Non-small cell lung cancer (NSCLC) represents 80–85%
of all lung cancer cases, making it a notable contributor to cancer
morbidity and mortality worldwide (1). The incidence of NSCLC is increasing,
particularly in countries such as China, where it is the leading
cause of cancer-associated mortality (2). Risk factors for NSCLC include tobacco
use, family history and environmental exposure, alongside chronic
lung diseases such as chronic obstructive pulmonary disease, which
further exacerbate the risk (3).
The clinical importance of NSCLC lies not only in its high
prevalence but also in its poor prognosis, with numerous patients
diagnosed at advanced stages when treatment options are limited,
and survival rates are low (4).
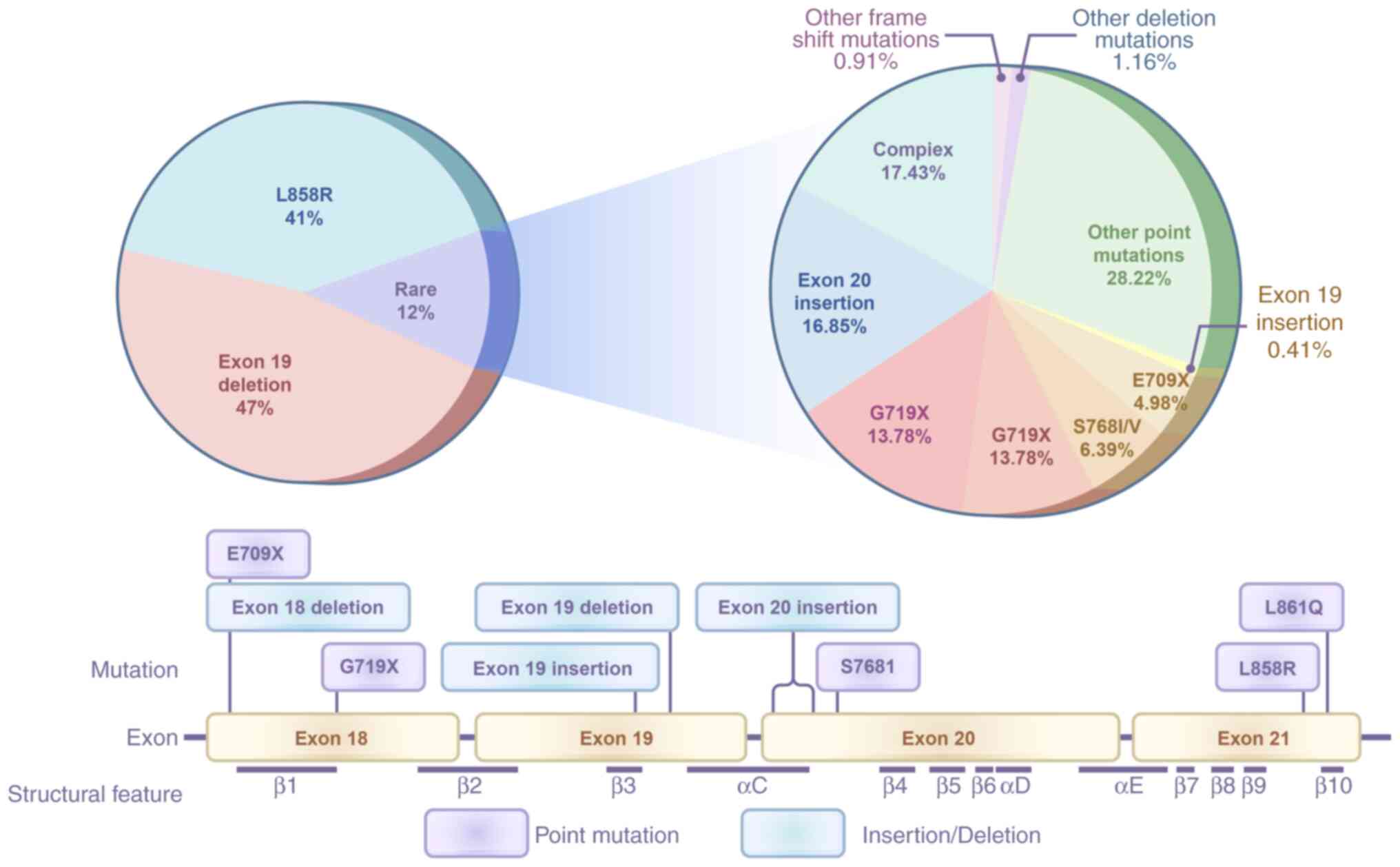
Mutations in the EGFR gene (mEGFRs) are prevalent in
NSCLC, occurring in ~40% of patients in Asian populations and ~20%
in non-Asian populations (5). These
mutations serve a key role in the pathogenesis of NSCLC, leading to
uncontrolled cell proliferation and survival (4). Among mutations, the EGFR T790M
mutation is a key driver of resistance to first- and
second-generation EGFR-tyrosine kinase inhibitors (TKIs) (6). Understanding the biological role of
mEGFRs is crucial, as they not only dictate the therapeutic
approach but also influence patient outcomes and survival rates
(7).
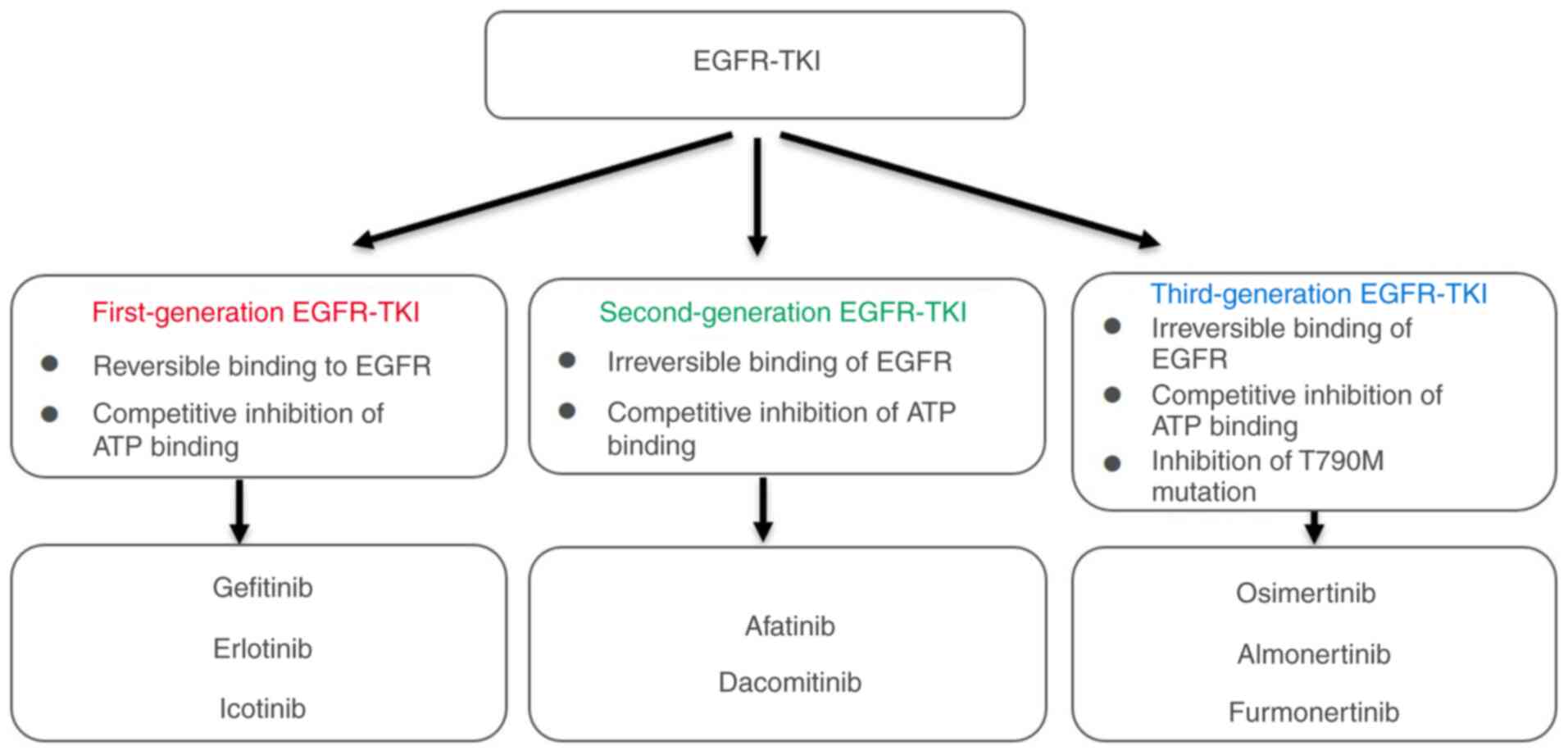
Third-generation EGFR-TKIs (such as osimertinib) are
designed to selectively target both activating mutations [such as
L858R and exon 19 deletions (19del)] and the T790M mutation via
irreversible binding to the EGFR kinase domain. Their enhanced
selectivity for mutant over wild-type EGFR decreases off-target
toxicity, making them preferable first-line options for advanced
NSCLC. However, the emergence of resistance mutations (such as
C797S) is a challenge.
