Introduction
Acute myeloid leukemia (AML) is an aggressive cancer
marked by the rapid growth of immature myeloid cells, which
disrupts normal blood cell production (1). Despite advancements in treatments such
as chemotherapy and stem cell transplantation (2), the lack of understanding of the tumor
microenvironment (TME) has hindered the effectiveness of
immunotherapies. The TME includes tumor cells, immune cells,
fibroblasts, blood vessels and cytokines, all interacting to
influence tumor progression. Immunotherapy aims to boost antitumor
responses whilst reducing immunosuppressive effects (3). In leukemia, the bone marrow is the
primary site for leukemia stem cells, with secondary lymphoid
organs also part of the TME. Current therapies such as checkpoint
inhibitors combined with chemotherapy and hypomethylating agents,
show promise. Further research into the immune microenvironment of
AML is essential to develop more effective immunotherapies
(4).
Unlike in solid tumors, the AML TME is primarily in
the bone marrow, with unique anatomical, cellular and metabolic
features. It includes hematopoietic stem/progenitor, stromal,
endothelial and immune regulatory cells, but lacks tumor-associated
fibroblasts and persistent antigenic stimulation typical of solid
tumors (5). The AML TME is highly
immunosuppressive, marked by increased regulatory T cells (Tregs),
myeloid-derived suppressor cells (MDSCs) and exhausted T cells,
which limit immune responses (6).
Leukemia cells remodel the TME by altering C-X-C motif chemokine
ligand 12 (CXCL12) expression in stromal cells, suppressing normal
hematopoiesis and promoting their survival (7). The metabolic profile, hypoxia and
immune composition of AML differ from solid tumors, further
reducing immunotherapy efficacy (8). Recent studies have highlighted that
AML TME characteristics are associated with disease progression and
serve as a key factor for patient risk stratification (9,10).
Therefore, understanding AML-specific TME structure and function is
crucial for optimizing immunotherapy strategies.
Furthermore, T cells are crucial in antitumor
immunity, especially CD8+ cytotoxic T lymphocytes
(CTLs), which kill tumor cells via granzyme B, perforin and
interferon-γ (IFN-γ), improving prognosis (6). Helper T cells (Th) 1, 2 and 17 also
serve roles: Th1 enhances CTL activity through IFN-γ and IL-2; Th2
recruits eosinophils and macrophages; and Th17 can have pro or
antitumor effects (11).
Conversely, Tregs suppress effector T cells (Teffs), often
associated with poor prognosis (12). In numerous cancers, Tregs are more
abundant in tumor tissues than in adjacent healthy tissues. Their
presence in the TME inhibits effector T cell function, promoting
disease progression and poor outcomes, as observed in colorectal
cancer (11).
Cells require nutrients for normal function, and
immune cells rely on nutrient uptake to regulate their activities
(13). T cell activation drives
metabolic shifts, boosting glycolysis to meet energy needs for
proliferation. Unlike cancer cells, which experience dysregulated
metabolism, T cells respond normally to these changes (14). Lipid and amino acid metabolism are
also crucial; lipid biosynthesis affects mTOR, a key regulator of
metabolic reprogramming (15),
whilst amino acid availability, particularly L-arginine, influences
T cell survival and adaptability (16). In the TME, tumor cells compete with
T cells for essential nutrients such as glucose, glutamine and
arginine, impairing T cell function and promoting tumor
progression. Through the Warburg effect, tumor cells outcompete T
cells for glucose, whilst programmed death-1 (PD-1) expression
further inhibits glycolysis. Fatty acid accumulation disrupts
mitochondrial function, driving T cell exhaustion. Tumor cells also
deplete arginine and glutamine, reducing their availability for T
cells. Other immune cells, such as dendritic cells, MDSCs and
tumor-associated macrophages, overexpress enzymes such as arginase
and indoleamine 2,3-dioxygenase (IDO), depleting essential amino
acids and altering T cell activation and differentiation. The
accumulation of immunosuppressive Tregs further exacerbates T cell
exhaustion (17).
The AML TME is marked by high glucose metabolism and
lactic acid (LA) accumulation, creating an acidic environment that
suppresses T cell function (18).
Neutralizing acidity with sodium bicarbonate (NaBi) has been
reported to enhance CD8+ T cell immunotherapy efficacy
(19). The fms-related tyrosine
kinase 3-internal tandem duplication mutation increases glycolytic
activity, contributing to chemotherapy resistance and impaired T
cell function (20). Elevated
glucose metabolism in AML cells further drives chemoresistance.
Excessive glucose consumption and high LA levels in AML cells
inhibit T cell function, weakening antitumor immunity (21,22).
Lipid metabolism also serves a key role in tumor progression
(23), immune evasion and drug
resistance (24). Dysregulated
lipid metabolism alters the balance between Tregs and Teffs in the
TME (13).
Additionally, recent studies have highlighted the
critical role of T-cell metabolic reprogramming in the AML TME. The
abnormal metabolic features of the AML microenvironment, such as
high LA levels, hypoxia and nutrient competition, directly suppress
T cell effector functions and drive exhaustion by altering
metabolic pathways such as glycolysis and fatty acid oxidation
(FAO) (25–27). These factors notably contribute to
immunotherapy failure. The present review assesses the molecular
mechanisms of T cell metabolic reprogramming in the AML TME,
focusing on the way metabolic imbalances impair T cell
functionality. It also explores potential strategies for combining
metabolic interventions with immunotherapy. By synthesizing current
research, the current review aims to provide a theoretical
foundation for developing novel AML therapies based on metabolic
regulation.
Factors in the TME that influence T cell
metabolism in AML
High levels of LA
A defining feature of the AML TME is the production
of excessive LA, creating a highly acidic environment (18). A study reported that LA inhibits T
cell growth and proliferation, and its accumulation in the TME
disrupts LA efflux from T cells, impairing their metabolism,
function and antitumor immunity (28). Additionally, LA downregulates
perforin and granzyme B, which are essential for T cell-mediated
tumor cell killing and proliferation (29,30).
Dichloroacetic acid (DCA) targets pyruvate dehydrogenase kinase
(PDK), an enzyme that drives glucose metabolism toward glycolysis
instead of oxidative phosphorylation (OXPHOS) in tumor cells. By
inhibiting PDK, DCA offers a potential strategy to modulate glucose
metabolism in the AML TME (31).
DCA can inhibit the shift toward glycolysis, reducing LA levels and
notably enhancing T cell proliferation, and cytokine production and
function whilst decreasing apoptosis (32). Tregs use monocarboxylate transporter
1 to uptake LA, which activates signaling pathways that promote
nuclear factor of activated T cells 1 translocation to the nucleus,
upregulating PD-1 expression (Fig.
1) (33). Therefore, high LA
levels increase PD-1 expression in Tregs, amplifying their
immunosuppressive effects. Additionally, LA accumulation and the
resulting acidic pH in the TME inhibit CD4+ T cell
activity, particularly the secretion of key cytokines such as IL-2
and IFN-γ. These cytokines are crucial for immune responses, and
their reduction weakens CD4+ T cell proliferation and
helper function, further promoting immunosuppression (34). In summary, high LA levels in the AML
TME are a major contributor to T cell dysfunction, impairing
antitumor immunity through metabolic disruption, cytotoxic
inhibition and induction of exhaustion.
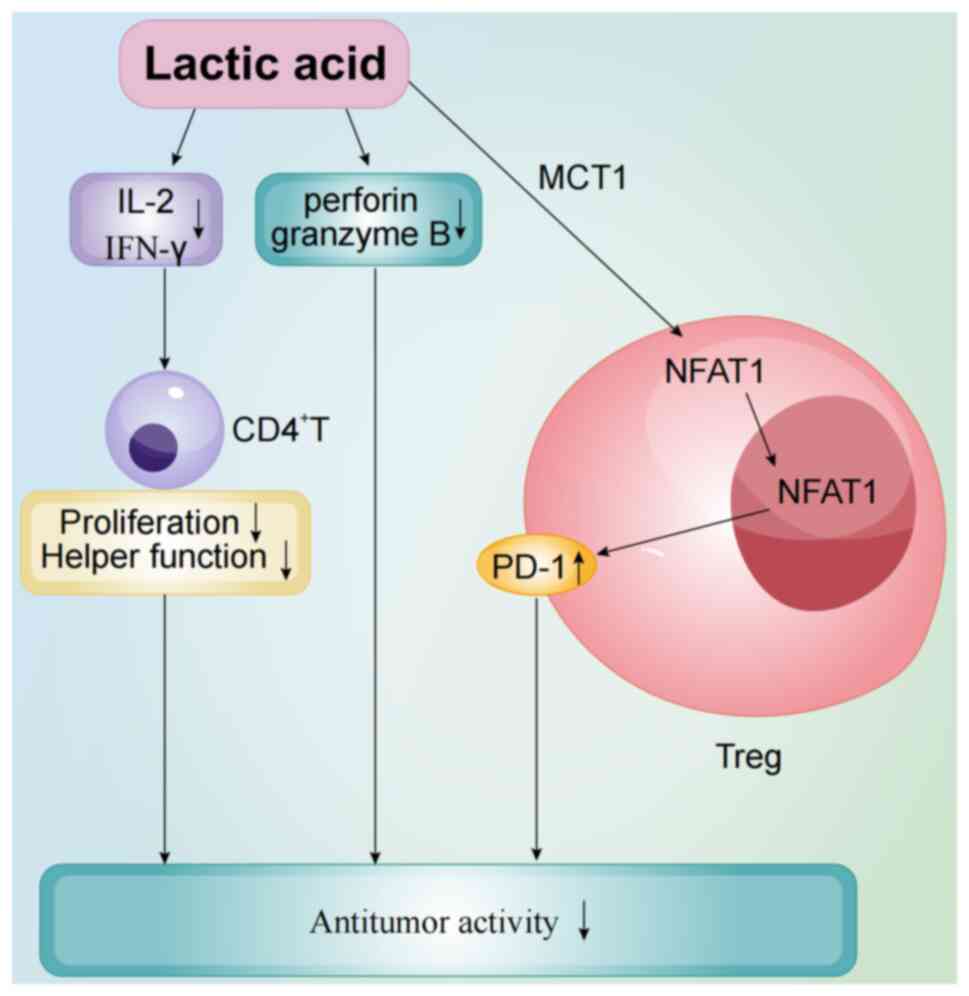 | Figure 1.Acute myeloid leukemia tumor
microenvironment is characterized by high lactic acid levels, which
create an acidic milieu that impairs T cell function. Lactic acid
inhibits perforin and granzyme B, essential for T cell-mediated
tumor cell killing, and reduces the secretion of cytokines such as
IL-2 and IFN-γ, critical for CD4+ T cell proliferation.
