OC is associated with the highest mortality rates
among female reproductive system malignancies, with a 5-year
survival rate of ~47% (1,2). Annually, there are >239,000 new
cases of OC worldwide and 152,000 deaths. Only 15% of cases of OC
are diagnosed clinically early, and the majority of patients are
already diagnosed with metastatic tumors upon receiving an OC
diagnosis, thereby exacerbating their unfavorable prognosis and
elevated mortality rate. Most cases of OC are treated with surgery
and cytoreduction (3); however, in
this context, drug resistance arising from repeated therapy may no
longer explain the low 5-year survival rates. In particular, OC
enhances chemotherapy resistance through its antioxidant capacity
by upregulating multiple key antioxidant systems, such as NAD(P)H
oxidoreductase 1 (4), the
SLC7A11/xCT antiporter (5) and the
NRF2 pathway (6), which can prevent
the formation of harmful reactive oxygen species (ROS) induced by
chemotherapy, particularly platinum-based therapies such as
cisplatin. Therefore, investigating new and potent biomarkers of OC
tumors, as well as elucidating the molecular mechanisms driving
cancer metastasis, is imperative to develop more targeted
therapeutic strategies for OC (7).
NPs are known to both contribute to and inhibit the
growth of cancer cells by controlling the cell cycle (8), stem cell development (9), exercise metabolism (10), the immune response (11) and DNA damage and repair (12,13).
Post-translational modifications (PTMs) of these proteins,
including phosphorylation, ubiquitin conjugation, SUMOylation and
poly(ADP-ribosyl)ation, are known to control these various
biological processes (14,15). Nuclear proteins (NPs) can be altered
in different ways, including genomic mutations,
transcriptional/splicing variations and PTMs, depending on the
expression levels at their target sites. These alterations, acting
alone or in concert, allow NPs to exert distinct molecular and
biological impacts. For example, when mutated, the nuclear protein
p53, normally a tumor suppressor, acquires oncogenic
gain-of-function activity at high concentrations, driving
metastasis and chemoresistance (16). Similarly, as a core PEST-NP that
governs ovarian cancer (OC) progression, especially critical in
high-grade serous OC (HGSOC), where ~96% of cases harbor p53
mutations, p53 activation in response to DNA damage induces the
expression of ring finger protein 144A (RNF144A) (11). RNF144A exhibits broad cellular
distribution, primarily localizing to the plasma membrane and
perinuclear region; notably, as an E3 ubiquitin ligase, it is
directly involved in regulating the PTM of p53, a key process
controlling the stability, degradation and functional output of
this PEST-NP. However, the functional importance of the
interactions of RNF144A with DNA-dependent protein kinases, which
itself acts as an E3 ubiquitin ligase to mediate p53 degradation
during p53 autoubiquitination, remains currently unclear.
Therefore, as ubiquitination status of p53 directly dictates its
ability to inhibit OC cell proliferation, induce apoptosis or drive
chemoresistance, clarifying how DNA-dependent protein kinase
(DNA-PK) modulates RNF144A-mediated p53 degradation could uncover
novel regulatory mechanisms of PEST-NP function in OC, and
potentially identify new therapeutic targets to restore the
tumor-suppressive role of p53 in OC. Several NPs, including KU70,
PARP1, DNA ligase III and XRCC1, undergo degradation through
PAR-dependent alterations (17).
PTMs serve key roles in regulating various physiological processes
involving these proteins. For instance, ubiquitination controls the
stability of PEST-containing NPs (PCNPs) such as p53, while
phosphorylation at Ser15/Ser20 disrupts p53-MDM2 interactions to
stabilize p53 during DNA damage (18). Such PTM-dependent regulation is
often disrupted by p53 mutations in OC, driving chemoresistance
(19).
The present review summarized the current research
on NP biology and associated potential molecular targets involved
in various cellular processes that contribute to the development of
OC. Further research may elucidate the precise associations of NPs
and mechanisms of cancer progression that hold significant
potential for the wider utilization of NPs as diagnostic and
therapeutic targets in various types of OC.