The present review aimed to summarize mEGFRs in
NSCLC, the role of third-generation EGFR-TKIs and the challenges
associated with resistance mechanisms to highlight the importance
of targeted therapies in improving patient outcomes and elucidate
potential pathways for overcoming resistance to EGFR-TKIs.
Understanding these dynamics is key for optimizing treatment
strategies and enhancing the clinical management of patients with
NSCLC.
The presence of 19del is associated with a higher
sensitivity to EGFR-TKIs. This association has been reported in
several previous studies (12,13),
highlighting that patients with NSCLC harboring these specific
mutations exhibit improved clinical outcomes when treated with
EGFR-TKIs compared with those with other mutations, such as the
L858R mutation (14). For example,
patients with 19del had markedly longer PFS compared with patients
with the L858R mutation, indicating a more favorable response to
treatment (14).
Moreover, the coexistence of the T790M mutation,
which is known to confer resistance to first-generation TKIs, has
been reported to be more prevalent in patients with the L858R
mutation compared with patients with 19del (15). This suggests that 19del not only
predicts an improved response to initial treatment but may also be
less likely to be associated with resistance mutations that
complicate therapy (16). In
addition, previous research indicates that certain exon 19 variants
exhibit decreased ATP-binding affinity, which is associated with
increased sensitivity to specific TKIs, such as afatinib (17). Thus, understanding the specific
types of activating mutation is essential for tailoring
personalized therapeutic strategies in NSCLC.
Rare mEGFRs, such as G719X and S768I, represent a
unique subset of mEGFR alterations that influence treatment
outcomes. Although less frequent than L858R and 19del, these
mutations have been shown to confer sensitivity to EGFR-TKIs,
albeit with variable response rates (18–20).
Previous studies have highlighted that patients with
NSCLC exhibiting G719X and S768I mutations may experience favorable
outcomes when treated with second-generation TKIs, suggesting these
agents could be optimal choices for this patient population
(21,22). For example, the objective response
rate (ORR) for patients with G719X mutations treated with afatinib
reached 77.8%, with a median progression-free survival (PFS) of
13.8 months, and similar efficacy was observed for the S768I
mutation (11). By contrast, the
ORR for G719X patients using first-generation TKIs (such as
gefitinib or erlotinib) was only 42.9%, with a median PFS of 5.98
months (18). A retrospective study
further confirmed that the median PFS for patients treated with
afatinib who carry the S768I compound mutation (such as
S768I+L858R) reached 9.79 months, significantly better than
gefitinib's 4.6 months (p=0.049) (19). Furthermore, the presence of these
mutations complicates treatment decisions, as they may coexist with
other mutations, leading to variability in response to therapy
(23). For example, a previous
study demonstrated that patients with lung adenocarcinoma harboring
the G719X mutation often present compound mutations, such as G719X
combined with S768I or L861Q, which can alter the efficacy of TKIs
such as gefitinib and erlotinib (23). A retrospective analysis reported
that patients with the G719X mutation alone have a different
response rate compared with those with compound mutations involving
S768I: The objective response rate for patients with G719X-only
mutations is reported to be higher compared with patients with
G719X combined with other mutations (24).
A previous study reported a patient with both G719X
and S768I mutations who responded well to afatinib, maintaining
disease control for an extended period. For example, a case study
reported a 67-year-old male patient with G719X and S768I compound
mutations. After failing initial chemoradiotherapy, he received
afatinib treatment (40 mg/day), achieving partial remission and
maintaining disease control for up to 17 months (25). This result is consistent with the
conclusions of several multicenter studies: the G719X mutation
(especially when present in combination with S768I) is associated
with a longer PFS (26–28). Specifically, a real-world study in
Vietnam showed that patients with the G719X-S768I compound mutation
had a median time to treatment failure (TTF) of 23.2 months,
significantly better than other mutation types (12.3 months)
(26). A multicenter study in
Taiwan also confirmed that the G719X mutation (whether as a single
mutation or in combination) is an independent favorable predictor
of PFS (HR=0.578), with an overall median PFS of 17.3 months
(27). Furthermore, a retrospective
analysis in Spain further indicated that the objective response
rate (ORR) in the compound mutation group (including G719X+S768I)
reached 70%, significantly higher than in other mutation subgroups
(28). The clinical importance of
these rare mutations underscores the necessity for comprehensive
genetic testing in patients with lung cancer, as they may benefit
from targeted therapies that are typically reserved for more common
mutations. Further research is needed to elucidate the mechanisms
underlying the differential responses observed in patients with
these rare mutations, which may inform future therapeutic
development and clinical guidelines (8).
The T790M mutation is an acquired alteration in EGFR
and occurs in 50–60% of NSCLC cases (29). By contrast, the C797S mutation
represents a tertiary resistance mechanism specific to
third-generation EGFR-TKIs (such as osimertinib). Both mutations
are clinically notable markers of therapeutic resistance, and their
detection necessitates tailored treatment strategies (30).
The association between mEGFR frequency and clinical
features in NSCLC has been a subject of extensive research. Several
studies have demonstrated that the prevalence of mEGFRs is notably
influenced by demographic and clinical characteristics such as age,
sex, smoking status and histological subtype (11,15,18).
For example, a study involving 388 patients with NSCLC demonstrated
that mEGFRs are more prevalent in younger patients, particularly
those aged <80 years, with a mutation rate of 49.6% compared
with 24.1% in older patients (31).
This suggests that age may serve a key role in the mutation
landscape of NSCLC. Additionally, female patients had a higher
incidence of mEGFRs, aligning with findings from other cohorts that
report a strong association between female sex and mEGFR status
(32).
Smoking status is an important factor influencing
mEGFR frequency. Research has consistently reported that mEGFRs are
more common in never-smokers compared with smokers (26,33).
For example, a cohort study from India reported a notable
association between mEGFRs and non-smoking status, with EGFR
mutations being statistically significant among non-smokers
compared to smokers (58.7% vs. 18.4%) (32). This trend is echoed in other
studies, reinforcing the hypothesis that smoking may be negatively
associated with the likelihood of mEGFRs in patients with NSCLC
(34,35).
Histological subtype also affects mEGFR frequency.
Adenocarcinoma, the most common subtype of NSCLC, is associated
with a higher rate of EGFR mutations compared with squamous cell
carcinoma. In 697 Chinese patients, mEGFRs were detected in 52.9%
of adenocarcinomas, while 14.5% of squamous cell carcinomas
exhibited similar mutations (36).
This highlights the value of histological classification in
understanding the mutation profile of NSCLC.
The types of mEGFR impact the prognosis of patients
with lung adenocarcinoma. For example, a meta-analysis indicated
that patients with the 19del mutation exhibit a higher objective
response rate and longer PFS compared with those with the L858R
mutation in exon 21 (37). This
suggests the 19del mutation may serve as a more effective clinical
marker for predicting responses to EGFR-TKIs in patients with NSCLC
(37). Furthermore, the presence of
concomitant mutations alongside the primary mEGFRs also impacts
prognosis. Patients with both 19del and L858R mutations had
different sensitivity to EGFR-TKIs, with 19del showing a more
favorable response (38).
Moreover, the prognostic value of TP53 mutations in
conjunction with mEGFRs has been explored: TP53 mutations are
associated with worse overall survival in advanced NSCLC,
particularly when coupled with specific mEGFR types (39). This highlights the complexity of the
molecular landscape in NSCLC and the need for comprehensive genetic
profiling to improve prediction of patient outcomes.
The role of mEGFR in NSCLC extends beyond its
function as a driver of tumor growth; it notably influences the
TME. The TME is a complex ecosystem composed of several cell types,
including cancer-associated fibroblasts (CAFs), tumor-associated
macrophages (TAMs) and immune cells, which interact dynamically
with cancer cells (40,41). A key mechanism by which mEGFR
affects the TME is the modulation of CAFs. CAFs promote tumor
progression and drug resistance by creating an immunosuppressive
environment. Studies have demonstrated that mEGFR signaling
enhances the recruitment and activation of CAFs, leading to
increased secretion of pro-tumorigenic factors that further support
cancer cell survival and proliferation (42,43).
This not only facilitates tumor growth but also contributes to the
remodeling of the extracellular matrix (ECM), which can hinder the
efficacy of therapeutic agents. The aberrant deposition of ECM
components (such as collagen and laminin) establishes a physical
barrier that impedes drug penetration, limiting the efficacy of
EGFR-TKIs such as osimertinib in tumor cores (44). Concurrently, EGFR activation
upregulates MMP2/MMP9, which not only degrade the basement membrane
to facilitate metastasis but also liberate ECM-sequestered growth
factors (e.g., TGF-β and VEGF) to reactivate PI3K/AKT and MAPK
signaling pathways, thereby bypassing targeted therapy (45,46).
Integrin-mediated interactions with remodeled ECM components (e.g.,
fibronectin) further activate FAK/SRC/STAT3 survival cascades,
fostering cell adhesion-mediated drug resistance (44).
