software, PBC). Least Absolute
Shrinkage and Selection Operator (LASSO) regression analysis was
performed using the ‘glmnet’ package (RStudio; Posit Software,
PBC), culminating in a predictive model after 10-fold
cross-validation. The formula was as follows:
The survival correlation regression coefficient was
denoted by Coefi. The expression value of each
m7G-related lncRNA was denoted by Expi.
Application of the prediction model in
CSCC prognosis
The ‘Survival’ package was employed to generate the
overall survival (OS) curve for CSCC. The ‘pROC’ package
facilitated the appraisal of clinicopathological characteristics
and prognostic implications through the computation of the area
under the curve (AUC) and utilizing the ‘rms’ package, a nomogram
was constructed and calibration curves were plotted to gauge the
predictive efficacy of the model across the aggregate sample.
GSEA and Kyoto Encyclopedia of Genes
and Genomes (KEGG) enrichment analysis
The ‘msigdbr’ package (RStudio; Posit Software, PBC)
was utilized for GSEA, with minimum and maximum values of gene
expression profiles set at 10 and 500 respectively, and 1,000
resampling iterations conducted. An FDR <0.25 and P<0.05 were
considered to indicate statistical significance. Pathway enrichment
analysis was performed using the ‘clusterProfiler’ package in
conjunction with KEGG analysis (http://www.genome.jp/kegg/). Visualization was
achieved through the use of the ‘ggplot2’ package (RStudio; Posit
Software, PBC).
Immune feature analysis
The ‘CIBERSORT’ package (RStudio; Posit Software,
PBC) was used to integrate transcriptomic data with the expression
of immune cell marker genes, which yielded an infiltrative
distribution score of immune cells within tumor tissues. Employing
1,000 permutations of the default matrix to ascertain P-values for
each specimen, the infiltration of immune cells in the cohort was
evaluated, with P<0.05 considered statistically significant. The
‘ggplot2’ and ‘barplot’ packages (Posit Software, PBC) were
utilized to visually represent the data.
Drug sensitivity evaluation
The ‘pRRophetic’ package (RStudio; Posit Software,
PBC) was used to evaluate the treatment efficacy for patients with
CSCC in high- and low-risk subgroups based on the IC50.
Subsequently, data visualization was performed using R packages
such as ‘ggplot2’ and ‘barplot’ (Posit Software, PBC).
RNA isolation and reverse
transcription-quantitative PCR (RT-qPCR) (26)
Human cervical cancer cell lines (SiHa and HeLa
cells) and the human cervical epithelial cell line H8 (cat. no.
BFN607200572) were obtained from the Shanghai Cell Bank (http://www.bluefcell.com). Cels were cultured in 1640
medium (containing 10% FBS and 1% streptomycin), at 37°C and 5%
CO2, in a humidified incubator with saturated humidity.
RT-qPCR was conducted to validate the expression levels of the
identified m7G-related lncRNAs. TRIzol® (cat.
no. 262307; Thermo Fisher Scientific, Inc.) was used to extract
total cellular RNA. Subsequently, total RNA was reverse-transcribed
using the PrimeScript™ RT reagent kit (cat. no. RR037A; Takara Bio,
Inc.). The thermocycling conditions used were as follows: 37°C for
15 min, 85°C for 5 sec and 4°C indefinitely. Amplification was
performed using TB Green™ Premix Ex Taq™ II (cat. no. RR820A;
Takara Bio, Inc.) on an ABI 7,500 detection system (Thermo Fisher
Scientific, Inc.). The thermocycling conditions used were as
follows: 95°C for 30 sec; 40 cycles of 95°C for 5 sec, 60°C for 34
sec; and 95°C for 15 sec, 60°C for 1 min and a final extension of
95°C for 15 sec. β-actin was used as the internal reference and the
relative expression levels of the target genes were calculated
using the 2−ΔΔCq method (26). Primer sequences are listed in
Table I.
 | Table I.Primer sequences for reverse
transcription-quantitative PCR. |
Table I.
Primer sequences for reverse
transcription-quantitative PCR.
| Gene | Sequence
(5′-3′) |
|---|
| Family with
sequence similarity 13 member A AS.1 | F:
CAAATATGGGTAAGGAGG |
|
| R:
GTTTAGAACTATGAGGGACT |
| Family with
sequence similarity 27 member E3 AS1 | F:
CACTTGAGAAACAGACCGTATTGT |
|
| R:
CTAGGATCAAGATGAACACACTGC |
| Fibroblast growth
factor 13 AS1 | F:
AAGAATGGCGGGGGCATTTA |
|
| R:
CCCCTCCCCCATACTCTTCA |
| Long intergenic
non-protein coding RNA 1089 | F:
TTTTGCCTACCCAACCCTGG |
|
| R:
CCTGCCGTTGACAGAAGGAA |
| RBAK downstream
neighbor | F:
TGGCTGTATTGATGGGGCTG |
|
| R:
ACAGGGAAAGCCCCATGTTC |
| Solute carrier
family 8 member A1 AS1 | F:
GCATATGTTGATGAGCAGGCA |
|
| R:
AGACTCAGTGACAGGGCTCA |
| β-actin | F:
AGCGAGCATCCCCCAAAGTT |
|
| R:
GGGCACGAAGGCTCATCATT |
Statistical analysis
Data analysis was primarily conducted using R
(version 4.3.2; Posit Software, PBC). LncRNAs closely associated
with m7G-related genes were defined as
m7G-related lncRNAs based on Pearson's correlation
coefficient analysis (r2), with the criteria of |Pearson
R|>0.6 and P<0.05. The present study employed univariate Cox
regression, LASSO regression, log-rank test, receiver operating
characteristic and principal component analysis (PCA) analysis,
with unpaired t-tests used for comparisons between high- and
low-risk groups. All experiments were repeated three times. One-way
ANOVA was used for the analysis of lncRNAs in multiple group
comparisons at different tumor staging, while Dunnett's post hoc
test after one-way ANOVA was used for the analysis of lncRNAs
between different cell lines. Data are presented as the mean ± SD.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Identification of 16
m7G-related lncRNAs in CSCC
In the present study, data from 260 samples were
collected from the TCGA and GTEx databases, comprising 248 tumor
samples and 12 control samples. PCA demonstrated a distinct
separation between the groups (Fig.
1A). Integrating data from GSEA database and prior literature
(27), 35 m7G-related
genes were included (Table SI).
Consequently, 16 m7G-related lncRNAs for CSCC were
identified (Fig. 1B), with 8
upregulated DEGs (NUDT1, NCBP3, WDR4, NCBP2, NCBP1, NUDT5, METTL1
and NUDT7) and 8 downregulated DEGs (EIF4A1, NUDT3, NUDT4, LSM1,
NUDT10, SNUPN, EIF4E and NUDT16; P<0.05). lncRNA information was
extracted from TCGA and GTEx databases and 1,382 differentially
expressed lncRNAs (DELs) were obtained based on the criteria of
|log2(FC)|>2 and P<0.05 (Table
SII). Heatmaps were generated to display the top 20 upregulated
and downregulated DELs (Fig. 1C).
Correlation analysis of the DEGs and DELs was performed to identify
m7G-related lncRNAs. Pearson's correlation analysis was
used to identify 203 DELs that were significantly correlated
(P<0.05) with m7G-related lncRNAs (Table SIII; Fig. 1D and E).