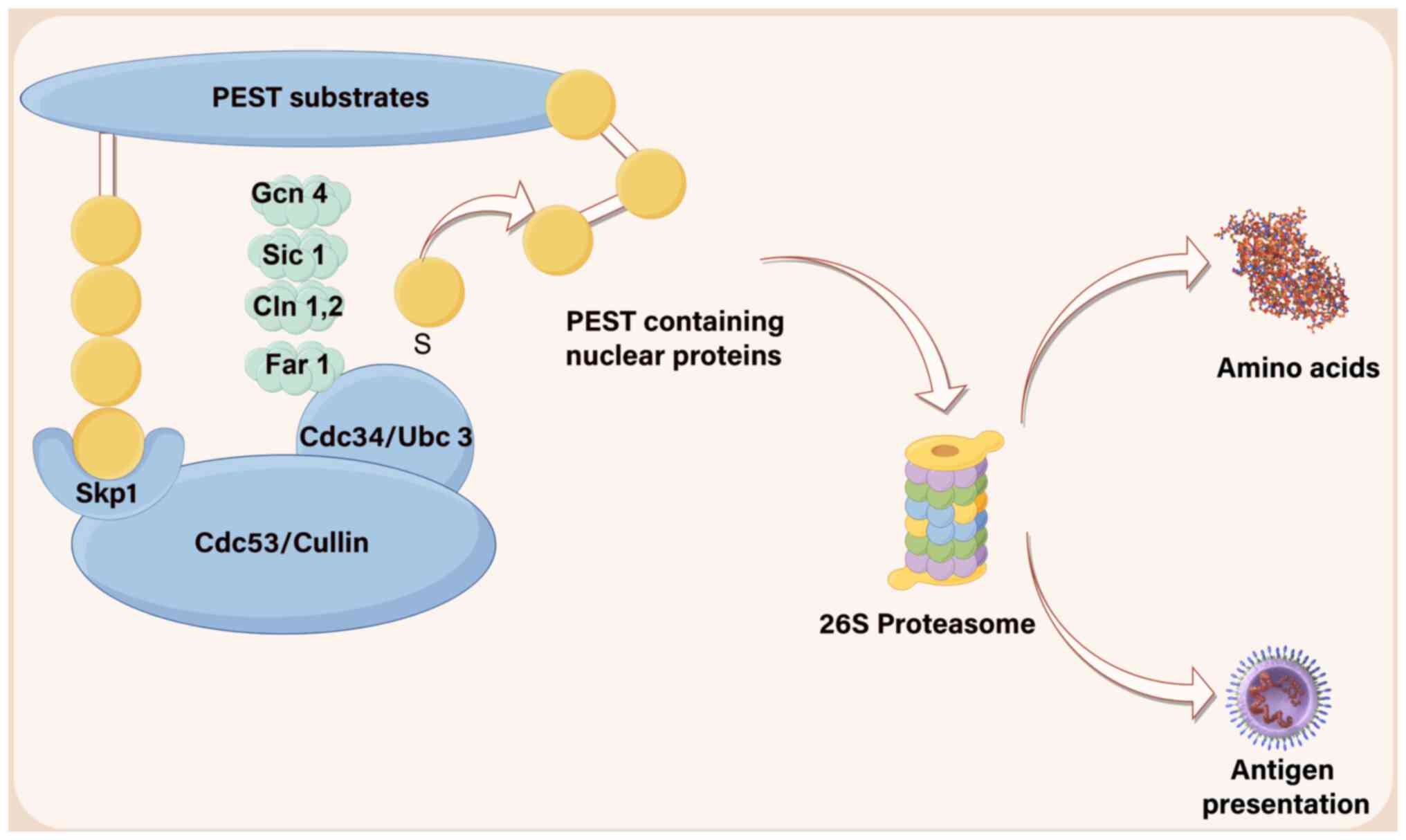
The PEST sequence is a peptide sequence rich in P,
E, S and T. PEST sequences are commonly found in proteins with a
short intracellular half-life, acting as a recognition motif for
degradation-related enzymes and thus influencing protein stability
and turnover in cellular processes. Further research has
demonstrated that PEST sequence-enriched NPs are numerous, widely
dispersed and involved in various physiological and cellular
processes. Often referred to as ‘cell guardians’, PEST-NPs regulate
pathways such as the ubiquitin-proteasome system, nuclear pore
glycosylation and the hexosamine biosynthetic pathway (20). PEST-NPs participate in cell cycle
control, cyclic nucleotide signaling, nucleocytoplasmic transport
and the nutritional regulation of cellular metabolism and
physiology (21). In cancer
biology, several PEST-NPs regulate the tumor cell cycle, either
directly by binding to DNA or indirectly by ubiquitinating cyclins
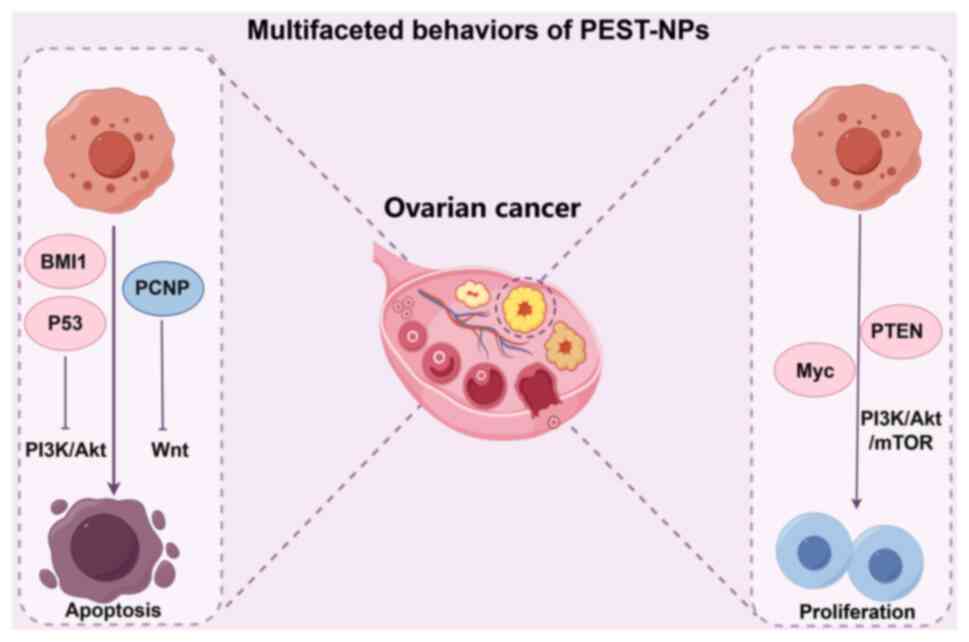
D1 and E1, leading to G1 arrest (Fig. 1). PEST-NPs can also modulate the
cancer-immune system through apoptosis and autophagy induction and
regulate cancer metabolism via the phosphatidylinositol 3-kinase
(PI3K), mTOR and MAPK signaling pathways. The biological activity
of PEST-NPs is influenced by their intracellular distribution and
abundance at target sites, with PTMs such as phosphorylation and