Moreover, mEGFR influences the polarization of TAMs
within the TME. Elevated levels of mEGFRs are associated with an
increase in M2-polarized macrophages, which are known to support
tumor growth and metastasis (47).
This shift in macrophage polarization is mediated by several
signaling pathways, including the STAT5A-indoleamine
2,3-dioxygenase 1 axis, which promotes an immunosuppressive
environment in NSCLC (48). By
enhancing the presence of M2 macrophages, mEGFR contributes to
immune evasion, allowing tumor cells to escape detection and
destruction by the immune system.
Additionally, the interaction between mEGFR and
immune checkpoint molecules, such as programmed death-ligand 1
(PD-L1), is key in shaping the TME. mEGFR signaling can upregulate
PD-L1 expression on tumor cells, which inhibits T cell activation
and proliferation, further exacerbating the immunosuppressive
landscape of the TME (49). This
dynamic interaction highlights the potential for combining mEGFR
inhibitors with immune checkpoint inhibitors to enhance antitumor
immunity and improve patient outcomes.
Despite the initial success of these therapies,
resistance to mEGFR-targeted treatments remains a challenge.
Mechanisms of resistance can include secondary mEGFRs, such as
T790M, which can diminish the effectiveness of first-generation
TKIs (55). This has led to the
development of third-generation inhibitors, such as osimertinib,
which are designed to target both the primary mEGFRs and the T790M
mutation (55). In addition to
TKIs, combination therapies that may enhance the efficacy of
mEGFR-targeted treatments. For example, combining TKIs with immune
checkpoint inhibitors or other targeted agents may provide
synergistic effects and overcome resistance mechanisms (56). Furthermore, the identification of
predictive biomarkers, such as specific serum metabolites, may aid
in stratifying patients who are most likely to benefit from
mEGFR-targeted therapy (54).
Second-generation EGFR-TKIs, such as afatinib and
dacomitinib, have demonstrated improved efficacy compared with
first-generation agents such as gefitinib and erlotinib (57). However, the introduction of
third-generation EGFR-TKIs, particularly osimertinib, has advanced
treatment options, especially for patients who develop resistance
due to the T790M mutation (58,59).
Third-generation EGFR-TKIs are designed to selectively target both
the activating mutations and the T790M mutation, providing a
clinical advantage over second-generation agents (60). For example, osimertinib has shown a
median overall survival of 38.6 months in patients with NSCLC,
which was significantly better than the control group (31.8 months)
(58). Furthermore,
third-generation inhibitors have a more favorable safety profile,
with fewer adverse effects compared with their predecessors
(61).
Despite these advancements, resistance to
third-generation TKIs is a challenge, with the emergence of
tertiary mutations such as C797S (62). This has prompted research into
fourth-generation EGFR-TKIs aimed at overcoming these novel
resistance mechanisms (63). By
contrast, second-generation TKIs, while effective, often lead to
resistance through several pathways, including the activation of
alternative signaling routes (64).
The relationship between drug selectivity and
resistance in the context of EGFR-TKIs is complex and multifaceted.
Third-generation TKIs, such as osimertinib, are designed with
enhanced selectivity for mutant over wild-type EGFR, which
decreases the likelihood of adverse effects associated with
off-target inhibition (65).
However, the emergence of resistance is a notable clinical
challenge. Studies indicate that while the selective pressure
exerted by these drugs can initially lead to substantial tumor
regression, it can also promote the selection of resistant clones
that harbor secondary mutations, such as C797S or alterations in
the MET proto-oncogene, receptor tyrosine kinase (MET) pathway
(66,67). This underscores the importance of
understanding the genetic landscape of tumors when administering
third-generation TKIs. Additionally, combination therapies that
target multiple pathways may be necessary to mitigate resistance
and prolong the effectiveness of treatment. Ongoing research aims
to identify biomarkers that can predict resistance and tailor
treatment strategies, thereby improving outcomes for patients with
mEGFR-NSCLC (68,69).
The secondary mEGFR T790M is caused by the
substitution of threonine with methionine in the tyrosine kinase
domain of EGFR. This mutation increases the affinity of the ATP
binding site, leading to spatial hindrance that interferes with the
binding of TKIs, resulting in resistance (70). The C797S mutation is caused by the
substitution of cysteine with serine at the C797 site of EGFR. This
mutation eliminates the covalent binding site between TKIs and
EGFR, rendering third-generation TKIs such as osimertinib
ineffective (71). In addition to
the T790M and C797S mutations, the L718Q mutation is also
noteworthy. Studies have demonstrated that the L718Q mutation can
coexist with other mutations such as C797S, further enhancing
resistance to osimertinib (72,73).
To address these challenges, researchers are developing
fourth-generation EGFR-TKIs, such as BLU-945, specifically
targeting T790M/C797S mutations. These novel inhibitors overcome
resistance issues through higher selectivity and specificity
(74). Additionally, dual-target
inhibitors such as aurora kinase B (AURKB)/EGFR inhibitors enhance
therapeutic effects and delay the onset of resistance by
simultaneously targeting multiple signaling pathways (MAPK and PI3K
pathways) (8). Highly selective
allosteric inhibitors represent another promising strategy. These
inhibitors suppress EGFR activity by altering its conformation
without directly competing with the ATP binding site, potentially
decreasing the impact on wild-type EGFR and lowering toxicity. This
strategy shows promise in overcoming resistance caused by EGFR
T790M and C797S mutations (75).
EGFR amplification compensates for the inhibitory
effects of EGFR-TKIs by upregulating EGFR levels, leading to
resistance to EGFR-TKIs. This resistance mechanism has been
reported in multiple studies (76,77).
EGFR amplification can counteract the inhibitory effects of TKIs by
enhancing the activity of the EGFR signaling pathway. For example,
amplification of the wild-type EGFR allele is sufficient to confer
acquired resistance to selective mutant EGFR-TKIs (78). This indicates that even in the
context of mEGFRs, signaling from wild-type EGFR serves a notable
role in resistance. Secondly, EGFR amplification is not limited to
the EGFR signaling pathway but may also involve the activation of
other bypass signaling pathways. For example, MET amplification and
MET receptor activation are also mechanisms of EGFR-TKI resistance
(79). The activation of these
bypass signaling pathways maintains cell proliferation and survival
through different mechanisms, such as cross-activation of receptor
TKs. Additionally, EGFR amplification may interact with other
genetic alterations to enhance resistance. For example, in patients
with advanced NSCLC, EGFR amplification coexists with the T790M
mutation, and when treated with third-generation EGFR-TKIs (such as
Osimertinib), patients experience improved PFS compared to those
without acquired EGFR amplification (80). This suggests that EGFR amplification
may synergize with other resistance mechanisms, affecting treatment
outcomes.
A mechanism involved in non-EGFR-dependent
resistance is bypass signal activation. In addition to the T790M
mEGFR, MET gene amplification is an important mechanism of
resistance to first- or second-generation EGFR-TKIs (81). In certain studies (81,82),
MET amplification has been identified as a key mechanism of
acquired resistance to third-generation EGFR-TKIs (such as
osimertinib), particularly when used as first-line treatment. In
NSCLC with mEGFRs, MET amplification or protein overactivation may
lead to escape from EGFR inhibition via the PI3K/AKT/mTOR pathway
(83). To overcome this resistance,
previous research suggests combining osimertinib with MET or MEK
inhibitors (81). Furthermore, HER2
amplification is also regarded as an acquired resistance mechanism
to EGFR inhibition (84). First,
HER2 amplification can mediate resistance by enhancing the activity
of the EGFR signaling pathway. Both EGFR and HER2 belong to the
ErbB receptor family, and the overexpression or amplification of
HER2 can activate downstream signaling pathways by forming
heterodimers, thereby counteracting the inhibitory effects of
EGFR-TKIs (84). Secondly, HER2
amplification may also promote resistance by affecting other
signaling pathways. For example, studies have found that HER2
amplification can activate the PI3K/Akt signaling pathway, which
plays a key role in cell proliferation and survival (85). Previous studies have reported that
HER2 amplification and mEGFR (T790M) are mutually exclusive,
revealing a mechanism of resistance to EGFR-TKIs and providing a
rationale for assessing HER2 status and potentially targeting HER2
(76,84). Although there are currently limited
standard treatment options for HER2 abnormalities, previous studies
have demonstrated that the third-generation TKI osimertinib
exhibits strong antitumor efficacy in lung cancer with HER2
abnormalities, providing a strong basis for future clinical trials
(86,87).
Another mechanism involved in non-EGFR-dependent
resistance is epithelial-mesenchymal transition (EMT). In NSCLC,
zinc finger E-box binding homeobox 1 (ZEB1) and Twist family BHLH
transcription factor 1 (TWIST1) are key transcription factors that
serve important roles in the EMT process. ZEB1 is a notable
regulator of EMT, promoting cell mesenchymalization by inhibiting
the expression of E-cadherin, thereby enhancing the invasive and
migratory ability of cells (88).
Studies have demonstrated that the upregulation of ZEB1 is
associated with the development of TKI resistance, especially in
mEGFR NSCLC cells (89,90). Additionally, ZEB1 promotes the
occurrence of EMT through interactions with other signaling
pathways, such as the TGF-β signaling pathway (91). TWIST1 is also an important regulator
of EMT, facilitating EMT by regulating genes (such as E-cadherin)
associated with cytoskeletal reorganization and cell adhesion
(92). Furthermore, TWIST1 enhances
the effects of EMT through interactions with other signaling
molecules, such as STAT3 (93).