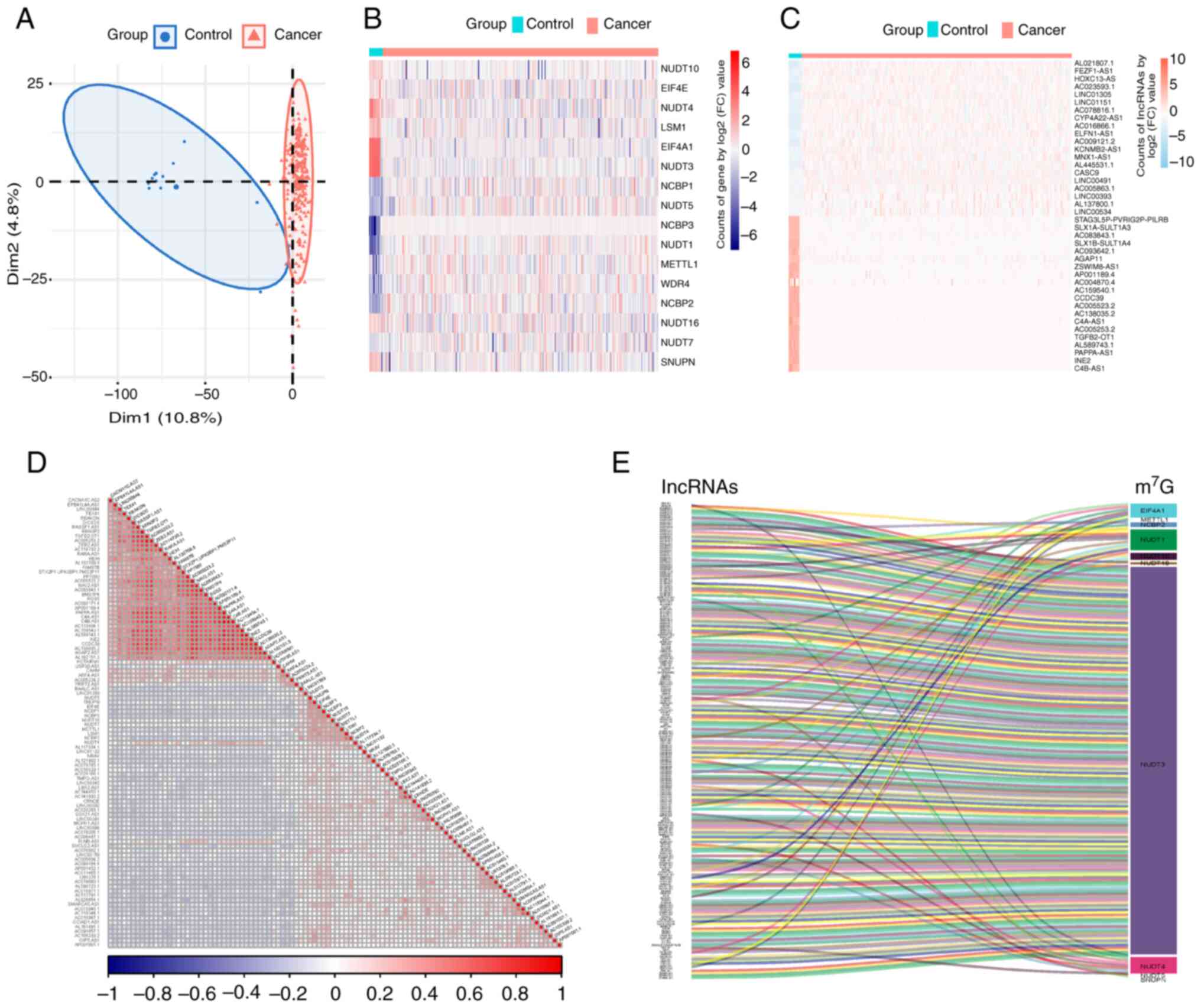 | Figure 1.Identification of
m7G-related lncRNAs in cervical squamous cell carcinoma.
(A) Principal component analysis clustering all selected samples,
with tumor tissue samples in red and samples from the healthy
control group in blue. (B) Heatmap of 16 m7G-associated
DEGs, with higher expression in red and lower expression in blue.
(C) Heatmap of DELs showcasing the top 20 DELs, with higher
expression in red and lower expression in blue. (D) Correlation
heatmap of DEGs and DELs, with positive correlations in red and
negative correlations in blue. (E) Sankey diagram showing the 204
DELs significantly correlated with the m7G genes. DEG,
differentially expressed gene; lncRNA, long non-coding RNA; DEL,
differentially expressed lncRNAs; m7G,
N7-methylguanosine; Dim1, dimension 1 formed after data
dimensionality reduction; Dim2, dimension 2 formed after data
dimensionality reduction. |
Establishment of a prediction model
based on m7G-related lncRNAs
A univariate Cox analysis of 204
m7G-related lncRNAs and survival data was conducted,
where 22 lncRNAs with independent predictive efficacy were
identified and shown in a forest plot (Fig. 2A). Following LASSO regression
analysis, 6 significant m7G-related lncRNAs were
identified: Family with sequence similarity 13 member A antisense
RNA 1 (FAM13A.AS1), family with sequence similarity 27 member E3
(FAM27E3), fibroblast growth factor 13 antisense RNA 1 (FGF13.AS1),
long intergenic non-protein coding RNA 1089 (LINC01089), RBAK
downstream neighbor (RBAKDN) and solute carrier family 8 member A1
antisense RNA 1 (SLC8A1.AS1; Fig.
2B). The predictive model was constructed with the following
formula: Risk score=(3.24 × FAM13A.AS1) + (−3.07 × FAM27E3) + (2.94
× FGF13.AS1) + (−3.49 × LINC01089) + (3.23 × RBAKDN) + (2.86 ×
SLC8A1.AS1). Sample risk scores were calculated using this formula
and samples were grouped into high- and low-risk groups according
to the median score of 1.181 (Fig.
2C). The OS of patients was analyzed using Kaplan-Meier
analysis and survival curves were plotted. The outcome demonstrated
that the prognosis of the low-risk group significantly improved
compared with that of the high-risk group (P<0.0001; Fig. 2D).
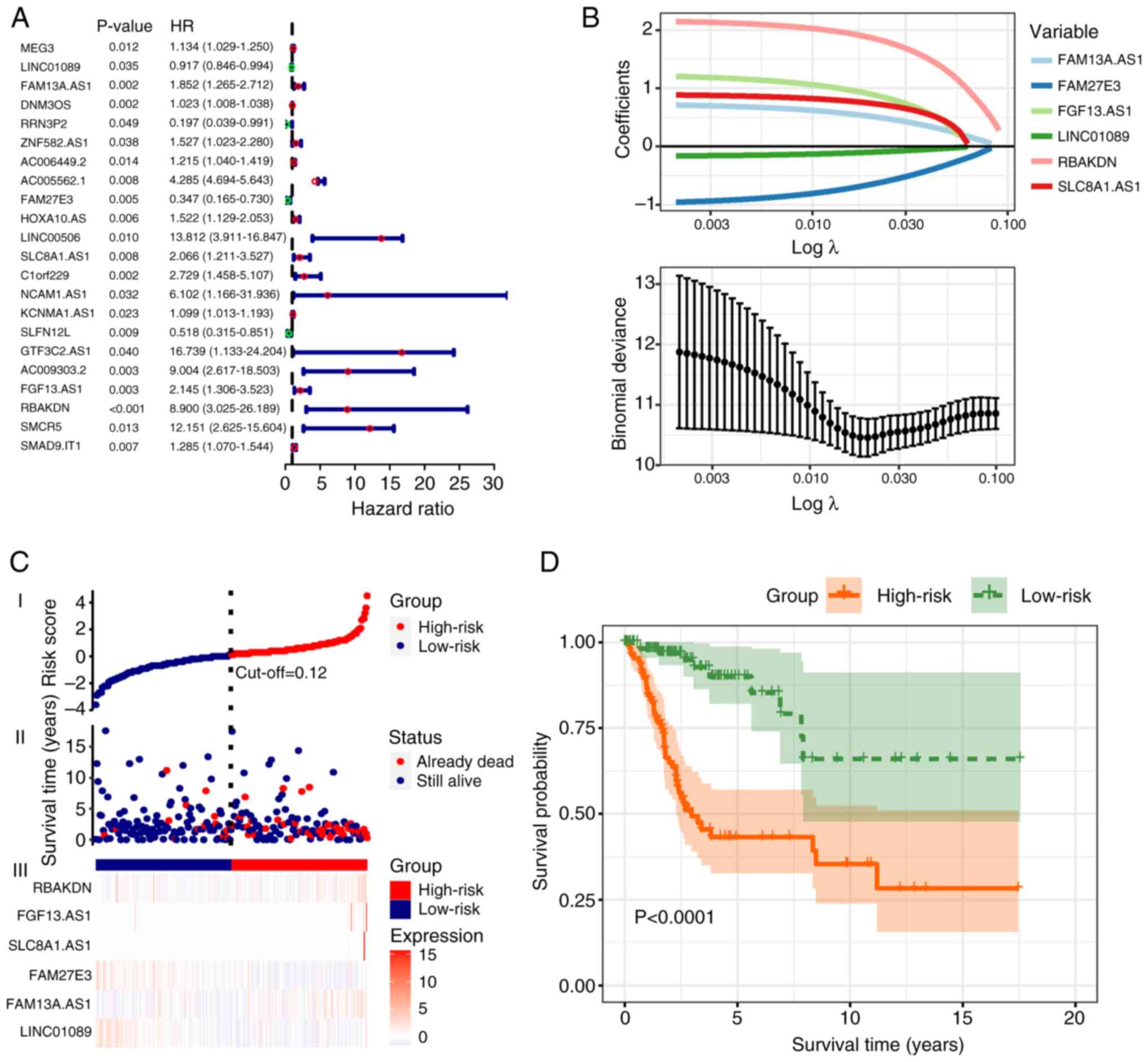 | Figure 2.Establishment of a predictive model
based on m7G-related lncRNAs. (A) Forest plot
illustrating the univariate Cox analysis used to identify 22
m7G-related lncRNAs with independent prognostic
prediction capabilities. The results are expressed using hazard
ratios (95% CI). (B) Least Absolute Shrinkage and Selection
Operator regression plot. Binomial deviances are expressed as mean
± SD. (C) Scatter plot and risk heatmap. The optimal cut-off value
was determined to be 0.12, which distinguished the high-risk group
(red) from the low-risk group (blue). (D) Kaplan-Meier survival
curves. The green curve represents the low-risk group, whereas the