Lactic acid accumulation in Tregs is facilitated by MCT1,
activating NFAT1 signaling and upregulating PD-1, enhancing
Treg-mediated immunosuppression. IFN-γ, interferon-γ; Treg,
regulatory T cell; MCT1, monocarboxylate transporter 1; NFAT1,
nuclear factor of activated T cells 1; PD-1, programmed
death-1. |
Hypoxia
Hypoxia is a defining feature of the bone marrow
TME, markedly impacting AML cell growth, metabolic reprogramming
and immune interactions. Studies have reported that patients with
higher hypoxia risk scores tend to have shorter overall survival
rates, linking hypoxia to poor prognosis in AML. Elevated hypoxia
risk scores are also strongly associated with disease progression
and the immunosuppressive TME (35,36).
Under hypoxic conditions, hypoxia-inducible factor 1α (HIF-1α) is
activated and directly upregulates programmed death-ligand 1
(PD-L1) expression. High PD-L1 levels bind to PD-1 on T cells,
inhibiting their activation and signaling, further suppressing
antitumor immunity (37).
Nutrition competition
AML tumor cells and immune cells compete for
essential nutrients such as glucose and amino acids, which are
vital for both rapid tumor growth and normal immune function. Tumor
cells, with their high metabolic demands, prioritize the uptake of
glucose, glutamine and other substrates for energy and
biosynthesis, depriving immune cells of these resources. For
instance, AML cells depend on glutamine for OXPHOS, and the
inhibition of glutaminase-1 has been reported to suppress AML
development in mouse models (38).
Glutamine is also critical for T cell activation, proliferation and
cytokine production (39).
Glutamine deficiency in the TME promotes the generation of Tregs,
further exacerbating immunosuppression (40). Glutamine deficiency not only
restricts effector T cell proliferation and function, but also
supports the metabolic adaptation of Tregs, enhancing their
immunosuppressive activity. Studies have highlighted the antisense
non-coding RNA at the INK4 locus (ANRIL) as a key regulator of
glucose metabolism in AML, where it is notably upregulated. ANRIL
modulates glucose metabolism via the AMP-activated protein kinase
(AMPK)/ sirtuin 1 pathway, promoting AML cell survival. Its
knockdown reduces glucose uptake and inhibits AML cell maintenance
(41,42). Under oxygen-sufficient conditions,
AML cells rely heavily on glucose as their primary metabolic
substrate, rapidly converting it to LA through glycolysis to meet
energy demands (43). A study by
Cunningham and Kohno (44) using
18FDG labeling in 124 patients reported consistently high glucose
uptake in AML bone marrow, highlighting the glucose dependence of
AML cells. This excessive glucose consumption by AML cells
suppresses T cell activation, induces exhaustion and drives
leukemia progression.
Chemokines and cytokines
The AML TME shapes T cell metabolism and function by
secreting chemokines and cytokines, influencing tumor progression
(45,46). Studies have reported that chemokines
such as chemokine CCL3 and CXCL12 in the AML microenvironment
promote Treg accumulation, which competitively inhibits Teffs and
indirectly affects their metabolic activity (47,48).
Whilst research does not directly address whether chemokines
regulate T cell metabolic pathways, it highlights the potential of
blocking Treg migration to delay disease progression. Inhibiting
these chemokines has been reported to slow AML progression in mouse
models (45).
Accumulation of other metabolic waste
products
Potassium ions (K+), abundant in
intracellular fluid, are essential electrolytes that regulate
immune cell function and several cellular processes (49,50).
Tumor cell necrosis releases large amounts of K+ into
the extracellular fluid of the TME. Elevated extracellular
K+ concentrations impair T cell receptor (TCR)-mediated
Akt-mTOR phosphorylation, hindering effector T cell activation and
function (51). Conversely, higher
K+ levels can enhance T cell stemness, maintaining their
undifferentiated state (52). This
indicates that K+ dynamics in the TME influence both
immediate T cell effector functions and their long-term survival
and potential. However, research on the role of K+ in
the AML TME remains limited.
Changes in T cell metabolic pathways in the
AML microenvironment
Glycolysis and OXPHOS
Before antigen exposure, naive T cells remain in a
quiescent state maintained by IL-7. As they do not require clonal
expansion or high cytokine production, their reliance on anabolic
pathways for DNA, protein and molecule synthesis is minimal.
Instead, they generate ATP primarily through mitochondrial OXPHOS
(53). By contrast, tumor cells
undergo a notable metabolic shift, favoring glycolysis over OXPHOS
despite its lower ATP yield. This adaptation supports their rapid
proliferation and survival by quickly meeting energy and metabolic
intermediate demands (54). Tregs
derived from CD4+ T cells serve a dual role: They
maintain immune homeostasis and prevent autoimmune diseases;
however, during AML progression, they suppress CTL activity,
weakening antitumor immunity and promoting immune evasion (55). Studies have reported that AML blasts
promote T cell differentiation into a Treg phenotype by expressing
inducible T cell co-stimulator ligand, markedly expanding the Treg
population (56,57). At the AML disease site, Tregs
suppress CTL activity, limiting their proliferation and hindering
the expansion of adoptively transferred CTLs in vivo. This
suppression weakens CTL-mediated antitumor effects, further
compromising immune responses against AML (58). Research indicates that mTOR and
glucose transporter-1 (GLUT-1) regulate CD4+ T cell
activation by influencing glycolysis. Increased glycolysis in
CD4+ T cells enhances their activation, proliferation
and survival whilst promoting effector T cell differentiation and
inhibiting Treg development, which suppresses immune responses
(59). Similarly, the
differentiation of CD8+ T cells from naive to effector
states requires upregulated glucose metabolism as glycolysis
provides the energy needed for their immune effector functions
(60). In mouse CD8+ T
cells, branched-chain amino acid (BCAA) accumulation boosts glucose
transporter 1 (GLUT1) levels via a forkhead box protein
O1-dependent mechanism, enhancing glycolysis and OXPHOS to
strengthen antitumor immunity. BCAA supplementation also improves
the efficacy of PD-1 immunotherapy in tumors (61). Thus, increased glycolysis serves a
crucial role in enhancing antitumor immune responses.
In AML, leukemia cells rely heavily on glycolysis
(Warburg effect) for energy, resulting in the accumulation of
glycolytic byproducts such as methylglyoxal. These reactive
compounds react non-enzymatically with proteins, lipids and DNA,
forming advanced glycosylation end products (AGEs) (62). AGEs bind to the receptor for AGEs
(RAGE), activating signaling pathways such as NF-κB, MAPK and
phosphoinositide 3-kinase (PI3K)/Akt, which drive pro-inflammatory
and pro-survival responses, influencing cellular functions and
disease progression (63,64). AGE-RAGE signaling promotes the
proliferation of AML cell lines, such as HL60 and HEL, by
inhibiting apoptosis and autophagy, enhancing cancer cell survival
and invasiveness. This mechanism contributes to the progression of
several tumors, including AML (64). Consequently, the AGE-RAGE axis
represents both a hallmark of AML metabolic reprogramming and a
potential diagnostic and therapeutic target.