In response to EMT-mediated resistance, researchers
have proposed potential therapeutic strategies. For example,
inhibiting the EMT-associated transcription factor TWIST1 enhances
the efficacy of EGFR-TKIs (92).
Studies have demonstrated that genetic silencing of TWIST1 or the
use of TWIST1 inhibitors can suppress the growth of mEGFR NSCLC and
induce apoptosis, thereby overcoming resistance (92,94).
Additionally, glycogen synthase kinase-3 (GSK-3) inhibitors reverse
EMT-associated resistance, particularly in resistant cells with a
mesenchymal phenotype, where GSK-3 inhibitors markedly inhibit cell
proliferation and induce apoptosis (95). Another strategy is the combined use
of EGFR and fibroblast growth factor receptor (FGFR) inhibitors to
suppress the survival and proliferation of resistant cells in mEGFR
NSCLC. Dual inhibition of EGFR and FGFR can suppress proliferation
of resistant cells over an extended period, preventing the
development of fully resistant cancer (96). Dual inhibition of EGFR and FGFR can
suppress cancer cell proliferation by blocking the cross-activation
of these two signaling pathways. For example, in NSCLC, resistance
to EGFR inhibitors is often associated with the activation of the
FGFR signaling pathway. By simultaneously inhibiting EGFR and FGFR,
cancer cell proliferation and survival is reduced (97). Secondly, dual inhibition of EGFR and
FGFR can enhance apoptosis by inhibiting downstream signaling
pathways such as the MEK/ERK and mTOR pathways. Studies have shown
that feedback activation of EGFR and FGFR limits the efficacy of
single FGFR inhibitors, while inhibition of EGFR can enhance the
effects of FGFR inhibitors, leading to more significant apoptosis
and tumor shrinkage (97).
Additionally, dual inhibition of EGFR and FGFR can limit the
development of resistance by reducing the frequency of tumor stem
cells. Dual inhibition of EGFR and FGFR can decrease the frequency
of tumor-initiating cells, thereby enhancing the response to
radiotherapy and other treatments (98). These findings provide a theoretical
basis for developing novel treatment regimens and offer new insight
for overcoming EMT-mediated resistance.
TME remodeling is also involved in
non-EGFR-dependent resistance. TAMs serve an important role in the
TME, as they weaken the therapeutic effect of EGFR-TKIs by
inhibiting the activation of CD8+ T cells (99). In addition, the ECM in the TME is an
important factor in EGFR-TKI resistance. Even in the absence of
genetic changes, ECM can immediately confer resistance to EGFR-TKIs
in tumor cells through its interaction with integrin β1. This
effect is dose-dependent and reversible, suggesting that targeting
the ECM and integrin β1 may be a potential strategy for treating
resistance (100). Moreover,
immune evasion mechanisms in the TME may lead to EGFR-TKI
resistance. EGFR-TKI resistance may promote immune evasion by
upregulating the expression of PD-L1. Studies have found that the
expression of PD-L1 is significantly increased in NSCLC cells
resistant to EGFR-TKIs, which may facilitate tumor immune evasion
by inhibiting lymphocyte activation and cytotoxicity (101,102). EGFR-TKI resistance may enhance the
immunosuppressive microenvironment by inhibiting the anti-tumor
function of CD8+ T cells. Furthermore, RNA methylation
modifications may also play a role in the immune evasion associated
with EGFR-TKI resistance. Research indicates that in EGFR-TKI
resistant NSCLC, the inhibition of the m6A RNA demethylase ALKBH5
leads to the upregulation of CD47, thereby triggering
immunosuppression (103). This
mechanism may promote tumor immune evasion by inhibiting the
phagocytic activity of dendritic cells and the cytotoxicity of
CD8+ T cells. Finally, EGFR-TKI resistance may also
reshape the tumor microenvironment from a non-inflammatory state to
an inflammatory state, thereby affecting the infiltration and
function of immune cells. Studies have found that after EGFR-TKI
treatment, pro-inflammatory signals such as interferon-γ and
inflammatory responses are significantly enriched in tumor samples,
which may be associated with upregulation of effector cells after
resistance (104).
Epigenetic regulation also serves a key role in
EGFR-TKI resistance, including changes in histone modification, DNA
methylation and non-coding RNA (ncRNA), which provide potential
therapeutic targets and biomarkers for overcoming drug resistance
(105). In the mechanisms of
EGFR-independent resistance, epigenetic regulation affects the
survival and proliferation of cancer cells through several
pathways. For example, the loss of the histone methyltransferase
enhancer of zeste homolog 2 (EZH2) is associated with EGFR-TKI
resistance, as it influences cell proliferation and resistance by
regulating the expression and phosphorylation of MET (106). Additionally, changes in the
expression of ncRNAs, such as nc886. In the study of the HCC827
NSCLC cell line, it was found that the expression of nc886 is not
related to EMT (epithelial-mesenchymal transition), but the
CRISPR/Cas9-mediated disruption of its sequence can increase the
sensitivity of cells to TKIs. Furthermore, the disruption of the
nc886 sequence hinders the activation of MET RTK, which is a
mechanism of TKI resistance with EMT as the endpoint (107). To address the resistance induced
by these epigenetic regulations, researchers have proposed various
therapeutic strategies: Combination therapy with PI3K/AKT
inhibitors and EGFR-TKIs breaks the MET-AKT-EZH2 feedback loop,
thereby enhancing tumor suppression effects (106). Furthermore, targeted therapies
against ncRNAs are potential strategies to overcome resistance by
modulating their expression or function to restore drug sensitivity
(107).
The advent of targeted therapies has revolutionized
the management of NSCLC, particularly in patients harboring mEGFRs.
These mutations are key in dictating the responsiveness to TKIs,
such as gefitinib and erlotinib. Personalized treatment strategies
that consider the specific type of mEGFR are key for optimizing
therapeutic outcomes. For example, 19del and the L858R point
mutation exhibit distinct responses to TKIs, necessitating tailored
approaches (108). Recent studies
have underscored the importance of comprehensive genomic profiling
prior to treatment initiation, which can guide clinicians in
selecting the most effective agent and dosage for individual
patients, thereby enhancing efficacy and minimizing adverse effects
(109,110). Furthermore, Combination therapy,
where TKIs are paired with immunotherapies or chemotherapy, aims to
overcome resistance mechanisms that often develop during treatment
(111). The integration of
artificial intelligence in analyzing patient data and predicting
treatment responses presents a promising avenue for future
research, potentially leading to more refined and effective
personalized treatment protocols (112).
Previous studies have highlighted the importance of
understanding the molecular mechanisms of NSCLC to identify
potential therapeutic targets (113,114). For example, the discovery of
mEGFRs and mutations in genes such as anaplastic lymphoma kinase
(115) and KRAS (116) has led to the development of
targeted agents that specifically inhibit these oncogenic drivers.
The implementation of these therapies has shown promising results
in clinical trials, demonstrating improved response rate and
survival outcomes compared with conventional treatment (117,118). Furthermore, the integration of
immunotherapy into the treatment paradigm harnesses the immune
system to combat cancer cells, offering hope for patients with
advanced disease (119).
Despite these advancements, challenges remain in the
clinical application of targeted therapies for NSCLC. A notable
hurdle is the development of resistance to these agents, which can
occur through various mechanisms, including secondary mutations and
activation of alternative signaling pathways. Understanding these
resistance mechanisms is key for developing strategies to overcome
them, such as combination therapies that target multiple pathways
simultaneously (120,121). Additionally, the identification of
robust predictive biomarkers is key for patient stratification,
ensuring that individuals most likely to benefit from specific
therapies are selected for treatment (121,122).
There is a need for clinical trials to evaluate the
efficacy of new targeted agents and combination therapy,
particularly in biomarker-selected populations. Secondly, the
exploration of novel molecular targets and therapeutic strategies,
including the potential repurposing of existing drugs, may provide
additional avenues for treatment (123,124). Lastly, enhancing the understanding
of the TME and its role in cancer progression is key for developing
innovative therapies that effectively combat NSCLC (125).
mEGFRs serve a key role in the molecular
classification and treatment of NSCLC. Activating mutations
(L858R/19del) are sensitive to EGFR-TKIs but prone to secondary
T790M/C797S resistance; rare mutations (G719X/S768I) require
second-generation TKIs or combination therapy. mEGFRs are also
involved in complex resistance mechanisms. EGFR-dependent
resistance (such as T790M/C797S) diminishes TKI efficacy through
steric hindrance or covalent binding site disruption, whereas
EGFR-independent resistance (MET/HER2 amplification, EMT, TME
remodeling) relies on bypass activation or immune evasion,
necessitating multi-target intervention (104). The present review highlighted the
innovative treatment strategies for NSCLC. Fourth-generation TKIs
(BLU-945) and dual-target inhibitors (AURKB/EGFR) target resistant
mutations and combining immune checkpoint inhibitors or epigenetic
modulators (such as histone deacetylase inhibitors) can reverse
resistance (126). Future studies
should integrate liquid biopsy, artificial intelligence
classification and multi-omics data to achieve dynamic monitoring
of resistance mechanisms. In addition, evaluating the immune
interactions of TME and developing new targeted drugs (such as CD47
inhibitors) may be key to precision treatment for NSCLC.