ubiquitination serving key roles in regulating their localization,
activation and expression levels. For instance, the ubiquitination
of the PEST-domain of p53 by MDM2 controls its nucleocytoplasmic
shuttling and stability, dictating apoptotic vs. pro-survival
outcomes in OC (18).
As a structural recognition element, the PEST motif
primarily governs substrate degradation primarily through
mechanisms such as proteasomal turnover (such as endocytosis of the
yeast a-factor receptor STE3) (22)
or lysosomal degradation (such as human calcium receptors on the
cell surface) (23). For example,
Ck2-mediated phosphorylation leads to the degradation of IkB and
IKK, whereas cAMP-dependent protein kinase breaks down proteins
involved in cyclic nucleotide metabolism. Phosphorylation of the
carboxyl terminus of the PEST domain promotes the degradation of
substrate proteins such as PCNP and MeCP2, such as PCNP and MeCP2,
which are also heavily ubiquitinated (24,25).
Additionally, IKK undergoes degradation via calpain-mediated
modification of Bcl6 and P300-mediated acetylation of Bcl6
(26,27). The stability and MAPK-induced
activation of VEGF2 are mediated by the phosphorylation of serine
1,188 within its PEST sequence (28). Under normal conditions, PEST-NPs
maintain short half-lives due to PEST-regulated proteasomal
degradation, ubiquitin conjugation and ligase-mediated control of
their cellular levels (Fig. 2).