FAO
Fatty acid uptake and metabolism are essential for
AML cell proliferation, providing energy and metabolic
intermediates whilst inhibiting apoptosis and conferring resistance
to cytotoxic drugs. AML cells thus depend on fatty acid metabolism
for survival (65). Lipid
metabolism also serves a key role in T cell metabolic
reprogramming, supporting membrane expansion through the production
of phospholipids and cholesterol. Naive CD8+ T cells in
the lymphatic system primarily rely on FAO for energy (66). The differentiation of Teffs compared
with memory T cells depends on the strength of signals from
co-stimulatory molecules, cytokines and antigen presentation
(67). Strong stimulation leads to
the generation of short-lived terminally differentiated effector
cells, whilst weaker stimulation promotes the differentiation of
memory precursor cells, which further transform into long-lived
memory cells, providing protection against re-infection (68). Certain memory T cells also arise
from Teffs that survive apoptosis at the end of an immune response
(69). In the AML TME, memory T
cells exhibit markedly reduced metabolic adaptability and
persistence. Research indicates that these cells fail to sustain
critical metabolic pathways such as FAO and mitochondrial OXPHOS
resulting in energy deficiency that compromises their survival and
long-term effector function (6,70). AML
cells exacerbate this dysfunction by competing for essential
nutrients such as glucose and glutamine, further suppressing T cell
metabolism. Prolonged nutrient deprivation not only impairs memory
T cell function, but also promotes their exhaustion, characterized
by upregulated expression of exhaustion markers such as PD-1 and
thymocyte selection-associated high mobility group box protein, and
diminished cytotoxic capacity (56,71).
Notably, memory T cells also serve a role in modulating the
response of hematological malignancies to PD-1 blockade therapy
(72). Studies have reported that
TNF receptor-associated factor 6 (TRAF6) influences CD8+
memory T cells through lipid metabolism regulation. Mice with T
cell-specific TRAF6 deficiency exhibit strong effector T cell
responses but fail to form memory T cells effectively (73,74).
IL-15 regulates mitochondrial spare respiratory capacity and
oxidative metabolism by modulating mitochondrial biogenesis and the
key enzyme carnitine O-palmitoyl transferase 1 (CPT1)a. CPT1a is
critical for the rate-limiting step in mitochondrial FAO (75). Enhancing FAO through AMPK activation
or mTOR inhibition can notably increase memory T cell numbers
(73,76). These findings emphasize the pivotal
role of lipid metabolism in T cell metabolic reprogramming.
Amino acid metabolism
Studies have reported that amino acids regulate
immune responses by influencing T cell activation, cytokine
production and other immune functions (77). Glutamine, the most abundant amino
acid in serum, is vital for maintaining metabolic balance and cell
function. Its absence in culture medium markedly impairs naive T
cell activation, proliferation and cytokine production (39). In the hypoxic TME, glutamine acts as
a primary carbon source, supporting the energy and metabolic needs
of tumor cells (78). Amino acids
and their metabolites are essential in regulating both tumor and
immune cell proliferation within the TME. For instance, glutamine
and its metabolic pathways are crucial for tumor cell glycolysis.
Using glutamine antagonists can effectively inhibit glycolysis in
tumor cells, suppressing their growth and enhancing the antitumor
immune response by altering the immunosuppressive TME, thereby
overcoming immune evasion (79).
Besides glutamine, the metabolism of arginine and tryptophan also
serves a pivotal role in immune regulation within the TME. Studies
have reported that monocytes, under macrophage-stimulating factor
influence, rapidly degrade tryptophan through increased IDO
activity, thereby suppressing T cell proliferation. This mechanism
aids tumor cells and macrophages in immune evasion. Furthermore,
extracellular arginine availability in the TME directly influences
T cell function and antitumor responses (80,81).
Arginine deprivation leads T cells to initiate autophagy,
downregulate the CD3ζ chain and ultimately undergo apoptosis. In
AML, this effect is more pronounced, as AML blasts express and
secrete arginase II, a key enzyme for arginine metabolism, whilst
arginase I is typically low and only detectable under specific
conditions. This metabolic regulation further impairs T cell
function, aiding AML cells in evading immune surveillance (70). These findings highlight the crucial
association between amino acid metabolism in the TME and T
cell-mediated antitumor immunity.
Nucleotide metabolism
Nucleotides, as essential components of genetic
material, are critical for highly proliferating cells, particularly
purine and pyrimidine nucleotides. Consequently, nucleotide
metabolism presents a potential target for cancer therapy (82,83).
Drugs such as methotrexate, which target nucleotide metabolism,
have been reported to be effective in treating acute lymphoblastic
leukemia (ALL). However, non-specific targeting of nucleotide
metabolism can inhibit normal cell processes, leading to severe
side effects (84,85). In the AML TME, high concentrations
of adenosine act as an immunosuppressive metabolite. Elevated
adenosine levels suppress T cell activity by inhibiting activation,
proliferation and cytokine secretion through adenosine receptor
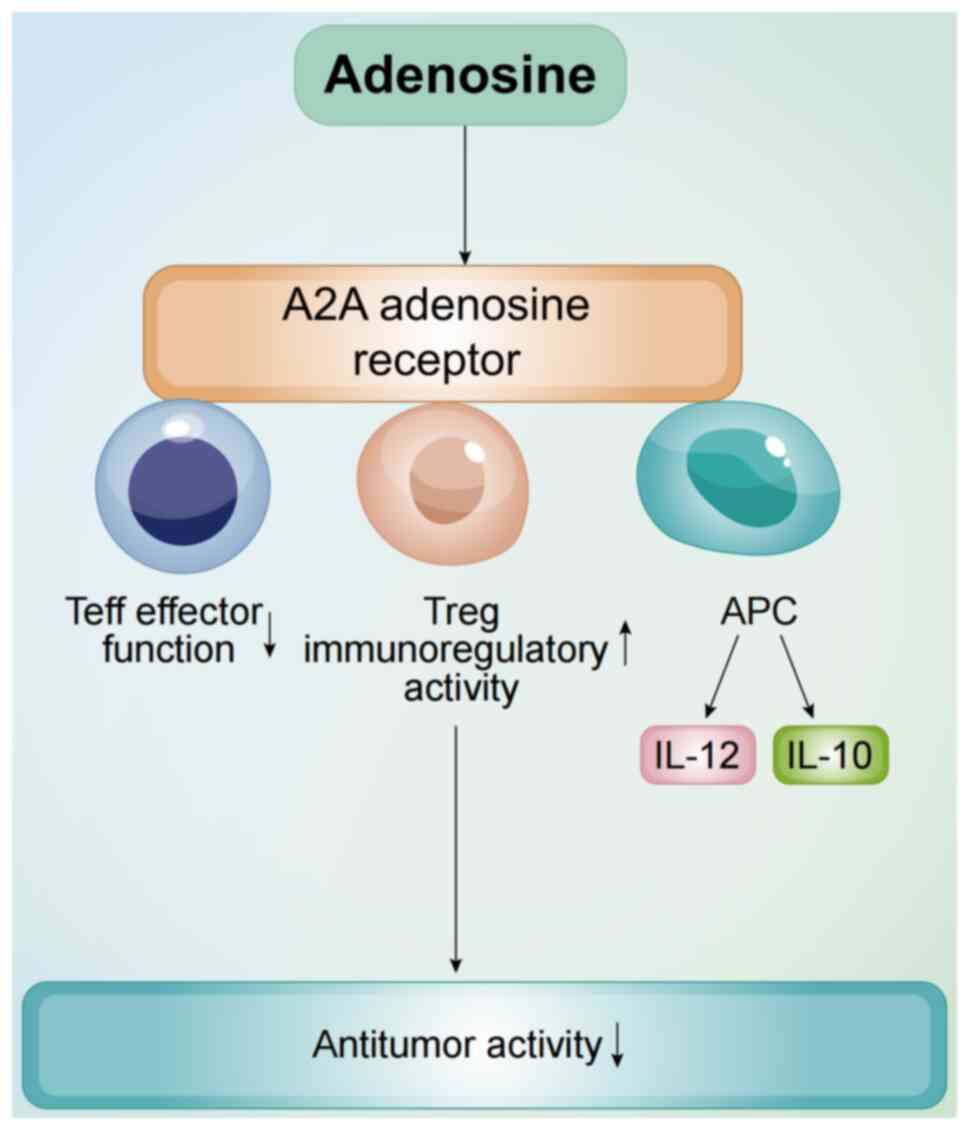
binding such as A2A receptors (56). The A2A adenosine receptor signaling
pathway markedly inhibits T lymphocyte proliferation, activation
and cytokine production. Additionally, this pathway activates
immunosuppressive cells, such as Tregs and MDSCs, further impairing
effector immune cell function (86). Activation of A2A receptors not only
suppresses effector T cell activity, but also enhances Treg cell
immunoregulatory function by upregulating key molecules and
metabolic pathways, thus promoting immune suppression (Fig. 2) (87). Furthermore, studies have reported
that LA treatment reduces nucleotide abundance in T cells,
impairing proliferation and cell cycle activity. LA also disrupts
several metabolic pathways, including amino acid biosynthesis and
pyrimidine metabolism. NaBi itself can serve as a substrate for
multiple carboxylase reactions such as pyrimidine metabolism
(88,89). The application of NaBi can reverse
these changes, pyrimidine metabolism increased in T cells rescued
with NaBi (19).