Not applicable.
The present study was supported by National Natural Science
Foundation of China (grant no. 81860021), Guangxi Natural Science
Foundation (grant no. 2021GXNSFAA325003), Baise Regional
Multimorbidity Joint Special Plan [grant no. BaiKe Zi (2022) no.
41], Guangxi University Young and Middle-aged Teachers Research
Basic Ability Promotion Project (grant no. 2023KY0573), Guangxi
Medical and Health Key Discipline Construction Project and Baise
Scientific Research and Technology Development Program Project
(grant no. Baike no. 20232086).
Not applicable.
ZT, LC and FW conceived the study, performed the
literature review and wrote the manuscript. JueD, YuH and YiH
performed the literature review and revised the manuscript. ZW and
JunD wrote the manuscript. YJ reviewed the manuscript. Data
authentication is not applicable. All authors have read and
approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Suster DI and Mino-Kenudson M: Molecular
pathology of primary non-small cell lung cancer. Arch Med Res.
51:784–798. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen P, Liu Y, Wen Y and Zhou C: Non-small
cell lung cancer in China. Cancer Commun (Lond). 42:937–970. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Houghton AM: Common mechanisms linking
chronic obstructive pulmonary disease and lung cancer. Ann Am
Thorac Soc. 15 (Suppl 4):S273–S277. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Duréndez-Sáez E, Torres-Martinez S,
Calabuig-Fariñas S, Meri-Abad M, Ferrero-Gimeno M and Camps C:
Exosomal microRNAs in non-small cell lung cancer. Transl Cancer
Res. 10:3128–3139. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gelatti ACZ, Drilon A and Santini FC:
Optimizing the sequencing of tyrosine kinase inhibitors (TKIs) in
epidermal growth factor receptor (EGFR) mutation-positive non-small
cell lung cancer (NSCLC). Lung Cancer. 137:113–122. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mosca M, Conci N, Di Federico A, Tateo V,
Favorito V, Zappi A, Gelsomino F and De Giglio A: First-generation
epidermal growth factor receptor inhibitors plus antiangiogenic
drugs versus third-generation epidermal growth factor receptor
inhibitors in advanced non-small-cell lung cancer: A meta-analysis.
JCO Precis Oncol. 7:e23000732023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qu F, Zhou Y and Yu W: A review of
research progress on mechanisms and overcoming strategies of
acquired osimertinib resistance. Anticancer Drugs. 33:e76–e83.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kurup S, Gesinski D, Assaad K and Reynolds
A: Design, synthesis, and evaluation of dual EGFR/AURKB inhibitors
as anticancer agents for non-small cell lung cancer. Bioorg Med
Chem Lett. 100:1296122024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Costa DB, Nguyen KSH, Cho BC, Sequist LV,
Jackman DM, Riely GJ, Yeap BY, Halmos B, Kim JH, Jänne PA, et al:
Effects of erlotinib in EGFR mutated non-small cell lung cancers
with resistance to gefitinib. Clin Cancer Res. 14:7060–7067. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Maione P, Rossi A, Bareschino M, Sacco PC,
Schettino C, Casaluce F, Sgambato A and Gridelli C: Irreversible
EGFR inhibitors in the treatment of advanced NSCLC. Curr Pharm Des.
20:3894–3900. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Choi YW and Choi JH: Does the efficacy of
epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor
differ according to the type of EGFR mutation in non-small cell
lung cancer? Korean J Intern Med. 32:422–428. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li J, Qu L, Wei X, Gao H, Wang W, Qin H,
Tang C, Guo W, Wang H and Liu X: Clinical observation of EGFR-TKI
as a first-line therapy on advanced non-small cell lung cancer.
Zhongguo Fei Ai Za Zhi. 15:299–304. 2012.(In Chinese). PubMed/NCBI
|
|
13
|
Vidal ÓJ: Afatinib as first-line therapy
in mutation-positive EGFR. Results by type of mutation. Med Clin
(Barc). 146 (Suppl 1):S12–S18. 2016.(In Spanish). View Article : Google Scholar
|
|
14
|
Won YW, Han JY, Lee GK, Park SY, Lim KY,
Yoon KA, Yun T, Kim HT and Lee JS: Comparison of clinical outcome
of patients with non-small-cell lung cancer harbouring epidermal
growth factor receptor exon 19 or exon 21 mutations. J Clin Pathol.
64:947–952. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu JY, Yu SF, Wang SH, Bai H, Zhao J, An
TT, Duan JC and Wang J: Clinical outcomes of EGFR-TKI treatment and
genetic heterogeneity in lung adenocarcinoma patients with EGFR
mutations on exons 19 and 21. Chin J Cancer. 35:302016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen LY, Molina-Vila MA, Ruan SY, Su KY,
Liao WY, Yu KL, Ho CC, Shih JY, Yu CJ, Yang JC, et al: Coexistence
of EGFR T790M mutation and common activating mutations in
pretreatment non-small cell lung cancer: A systematic review and
meta-analysis. Lung Cancer. 94:46–53. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
van Alderwerelt van Rosenburgh IK, Lu DM,
Grant MJ, Stayrook SE, Phadke M, Walther Z, Goldberg SB, Politi K,
Lemmon MA, Ashtekar KD and Tsutsui Y: Biochemical and structural
basis for differential inhibitor sensitivity of EGFR with distinct
exon 19 mutations. Nat Commun. 13:67912022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu J, Jin B, Chu T, Dong X, Yang H, Zhang
Y, Wu D, Lou Y, Zhang X, Wang H and Han B: EGFR tyrosine kinase
inhibitor (TKI) in patients with advanced non-small cell lung
cancer (NSCLC) harboring uncommon EGFR mutations: A real-world
study in China. Lung Cancer. 96:87–92. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Park S, Lee SY, Kim D, Sim YS, Ryu JS,
Choi J, Lee SH, Ryu YJ, Lee JH and Chang JH: Comparison of
epidermal growth factor receptor tyrosine kinase inhibitors for
patients with lung adenocarcinoma harboring different epidermal
growth factor receptor mutation types. BMC Cancer. 21:522021.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Singh V, Nambirajan A, Malik PS, Thulkar
S, Pandey RM, Luthra K, Arava S, Ray R, Mohan A and Jain D:
Spectrum of uncommon and compound epidermal growth factor receptor
mutations in non-small-cell lung carcinomas with treatment response
and outcome analysis: A study from India. Lung Cancer. 149:53–60.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chiu CH, Yang CT, Shih JY, Huang MS, Su
WC, Lai RS, Wang CC, Hsiao SH, Lin YC, Ho CL, et al: Epidermal
growth factor receptor tyrosine Kinase inhibitor treatment response
in advanced lung adenocarcinomas with G719X/L861Q/S768I mutations.
J Thorac Oncol. 10:793–799. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Watanabe M, Oizumi S, Kiuchi S, Yamada N,
Yokouchi H, Fukumoto S and Harada M: The effectiveness of afatinib
in a patient with advanced lung adenocarcinoma harboring rare G719X
and S768I mutations. Intern Med. 57:993–996. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Leventakos K, Kipp BR, Rumilla KM, Winters
JL, Yi ES and Mansfield AS: S768I mutation in EGFR in patients with
lung cancer. J Thorac Oncol. 11:1798–1801. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhu X, Bai Q, Lu Y, Qi P, Ding J, Wang J
and Zhou X: Response to tyrosine kinase inhibitors in lung
adenocarcinoma with the rare epidermal growth factor receptor
mutation S768I: A retrospective analysis and literature review.
Target Oncol. 12:81–88. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kutsuzawa N, Takahashi F, Tomomatsu K,
Obayashi S, Takeuchi T, Takihara T, Hayama N, Oguma T, Aoki T and
Asano K: Successful treatment of a patient with lung adenocarcinoma
harboring compound EGFR gene mutations, G719X and S768I, with
afatinib. Tokai J Exp Clin Med. 45:113–116. 2020.PubMed/NCBI
|
|
26
|
Pham VL, Le TA, Pham CP, Hoa Nguyen TT, Do
AT, Nguyen TK, Nguyen MH, Thu Hoang TA, Hao Vuong DT, Tam Nguyen
DN, et al: Real-world analysis of afatinib as a first-line
treatment for patients with advanced stage non-small-cell lung
cancer with uncommon EGFR mutations: A multicenter study in
Vietnam. Ther Adv Med Oncol. 16:175883592412429722024. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hsu PC, Lee SH, Chiu LC, Lee CS, Wu CE,
Kuo SC, Ju JS, Huang AC, Li SH, Ko HW, et al: Afatinib in untreated
stage IIIB/IV lung adenocarcinoma with major uncommon epidermal
growth factor receptor (EGFR) mutations (G719X/L861Q/S768I): A
multicenter observational study in Taiwan. Target Oncol.