orange curve represents the high-risk group. The survival rate of
the high-risk group was found to be lower compared to that of the
low-risk group. m7G, N7-methylguanosine;
lncRNA, long non-coding RNA; AS1, antisense RNA 1; FAM13A.AS1,
family with sequence similarity 13 member A AS.1; FAM27E3, family
with sequence similarity 27 member E3 AS1; FGF13.AS1, fibroblast
growth factor 13 AS1; LINC01089, long intergenic non-protein coding
RNA 1089; RBAKDN, RBAK downstream neighbor; SLC8A1.AS1 solute
carrier family 8 member A1 AS1. |
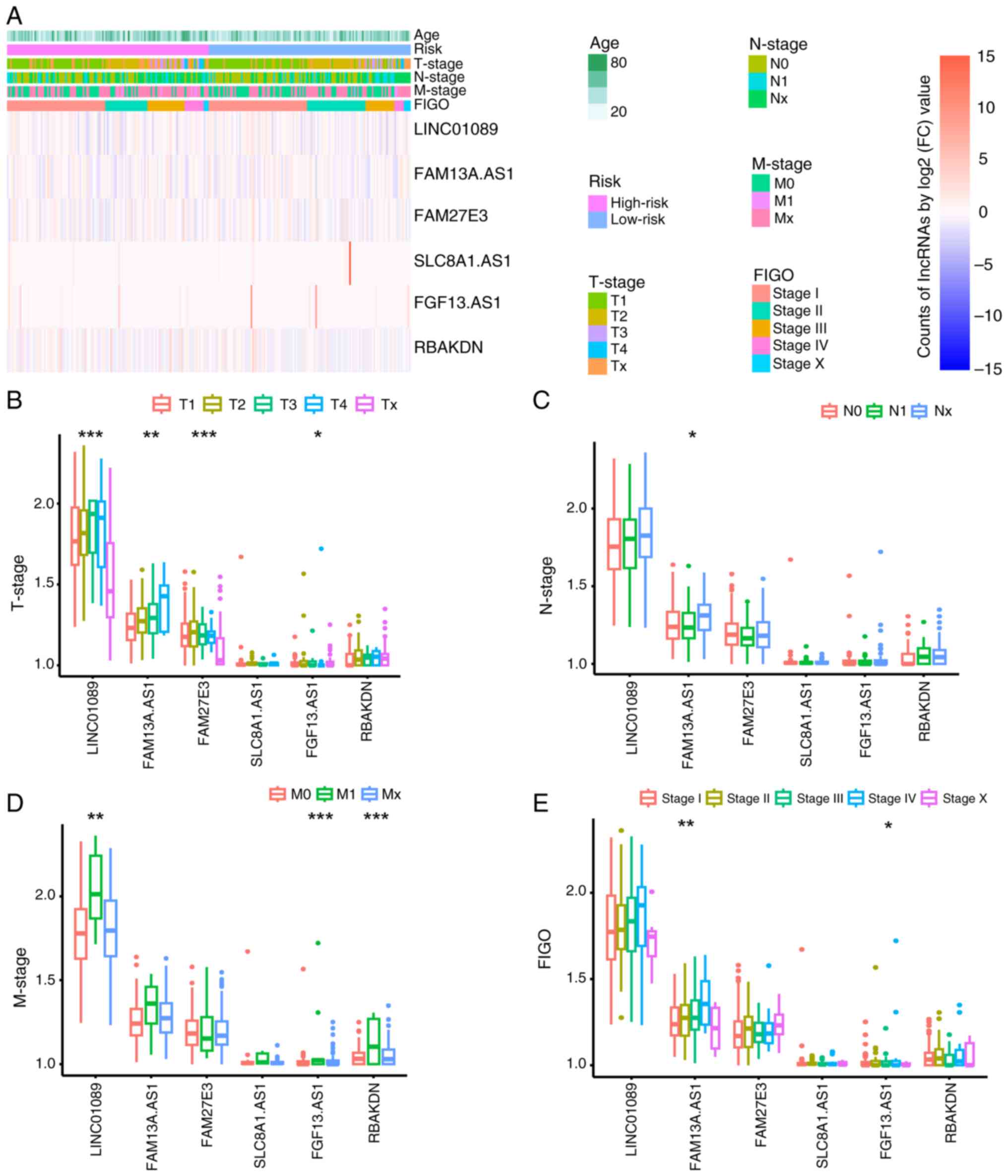
Comparison of clinical characteristics
between groups
One-way ANOVA indicated no significant differences
in age, TNM or International Federation of Gynecology and
Obstetrics (FIGO) staging between the groups (Fig. 3A). To further examine potential
associations between clinical factors and m7G-related
lncRNAs, one-way ANOVA was used. Significant differences were
observed in the expression levels of LINC01089, FAM13A.AS1,
FAM27E3, and FAM13.AS1 across different T stages (P<0.05), with
the expression levels of FAM13A.AS1 also showing significant
differences across different N stages (P<0.05) and the
expression levels of LINC01089, FAM13.AS1 and RBAKDN showing
significant differences across different M stages (P<0.05).
Additionally, FAM13A.AS1 and FAM13.AS1 expression exhibited
significant differences across different FIGO stages (P<0.05;
Fig. 3B-E). Based on these
findings, it was evident that FAM13A.AS1 and FAM13.AS1 exhibited
notable differences across various clinical parameters,
underscoring their pivotal roles as principal predictive
biomarkers.
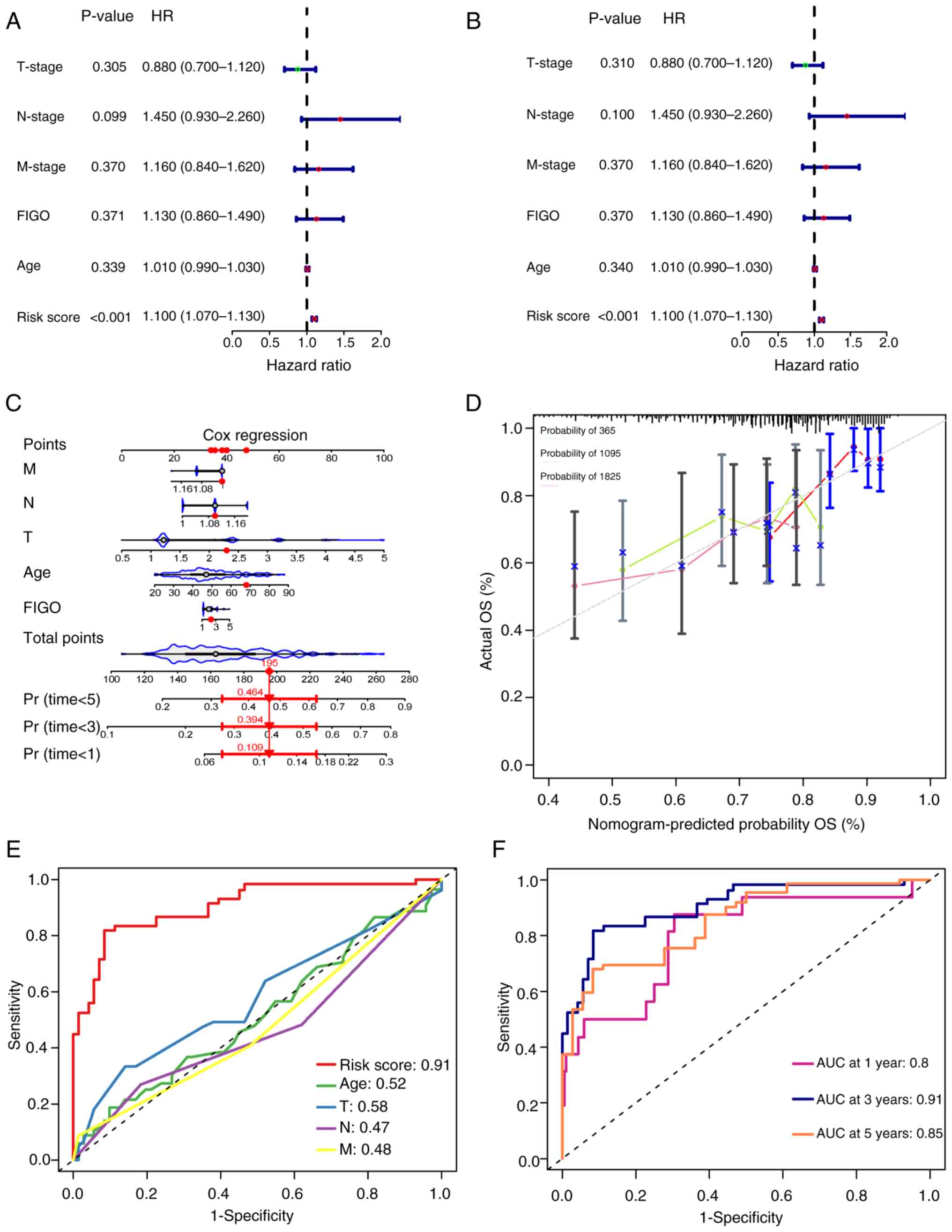
Prediction model serves as an
independent risk factor for the prognosis of CSCC
To validate whether the risk prediction model serves
as an independent prognostic factor for cervical cancer, univariate
and multivariate Cox regression analyses were conducted using age,
TNM stage, FIGO stage and model scores as covariates and the
prognostic outcomes as the independent variable. The results
demonstrated that the hazard ratio (HR) of the model score was 1.10
(P<0.001; Fig. 4A and B),
suggesting that the model score could be considered an independent
prognostic risk factor for CSCC. Furthermore, a nomogram was
constructed to evaluate the predictive efficiency of each factor
(Fig. 4C). The calibration curves
plotted demonstrated the normogram-predicted probability,
indicating good model calibration and reliability of the predictive
performance (Fig. 4D). Compared
with age or TNM staging, the model score achieves a higher AUC
value (AUC=0.91) (Fig. 4E). Based
on the prognostic times assessed using the model, the AUC for 1-,
3- and 5-year survival was 0.8, 0.91 and 0.85, respectively
(Fig. 4F), which indicated a
commendable predictive performance. These results suggested that
the risk prediction model, based on m7G-related lncRNAs,
exhibited high sensitivity and specificity in forecasting the
prognosis of CSCC.