Signaling pathways of T cell metabolic
remodeling in AML TME
mTOR signaling pathway
mTOR, a serine/threonine kinase, serves a pivotal
role in several cellular processes, including metabolism
regulation. It is a key regulator of cell metabolism and serves as
the catalytic subunit of both mTORC1 and mTORC2 complexes. mTORC1
supports effector T cell function by promoting glycolysis and
protein synthesis, whilst mTORC2 regulates T cell differentiation
and survival through the cytoskeleton and lipid metabolism
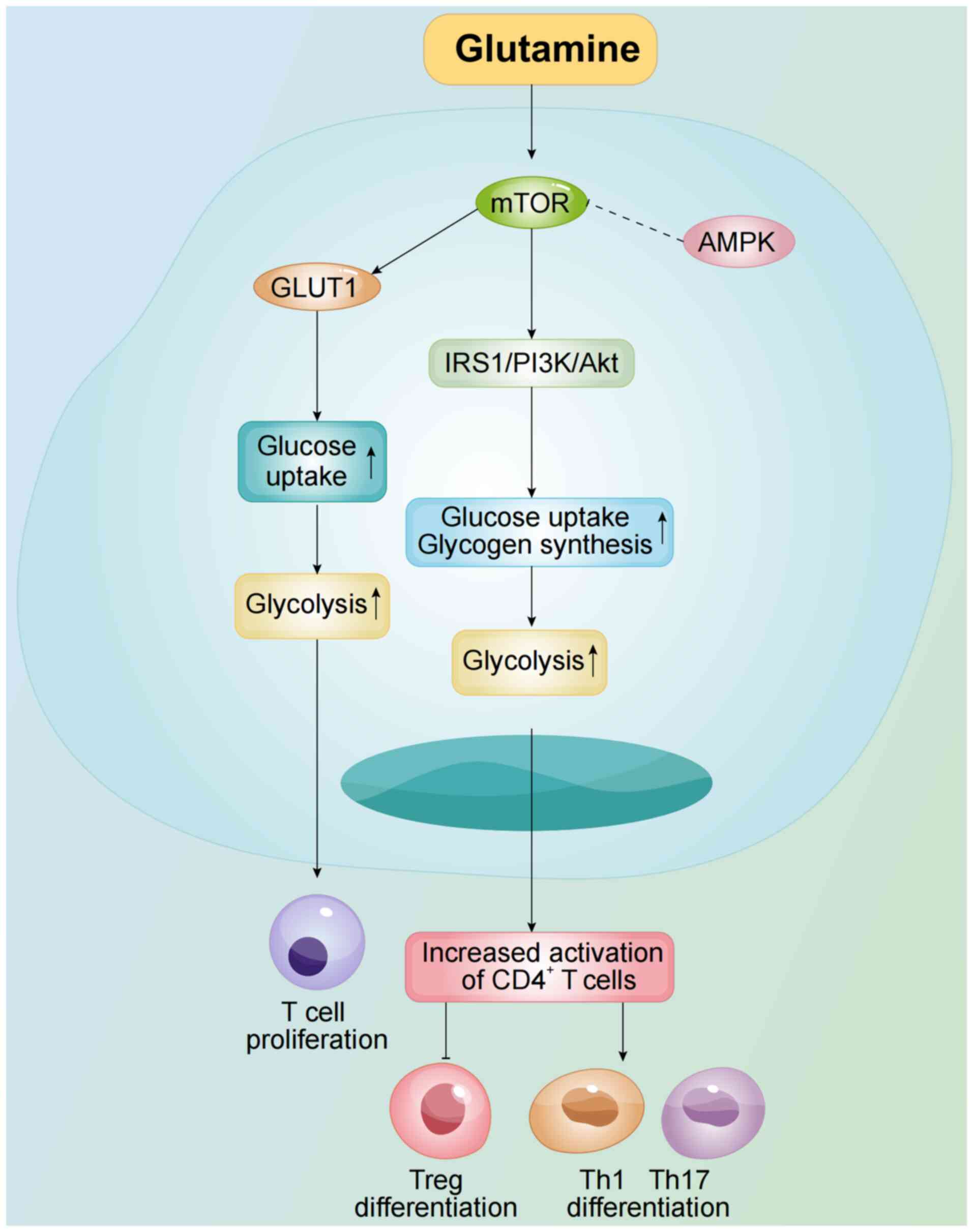
(90). mTOR enhances glucose uptake
and glycogen synthesis by modulating the insulin receptor substrate
1 (IRS1)/PI3K/Akt pathway, thereby boosting glycolysis. Inhibition
of mTOR activation or its downregulation in CD4+ T cells
reduce glycolysis, impairing their activation (31). mTOR activation also increases GLUT1
expression, promoting T cell proliferation and cytokine production
(Fig. 3) (91). Additionally, mTOR is a crucial
regulator of memory CD8+ T cell differentiation, with
the mTOR-specific inhibitor rapamycin, an immunosuppressive drug,
demonstrating an immunostimulatory effect on memory CD8+
T cells (87).
AMPK signaling pathway
In the AML TME, AMPK, as a key energy sensor, serves
a crucial role in regulating the metabolic state of immune cells,
enabling them to effectively maintain their activity and function.
It also serves an important role in T cell metabolic reprogramming.
A previous study reported that AMPK signaling promotes lipid
metabolism to generate functional memory CD8+ T cells
(92). Furthermore, as an upstream
inhibitor of mTOR activity, AMPK can inhibit mTOR through the AMPK
activator metformin, which helps reduce glycolysis in T cells
(Fig. 4). This, in turn, promotes
the generation of Tregs by suppressing Th1 and Th17 cells (93). Metformin also activates AMPK and
inhibits the proliferation of AML cell lines and primary AML cells
(94). However, future research
needs to further explore the specific molecular mechanisms of AMPK
activation and its potential for clinical translation.
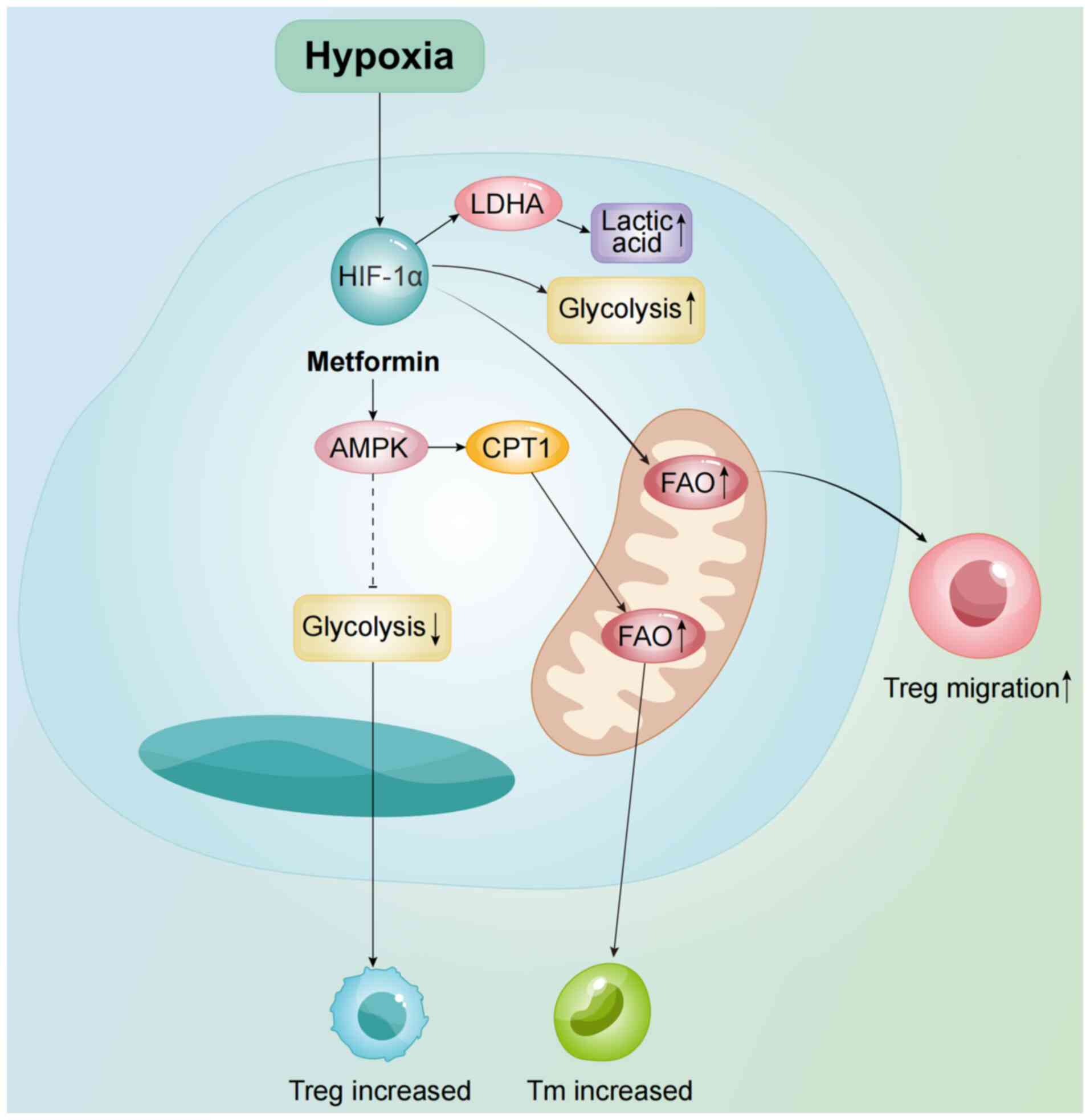 | Figure 4.As an mTOR inhibitor, AMPK suppresses
mTOR activity via metformin, reducing glycolysis and promoting Treg
generation whilst suppressing Th1/Th17 differentiation. AMPK also
upregulates CPT1, enhancing FAO and supporting T cell metabolic
reprogramming. Additionally, HIF-1α promotes Treg migration by
upregulating glycolysis and FAO. AMPK, AMP-activated protein
kinase; Th, Helper T cell; Treg, regulatory T cell; CPT1, carnitine
O-palmitoyl transferase 1; HIF-1α, hypoxia-inducible factor 1α;
LDHA, lactate dehydrogenase A; FAO, fatty acid oxidation; Tm,
memory T cell. |
Peroxisome proliferator-activated
receptor (PPAR) family of transcription factors
The PPAR family includes PPARα, PPARδ and PPARγ. The
nuclear receptor PPARγ serves an essential role in adipogenesis,
immune responses and the metabolism of lipids and carbohydrates.