18:195–207. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Moran T, Taus A, Arriola E, Aguado C,
Dómine M, Rueda AG, Calles A, Cedrés S, Viñolas N, Isla D, et al:
Clinical activity of afatinib in patients with non-small-cell lung
cancer harboring uncommon EGFR mutations: A Spanish retrospective
multicenter study. Clin Lung Cancer. 21:428–436.e2. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee SH, Kim EY, Kim A and Chang YS:
Clinical implication and usefulness of de novo EGFR T790M mutation
in lung adenocarcinoma with EGFR-tyrosine kinase inhibitor
sensitizing mutation. Cancer Biol Ther. 21:741–748. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Barsouk A, Elghawy O, Heidlauf A, Yu C,
Wang L, Yang D, Kurian M, Goel K, Rushkin L, Anran Huang A, et al:
Real-world outcomes of atypical EGFR-mutated metastatic non-small
cell lung cancer (mNSCLC) treated with osimertinib (osi) vs
afatinib or erlotinib. Lung Cancer. 195:1079262024. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nishii T, Yokose T, Miyagi Y, Daigo Y, Ito
H, Isaka T, Imai K, Murakami S, Kondo T, Saito H, et al:
Clinicopathological features and EGFR gene mutation status in
elderly patients with resected non-small-cell lung cancer. BMC
Cancer. 14:6102014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Thomas R, Balaram G, Varayathu H, Ghorpade
SN, Kowsik PV, Dharman B, Thomas BE, Ramaswamy V, Nanjaiah T, Patil
S, et al: Molecular epidemiology and clinical characteristics of
epidermal growth factor receptor mutations in NSCLC: A
single-center experience from India. J Cancer Res Ther.
19:1398–1406. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang T, Wan B, Zhao Y, Li C, Liu H, Lv T,
Zhan P and Song Y: Treatment of uncommon EGFR mutations in
non-small cell lung cancer: New evidence and treatment. Transl Lung
Cancer Res. 8:302–316. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wen L, Wang S, Xu W, Xu X, Li M, Zhang Y,
Du X and Liu S: Value of serum tumor markers for predicting EGFR
mutations in non-small cell lung cancer patients. Ann Diagn Pathol.
49:1516332020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim EY, Kim A, Lee G, Lee H and Chang YS:
Different mutational characteristics of the subsets of
EGFR-tyrosine kinase inhibitor sensitizing mutation-positive lung
adenocarcinoma. BMC Cancer. 18:12212018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lai Y, Zhang Z, Li J, Sun D, Zhou Y, Jiang
T, Han Y, Huang L, Zhu Y, Li X and Yan X: EGFR mutations in
surgically resected fresh specimens from 697 consecutive Chinese
patients with non-small cell lung cancer and their relationships
with clinical features. Int J Mol Sci. 14:24549–24559. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang H, Huang J, Yu X, Han S, Yan X, Sun S
and Zhu X: Different efficacy of EGFR tyrosine kinase inhibitors
and prognosis in patients with subtypes of EGFR-mutated advanced
non-small cell lung cancer: A meta-analysis. J Cancer Res Clin
Oncol. 140:1901–1909. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liang H, Li C, Zhao Y, Zhao S, Huang J,
Cai X, Cheng B, Xiong S, Li J, Wang W, et al: Concomitant mutations
in EGFR 19Del/L858R mutation and their association with response to
EGFR-TKIs in NSCLC patients. Cancer Manag Res. 12:8653–8662. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jiao XD, Qin BD, You P, Cai J and Zang YS:
The prognostic value of TP53 and its correlation with EGFR mutation
in advanced non-small cell lung cancer, an analysis based on
cBioPortal data base. Lung Cancer. 123:70–75. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Timperi E, Croizer H, Khantakova D, Rana
M, Molgora M, Guerriero JL, Mechta-Grigoriou F and Romano E: At the
interface of tumor-associated macrophages and fibroblasts:
Immune-suppressive networks and emerging exploitable targets. Clin
Cancer Res. 30:5242–5251. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tajaldini M, Saeedi M, Amiriani T,
Amiriani AH, Sedighi S, Mohammad Zadeh F, Dehghan M, Jahanshahi M,
Zanjan Ghandian M, Khalili P, et al: Cancer-associated fibroblasts
(CAFs) and tumor-associated macrophages (TAMs); where do they stand
in tumorigenesis and how they can change the face of cancer
therapy? Eur J Pharmacol. 928:1750872022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Suzuki J, Tsuboi M and Ishii G:
Cancer-associated fibroblasts and the tumor microenvironment in
non-small cell lung cancer. Expert Rev Anticancer Ther. 22:169–182.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Garvey CM, Lau R, Sanchez A, Sun RX, Fong
EJ, Doche ME, Chen O, Jusuf A, Lenz HJ, Larson B and Mumenthaler
SM: Anti-EGFR therapy induces EGF secretion by cancer-associated
fibroblasts to confer colorectal cancer chemoresistance. Cancers
(Basel). 12:13932020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Morgillo F, Woo JK, Kim ES, Hong WK and
Lee HY: Heterodimerization of insulin-like growth factor
receptor/epidermal growth factor receptor and induction of survivin
expression counteract the antitumor action of erlotinib. Cancer
Res. 66:10100–10111. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mitsudomi T and Yatabe Y: Epidermal growth
factor receptor in relation to tumor development: EGFR gene and
cancer. FEBS J. 277:301–308. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hirsch FR, Varella-Garcia M, Bunn PA Jr,
Di Maria MV, Veve R, Bremmes RM, Barón AE, Zeng C and Franklin WA:
Epidermal growth factor receptor in non-small-cell lung carcinomas:
Correlation between gene copy number and protein expression and
impact on prognosis. J Clin Oncol. 21:3798–3807. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Madeddu C, Donisi C, Liscia N, Lai E,
Scartozzi M and Macciò A: EGFR-mutated non-small cell lung cancer
and resistance to immunotherapy: Role of the tumor
microenvironment. Int J Mol Sci. 23:64892022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yang Y, Zheng X, Ni P, Li D, Dan Q, Wang
X, Wang Y, Sun Y, Liu K, Dong Z and Ge H: Targeting the STAT5A/IDO1
axis overcomes radioresistance and reverses the immunosuppressive
tumor microenvironment in NSCLC. Int J Oncol. 62:122023. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lin A, Wei T, Meng H, Luo P and Zhang J:
Role of the dynamic tumor microenvironment in controversies
regarding immune checkpoint inhibitors for the treatment of
non-small cell lung cancer (NSCLC) with EGFR mutations. Mol Cancer.
18:1392019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Burotto M, Ali SA and O'sullivan Coyne G:
Class act: Safety comparison of approved tyrosine kinase inhibitors
for non-small-cell lung carcinoma. Expert Opin Drug Saf. 14:97–110.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhang Y, Liu H, Liu X and Lang L:
Gefitinib induces apoptosis in NSCLC cells by promoting
glutaminolysis and inhibiting the MEK/ERK signaling pathway. Discov
Med. 36:836–841. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhao ZQ, Yu ZY, Li J and Ouyang XN:
Gefitinib induces lung cancer cell autophagy and apoptosis via
blockade of the PI3K/AKT/mTOR pathway. Oncol Lett. 12:63–68. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Li Q, Zhang D, Chen X, He L, Li T, Xu X
and Li M: Nuclear PKM2 contributes to gefitinib resistance via
upregulation of STAT3 activation in colorectal cancer. Sci Rep.
5:160822015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Han X, Luo R, Wang L, Zhang L, Wang T,
Zhao Y, Xiao S, Qiao N, Xu C, Ding L, et al: Potential predictive
value of serum targeted metabolites and concurrently mutated genes
for EGFR-TKI therapeutic efficacy in lung adenocarcinoma patients
with EGFR sensitizing mutations. Am J Cancer Res. 10:4266–4286.
2020.PubMed/NCBI
|
|
55
|
Lee SH, Kim EY, Kim T and Chang YS:
Compared to plasma, bronchial washing fluid shows higher diagnostic
yields for detecting EGFR-TKI sensitizing mutations by ddPCR in
lung cancer. Respir Res. 21:1422020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Masui K, Gini B, Wykosky J, Zanca C,
Mischel PS, Furnari FB and Cavenee WK: A tale of two approaches:
Complementary mechanisms of cytotoxic and targeted therapy
resistance may inform next-generation cancer treatments.
Carcinogenesis. 34:725–738. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Hou B, Lu X, Gao DC, Liu QX, Zhou D, Zheng
H and Dai JG: REPORT-clinical outcomes of using second-versus
first-generation EGFR-tkis for the first-line treatment of advanced
NSCLC patients with EGFR mutations: A meta-analysis. Pak J Pharm
Sci. 34:1459–1468. 2021.PubMed/NCBI
|
|
58
|
Du X, Yang B, An Q, Assaraf YG, Cao X and
Xia J: Acquired resistance to third-generation EGFR-TKIs and
emerging next-generation EGFR inhibitors. Innovation (Camb).