Two major genetic alterations drive carcinogenesis:
The activation of oncogenes and the loss of tumor suppressor
activity. In this context, protein modifications mediate
transcription, intracellular localization and degradation (29,30).
Overproduction or overactivation of transcription factors such as
Myc and NF-κB leads to uncontrolled growth and metastasis in
various types of cancer (31). For
example, PTEN activity, localization and stability are regulated by
SUMOylation (mono- or poly-ubiquitination of SUMO1 and SUMO2/3)
(32,33). Similarly, in NF-κB signaling,
phosphorylation of the PEST motif and subsequent proteolytic
degradation of the inhibitor of NF-κB (IκB) kinase initiates NF-κB
nuclear translocation, transcriptional activation and DNA damage.
Overall, PTMs are responsible for regulating activation and
inhibition, maintaining protein stability and degradation, and
determining the intracellular localization of PEST-NPs (34,35).
p53, a PEST-NP with anticancer properties, induces
apoptosis by increasing mitochondrial permeability. Under stress,
nuclear p53 interacts with the cytoplasmic Bcl-xl-p53 complex,
releasing the p53-upregulated modulator of apoptosis (PUMA) to
initiate mitochondria-dependent apoptosis (36,37).
Following apoptosis, p53 is rapidly degraded in both the cytoplasm
and nucleus: Nuclear p53 is conjugated with ubiquitin, translocates
across the nuclear membrane and is degraded by the ubiquitin ligase
mouse double minute 2 (MDM2) in the cytoplasm (38,39).
This PTM-mediated activation (via stress-induced conformational
changes) and inhibition (via MDM2-dependent ubiquitination)
exemplify how PTMs control p53 function.
PEST motif modifications are critical for PEST-NP
stability, activation and deactivation. The PEST-containing
transcription factor BMI1 proto-oncogene, polycomb ring finger
(Bmi1, a PEST-NP) interacts with O-GlcNAcylation transferase at
serine 255, increasing its stability and oncogenic activity by
suppressing p53, PTEN and CDKN1A/CDKN2A (40). Similarly, tankyrase-mediated
ADP-ribosylation of PTEN (a PEST-NP with two PEST sequences)
promotes its ubiquitination and degradation by the E3 ligase
RNF146, reducing its tumor suppressor activity (33). These examples illustrate how PTMs
directly modulate PEST-NP turnover to influence carcinogenesis.
PTMs of the PEST motif also regulate the
localization, a key determinant of their pharmacological and
carcinogenic effects, through modifications that impact protein
activation, stability and subcellular distribution (35). The nuclear localization sequences
(NLSs) and nuclear export sequences (NES) found on cargo proteins
are recognized by importins and export proteins, which mediate
directional transport across the nuclear envelope (41,42).
The imbalanced shuttling of cargo proteins between the nucleus and
cytoplasm, often driven by mutations in NLS/NES regions or
dysregulation of shuttle determinants like PSPC1, is closely
related to tumorigenesis (43,44).
Unlike the cytoplasmic form of p53, which induces apoptosis through
interactions with mitochondrial Bcl-2 family proteins (such as
Bax), the nucleolar form of p53 is activated in response to
nucleolar stress and participates in the DNA damage response
(45). The antiapoptotic protein
Bcl-xL, by contrast, binds to cytoplasmic p53. Unless PUMA releases
the protein trapped in p53, the mitochondrial membrane cannot be
opened to carry out anticancer actions. Numerous transporter
inhibitors are being studied for their potential anticancer
properties, such as withacnistin which decreases the levels of
antiapoptotic proteins, and prevents signal transducer and
activator of STAT3 from being localized in the nucleus (46). As a result, the PEST motif
contributes to the diverse properties of NPs, giving PEST-NPs a
wide range of molecular and physiological functions.