Fatty acids can also act as ligands for PPARγ (95,96).
Study have reported that in chronic lymphocytic leukemia (CLL),
high doses of glucocorticoids induce the activation of PPARα and
downstream FAO, leading to drug resistance (97). Moreover, it has been reported that
activation of PPAR promotes the proliferation of CD8+ T
cells, increasing the number of functional Teffs. The activation of
the PPAR pathway can also rescue PD-1 blockade-induced T cell
apoptosis by upregulating anti-apoptotic proteins such as Bcl2,
baculoviral IAP repeat containing 3 and apoptosis inhibitor 5.
Additionally, PPAR activation can reprogram CTL energy metabolism
and overcome the reduction in the number of functional Teffs
associated with PD-1 blockade by reducing apoptosis or increasing
proliferation (98). Therefore,
targeting the PPAR signaling pathway, such as by using PPAR
agonists, may serve as a potential therapeutic target for AML.
HIF-1α and hypoxic response
HIF-1α is a central transcription factor in hypoxic
cells and a hallmark of TME. It is also a downstream target of
GLUT-1 (99). It facilitates Treg
migration by promoting glycolysis and FAO. Elevated glucose uptake
by cancer cells stabilizes HIF-1α, thereby suppressing antitumor
immunity (100,101). Moreover, HIF-1α-driven
transcription enhances glycolysis in T cells, supporting Th17
differentiation whilst inhibiting Tregs (91). Mitochondrial dysfunction and
HIF-1α-mediated metabolic reprogramming contribute to T cell
exhaustion, a process reversible through glycolysis inhibition
(102). Treatment with digoxin or
acriflavine, both inhibitors of HIF-1 expression and function, in
subcutaneous tumor mice has been reported to limit tumor growth
(103). Therefore, targeting HIF-1
in the TME may be an effective therapeutic strategy for AML.
Metabolic features of the AML
microenvironment and their impact on immunotherapy
Immune checkpoint (IC) inhibitors
ICs are molecular mechanisms that regulate immune
system activity, comprising co-stimulatory receptors such as CD40
and CD80 and inhibitory receptors such as cytotoxic
T-lymphocyte-associated protein 4 and PD-1. These checkpoints
maintain immune tolerance, protect normal tissues from excessive
immune responses, or, in certain cases, enable cancer cells to
evade immune surveillance (34).
The advent of IC inhibitors has improved the prognosis for numerous
solid tumors and certain lymphomas by blocking inhibitory signals
such as the PD-1/PD-L1 pathway, thereby enhancing antitumor
immunity (104). However, their
efficacy in AML remains limited, particularly with PD-1/PD-L1
inhibitors (105), for reasons
that are not yet fully understood. In the AML TME, competition for
nutrients between AML cells and T cells restricts T cell access to
glucose and glutamine, impairing their metabolic function and
antitumor response. Consequently, IC inhibitors fail to enhance T
cell-mediated antitumor effects (22). Furthermore, the PD-1/PD-L1
interaction serves a critical immunosuppressive role in the TME,
promoting regulatory T cell function and inhibiting the activation
and proliferation of Teffs, thereby further dampening the antitumor
immune response (106).
Limitations of adoptive T cell therapy
(ACT)
ACT enhances immune responses against tumors or
infections by modifying or expanding autologous or donor-derived T
cells ex vivo. This includes engineering T cells with
chimeric antigen receptors (CARs) or TCRs to recognize specific
tumor antigens. Following expansion, these Teffs are re-infused to
mediate targeted immune responses (107,108). However, unlike in ALL, CAR-T cell
therapy shows limited efficacy in AML, largely due to the
immunosuppressive TME (29).
Lymphodepleting chemotherapy prior to CAR-T cell infusion can
enhance therapeutic outcomes by reducing Tregs in the TME and
alleviating their suppressive effects, thereby improving CAR-T cell
proliferation and persistence in vivo (109). In AML, expanded Tregs secrete
immunosuppressive cytokines such as IL-10 and TGF-β, which impair
CAR-T cell function (57).
Additionally, AML cells secrete arginase II, disrupting T cell
metabolism and promoting immune evasion (41). Inhibition of arginine metabolism has
been reported to enhance the efficacy of CD33-CAR T cells in
preclinical AML models (110).
Similarly, blocking the adenosine A2A receptor (A2AR), a downstream
mediator of adenosine signaling, improves CAR-T cell efficacy in
solid tumors (111). Adenosine
suppresses T cell proliferation, activation and effector function
via A2AR, whilst promoting Treg expansion, thereby dampening
antitumor immunity (112).
However, whether this mechanism extends to hematologic malignancies
such as AML and CLL remains unclear.
Potential therapeutic strategies for
metabolic regulation
In patients with AML, elevated intracellular and
plasma arginase activity markedly inhibits T cell proliferation,
contributing to immune dysfunction. This effect is largely mediated
by increased expression and activity of arginase II in AML cells,
identifying it as a potential biomarker for immune status and
disease progression. Inhibition of arginase II has been reported to
restore T cell proliferation and enhance antitumor immunity
(110,113). When PD-1 binds to its ligand
PD-L1, activated T cells are unable to continue glycolysis and
normal amino acid metabolism, which results in insufficient energy
production to support their effector functions. In addition to
inhibiting glycolysis and amino acid metabolism, PD-1 may also
impair T cell oxidative detoxification capacity, reducing their
ability to cope with oxidative stress (114). Thus, elucidating the PD-1
signaling axis is crucial for understanding T cell dysfunction and
identifying novel therapeutic targets.
Future research directions and
challenges
Key areas for further exploration in T
cell metabolism in AML TME
Although the role of T cell metabolism in the AML
TME has been preliminarily elucidated, further in-depth exploration
is needed in the following key areas.
Metabolic reprogramming and
personalized therapy
In future research, individualized treatments
targeting T cell metabolic reprogramming in the TME of patients
with AML should focus on the impact of metabolic heterogeneity on
treatment responses. Differences in metabolic characteristics
between patients with AML may profoundly influence the metabolic
state of T cells and their antitumor capabilities. For example,
variations in glycolysis, FAO or amino acid metabolism across
different patients could lead to notable differences in therapeutic
efficacy. Utilizing metabolomics and single-cell analysis
techniques could uncover individual differences in T cell metabolic
reprogramming and provide a basis for precise metabolic
interventions. However, this approach faces several challenges,
such as the ways to integrate multidimensional data to accurately
identify key metabolic nodes, implement personalized metabolic
regulation of targets in clinical applications and minimize
potential side effects of metabolic interventions on systemic
metabolic homeostasis. In the future, combining advanced
technological methods and large-scale clinical studies will be
necessary to explore the feasibility of personalized metabolic
interventions, with the goal of achieving precision treatment for
patients with AML.
Application of emerging
technologies
In future research on T cell metabolic reprogramming
within the AML TME, emerging technologies will provide crucial
support for uncovering metabolic regulatory mechanisms and
therapeutic potential. The application of single-cell metabolomics
and spatial metabolomics can capture the dynamic changes and
spatial heterogeneity of T cell metabolism in the AML
microenvironment with high resolution, offering a new perspective
on the role of metabolic reprogramming in tumor immune evasion.
Moreover, metabolic flux analysis can track the dynamic changes in
key metabolic pathways within T cells in real-time, revealing the
flow and regulation patterns of metabolites under different
conditions. By contrast, CRISPR screening technology can precisely
identify key genes and metabolic nodes involved in T cell metabolic
reprogramming, providing specific targets for developing
intervention strategies. The combination of these technologies will
not only deepen the understanding of T cell metabolic regulation
mechanisms but also advance the design of personalized metabolic
treatment plans. However, the application of the aforementioned
technologies in AML still faces challenges, such as integrating
multidimensional data, high costs and unclear clinical translation
pathways. Future multi-disciplinary collaborations will be required
to further optimize their application.