2:1001032021.PubMed/NCBI
|
|
59
|
Tan CS, Kumarakulasinghe NB, Huang YQ, Ang
YLE, Choo JRE, Goh BC and Soo RA: Third generation EGFR TKIs:
Current data and future directions. Mol Cancer. 17:292018.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Pirker R: Third-generation epidermal
growth factor receptor tyrosine kinase inhibitors in advanced
nonsmall cell lung cancer. Curr Opin Oncol. 28:115–121. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wang S and Li J: Second-generation EGFR
and ErbB tyrosine kinase inhibitors as first-line treatments for
non-small cell lung cancer. Onco Targets Ther. 12:6535–6548. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wang S, Tsui ST, Liu C, Song Y and Liu D:
EGFR C797S mutation mediates resistance to third-generation
inhibitors in T790M-positive non-small cell lung cancer. J Hematol
Oncol. 9:592016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Scalvini L, Castelli R, La Monica S, Tiseo
M and Alfieri R: Fighting tertiary mutations in EGFR-driven
lung-cancers: Current advances and future perspectives in medicinal
chemistry. Biochem Pharmacol. 190:1145432021. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Nagano T, Tachihara M and Nishimura Y:
Mechanism of resistance to epidermal growth factor
receptor-tyrosine kinase inhibitors and a potential treatment
strategy. Cells. 7:2122018. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Shi P, Zhang S, Zhu L, Qian G, Ren H,
Ramalingam SS, Chen M and Sun SY: The third-generation EGFR
inhibitor, osimertinib, promotes c-FLIP degradation, enhancing
apoptosis including TRAIL-induced apoptosis in NSCLC cells with
activating EGFR mutations. Transl Oncol. 12:705–713. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Venkatesan S, Swanton C, Taylor BS and
Costello JF: Treatment-induced mutagenesis and selective pressures
sculpt cancer evolution. Cold Spring Harb Perspect Med.
7:a0266172017. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Vander Velde R, Yoon N, Marusyk V, Durmaz
A, Dhawan A, Miroshnychenko D, Lozano-Peral D, Desai B, Balynska O,
Poleszhuk J, et al: Resistance to targeted therapies as a
multifactorial, gradual adaptation to inhibitor specific selective
pressures. Nat Commun. 11:23932020. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Li K, Peng ZY, Wang R, Li X, Du N, Liu DP,
Zhang J, Zhang YF, Ma L, Sun Y, et al: Enhancement of TKI
sensitivity in lung adenocarcinoma through m6A-dependent
translational repression of Wnt signaling by circ-FBXW7. Mol
Cancer. 22:1032023. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Pang LL, Zhuang WT, Huang YH, Liao J, Li
MZ, Lv Y, Zhang L and Fang WF: Uncommon de novo
EGFRT790M-Mutant NSCLC characterized with unique genetic
features: Clinical response and acquired resistance to the
third-generation EGFR-TKIs treatment. Lung Cancer. 190:1075282024.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Denis MG, Vallée A and Théoleyre S: EGFR
T790M resistance mutation in non-small-cell lung carcinoma. Clin
Chim Acta. 444:81–85. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Shaikh M, Shinde Y, Pawara R, Noolvi M,
Surana S, Ahmad I and Patel H: Emerging approaches to overcome
acquired drug resistance obstacles to osimertinib in non-small-cell
lung cancer. J Med Chem. 65:1008–1046. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Nishino M, Suda K, Kobayashi Y, Ohara S,
Fujino T, Koga T, Chiba M, Shimoji M, Tomizawa K, Takemoto T and
Mitsudomi T: Effects of secondary EGFR mutations on resistance
against upfront osimertinib in cells with EGFR-activating mutations
in vitro. Lung Cancer. 126:149–155. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Ding K, Peng Z and Xu Y: Triplet therapy
overcomes 3rd-EGFR TKI-resistant EGFR-L858R/T790M/C797S in trans
and in cis/L718Q mutation. Genes Dis. 12:1014082024. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Lim SM, Schalm SS, Lee EJ, Park S, Conti
C, Millet YA, Woessner R, Zhang Z, Tavera-Mendoza LE, Stevison F,
et al: BLU-945, a potent and selective next-generation EGFR TKI,
has antitumor activity in models of osimertinib-resistant
non-small-cell lung cancer. Ther Adv Med Oncol.
16:175883592412806892024. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ahmadi A, Mohammadnejadi E and
Razzaghi-Asl N: Gefitinib derivatives and drug-resistance: A
perspective from molecular dynamics simulations. Comput Biol Med.
163:1072042023. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Ercan D, Zejnullahu K, Yonesaka K, Xiao Y,
Capelletti M, Rogers A, Lifshits E, Brown A, Lee C, Christensen JG,
et al: Amplification of EGFR T790M causes resistance to an
irreversible EGFR inhibitor. Oncogene. 29:2346–2356. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Kubo T, Yamamoto H, Lockwood WW, Valencia
I, Soh J, Peyton M, Jida M, Otani H, Fujii T, Ouchida M, et al: MET
gene amplification or EGFR mutation activate MET in lung cancers
untreated with EGFR tyrosine kinase inhibitors. Int J Cancer.
124:1778–1784. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Nukaga S, Yasuda H, Tsuchihara K, Hamamoto
J, Masuzawa K, Kawada I, Naoki K, Matsumoto S, Mimaki S, Ikemura S,
et al: Amplification of EGFR wild-type alleles in non-small cell
lung cancer cells confers acquired resistance to mutation-selective
EGFR tyrosine kinase inhibitors. Cancer Res. 77:2078–2089. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Presutti D, Santini S, Cardinali B, Papoff
G, Lalli C, Samperna S, Fustaino V, Giannini G and Ruberti G: MET
gene amplification and MET receptor activation are not sufficient
to predict efficacy of combined MET and EGFR inhibitors in EGFR
TKI-resistant NSCLC cells. PLoS One. 10:e01433332015. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zhang Y, Xu Y, Xu J, Zhong H, Xia J and
Zhong R: Osimertinib for EGFR-mutant NSCLC patients with acquired
T790M and EGFR amplification after first-generation EGFR-TKI
resistance. Cancer Sci. 116:753–763. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zhang Z, Yang S and Wang Q: Impact of MET
alterations on targeted therapy with EGFR-tyrosine kinase
inhibitors for EGFR-mutant lung cancer. Biomark Res. 7:272019.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Nie N, Li J, Zhang J, Dai J, Liu Z, Ding
Z, Wang Y, Zhu M, Hu C, Han R, et al: First-line osimertinib in
patients with EGFR-mutated non-small cell lung cancer:
Effectiveness, resistance mechanisms, and prognosis of different
subsequent treatments. Clin Med Insights Oncol.
16:117955492211347352022. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Turke AB, Zejnullahu K, Wu YL, Song Y,
Dias-Santagata D, Lifshits E, Toschi L, Rogers A, Mok T, Sequist L,
et al: Preexistence and clonal selection of MET amplification in
EGFR mutant NSCLC. Cancer Cell. 17:77–88. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Takezawa K, Pirazzoli V, Arcila ME, Nebhan
CA, Song X, de Stanchina E, Ohashi K, Janjigian YY, Spitzler PJ,
Melnick MA, et al: HER2 amplification: a potential mechanism of
acquired resistance to EGFR inhibition in EGFR-mutant lung cancers
that lack the second-site EGFR T790M mutation. Cancer Discov.
2:922–933. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Ding D, Zhang J, Luo Z, Wu H, Lin Z, Liang
W and Xue X: Analysis of the lncRNA-miRNA-mRNA network reveals a
potential regulatory mechanism of EGFR-TKI resistance in NSCLC.
Front Genet. 13:8513912022. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Liu S, Li S, Hai J, Wang X, Chen T, Quinn
MM, Gao P, Zhang Y, Ji H, Cross DAE and Wong KK: Targeting HER2
aberrations in non-small cell lung cancer with osimertinib. Clin
Cancer Res. 24:2594–2604. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Jonsdottir G, Smith M, Wood S, Hejleh TA
and Furqan M: Activity of osimertinib in a patient with stage IV
non-small cell lung cancer harboring HER2 exon 19, p.L755P
mutation: Case report. Transl Lung Cancer Res. 12:927–932. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Rastogi I, Rajanna S, Webb A, Chhabra G,
Foster B, Webb B and Puri N: Mechanism of c-Met and EGFR tyrosine
kinase inhibitor resistance through epithelial mesenchymal
transition in non-small cell lung cancer. Biochem Biophys Res
Commun. 477:937–944. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Soucheray M, Capelletti M, Pulido I, Kuang
Y, Paweletz CP, Becker JH, Kikuchi E, Xu C, Patel TB, Al-Shahrour
F, et al: Intratumoral heterogeneity in EGFR-mutant NSCLC results
in divergent resistance mechanisms in response to EGFR tyrosine
kinase inhibition. Cancer Res. 75:4372–4383. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Lu Y, Liu Y, Oeck S, Zhang GJ, Schramm A
and Glazer PM: Hypoxia induces resistance to EGFR inhibitors in
lung cancer cells via upregulation of FGFR1 and the MAPK pathway.