The present review outlined how PTMs of PEST NPs
affect their activation/inhibition, stability/degradation and
intracellular localization to induce a tumor-promoting or
tumor-suppressive response. The emphasis on molecular-level
protein-protein interactions enables supports the understanding of
the complex behavior of PEST NPs in cancer biology. Although
numerous NPs exist, the most significant model PEST NPs, including
p53, PTEN, Bmi1, c-Myc and PCNP, were examined as model
PEST-containing proteins. The structural composition of these
proteins determines their genesis, mode of action and ultimate
fate. The computer-aided application ‘PEST Discover’ defines
authentic PEST domains by calculating a ‘PEST score’, a metric
combining residue composition (P, E, S and T) and hydrophobicity to
predict proteolytic signals. This score [PEST-FIND=0.5 × (mole
percent of P/E/D/S/T)-0.5 × (hydrophobicity index)] discriminates
degradation-prone regions (score >0.5) and enables structural
comparisons of PEST-containing proteins. The computational
framework aligns with tools like PESTfind (http://emb1.bcc.univie.ac.at/embnet/tools/bio/PESTfind),
which implements this algorithm (Table
I).
A number of malignancies (50%) have a mutation in
the p53 gene, which can result in either loss of function or
carcinogenicity (47).
Loss-of-function mutations in the p53 tumor suppressor have the
potential to cause chromosomal instability, loss of cell cycle
arrest, loss of apoptosis, loss of senescent growth arrest and
ineffective DNA base excision repair (48). These modifications enable the
avoidance of key cell cycle pathways, promoting cancer growth. By
contrast, p53 oncogenic mutations with gain-of-function effects can
accelerate tumor metastasis, alter the transcriptional activity of
target genes, inhibit apoptosis and increase chemotherapeutic
resistance (49). A large
proportion (96%) of patients with HGSOC have p53 mutations that
span both gain-of-function (oncogenic) and loss-of-function (loss
of p53 activity) events (50–52).
Human p53 functions as an active homotetramer, with
each subunit consisting of 393 amino acids. This protein has a
complex domain structure, including an N-terminal region with a
ubiquitin ligase (MDM2) binding site, an amino-terminal
transactivation domain (TAD; comprising TAD1 and TAD2), a
DNA-binding domain (DBD), a tetramerization domain, a linker
region, a proline-rich domain and a carboxyl-terminal domain (CTD)
(41,53). The N-terminal segment features five
repeats of proline-XX-proline sequences, where ‘X’ represents an
amino acid that varies across species, as do several
phosphorylation sites between residues 62 and 94 (53). The MDM2 binding site overlaps with
the TAD, specifically TAD1 (residues 1–39) and TAD2 (residues
40–61). The N-terminal region, particularly TAD2, contains PEST
motifs, which can interact with the DBD, potentially inhibiting DNA
binding. The DBD regulates the nuclear and cytoplasmic functions of
p53, as it is critical for mediating DNA-dependent nuclear
activities and indirectly influencing cytoplasmic processes through
its structural role in maintaining p53 functional integrity
(54). Missense mutations in the
DBD (R175, Y220, G245, R248, R249, R273 or R282) account for 62% of
all p53 mutations that are associated with cancer (82% of HGSOC
cases). These mutations eliminate DNA-binding capabilities either
directly or indirectly (53–55).
According to a previous study, the amino acid arginine at position
273 (R273) in the DBD is the most frequently altered, with
histidine (R273H) accounting for 46.6% of the R273 mutations and
cysteine (R273C) accounting for 39.1% (19). It has been proposed that these DBD
mutations lead to a structural shift that affects p53 stability,
making the protein structure stiffer compared with wild-type (WT)
p53 (56). These important DBD
mutations interfere with the ability of p53 ability to regulate
apoptosis, causing malignant cells to proliferate unrestrained. The
CTD carries three nuclear localization signals that allow p53 to
localize to the nucleus (57).
As a transcription factor, p53 interacts with
thousands of genes and is essential for preserving cellular
homeostasis (58,59). In normal cells, p53 is maintained at
a tightly regulated, physiological level; this strict control is
necessary because p53 acts as a potent inducer of apoptosis, and
unregulated p53 activity would trigger excessive cell death that
disrupts normal tissue homeostasis (60). Under normal circumstances, the
cellular expression level of the p53 protein, similar to that of
other PEST-NPs, is very low because of its short half-life of ~620
min. PEST motifs, which are attached to the COOH terminus of
residual proteins, act as signals for PTMs of p53 (phosphorylation
and ubiquitination) and regulate not only proteasomal degradation
(57). The PEST motifs attached to
the CTD of p53 function as signals for its PTMs, including
phosphorylation and ubiquitination; these motifs not only regulate
the proteasomal degradation of p53 but also modulate its
intracellular localization (53).