Challenge of clinical translation
There are still notable obstacles and challenges in
translating the research on T cell metabolic reprogramming within
the AML TME into clinical application. A key obstacle from basic
research to clinical use is the way to simplify complex metabolic
mechanism studies into clear clinical targets, whilst ensuring
these targets have broad applicability across heterogeneous patient
populations. Moreover, although certain preliminary progress has
been made in clinical trials of metabolic interventions in AML,
such as improving the immune microenvironment through the
regulation of glycolysis, FAO or amino acid metabolism, issues such
as individual variability in efficacy, long-term treatment side
effects and resistance remain prominent. Metabolic interventions
may affect systemic metabolic homeostasis leading to unpredictable
toxic reactions, and the adaptive metabolic mechanisms of tumor
cells may induce resistance. Therefore, future research needs more
precise targeting strategies to achieve efficient regulation within
specific metabolic pathways, whilst minimizing systemic effects.
The combination of advanced technologies, such as metabolomics and
single-cell analysis for personalized treatment design, along with
the development of combination therapies to mitigate resistance,
may be key approaches to overcoming these challenges.
Conclusions
The present review explored the key mechanisms of T
cell metabolic reprogramming in the TME of AML and its notable
impact on antitumor immune responses. The AML microenvironment,
through the synergistic effects of high LA levels, hypoxia,
nutrient competition and chemokines, markedly suppresses critical
metabolic pathways in T cells, such as glycolysis, lipid metabolism
and amino acid metabolism, weakening their proliferation, effector
functions and antitumor capabilities. Additionally, the
accumulation of metabolic waste products from AML cells, as well as
abnormalities in adenosine and potassium ion metabolism, further
promotes the establishment of an immunosuppressive state.
Furthermore, although the mechanisms of T cell metabolic
reprogramming are preliminarily understood, designing personalized
treatment strategies based on the metabolic characteristics of
patients remains a major challenge. Emerging technologies, such as
single-cell metabolomics and metabolic flux analysis, provide new
research directions for uncovering metabolic mechanisms and
developing metabolic-targeted therapies. At the same time, future
clinical translation needs to balance efficacy with side effects,
optimizing metabolic intervention strategies to enhance the
effectiveness of immunotherapy.
In summary, in-depth research on T cell metabolic
reprogramming in the AML microenvironment will provide important
theoretical support for improving AML immunotherapy strategies,
whilst also offering new insights for the clinical application of
metabolic intervention therapies.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
XYZ designed and conceived the study. YHL wrote the
manuscript. JL and MY revised the manuscript. Data authentication
is not applicable. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Döhner H, Weisdorf DJ and Bloomfield CD:
Acute myeloid leukemia. N Engl J Med. 373:1136–1152. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu H: Emerging agents and regimens for
AML. J Hematol Oncol. 14:492021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Peng C, Xu Y, Wu J, Wu D, Zhou L and Xia
X: TME-related biomimetic strategies against cancer. Int J
Nanomedicine. 19:109–135. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bawek S, Gurusinghe S, Burwinkel M and
Przespolewski A: Updates in novel immunotherapeutic strategies for
relapsed/refractory AML. Front Oncol. 14:13749632024. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Menter T and Tzankov A: Tumor
microenvironment in acute myeloid leukemia: Adjusting niches. Front
Immunol. 13:8111442022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lamble AJ and Lind EF: Targeting the
immune microenvironment in acute myeloid leukemia: A focus on T
cell immunity. Front Oncol. 8:2132018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Korn C and Méndez-Ferrer S: Myeloid
malignancies and the microenvironment. Blood. 129:811–822. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rieger CT and Fiegl M: Microenvironmental
oxygen partial pressure in acute myeloid leukemia: Is there really
a role for hypoxia? Exp Hematol. 44:578–582. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu S and Jiang J: Immune
infiltration-related genes regulate the progression of AML by
invading the bone marrow microenvironment. Front Immunol.
15:14099452024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zeng T, Cui L, Huang W, Liu Y, Si C, Qian
T, Deng C and Fu L: The establishment of a prognostic scoring model
based on the new tumor immune microenvironment classification in
acute myeloid leukemia. BMC Med. 19:1762021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chraa D, Naim A, Olive D and Badou A: T
lymphocyte subsets in cancer immunity: Friends or foes. J Leukoc
Biol. 105:243–255. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Plitas G and Rudensky AY: Regulatory T
cells: Differentiation and function. Cancer Immunol Res. 4:721–725.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
MacIver NJ, Michalek RD and Rathmell JC:
Metabolic regulation of T lymphocytes. Annu Rev Immunol.
31:259–283. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lochner M, Berod L and Sparwasser T: Fatty
acid metabolism in the regulation of T cell function. Trends
Immunol. 36:81–91. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Endo Y, Kanno T and Nakajima T: Fatty acid
metabolism in T-cell function and differentiation. Int Immunol.
34:579–587. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Geiger R, Rieckmann JC, Wolf T, Basso C,
Feng Y, Fuhrer T, Kogadeeva M, Picotti P, Meissner F, Mann M, et
al: L-arginine modulates T cell metabolism and enhances survival
and anti-tumor activity. Cell. 167:829–842.e13. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang J, He Y, Hu F, Hu C, Sun Y, Yang K
and Yang S: Metabolic reprogramming of immune cells in the tumor
microenvironment. Int J Mol Sci. 25:122232024. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Halestrap AP: The monocarboxylate
transporter family-structure and functional characterization. IUBMB
Life. 64:1–9. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Uhl FM, Chen S, O'Sullivan D,
Edwards-Hicks J, Richter G, Haring E, Andrieux G, Halbach S,
Apostolova P, Büscher J, et al: Metabolic reprogramming of donor T
cells enhances graft-versus-leukemia effects in mice and humans.
Sci Transl Med. 12:eabb89692020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ju HQ, Zhan G, Huang A, Sun Y, Wen S, Yang
J, Lu WH, Xu RH, Li J, Li Y, et al: ITD mutation in FLT3 tyrosine
kinase promotes Warburg effect and renders therapeutic sensitivity
to glycolytic inhibition. Leukemia. 31:2143–2150. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Herst PM, Howman RA, Neeson PJ, Berridge
MV and Ritchie DS: The level of glycolytic metabolism in acute
myeloid leukemia blasts at diagnosis is prognostic for clinical
outcome. J Leukoc Biol. 89:51–55. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Herst PM, Hesketh EL, Ritchie DS and
Berridge MV: Glycolytic metabolism confers resistance to combined
all-trans retinoic acid and arsenic trioxide-induced apoptosis in
HL60rho0 cells. Leuk Res. 32:327–333. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jones RG and Thompson CB: Tumor
suppressors and cell metabolism: A recipe for cancer growth. Genes
Dev. 23:537–548. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Röhrig F and Schulze A: The multifaceted
roles of fatty acid synthesis in cancer. Nat Rev Cancer.
16:732–749. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Heintzman DR, Fisher EL and Rathmell JC:
Microenvironmental influences on T cell immunity in cancer and
inflammation. Cell Mol Immunol. 19:316–326. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zha C, Yang X, Yang J, Zhang Y and Huang
R: Immunosuppressive microenvironment in acute myeloid leukemia:
Overview, therapeutic targets and corresponding strategies. Ann
Hematol. 103:4883–4899. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Böttcher M, Baur R, Stoll A, Mackensen A
and Mougiakakos D: Linking immunoevasion and metabolic
reprogramming in B-cell-derived lymphomas. Front Oncol.
10:5947822020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fischer K, Hoffmann P, Voelkl S,
Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G,
Hoves S, et al: Inhibitory effect of tumor cell-derived lactic acid
on human T cells. Blood. 109:3812–3819. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen Y, Feng Z, Kuang X, Zhao P, Chen B,
Fang Q, Cheng W and Wang J: Increased lactate in AML blasts
upregulates TOX expression, leading to exhaustion of CD8+ cytolytic
T cells. Am J Cancer Res. 11:5726–6742. 2021.PubMed/NCBI
|
|
30
|
Voskoboinik I, Whisstock JC and Trapani
JA: Perforin and granzymes: Function, dysfunction and human
pathology. Nat Rev Immunol. 15:388–400. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sradhanjali S and Reddy MM: Inhibition of
pyruvate dehydrogenase kinase as a therapeutic strategy against
cancer. Curr Top Med Chem. 18:444–453. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rostamian H, Khakpoor-Koosheh M,
Jafarzadeh L, Masoumi E, Fallah-Mehrjardi K, Tavassolifar MJ, M
Pawelek J, Mirzaei HR and Hadjati J: Restricting tumor lactic acid
metabolism using dichloroacetate improves T cell functions. BMC
Cancer. 22:392022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kumagai S, Koyama S, Itahashi K,
Tanegashima T, Lin YT, Togashi Y, Kamada T, Irie T, Okumura G, Kono
H, et al: Lactic acid promotes PD-1 expression in regulatory T
cells in highly glycolytic tumor microenvironments. Cancer Cell.
40:201–218.e9. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang P, Sun Y, Zhang M, Hu L, Wang X, Luo
L, Qiao C, Wang J, Xiao H, Li X, et al: The inhibition of
CD4+ T cell proinflammatory response by lactic acid is
independent of monocarboxylate transporter 1. Scand J Immunol.