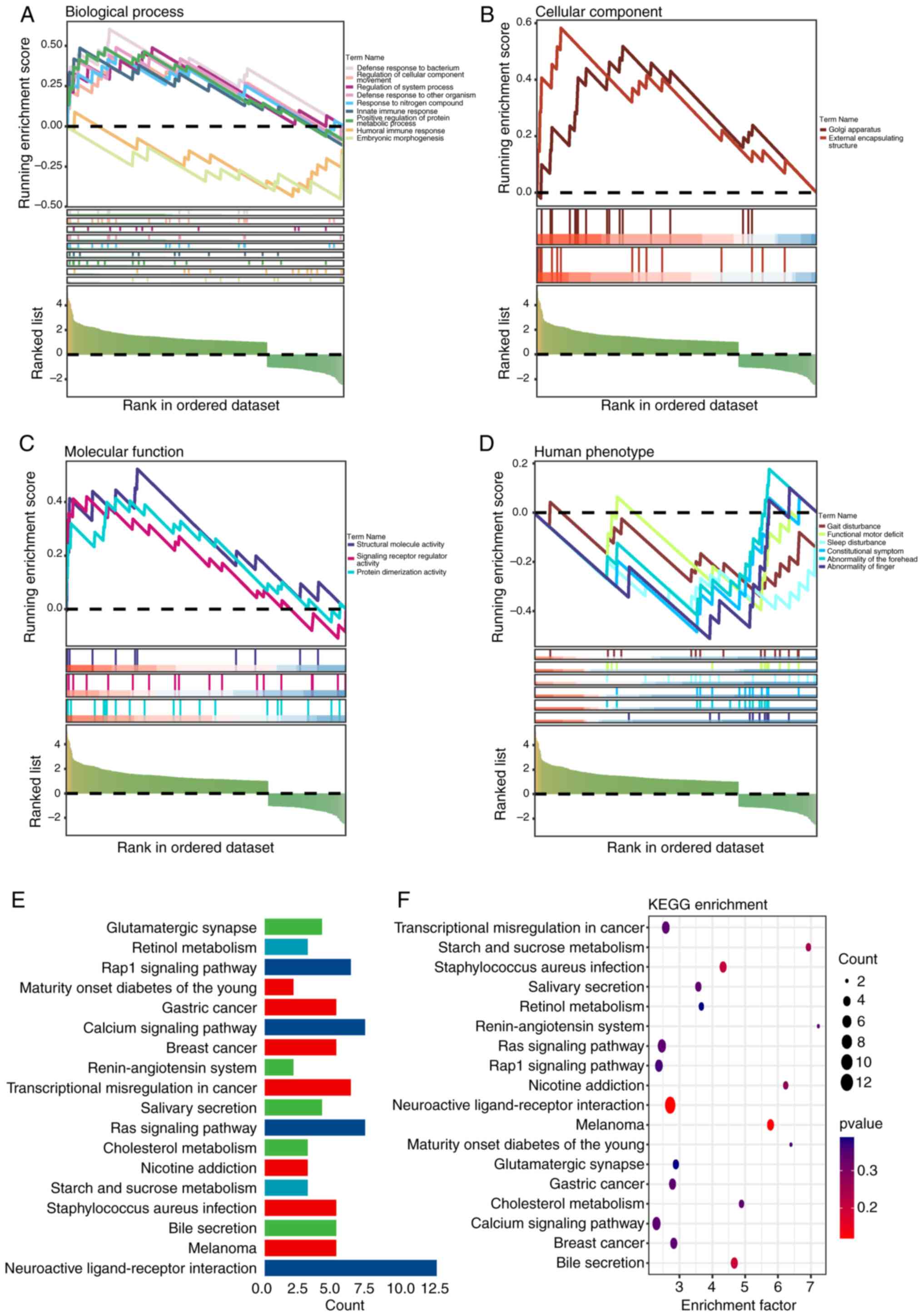
GSEA and KEGG enrichment analysis
To elucidate the enrichment processes of DEGs, GSEA
was conducted. The analysis indicated disparities in immunological
processes such as the defense response to bacteria (enrichment
score=1.943; P=0.005), innate immune response (enrichment
score=1.668; P=0.037) and humoral immune response (enrichment
score=1.646; P=0.045) between groups (Fig. 5A-D). Further exploration through
KEGG analysis highlighted pathways of interest, including
transcriptional misregulation in cancer, Ras, Ras-related protein 1
(Rap1) and calcium (Fig. 5E and
F).
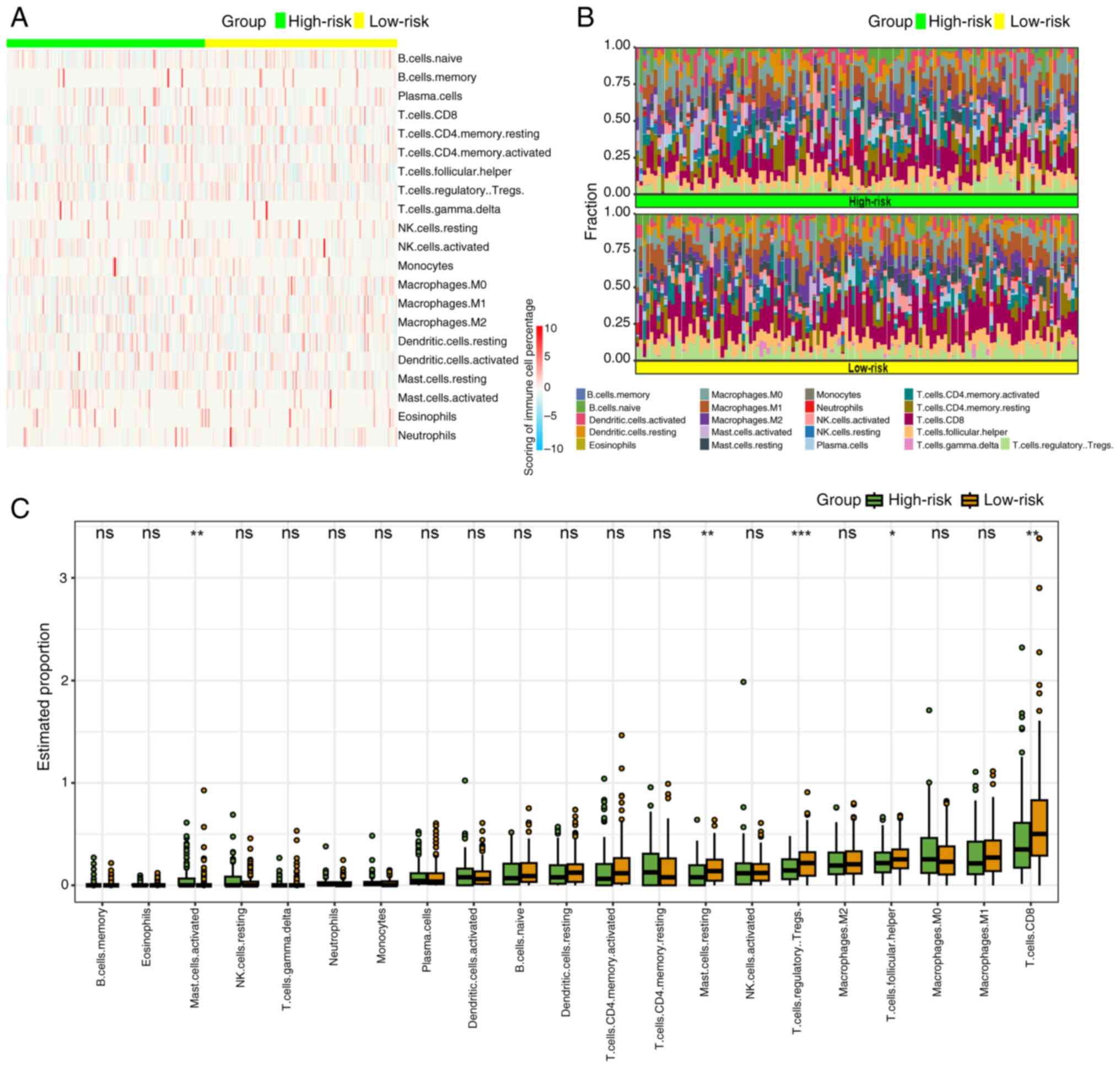
Immune infiltration landscape
The results of GSEA suggested that the progression
of CSCC was linked to anomalies in immune responses, particularly
innate and humoral immune reactions. Utilizing the ‘CIBERSORT’
algorithm, the tumor immune microenvironment was compared between
high- and low-risk groups. Given the absence of CD4 naïve T cells
in any group, the distribution differences of the remaining 21
types of immune cells were examined. Heatmaps and percentage plots
demonstrated distinct distributions of immune cells between the
groups (Fig. 6A and B).