Cancer Res. 80:4655–4667. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Comaills V, Kabeche L, Morris R, Buisson
R, Yu M, Madden MW, LiCausi JA, Boukhali M, Tajima K, Pan S, et al:
Genomic instability is induced by persistent proliferation of cells
undergoing epithelial-to-mesenchymal transition. Cell Rep.
17:2632–2647. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Yochum ZA, Cades J, Wang H, Chatterjee S,
Simons BW, O'Brien JP, Khetarpal SK, Lemtiri-Chlieh G, Myers KV,
Huang EH, et al: Targeting the EMT transcription factor TWIST1
overcomes resistance to EGFR inhibitors in EGFR-mutant
non-small-cell lung cancer. Oncogene. 38:656–670. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Chen K, Xu J, Tong YL, Yan JF, Pan Y, Wang
WJ, Zheng L, Zheng XX, Hu C, Hu X, et al: Rab31 promotes metastasis
and cisplatin resistance in stomach adenocarcinoma through
Twist1-mediated EMT. Cell Death Dis. 14:1152023. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Burns TF, Dobromilskaya I, Murphy SC,
Gajula RP, Thiyagarajan S, Chatley SN, Aziz K, Cho YJ, Tran PT and
Rudin CM: Inhibition of TWIST1 leads to activation of
oncogene-induced senescence in oncogene-driven non-small cell lung
cancer. Mol Cancer Res. 11:329–338. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Fukuda K, Takeuchi S, Arai S, Kita K,
Tanimoto A, Nishiyama A and Yano S: Glycogen synthase kinase-3
inhibition overcomes epithelial-mesenchymal transition-associated
resistance to osimertinib in EGFR-mutant lung cancer. Cancer Sci.
111:2374–2384. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Raoof S, Mulford IJ, Frisco-Cabanos H,
Nangia V, Timonina D, Labrot E, Hafeez N, Bilton SJ, Drier Y, Ji F,
et al: Targeting FGFR overcomes EMT-mediated resistance in EGFR
mutant non-small cell lung cancer. Oncogene. 38:6399–6413. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Wu Q, Zhen Y, Shi L, Vu P, Greninger P,
Adil R, Merritt J, Egan R, Wu MJ, Yin X, et al: EGFR inhibition
potentiates FGFR inhibitor therapy and overcomes resistance in
FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov.
12:1378–1395. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Hu S, Fu W, Li T, Yuan Q, Wang F, Lv G, Lv
Y, Fan X, Shen Y, Lin F, et al: Antagonism of EGFR and Notch limits
resistance to EGFR inhibitors and radiation by decreasing
tumor-initiating cell frequency. Sci Transl Med. 9:eaag03392017.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Lin Z, Wang Q, Jiang T, Wang W and Zhao
JJ: Targeting tumor-associated macrophages with STING agonism
improves the antitumor efficacy of osimertinib in a mouse model of
EGFR-mutant lung cancer. Front Immunol. 14:10772032023. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Wang Y, Zhang T, Guo L, Ren T and Yang Y:
Stromal extracellular matrix is a microenvironmental cue promoting
resistance to EGFR tyrosine kinase inhibitors in lung cancer cells.
Int J Biochem Cell Biol. 106:96–106. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Peng S, Wang R, Zhang X, Ma Y, Zhong L, Li
K, Nishiyama A, Arai S, Yano S and Wang W: EGFR-TKI resistance
promotes immune escape in lung cancer via increased PD-L1
expression. Mol Cancer. 18:1652019. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Hsu KH, Huang YH, Tseng JS, Chen KC, Ku
WH, Su KY, Chen JJW, Chen HW, Yu SL, Yang TY and Chang GC: High
PD-L1 expression correlates with primary resistance to EGFR-TKIs in
treatment naïve advanced EGFR-mutant lung adenocarcinoma patients.
Lung Cancer. 127:37–43. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Zhang W, Wang J, Liang J, He Z, Wang K and
Lin H: RNA methylation of CD47 mediates tumor immunosuppression in
EGFR-TKI resistant NSCLC. Br J Cancer. 132:569–579. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Chen Q, Xia L, Wang J, Zhu S, Wang J, Li
X, Yu Y, Li Z, Wang Y, Zhu G and Lu S: EGFR-mutant NSCLC may
remodel TME from non-inflamed to inflamed through acquiring
resistance to EGFR-TKI treatment. Lung Cancer. 192:1078152024.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Yao X, Gao C, Sun C, Chen ZS and Zhuang J:
Epigenetic code underlying EGFR-TKI resistance in non-small cell
lung cancer: Elucidation of mechanisms and perspectives on
therapeutic strategies. Drug Discov Today. 30:1043212025.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Quan C, Chen Y, Wang X, Yang D, Wang Q,
Huang Y, Petersen RB, Liu X, Zheng L, Li Y and Huang K: Loss of
histone lysine methyltransferase EZH2 confers resistance to
tyrosine kinase inhibitors in non-small cell lung cancer. Cancer
Lett. 495:41–52. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Bui VNV, Daugaard TF, Sorensen BS and
Nielsen AL: Expression of the non-coding RNA nc886 facilitates the
development of tyrosine kinase inhibitor resistance in EGFR-mutated
non-small-cell lung cancer cells. Biochem Biophys Res Commun.
731:1503952024. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Hong W, Wu Q, Zhang J and Zhou Y:
Prognostic value of EGFR 19-del and 21-L858R mutations in patients
with non-small cell lung cancer. Oncol Lett. 18:3887–3895.
2019.PubMed/NCBI
|
|
109
|
Soleimani-Meigooni DN and Rabinovici GD:
Tau PET visual reads: Research and clinical applications and future
directions. J Nucl Med. 64:822–824. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
George RS, Htoo A, Cheng M, Masterson TM,
Huang K, Adra N, Kaimakliotis HZ, Akgul M and Cheng L: Artificial
intelligence in prostate cancer: Definitions, current research, and
future directions. Urol Oncol. 40:262–270. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Luo Q, Zhang L, Luo C and Jiang M:
Emerging strategies in cancer therapy combining chemotherapy with
immunotherapy. Cancer Lett. 454:191–203. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Rahmat K, Mumin NA, Hamid MTR, Hamid SA
and Ng WL: MRI breast: Current imaging trends, clinical
applications, and future research directions. Curr Med Imaging.
18:1347–1361. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Bajbouj K, Al-Ali A, Ramakrishnan RK,
Saber-Ayad M and Hamid Q: Histone modification in NSCLC: Molecular
mechanisms and therapeutic targets. Int J Mol Sci. 22:117012021.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Reungwetwattana T, Weroha SJ and Molina
JR: Oncogenic pathways, molecularly targeted therapies, and
highlighted clinical trials in non-small-cell lung cancer (NSCLC).
Clin Lung Cancer. 13:252–266. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Chia PL, Dobrovic A, Dobrovic A and John
T: Prevalence and natural history of ALK positive non-small-cell
lung cancer and the clinical impact of targeted therapy with ALK
inhibitors. Clin Epidemiol. 6:423–432. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Hsu J, Annunziata JF, Burns E, Bernicker
EH, Olsen RJ and Thomas JS: Molecular signatures of KRAS-mutated
lung adenocarcinoma: Analysis of concomitant EGFR, ALK, STK11, and
PD-L1 status. Clin Pathol. 15:2632010X2211020542022. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Gkolfinopoulos S and Mountzios G: Beyond
EGFR and ALK: Targeting rare mutations in advanced non-small cell
lung cancer. Ann Transl Med. 6:1422018. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Hirsch FR, Suda K, Wiens J and Bunn PA Jr:
New and emerging targeted treatments in advanced non-small-cell
lung cancer. Lancet. 388:1012–1024. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Weishan H, Donglin Z, Guangmei D, Wenya L,
Fasheng W and Jibing C: Immunoradiotherapy for NSCLC: Mechanisms,
clinical outcomes, and future directions. Clin Transl Oncol.
26:1063–1076. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Selzer E: Impact of molecular targets in
cancer drug development: Historical influence and future
perspectives. Expert Rev Clin Pharmacol. 3:161–163. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Amaravadi RK, Kimmelman AC and Debnath J:
Targeting autophagy in cancer: Recent advances and future
directions. Cancer Discov. 9:1167–1181. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Black A and Morris D: Personalized
medicine in metastatic non-small-cell lung cancer: Promising
targets and current clinical trials. Curr Oncol. 19 (Suppl
1):S73–S85. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Saxena A, Becker D, Preeshagul I, Lee K,
Katz E and Levy B: Therapeutic effects of repurposed therapies in
non-small cell lung cancer: What is old is new again. Oncologist.
20:934–945. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Harmey D, Griffin PR and Kenny PJ:
Development of novel pharmacotherapeutics for tobacco dependence:
Progress and future directions. Nicotine Tob Res. 14:1300–1318.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Ciardiello D, Elez E, Tabernero J and
Seoane J: Clinical development of therapies targeting TGFβ: Current
knowledge and future perspectives. Ann Oncol. 31:1336–1349. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Kannan K and Mohan S: Targeting exon
mutations in NSCLC: Clinical insights into LAG-3, TIM-3 pathways,
and advances in fourth-generation EGFR-TKIs. Med Oncol. 42:1962025.
View Article : Google Scholar : PubMed/NCBI
|