94:e131032021. View Article : Google Scholar
|
|
35
|
Jiang F, Mao Y, Lu B, Zhou G and Wang J: A
hypoxia risk signature for the tumor immune microenvironment
evaluation and prognosis prediction in acute myeloid leukemia. Sci
Rep. 11:146572021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu X, Wang L, Kang Q, Feng C and Wang J:
A hypoxia-related genes prognostic risk model, and mechanisms of
hypoxia contributing to poor prognosis through immune
microenvironment and drug resistance in acute myeloid leukemia.
Front Pharmacol. 15:13394652024. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Augustin RC, Delgoffe GM and Najjar YG:
Characteristics of the tumor microenvironment that influence immune
cell functions: Hypoxia, oxidative stress, metabolic alterations.
Cancers (Basel). 12:38022020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jacque N, Ronchetti AM, Larrue C, Meunier
G, Birsen R, Willems L, Saland E, Decroocq J, Maciel TT, Lambert M,
et al: Targeting glutaminolysis has antileukemic activity in acute
myeloid leukemia and synergizes with BCL-2 inhibition. Blood.
126:1346–1356. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Carr EL, Kelman A, Wu GS, Gopaul R,
Senkevitch E, Aghvanyan A, Turay AM and Frauwirth KA: Glutamine
uptake and metabolism are coordinately regulated by ERK/MAPK during
T lymphocyte activation. J Immunol. 185:1037–1044. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Klysz D, Tai XG, Robert PA, Craveiro M,
Cretenet G, Oburoglu L, Mongellaz C, Floess S, Fritz V, Matias MI,
et al: Glutamine-dependent α-ketoglutarate production regulates the
balance between T helper 1 cell and regulatory T cell generation.
Sci Signal. 8:ra972015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sun LY, Li XJ, Sun YM, Huang W, Fang K,
Han C, Chen ZH, Luo XQ, Chen YQ and Wang WT: LncRNA ANRIL regulates
AML development through modulating the glucose metabolism pathway
of AdipoR1/AMPK/SIRT1. Mol Cancer. 17:1272018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Balihodzic A, Barth DA, Prinz F and
Pichler M: Involvement of long non-coding RNAs in glucose
metabolism in cancer. Cancers (Basel). 13:9772021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pavlova NN, Zhu J and Thompson CB: The
hallmarks of cancer metabolism: Still emerging. Cell Metab.
34:355–377. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cunningham I and Kohno B: 18 FDG-PET/CT:
21st century approach to leukemic tumors in 124 cases. Am J
Hematol. 91:379–384. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang R, Feng W, Wang H, Wang L, Yang X,
Yang F, Zhang Y, Liu X, Zhang D, Ren Q, et al: Blocking migration
of regulatory T cells to leukemic hematopoietic microenvironment
delays disease progression in mouse leukemia model. Cancer Lett.
469:151–161. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bakker E, Qattan M, Mutti L, Demonacos C
and Krstic-Demonacos M: The role of microenvironment and immunity
in drug response in leukemia. Biochim Biophys Acta. 1863:414–426.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang M, Yang Y, Liu J, Guo L, Guo Q and
Liu W: Bone marrow immune cells and drug resistance in acute
myeloid leukemia. Exp Biol Med (Maywood). 250:102352025. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ciciarello M, Corradi G, Forte D, Cavo M
and Curti A: Emerging bone marrow microenvironment-driven
mechanisms of drug resistance in acute myeloid leukemia: Tangle or
chance? Cancers (Basel). 13:53192021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Feske S, Colucci F and Coetzee WA: Do
KATP channels have a role in immunity? Front Immunol.
15:14849712024. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Feske S, Wulff H and Skolnik EY: Ion
channels in innate and adaptive immunity. Annu Rev Immunol.
33:291–353. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Eil R, Vodnala SK, Clever D, Klebanoff CA,
Sukumar M, Pan JH, Palmer DC, Gros A, Yamamoto TN, Patel SJ, et al:
Ionic immune suppression within the tumour microenvironment limits
T cell effector function. Nature. 537:539–543. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Vodnala SK, Eil R, Kishton RJ, Sukumar M,
Yamamoto TN, Ha NH, Lee PH, Shin M, Patel SJ, Yu Z, et al: T cell
stemness and dysfunction in tumors are triggered by a common
mechanism. Science. 363:eaau01352019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Almeida L, Lochner M, Berod L and
Sparwasser T: Metabolic pathways in T cell activation and lineage
differentiation. Semin Immunol. 28:514–524. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Fukushi A, Kim HD, Chang YC and Kim CH:
Revisited metabolic control and reprogramming cancers by means of
the warburg effect in tumor cells. Int J Mol Sci. 23:100372022.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Riether C: Regulation of hematopoietic and
leukemia stem cells by regulatory T cells. Front Immunol.
13:10493012022. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Epperly R, Gottschalk S and Velasquez MP:
A bump in the road: how the hostile AML microenvironment affects
CAR T cell therapy. Front Oncol. 10:2622020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Han Y, Dong Y, Yang Q, Xu W, Jiang S, Yu
Z, Yu K and Zhang S: Acute myeloid leukemia cells express ICOS
ligand to promote the expansion of regulatory T cells. Front
Immunol. 9:22272018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhou Q, Bucher C, Munger ME, Highfill SL,
Tolar J, Munn DH, Levine BL, Riddle M, June CH, Vallera DA, et al:
Depletion of endogenous tumor-associated regulatory T cells
improves the efficacy of adoptive cytotoxic T-cell immunotherapy in
murine acute myeloid leukemia. Blood. 114:3793–3802. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Liu SY, Liao S, Liang L, Deng J and Zhou
Y: The relationship between CD4+ T cell glycolysis and
their functions. Trends Endocrinol Metab. 34:345–360. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Cao J, Liao S, Zeng F, Liao Q, Luo G and
Zhou Y: Effects of altered glycolysis levels on CD8+ T
cell activation and function. Cell Death Dis. 14:4072023.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yao CC, Sun RM, Yang Y, Zhou HY, Meng ZW,
Chi R, Xia LL, Ji P, Chen YY, Zhang GQ, et al: Accumulation of
branched-chain amino acids reprograms glucose metabolism in
CD8+ T cells with enhanced effector function and
anti-tumor response. Cell Rep. 42:1121862023. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Rabbani N and Thornalley PJ:
Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino
Acids. 42:1133–1142. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Palanissami G and Paul SFD: AGEs and RAGE:
Metabolic and molecular signatures of the glycation-inflammation
axis in malignant or metastatic cancers. Explor Target Antitumor
Ther. 4:812–849. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Waghela BN, Vaidya FU, Ranjan K, Chhipa
AS, Tiwari BS and Pathak C: AGE-RAGE synergy influences programmed
cell death signaling to promote cancer. Mol Cell Biochem.
476:585–598. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Bakhtiyari M, Liaghat M, Aziziyan F,
Shapourian H, Yahyazadeh S, Alipour M, Shahveh S,
Maleki-Sheikhabadi F, Halimi H, Forghaniesfidvajani R, et al: The
role of bone marrow microenvironment (BMM) cells in acute myeloid
leukemia (AML) progression: Immune checkpoints, metabolic
checkpoints, and signaling pathways. Cell Commun Signal.
21:2522023. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wang R, Liu Z, Fan Z and Zhan H: Lipid
metabolism reprogramming of CD8+ T cell and therapeutic
implications in cancer. Cancer Lett. 567:2162672023. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Jameson SC and Masopust D: Understanding
subset diversity in T cell memory. Immunity. 48:214–226. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Kaech SM and Cui W: Transcriptional
control of effector and memory CD8+ T cell differentiation. Nat Rev
Immunol. 12:749–761. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
D'Cruz LM, Rubinstein MP and Goldrath AW:
Surviving the crash: Transitioning from effector to memory CD8+ T
cell. Semin Immunol. 21:92–98. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Mougiakakos D: The induction of a
permissive environment to promote T cell immune evasion in acute
myeloid leukemia: The metabolic perspective. Front Oncol.
9:11662019. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Noviello M, Manfredi F, Ruggiero E, Perini
T, Oliveira G, Cortesi F, De Simone P, Toffalori C, Gambacorta V,
Greco R, et al: Bone marrow central memory and memory stem T-cell
exhaustion in AML patients relapsing after HSCT. Nat Commun.
10:10652019. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Abbas HA, Hao D, Tomczak K, Barrodia P, Im
JS, Reville PK, Alaniz Z, Wang W, Wang R, Wang F, et al: Single
cell T cell landscape and T cell receptor repertoire profiling of
AML in context of PD-1 blockade therapy. Nat Commun. 12:60712021.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Pearce EL, Walsh MC, Cejas PJ, Harms GM,
Shen H, Wang LS, Jones RG and Choi Y: Enhancing CD8 T-cell memory
by modulating fatty acid metabolism. Nature. 460:103–107. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Raud B, McGuire PJ, Jones RG, Sparwasser T
and Berod L: Fatty acid metabolism in CD8+ T cell
memory: Challenging current concepts. Immunol Rev. 283:213–231.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
van der Windt GJW, Everts B, Chang CH,
Curtis JD, Freitas TC, Amiel E, Pearce EJ and Pearce EL:
Mitochondrial respiratory capacity is a critical regulator of CD8+
T cell memory development. Immunity. 36:68–78. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Araki K, Turner AP, Shaffer VO, Gangappa
S, Keller SA, Bachmann MF, Larsen CP and Ahmed R: mTOR regulates
memory CD8 T-cell differentiation. Nature. 460:108–112. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Li P, Yin YL, Li D, Woo Kim S and Wu G:
Amino acids and immune function. Br J Nutr. 98:237–252. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Wang Y, Bai C, Ruan Y, Liu M, Chu Q, Qiu
L, Yang C and Li B: Coordinative metabolism of glutamine carbon and
nitrogen in proliferating cancer cells under hypoxia. Nat Commun.