Specifically, mast activated cells exhibited a significantly
increased infiltration in the high-risk group (P<0.05), whereas
mast resting cells, T regulatory cells, T follicular cells and
CD8+ T cells showed significantly higher infiltration in
the low-risk group (P<0.01, P<0.001, P<0.05 and P<0.01,
respectively; Fig. 6C).
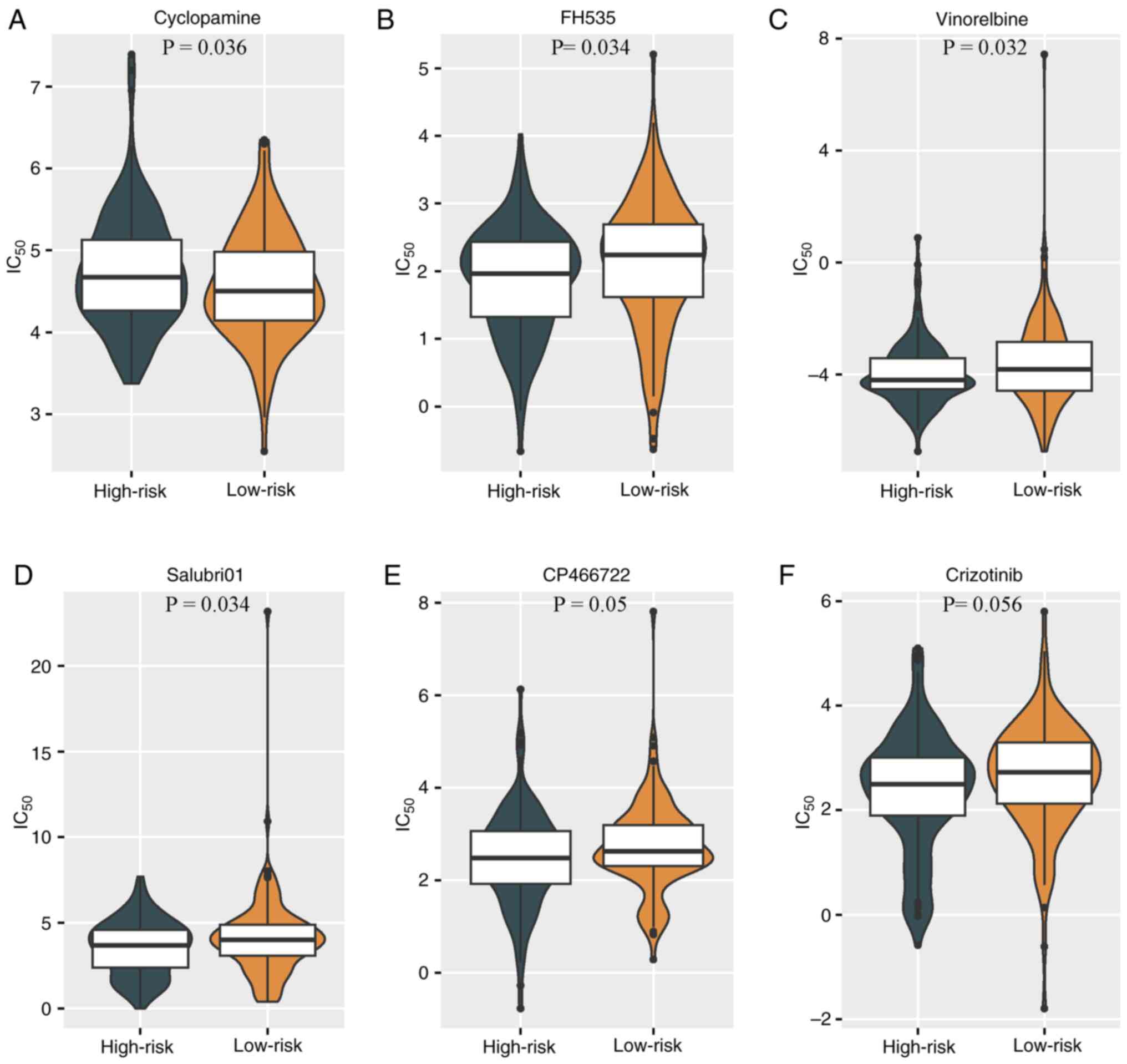
Clinical translational value of
prediction models
To further appraise the clinical applicability and
the potential for future clinical translation of the predictive
model, the differences in drug sensitivity between groups were
analyzed. GSEA and KEGG pathway enrichment analysis results
suggested a connection between cervical cancer and abnormalities in
transcriptional dysregulation, Ras signaling and other cell cycle
proteins. The analysis of drug sensitivity differences demonstrated
varying tendencies in the response to drug treatments among
different CSCC groups. The high-risk group exhibited a
significantly improved responsiveness to the cyclopamine
(P<0.05; Fig. 7A), while the
low-risk group exhibited higher sensitivity to the Wnt signaling
pathway inhibitor FH535, the cell cycle inhibitor vinorelbine, the
protein phosphatase 1 (PP1) inhibitor Salubri01, the
serine/threonine protein kinase inhibitor CP-466722 and the
tyrosine kinase receptor inhibitor crizotinib (Fig. 7B-F).
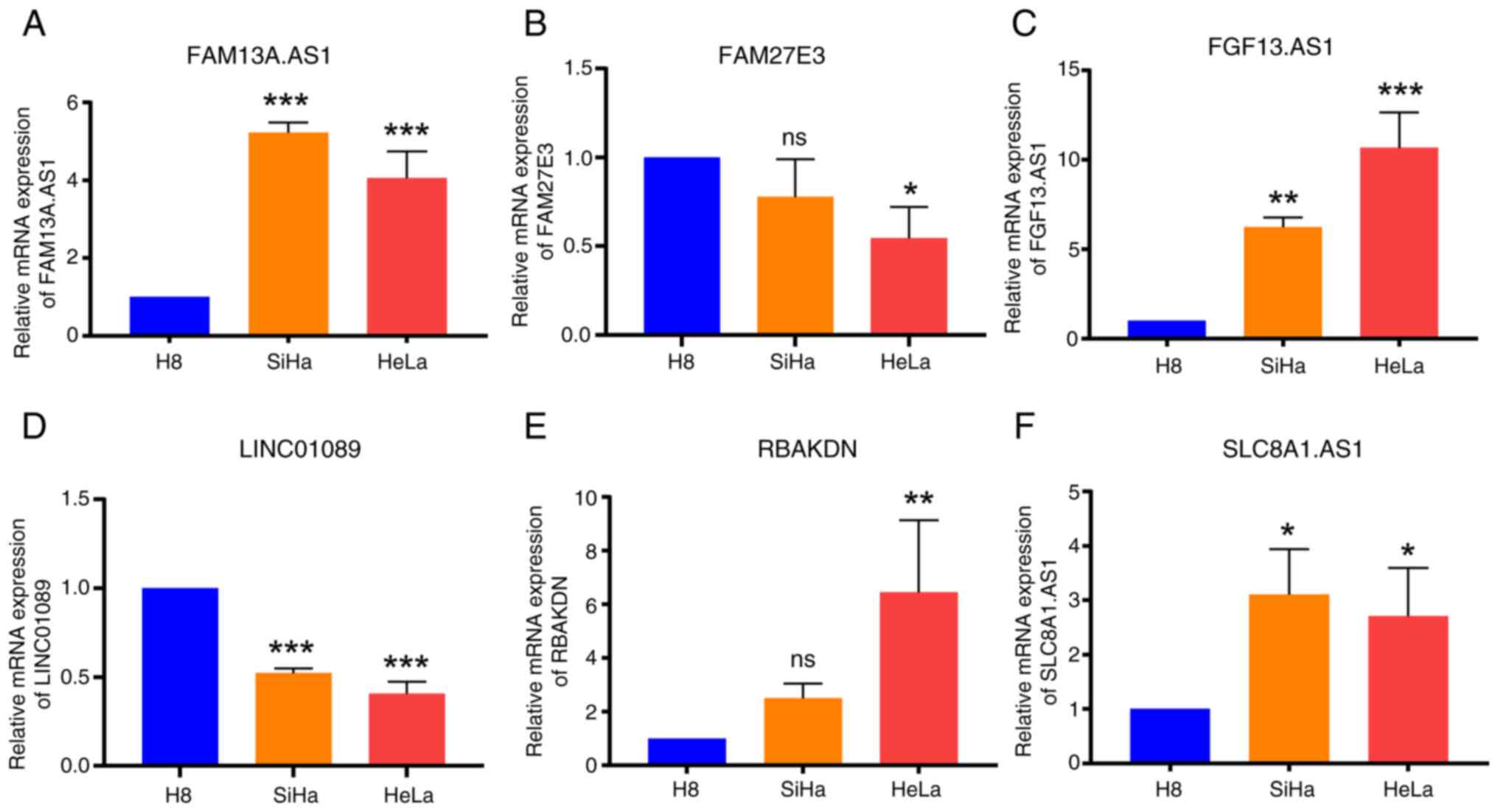
Validation of model-constructing
lncRNAs through RT-qPCR
Through analysis of the risk prediction model in
terms of clinical data, immune infiltration and drug sensitivity,
its substantial applicability in the context of CSCC was discerned.