The protein p53 mediates distinct responses depending on its
subcellular localization: Cytoplasmic accumulation of p53 promotes
mitochondrial outer membrane permeability and initiates
mitochondria-dependent apoptosis (61), whereas nuclear localization enables
p53 to trigger the DNA damage response and transcribe target genes
involved in cell cycle regulation (54,62).
Upon cellular stress, p53 is activated via phosphorylation and
other PTMs; this activation enhances its stability and DNA-binding
capacity, thereby increasing the expression of target genes
associated with cell cycle arrest and DNA repair (62). In the cytoplasm and mitochondria,
monomeric or homodimeric p53 interacts with Bcl-xL (antiapoptotic)
and Bak (apoptotic regulator) to control autophagy and apoptosis
(60). In the cytoplasm and
mitochondria, monomeric or homodimeric p53 interacts with Bcl-xL
(antiapoptotic) and Bak (apoptotic regulator) to control autophagy
and apoptosis (61–63).
Inactivation of p53 activity can occur directly
through mutation or indirectly through the aggregation or
disruption of regulatory proteins (64). The E3 ubiquitin ligase MDM2
typically maintains low levels of p53 through ubiquitin-mediated
proteasomal degradation, a process facilitated by the PEST motifs
located in the CTD of p53 (65).
Overall, two basic pathways for p53 inactivation have been
proposed: p53 mutation and WT p53 degradation through negative
regulatory proteins, such as MDM2 or MDM4 (41). The ubiquitin ligases MDM2 and MDM4
facilitate the targeting of p53 for proteasomal degradation
(65). In malignancies such as
epithelial (EOC) and HGSOC, the most aggressive and common subtypes
of OC (52), the upregulation of
MDM2 and MDM4 (inhibitors of p53) can lead to the downregulation of
p53, in addition to p53 mutations (57,65).
The upregulation of MDM2 and MDM4 inhibitors can lead to the
downregulation of p53, in addition to p53 mutations. Thus, a
primary focus of treatment strategies for malignancies with
wild-type p53 has been the development of antagonists for MDM2 and
MDM4 (65).
The overexpression of the PI3K pathway occurs in
40–70% of OCs and is thus another significant mechanism for p53
regulation (66). The suppression
of PI3K has been shown to lead to the regression of OC both in
vivo and in vitro (67).
Additionally, PTEN, a negative regulator of PI3K, is often found to
be absent in OC. Furthermore, owing to the exposure of a structural
‘sticky’ region, typical p53 mutations can lead to protein
aggregation (57). These distinct
yet related pathways highlight the complexity of p53 regulation,
several mechanisms of which are currently poorly understood. Cancer
cells have developed the capacity to modify and inactivate p53,
dubbed ‘the guardian of the genome’ for its role in maintaining
genomic integrity (60), to evade
tumor suppression and promote proliferation; p53 mutations are
nearly ubiquitous in HGSOC, with reported frequencies exceeding 90%
in clinical cohorts (53). Notably,
however, there are currently no Food and Drug administration
approved p53-based therapeutics for HGSOC. While WT p53 and
p53-based therapeutics, which may be administered via drug delivery
systems such as nanoparticles, viruses, polymers and liposomes,
have been a focus of extensive study (68–73),
these innovative therapeutic approaches have yet to be fully
developed and optimized.