10:2012019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Leone RD, Zhao L, Englert JM, Sun IM, Oh
MH, Sun IH, Arwood ML, Bettencourt IA, Patel CH, Wen J, et al:
Glutamine blockade induces divergent metabolic programs to overcome
tumor immune evasion. Science. 366:1013–1021. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Munn DH, Shafizadeh E, Attwood JT,
Bondarev I, Pashine A and Mellor AL: Inhibition of T cell
proliferation by macrophage tryptophan catabolism. J Exp Med.
189:1363–1372. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Murray PJ: Amino acid auxotrophy as a
system of immunological control nodes. Nat Immunol. 17:132–139.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Di Marcantonio D, Martinez E, Kanefsky JS,
Huhn JM, Gabbasov R, Gupta A, Krais JJ, Peri S, Tan Y, Skorski T,
et al: ATF3 coordinates serine and nucleotide metabolism to drive
cell cycle progression in acute myeloid leukemia. Mol Cell.
81:2752–2764.e6. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Yabushita T and Goyama S: Nucleic acid
metabolism: The key therapeutic target for myeloid tumors. Exp
Hematol. 142:1046932025. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Capelletti MM, Montini O, Ruini E,
Tettamanti S, Savino AM and Sarno J: Unlocking the heterogeneity in
acute leukaemia: Dissection of clonal architecture and metabolic
properties for clinical interventions. Int J Mol Sci. 26:452024.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Wu HL, Gong Y, Ji P, Xie YF, Jiang YZ and
Liu GY: Targeting nucleotide metabolism: A promising approach to
enhance cancer immunotherapy. J Hematol Oncol. 15:452022.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wang H, Wei Y and Wang N: Purinergic
pathways and their clinical use in the treatment of acute myeloid
leukemia. Purinergic Signal. Mar 6–2024.(Epub ahead of print).
View Article : Google Scholar
|
|
87
|
Ohta A: A metabolic immune checkpoint:
Adenosine in tumor microenvironment. Front Immunol. 7:1092016.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Evans DR and Guy HI: Mammalian pyrimidine
biosynthesis: Fresh insights into an ancient pathway. J Biol Chem.
279:33035–33038. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Santi A, Caselli A, Paoli P, Corti D,
Camici G, Pieraccini G, Taddei ML, Serni S, Chiarugi P and Cirri P:
The effects of CA IX catalysis products within tumor
microenvironment. Cell Commun Signal. 11:812013. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Pollizzi KN, Patel CH, Sun IH, Oh MH,
Waickman AT, Wen J, Delgoffe GM and Powell JD: mTORC1 and mTORC2
selectively regulate CD8+ T cell differentiation. J Clin
Invest. 125:2090–2108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Dabi YT, Andualem H, Degechisa ST and
Gizaw ST: Targeting metabolic reprogramming of T-cells for enhanced
anti-tumor response. Biologics. 16:35–45. 2022.PubMed/NCBI
|
|
92
|
Saravia J, Raynor JL, Chapman NM, Lim SA
and Chi H: Signaling networks in immunometabolism. Cell Res.
30:328–342. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Yan Y, Huang L, Liu Y, Yi M, Chu Q, Jiao D
and Wu K: Metabolic profiles of regulatory T cells and their
adaptations to the tumor microenvironment: Implications for
antitumor immunity. J Hematol Oncol. 15:1042022. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Castro I, Sampaio-Marques B and Ludovico
P: Targeting metabolic reprogramming in acute myeloid leukemia.
Cells. 8:9672019. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Ahmadian M, Suh JM, Hah N, Liddle C,
Atkins AR, Downes M and Evans RM: PPARγ signaling and metabolism:
The good, the bad and the future. Nat Med. 19:557–566. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Angela M, Endo Y, Asou HK, Yamamoto T,
Tumes DJ, Tokuyama H, Yokote K and Nakayama T: Fatty acid metabolic
reprogramming via mTOR-mediated inductions of PPARγ directs early
activation of T cells. Nat Commun. 7:136832016. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Tabe Y, Konopleva M and Andreeff M: Fatty
acid metabolism, bone marrow adipocytes, and AML. Front Oncol.
10:1552020. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Chowdhury PS, Chamoto K, Kumar A and Honjo
T: PPAR-induced fatty acid oxidation in T cells increases the
number of tumor-reactive CD8+ T cells and facilitates
anti-PD-1 therapy. Cancer Immunol Res. 6:1375–1387. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Zhang JZ, Behrooz A and Ismail-Beigi F:
Regulation of glucose transport by hypoxia. Am J Kidney Dis.
34:189–202. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Miska J, Lee-Chang C, Rashidi A, Muroski
ME, Chang AL, Lopez-Rosas A, Zhang P, Panek WK, Cordero A, Han Y,
et al: HIF-1α is a metabolic switch between glycolytic-driven
migration and oxidative phosphorylation-driven immunosuppression of
tregs in glioblastoma. Cell Rep. 27:226–237.e4. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Nagao A, Kobayashi M, Koyasu S, Chow CCT
and Harada H: HIF-1-dependent reprogramming of glucose metabolic
pathway of cancer cells and its therapeutic significance. Int J Mol
Sci. 20:2382019. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Wu H, Zhao X, Hochrein SM, Eckstein M,
Gubert GF, Knöpper K, Mansilla AM, Öner A, Doucet-Ladevèze R,
Schmitz W, et al: Mitochondrial dysfunction promotes the transition
of precursor to terminally exhausted T cells through
HIF-1α-mediated glycolytic reprogramming. Nat Commun. 14:68582023.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Pan F, Barbi J and Pardoll DM:
Hypoxia-inducible factor 1: A link between metabolism and T cell
differentiation and a potential therapeutic target. Oncoimmunology.
1:510–515. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Alatrash G, Daver N and Mittendorf EA:
Targeting immune checkpoints in hematologic malignancies. Pharmacol
Rev. 68:1014–1025. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Stahl M and Goldberg AD: Immune checkpoint
inhibitors in acute myeloid leukemia: Novel combinations and
therapeutic targets. Curr Oncol Rep. 21:372019. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Zhou Q, Munger ME, Highfill SL, Tolar J,
Weigel BJ, Riddle M, Sharpe AH, Vallera DA, Azuma M, Levine BL, et
al: Program death-1 signaling and regulatory T cells collaborate to
resist the function of adoptively transferred cytotoxic T
lymphocytes in advanced acute myeloid leukemia. Blood.
116:2484–2493. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Sadelain M, Brentjens R and Rivière I: The
basic principles of chimeric antigen receptor design. Cancer
Discov. 3:388–398. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Riddell SR, Jensen MC and June CH:
Chimeric antigen receptor-modified T cells: Clinical translation in
stem cell transplantation and beyond. Biol Blood Marrow Transplant.
19 (1 Suppl):S2–S5. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Suryadevara CM, Desai R, Farber SH, Choi
BD, Swartz AM, Shen SH, Gedeon PC, Snyder DJ, Herndon JE II, Healy
P, et al: Preventing Lck activation in CAR T cells confers treg
resistance but requires 4-1BB signaling for them to persist and
treat solid tumors in nonlymphodepleted hosts. Clin Cancer Res.
25:358–368. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Mussai F, Wheat R, Sarrou E, Booth S,
Stavrou V, Fultang L, Perry T, Kearns P, Cheng P, Keeshan K, et al:
Targeting the arginine metabolic brake enhances immunotherapy for
leukaemia. Int J Cancer. 145:2201–2208. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Beavis PA, Henderson MA, Giuffrida L,
Mills JK, Sek K, Cross RS, Davenport AJ, John LB, Mardiana S,
Slaney CY, et al: Targeting the adenosine 2A receptor enhances
chimeric antigen receptor T cell efficacy. J Clin Invest.
127:929–941. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Leone RD, Sun IM, Oh MH, Sun IH, Wen J,
Englert J and Powell JD: Inhibition of the adenosine A2a receptor
modulates expression of T cell coinhibitory receptors and improves
effector function for enhanced checkpoint blockade and ACT in
murine cancer models. Cancer Immunol Immunother. 67:1271–1284.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Mussai F, De Santo C, Abu-Dayyeh I, Booth
S, Quek L, McEwen-Smith RM, Qureshi A, Dazzi F, Vyas P and
Cerundolo V: Acute myeloid leukemia creates an arginase-dependent
immunosuppressive microenvironment. Blood. 122:749–758. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Patsoukis N, Bardhan K, Chatterjee P, Sari
D, Liu B, Bell LN, Karoly ED, Freeman GJ, Petkova V, Seth P, et al:
PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis
and promoting lipolysis and fatty acid oxidation. Nat Commun.
6:66922015. View Article : Google Scholar : PubMed/NCBI
|