To ascertain the precision and reliability of the lncRNAs
implicated in constructing the model, the aforementioned 6 lncRNAs
were evaluated using RT-qPCR analysis in H8, SiHa and HeLa cervical
cancer cell lines. The expression levels verified using RT-qPCR
aligned with the expression trends of lncRNAs in the datasets,
thereby affirming the high caliber and efficacy of RNA-seq data
from TCGA and GTEx databases. This corroboration further reiterated
the stability of the prognostic model (Fig. 8).
Discussion
CSCC represents a global public health challenge,
with a particularly onerous burden in numerous low- and
middle-income countries (28). An
IARC study conducted by Singh et al (28) compiled incidence and mortality rates
of cervical cancer for a decade. Their findings indicated that by
2020, there were an estimated 604,127 cases of cervical cancer
worldwide, with 341,831 fatalities (28). Squamous cell carcinoma remains the
most prevalent histological type of cervical cancer, accounting for
75–80% of cases, followed by adenocarcinoma, which accounts for
20–25% of cases (29).
Epigenetics refers to alterations in gene expression
without modifying the genetic sequence itself (30). Epigenetic modification mechanisms
are intimately linked to the development of cervical cancer, with
lncRNAs offering advantages for diagnostic and therapeutic
applications, rendering them promising targets. A previous study
reported a close association between lncRNA dysregulation and the
pathological processes underlying cervical intraepithelial
neoplasia (31). Hu et al
(32) reported that MIR210HG was
upregulated in cervical cancer cells, promoting proliferation and
migration through hypoxia-inducible factor 1α. The lncRNA DINO
activates the dormant tumor suppressor TP53 via the ATM/checkpoint
kinase 2 signaling pathway, thereby inhibiting cervical cancer cell
activity (33). In terms of
treatment, lncRNAs can influence the sensitivity of cervical cancer
to chemoradiotherapy. Zhao et al (34) found that LINC00958 could
downregulate the radiosensitivity of cervical cancer cells by
upregulating ribonucleotide reductase regulatory subunit M2.
RNA methylation modifications represent one of the
pivotal post-transcriptional regulatory mechanisms (35). Analysis of public databases by Ji
et al (36) indicated that
various m6A methylation modification-associated proteins
are upregulated, such as programmed cell death ligand 1, in
cervical cancer tissues, contributing to carcinogenesis and
correlating with elevated programmed death-ligand 1 expression. At
present, the m7G methylation modification regulatory
proteins METTL1 and WDR4, which have garnered considerable research
attention, are recognized for their role in modulating the course
of various tumors (8,12,37–40).
However, lncRNAs associated with m7G have yet to be
reported in the pathogenesis of cervical cancer. Consequently, the
present study focused on the potential of m7G-related
lncRNAs to serve as biomarkers in cervical cancer, which elucidated
their role and offered diagnostic and therapeutic insights, as well
as the identification of prospective targets for intervention.
Samples included in the present study were sourced
from TCGA and GTEx databases, where PCA demonstrated a clear
distinction between tumor and normal tissues. Drawing from the GSEA
database and extant literature, 35 m7G methylation
regulatory genes were identified. Following differential analysis
of these genes and lncRNAs, Pearson's correlation analysis yielded
204 m7G-related lncRNAs. Univariate Cox regression
analysis and LASSO regression analysis identified six
m7G-related lncRNAs (FAM13A.AS1, FAM27E3, FGF13.AS1,
LINC01089, RBAKDN and SLC8A1.AS1). Qiu et al (41) observed reduced FAM13A-AS1 expression
and elevated levels of miRNA-205-3p in cervical cancer tissues and
cell lines (SiHa and HeLa). Upregulation of FAM13A-AS1 expression
was found to inhibit the proliferation, migration and invasion of
SiHa and HeLa cells, while concurrently increasing apoptosis
(41). In renal cancer, lncRNA
FAM13A-AS1 can foster the onset of the disease through the
FAM13A-AS1/miR-141-3p/NIMA related kinase 6 axis (42). Bioinformatics studies have
identified lncRNA FAM13A-AS1 as a prognostic and drug resistance
marker in tumors such as neuroblastoma (43) and glioma (44). Although empirical validation is
pending, these findings pave the way for future research
directions. Previous studies have reported that LINC01089 exerts a
key protective effect in a variety of tumors, such as non-small
lung cancer (45–48). The predominant mechanisms are
largely associated with the competing endogenous RNA network,
principally involving pathways such as the LINC01089/miR-27a-3p/tet
methylcytosine dioxygenase 1 (45),
LINC01089/miR-152-3p/PTEN (46),
LINC01089/miR-27b-3p/HOXA10 (47)
and LINC01089/miR-27a/secreted frizzled related protein 1 (48) pathways. Among these, the
relationship between LINC01089 and miR-27a has been extensively
investigated. Li et al (49)
reported that the LINC01089/miR-27a-3p/BTG axis serves a pivotal
role in inhibiting the progression of cervical cancer.
Investigations have determined that RBAKDN is principally involved
in developmental processes (50).
Qin et al (51) also
identified RBAKDN as an immunologically relevant biomarker
characteristic of predicting early-stage CSCC. SLC8A1.AS1 is
closely associated with the biological processes of glioma
(52), thyroid carcinoma (53) and oral squamous cell carcinoma
(54). FAM27E3 and FGF13.AS1 are
still devoid of fundamental research and are primarily utilized in
the construction of predictive models. Subsequent validation
experiments indicated that, with the exception of a notable
decrease in LINC01089 expression, the remaining lncRNAs exhibited a
notable increase in cervical cancer cell lines. This finding
converges with the outcomes of the aforementioned studies, which
indirectly corroborate the results of the present study.
The predictive model constructed based on
m7G-related lncRNAs and clinical data was evaluated and
Kaplan-Meier analysis demonstrated that the low-risk group had a
significantly improved survival prognosis compared with the
high-risk group. Although no significant differences were observed
between the groups in terms of age, tumor stage or FIGO stage,
individual lncRNAs exhibited significant disparities in cervical
cancer TNM and FIGO stages, particularly FAM13A.AS1 and LINC01089.
This suggested that lncRNAs may be key prognostic indicators,
meriting focused attention in future foundational research on
cervical cancer. Furthermore, the present study evaluated whether
the risk score from the predictive model was an independent
prognostic factor for cervical cancer. The HR for the risk
prediction model score was 1.10, indicating that the risk
prediction model score could serve as an independent prognostic
risk factor for cervical cancer. In summary, the model exhibited
high sensitivity and specificity in forecasting the prognosis of
CSCC, offering valuable theoretical evidence for future clinical
applications.
Given the robustness of the present predictive
model, it was imperative to assess its clinical translational
potential. GSEA demonstrated that the progression of cervical
cancer in the high-risk group was closely associated with immune
dysregulation. This led to the conjecture that the onset of
cervical cancer is closely associated with aberrations in immune
responses. Activated mast cells exhibited higher infiltration in
the high-risk group, while resting mast cells showed higher
infiltration in the low-risk group. Studies have found that
activated mast cells in tumor tissues can promote tumor
angiogenesis and invasion by releasing classic pro-angiogenic
factors (VEGF, fibroblast growth factor 2, platelet-derived growth
factor and IL-6), non-classic pro-angiogenic factors (for example,
tryptase and chymase) and various matrix metalloproteinases
(55,56). The T regulatory cells (Tregs) were
higher in the low-risk group. While an increased number of
intratumoral Tregs is generally associated with poor prognosis in
most cancer types, such as breast cancer, lung cancer, ovarian
cancer and hepatocellular carcinoma, elevated Tregs are linked to
favorable prognosis in cancer types such as colorectal cancer,
estrogen receptor-negative breast cancer, esophageal squamous cell
carcinoma and ovarian cancer (57).
This discrepancy is primarily due to the phenotypic and functional
heterogeneity of Tregs in tumor tissues and studies associating
Tregs with favorable prognosis are often conducted in the context
of chronic inflammation (57,58).
Follicular T cells and CD8+ T cells showed higher
infiltration in the low-risk group compared with the high-risk
group. Notably, CD8+ T cells are key immune defense
cells and their exhaustion is often associated with tumor
malignancy (59). In conclusion,
findings from the present study may provide insights for future
cervical cancer immunotherapy strategies.