In response to active accumulation in specific
subcellular compartments, the p53 protein mediates distinct
cytoplasmic and nuclear responses. Cytoplasmic accumulation of p53
induces mitochondrial outer membrane permeability and apoptosis
through direct interactions with anti-apoptotic Bcl-2 family
proteins (60,61), whereas nuclear localization enables
p53 to initiate the DNA damage response and transcription of target
genes involved in cell cycle regulation and genome maintenance
(53,54). A p53 mutation, however, can suddenly
disrupt the normal processes of tightly regulated nucleocytoplasmic
shuttling. PTMs include phosphorylation at threonine 155, which
mediates nuclear export (74);
phosphorylation at serine 392, which mediates mitochondrial
localization (75); and kinase
inhibitors, which suppress nuclear export and are responsible for
p53 nuclear export (57). Another
alteration of p53 that results in activation, DNA binding and
nuclear localization is acetylation at lysine residues (76).
PTEN is a tumor suppressor nuclear protein initially
discovered in a mutant form in glioblastoma, prostate and breast
cancer cell lines and xenografts (77). PEST-NP PTEN (or MMACI/TEP1) has 9
exons, 1,212 nucleotides and 403 amino acids and has a molecular
mass of 47 kDa (78–80). The N-terminal phosphatase and
C-terminal domains, along with a short N-terminal tail, constitute
the major portion of the PDZ-binding C2 domain, which is
responsible for binding the phospholipid membrane and serves a key
role in the proper positioning of PTEN at the plasma membrane in
the C-terminal region. The protein also contains two PEST
sequences, which regulate its tumor-suppressive function (81,82).
The tail of the C-terminus comprises ~50 amino acids
and is responsible for active phosphorylation. The primary cause of
the anticancer activity of PTEN is its lipid phosphatase activity.
Loss of function is characterized by the inhibition of the
enzymatic activity of PTEN. Invasion and gene expression are
correlated with phosphatase activity. In addition to being
responsible for the ubiquitin-mediated proteasomal degradation of
proteins, the PEST region of PTEN also contributes to increased
protein stability, and its loss significantly decreases the
expression of the PTEN protein. The nuclear import,
tumor-suppressing properties and degradation of PTEN are controlled
by ubiquitination, specifically ubiquitination of its PEST motifs
(81–83). PTEN contains two PEST sequences in
its carboxyl-terminal region; these motifs not only contribute to
maintaining PTEN protein stability (81,82)
but also serve as key targets for ubiquitin ligases. This PEST
motif-specific ubiquitination directly modulates PTEN's nuclear
translocation, abrogates its tumor-suppressive activity and
promotes its proteasomal degradation (83).
KRAS, BRAF, PTEN and β-catenin mutations are present
in type I tumors, which are considered to be genetically stable
(84–86). KRAS and BRAF mutations, in
particular, are mutually exclusive, affect two-thirds of type I
OCs, and cause early neoplastic transformation by constitutively
activating the mitogen-activated protein kinase signal transduction
pathway (87). PTEN mutations can
coexist with KRAS mutations in the ovary, which has been found to
cause invasive and broadly metastatic EOC (86,87).
PTEN mutations can also occur independently, leading to aberrant
activation of the PI3K/Akt/mTOR pathway (88). According to several preclinical
models, one of several genetic modifications affecting the
fallopian tube is the deletion of PTEN (89–91).
The conditional homozygous PTEN deletion mediated by
PAX8-cre recombinase was sufficient to cause borderline serous OC
and endometrioid tumors in mouse models (92). Research suggests that HGSOC has a
unique etiology; the current HGSOC formation paradigm suggests that
it originates in the fallopian tube, which is supported by genomic,
transcriptomic and proteomic investigations demonstrating shared
molecular signatures between fallopian tube precursor lesions and
invasive HGSOC (93–95). Mutations in the p53 gene that cause
protein stabilization in the fallopian tube are crucial in this
process. Following the loss of other tumor suppressors or the
amplification of oncogenes (such as PTEN and KRAS), a serous tubal
intraepithelial carcinoma develops, which later metastasizes to the
ovary and peritoneum (94).