Drawing from the results of KEGG analysis, the
present study further examined the differences in drug sensitivity
between the two groups, which demonstrated that dysregulated
transcription, Ras, Rap1 and calcium signaling pathways, among
others, were implicated in the progression of high-risk group
cervical cancer. The high-risk group exhibited significantly
increased responsiveness to the cell cycle inhibitor cyclopamine,
whereas the low-risk group exhibited significantly decreased
sensitivity to a range of inhibitors, including the Wnt signaling
pathway inhibitor FH535, the cell cycle inhibitor vinorelbine, the
PP1 inhibitor Salubri01, the serine/threonine-protein kinase
inhibitor CP-466722 and the tyrosine kinase receptor inhibitor
crizotinib. Cyclopamine and vinorelbine are quintessential cell
cycle inhibitory drugs, while crizotinib, a tyrosine kinase
receptor inhibitor, has been noted for its relevance due to the
upregulation of tyrosine kinase receptors in cervical cancer
(60). Crizotinib serves as a
potential targeted therapy for cervical cancer (61). CP-466722 hinders ATM kinase activity
induced by ionizing radiation and this inhibition is rapidly and
fully reversible (62). FH535 acts
as a small molecule inhibitor of Wnt/β-catenin signaling and
concurrently antagonizes both PPARγ and δ, impeding the aggregation
of glutamate receptor interacting protein 1 with β-catenin
(63). Salubri01, a PP1 inhibitor,
fortifies cells against endoplasmic reticulum stress across various
model systems, synergizing markedly with proteasome inhibitors and,
to some extent, amplifying apoptosis (64). Notably, vinorelbine has reached a
mature stage of clinical application for cervical cancer and is one
of the key drugs in chemotherapy regimens for this disease
(65). Foundational research on the
tyrosine kinase receptor inhibitor crizotinib has demonstrated
anticancer activity in cervical cancer cells through the induction
of apoptosis (66). Furthermore,
the serine/threonine protein kinase inhibitor CP-466722 is known to
augment cancer cell sensitivity to radiotherapy, a modality on
which cervical cancer treatment is reliant (67). The remaining drugs have not yet been
investigated in the context of cervical cancer, underscoring the
potential of this predictive model to serve as a key guide in
future clinical applications. Overall, the risk model constructed
in the present study had notable clinical value. If patients with
CSCC can be risk stratified using m7G-related lncRNAs
before treatment, it could potentially guide clinicians in making
informed choices of therapeutic drugs in the future.
While the stability of the current risk model was
corroborated from multiple perspectives, the present study may
still harbor limitations. Since the transcriptome expression data
and clinical information of the present study subjects were
downloaded from the TCGA and GTEx databases, the difference in the
number of normal and tumor tissues is a potential limitation of the
present study, which may have introduced bias in the statistical
analysis of the results. Therefore, further validation through
expanded sample sizes in subsequent basic and clinical studies is
needed. Additionally, the lncRNAs have only been detected in
vitro, lacking confirmation through in vivo studies and
mechanistic experiments. The present study identified m7G-related
lncRNAs and developed prognostic, immune infiltration and
drug-sensitivity models, contributing to CSCC genotyping, diagnosis
and prognosis. In future studies, the complex and potential
molecular regulatory mechanisms involved should be explored.
Additionally, experiments interfering with the identified lncRNAs
in vitro to observe their effects on tumor biological
behavior should be performed in addition to the sequencing of
cervical cancer tissues prior to treatment to distinguish between
high-risk and low-risk groups for clinical drug trials and
validation of drug resistance mechanisms through in vitro
experiments.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by the Youth Training Program of
Inner Mongolia Medical University (grant no. YKD2021QN042), Science
and Technology Million Project Joint Project of Inner Mongolia
Medical University [grant no. YKD2020KJBW(LH)006], Construction of
Multi-disciplinary Comprehensive System of Clinical Medicine and
Tumor in 2023 (grant no. DC2300000607), General Project of Inner
Mongolia Medical University (grant no. YKD2021MS015), Inner
Mongolia Autonomous Region the Natural Science Foundation of Inner
Mongolia (grant no. 2023LHMS08060) and Inner Mongolia Autonomous
Region Science and Technology Planning Project (grant no.
2021GG0204).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
JZ, YB and YL designed the study and developed the
methodology. JZ and YB acquired, analyzed and interpreted the data.
JZ and YB performed the experiments. JZ wrote and revised the
original draft. ZY collected the data and revised the original
draft. YL and ZY confirmed the authenticity of all the raw data.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shanmugasundaram S and You J: Targeting
persistent human papillomavirus infection. Viruses. 9:2292017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu H, Ma H, Li Y and Zhao H: Advances in
epigenetic modifications and CC research. Biochim Biophys Acta Rev
Cancer. 1878:1888942023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang T, Kong S, Tao M and Ju S: The
potential role of RNA N6-methyladenosine in cancer progression. Mol
Cancer. 19:882020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhao F, Dong Z, Li Y, Liu S, Guo P, Zhang
D and Li S: Comprehensive analysis of molecular clusters and
prognostic signature based on m7G-related LncRNAs in esophageal
squamous cell carcinoma. Front Oncol. 12:8931862022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Luo Y, Yao Y, Wu P, Zi X, Sun N and He J:
The potential role of N7-methylguanosine (m7G) in cancer. J Hematol
Oncol. 15:632022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Alexandrov A, Martzen MR and Phizicky EM:
Two proteins that form a complex are required for 7-methylguanosine
modification of yeast tRNA. RNA. 8:1253–1266. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ying X, Liu B, Yuan Z, Huang Y, Chen C,
Jiang X, Zhang H, Qi D, Yang S, Lin S, et al: METTL1-m7
G-EGFR/EFEMP1 axis promotes the bladder cancer development. Clin
Transl Med. 11:e6752021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Okamoto M, Fujiwara M, Hori M, Okada K,
Yazama F, Konishi H, Xiao Y, Qi G, Shimamoto F, Ota T, et al: tRNA
modifying enzymes, NSUN2 and METTL1, determine sensitivity to
5-fluorouracil in HeLa cells. PLoS Genet. 10:e10046392014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang M, Long J, Yao Z, Zhao Y, Zhao Y,
Liao J, Lei K, Xiao H, Dai Z, Peng S, et al: METTL1-Mediated m7G
tRNA modification promotes lenvatinib resistance in hepatocellular
carcinoma. Cancer Res. 83:89–102. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cheng W, Gao A, Lin H and Zhang W: Novel
roles of METTL1/WDR4 in tumor via m7G methylation. Mol Ther
Oncolytics. 26:27–34. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Deng Y, Zhou Z, Ji W, Lin S and Wang M:
METTL1-mediated m7G methylation maintains pluripotency in human
stem cells and limits mesoderm differentiation and vascular
development. Stem Cell Res Ther. 11:3062020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang KC and Chang HY: Molecular mechanisms
of long noncoding RNAs. Mol Cell. 43:904–914. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schmitz SU, Grote P and Herrmann BG:
Mechanisms of long noncoding RNA function in development and
disease. Cell Mol Life Sci. 73:2491–2509. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Long Y, Wang X, Youmans DT and Cech TR:
How do lncRNAs regulate transcription? Sci Adv. 3:eaao21102017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guttman M and Rinn JL: Modular regulatory
principles of large non-coding RNAs. Nature. 482:339–346. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
He J, Huang B, Zhang K, Liu M and Xu T:
Long non-coding RNA in CC: From biology to therapeutic opportunity.
Biomed Pharmacother. 127:1102092020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cabili MN, Trapnell C, Goff L, Koziol M,
Tazon-Vega B, Regev A and Rinn JL: Integrative annotation of human
large intergenic noncoding RNAs reveals global properties and
specific subclasses. Genes Dev. 25:1915–1927. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mercer TR, Dinger ME, Sunkin SM, Mehler MF
and Mattick JS: Specific expression of long noncoding RNAs in the
mouse brain. Proc Natl Acad Sci USA. 105:716–721. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ravasi T, Suzuki H, Pang KC, Katayama S,
Furuno M, Okunishi R, Fukuda S, Ru K, Frith MC, Gongora MM, et al:
Experimental validation of the regulated expression of large
numbers of non-coding RNAs from the mouse genome. Genome Res.
16:11–19. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Iyer MK, Niknafs YS, Malik R, Singhal U,
Sahu A, Hosono Y, Barrette TR, Prensner JR, Evans JR, Zhao S, et
al: The landscape of long noncoding RNAs in the human
transcriptome. Nat Genet. 47:199–208. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brunner AL, Beck AH, Edris B, Sweeney RT,
Zhu SX, Li R, Montgomery K, Varma S, Gilks T, Guo X, et al:
Transcriptional profiling of long non-coding RNAs and novel
transcribed regions across a diverse panel of archived human
cancers. Genome Biol. 13:R752012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yan X, Hu Z, Feng Y, Hu X, Yuan J, Zhao
SD, Zhang Y, Yang L, Shan W, He Q, et al: Comprehensive genomic
characterization of long non-coding RNAs across human cancers.
Cancer Cell. 28:529–540. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Du Z, Fei T, Verhaak RG, Su Z, Zhang Y,
Brown M, Chen Y and Liu XS: Integrative genomic analyses reveal
clinically relevant long noncoding RNAs in human cancer. Nat Struct
Mol Biol. 20:908–913. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tomikawa C: 7-Methylguanosine
modifications in transfer RNA (tRNA). Int J Mol Sci. 19:40802018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Singh D, Vignat J, Lorenzoni V, Eslahi M,
Ginsburg O, Lauby-Secretan B, Arbyn M, Basu P, Bray F and
Vaccarella S: Global estimates of incidence and mortality of CC in
2020: A baseline analysis of the WHO Global CC elimination
initiative. Lancet Glob Health. 11:e197–e206. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Small W Jr, Bacon MA, Bajaj A, Chuang LT,
Fisher BJ, Harkenrider MM, Jhingran A, Kitchener HC, Mileshkin LR,
Viswanathan AN and Gaffney DK: Cervical cancer: A global health
crisis. Cancer. 123:2404–2412. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
John RM and Rougeulle C: Developmental
epigenetics: Phenotype and the flexible epigenome. Front Cell Dev
Biol. 6:1302018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gibb EA, Becker-Santos DD, Enfield KS,
Guillaud M, van Niekerk D, Matisic JP, Macaulay CE and Lam WL:
Aberrant expression of long noncoding RNAs in cervical
intraepithelial neoplasia. Int J Gynecol Cancer. 22:1557–1563.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hu XL, Huang XT, Zhang JN, Liu J, Wen LJ,
Xu X and Zhou JY: Long noncoding RNA MIR210HG is induced by
hypoxia-inducible factor 1α and promotes CC progression. Am J
Cancer Res. 12:2783–2797. 2022.PubMed/NCBI
|
|
33
|
Sharma S and Munger K: Expression of the
long noncoding RNA DINO in human papillomavirus-positive CC cells
reactivates the dormant TP53 tumor suppressor through ATM/CHK2
signaling. mBio. 11:e01190–e01120. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhao H, Zheng GH, Li GC, Xin L, Wang YS,
Chen Y and Zheng XM: Long noncoding RNA LINC00958 regulates cell
sensitivity to radiotherapy through RRM2 by binding to
microRNA-5095 in CC. J Cell Physiol. 234:23349–23359. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang J, Chew BL, Lai Y, Dong H, Xu L,
Balamkundu S, Cai WM, Cui L, Liu CF, Fu XY, et al: Quantifying the
RNA cap epitranscriptome reveals novel caps in cellular and viral
RNA. Nucleic Acids Res. 47:e1302019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ji H, Zhang JA, Liu H, Li K, Wang ZW and
Zhu X: Comprehensive characterization of tumor microenvironment and
m6A RNA methylation regulators and its effects on PD-L1 and immune
infiltrates in cervical cancer. Front Immunol. 13:9761072022.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen J, Li K, Chen J, Wang X, Ling R,
Cheng M, Chen Z, Chen F, He Q, Li S, et al: Aberrant translation
regulated by METTL1/WDR4-mediated tRNA N7-methylguanosine
modification drives head and neck squamous cell carcinoma
progression. Cancer Commun (Lond). 42:223–244. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ma J, Han H, Huang Y, Yang C, Zheng S, Cai
T, Bi J, Huang X, Liu R, Huang L, et al: METTL1/WDR4-mediated m7G
tRNA modifications and m7G codon usage promote mRNA translation and
lung cancer progression. Mol Ther. 29:3422–3435. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen Z, Zhu W, Zhu S, Sun K, Liao J, Liu
H, Dai Z, Han H, Ren X, Yang Q, et al: METTL1 promotes
hepatocarcinogenesis via m7 G tRNA modification-dependent
translation control. Clin Transl Med. 11:e6612021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu Y, Zhang Y, Chi Q, Wang Z and Sun B:
RETRACTED: Methyltransferase-like 1 (METTL1) served as a tumor
suppressor in colon cancer by activating 7-methyguanosine (m7G)
regulated let-7e miRNA/HMGA2 axis. Life Sci. 249:1174802020.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Qiu Z, He L, Yu F, Lv H and Zhou Y: LncRNA
FAM13A-AS1 regulates proliferation and apoptosis of cervical cancer
cells by targeting miRNA-205-3p/DDI2 axis. J Oncol.
2022:84119192022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang XJ, Li S, Fang J, Yan ZJ and Luo GC:
LncRNA FAM13A-AS1 promotes renal carcinoma tumorigenesis through
sponging miR-141-3p to upregulate NEK6 expression. Front Mol
Biosci. 9:7387112022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sugino RP, Ohira M, Mansai SP and Kamijo
T: Comparative epigenomics by machine learning approach for
neuroblastoma. BMC Genomics. 23:8522022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Roh J, Im M, Kang J, Youn B and Kim W:
Long non-coding RNA in glioma: Novel genetic players in
temozolomide resistance. Anim Cells Syst (Seoul). 27:19–28. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Guo X and Li M: LINC01089 is a
tumor-suppressive lncRNA in gastric cancer and it regulates
miR-27a-3p/TET1 axis. Cancer Cell Int. 20:5072020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang H, Zhang H, Li X, Huang S, Guo Q and
Geng D: LINC01089 functions as a ceRNA for miR-152-3p to inhibit
non-small lung cancer progression through regulating PTEN. Cancer
Cell Int. 21:1432021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Li M and Guo X: LINC01089 blocks the
proliferation and metastasis of colorectal cancer cells via
regulating miR-27b-3p/HOXA10 axis. Onco Targets Ther. 13:8251–8260.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li X, Lv F, Li F, Du M, Liang Y, Ju S, Liu
Z, Zhou B, Wang B and Gao Y: LINC01089 inhibits tumorigenesis and
epithelial-mesenchymal transition of non-small cell lung cancer via
the miR-27a/SFRP1/Wnt/β-catenin axis. Front Oncol. 10:5325812020.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Li S, Han Y, Liang X and Zhao M: LINC01089
inhibits the progression of CC via inhibiting miR-27a-3p and
increasing BTG2. J Gene Med. 23:e32802021. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Liu W, Zhao Y, Liu X, Zhang X, Ding J, Li
Y, Tian Y, Wang H, Liu W and Lu Z: A novel meiosis-related lncRNA,
Rbakdn, contributes to spermatogenesis by stabilizing Ptbp2. Front
Genet. 12:7524952021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Qin R, Cao L, Ye C, Wang J and Sun Z: A
novel prognostic prediction model based on seven immune-related
RNAs for predicting overall survival of patients in early cervical
squamous cell carcinoma. BMC Med Genomics. 14:492021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tomoo Y: Prognostic factors of ovarian
cancer at our department. Igaku Kenkyu. 57:154–164. 1987.(In
Japanese). PubMed/NCBI
|
|
53
|
Xin Y, Shang X, Sun X, Xu G and Liu Y and
Liu Y: SLC8A1 antisense RNA 1 suppresses papillary thyroid cancer
malignant progression via the FUS RNA binding protein (FUS)/NUMB
like endocytic adaptor protein (Numbl) axis. Bioengineered.
13:12572–12582. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Li Y, Cao X and Li H: Identification and
validation of novel long non-coding RNA biomarkers for early
diagnosis of oral squamous cell carcinoma. Front Bioeng Biotechnol.
8:2562020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Komi DEA and Redegeld FA: Role of mast
cells in shaping the tumor microenvironment. Clin Rev Allergy
Immunol. 58:313–325. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Liu X, Li X, Wei H, Liu Y and Li N: Mast
cells in colorectal cancer tumour progression, angiogenesis, and
lymphangiogenesis. Front Immunol. 14:12090562023. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Shan F, Somasundaram A, Bruno TC, Workman
CJ and Vignali DAA: Therapeutic targeting of regulatory T cells in
cancer. Trends Cancer. 8:944–961. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Tzankov A, Meier C, Hirschmann P, Went P,
Pileri SA and Dirnhofer S: Correlation of high numbers of
intratumoral FOXP3+ regulatory T cells with improved survival in
germinal center-like diffuse large B-cell lymphoma, follicular
lymphoma and classical Hodgkin's lymphoma. Haematologica.
93:193–200. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Dolina JS, Van Braeckel-Budimir N, Thomas
GD and Salek-Ardakani S: CD8+ T cell exhaustion in cancer. Front
Immunol. 12:7152342021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Muthusami S, Sabanayagam R, Periyasamy L,
Muruganantham B and Park WY: A review on the role of epidermal
growth factor signaling in the development, progression and
treatment of CC. Int J Biol Macromol. 194:179–187. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Boromand N, Hasanzadeh M, ShahidSales S,
Farazestanian M, Gharib M, Fiuji H, Behboodi N, Ghobadi N,
Hassanian SM, Ferns GA and Avan A: Clinical and prognostic value of
the C-Met/HGF signaling pathway in cervical cancer. J Cell Physiol.
233:4490–4496. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Guo K, Shelat AA, Guy RK and Kastan MB:
Development of a cell-based, high-throughput screening assay for
ATM kinase inhibitors. J Biomol Screen. 19:538–546. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Hsieh MJ, Weng CC, Lin YC, Wu CC, Chen LT
and Cheng KH: Inhibition of β-catenin activity abolishes LKB1
loss-driven pancreatic cystadenoma in mice. Int J Mol Sci.
22:46492021. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Lu W, Ni K, Li Z, Xiao L, Li Y, Jiang Y,
Zhang J and Shi H: Salubrinal protects against cisplatin-induced
cochlear hair cell endoplasmic reticulum stress by regulating
eukaryotic translation initiation factor 2α signalling. Front Mol
Neurosci. 15:9164582022. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Frenel JS, Mathiot L, Cropet C, Borcoman
E, Hervieu A, Coquan E, De La Motte Rouge T, Saada-Bouzid E,
Sabatier R, Lavaud P, et al: Durvalumab and tremelimumab in
combination with metronomic oral vinorelbine for recurrent advanced
cervical cancer: An open-label phase I/II study. J Immunother
Cancer. 13:e0107082025. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Varma DA and Tiwari M: Crizotinib-induced
anti-cancer activity in human cervical carcinoma cells via
ROS-dependent mitochondrial depolarization and induction of
apoptotic pathway. J Obstet Gynaecol Res. 47:3923–3930. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Jin MH and Oh DY: ATM in DNA repair in
cancer. Pharmacol Ther. 203:1073912019. View Article : Google Scholar : PubMed/NCBI
|