Research has shown that four out of twelve (33%)
serous intratubal carcinoma samples had PTEN deletion (95). These findings, in line with those of
endometrioid carcinomas of the uterus, where progressive loss of
PTEN and PAX2 expression characterizes early precancerous lesions
and carcinogenesis (96,97), led to the suggestion that variations
in PTEN and PAX2 expression may be involved in the early stages of
serous carcinogenesis. As observed in the endometrial model, it is
unknown whether these modifications occur before malignancy or
concurrently with its onset. Additionally, it has been demonstrated
that the loss of PTEN enables the formation of multicellular tumor
spheroids in the fallopian tube epithelium. These spheroids
colonize the ovary by adhering to the extracellular matrix that is
exposed after ovulation and may have seeded the ovary in patients
with HGSOC (96). PTEN loss has
been associated with increased cell transformation and
proliferation both in vitro and in vivo, as well as
with the stimulation of MUC1 expression, which is known to serve
key roles in cancer cell migration and metastasis (97).
Bmi1, a polycomb group protein (also known as PRC1),
is essential for the epigenetic regulation of several cellular
processes, including proliferation, differentiation, stem cell
self-renewal and chemoresistance to various cancer drugs. In EOC,
Bmi1 contributes to resistance against platinum-based
chemotherapeutic agents (such as cisplatin), Bmi1 encodes 326 amino
acids with a molecular weight of 44–46 kDa. The protein has two
domains: Bmi1, which associates with and partially encloses Ring1b,
and the tail of Ring1b, which wraps around Bmi1. The oncogene Bmi1
binds to the substrate protein and ubiquitinates the lysine of the
substrate protein, via the E3 ligase Ring1b, (97,98).
For example, Bmi1 reduces the stability of p53 by ubiquitinating it
via Ring Finger Protein 2/Ring1b (99,100).
Two NLSs, NLS1 and NLS2, are located in the protein
Bmi1 and are responsible for the nuclear localization of Bmi1. NLS2
is a key signal for the nuclear localization of Bmi1 (98). Bmi1 is split into three parts on the
basis of its function: An N-terminal conserved ring domain, a
central helix-turn-helix (HTH) domain and a carboxyl-terminal area
with a PEST-like domain. The ring domain of Bmi1 focuses on DNA
strand breaks in response to DNA damage, which helps cells escape
senescence due to the synergistic action between the N-terminal
ring domain and central HTH domain Bmi1 (101,102). As a result, it lengthens the time
that cells can replicate, leading to the accumulation of more cells
in the G2/M phase and thereby increasing cell
proliferation (103).
Bmi1 also increases the length of time that a tumor
cell can survive because by activating N-Myc, as it blocks the
transcription of tumor suppressor genes such as p16, p19, p53 and
PTEN; thus, Bmi1 has an oncogenic function (104,105). Bmi1 overexpression has been
documented in lymphomas (106),
prostate cancer (107), non-small
cell lung cancer (NSCLC) (108),
nasopharyngeal carcinoma (109)
and breast cancer. Specifically, in prostate cancer, Bmi1
upregulation correlates with downregulated p16(INK4A) and p14(ARF).
In NSCLC, high Bmi1 expression is associated with reduced p16 and
p14ARF levels. In colon cancer, Bmi1 overexpression coincides with
decreased p16INK4a/p14ARF expression. Through the suppression of
the p21CIP1 gene and the INK4A/ARF locus, Bmi1 is essential for
tumor cell growth and survival in brain cells (110).
The progression of neuroblastoma caused by Bmi1 is
facilitated by the upregulation of cyclin E (111). In prostate cancer cells and
mammary epithelial cells, Bmi1 also increases telomerase activity
(112,113). Bmi1 PTMs at various serine
residues control its INK4A/ARF-dependent and INK4A/ARF-independent
functions (114). Through an
INK4A/ARF-independent mechanism, phosphorylation of Bmi1 by AKT
promotes glioma and hepatic carcinogenesis (115,116) and reduces tumor growth through an
INK4A/ARF-dependent pathway. In accordance with the type of
residual substrate, alterations in the AKT kinase activity of Bmi1
alter its oncogenic characteristics (117).
MAPKAPK3, a MAPK-activated protein kinase, also
phosphorylates Bmi1. The activation or upregulation of MAPKAPK3
leads to the phosphorylation of Bmi1 and other Polycomb proteins,
the recruitment of substrate proteins from chromatin, the
activation of the INKA4/ARF locus induce cell cycle arrest and
senescence to inhibit cancer progression (118,119). Bmi1 is positively regulated by
numerous transcriptional and posttranscriptional regulators,
including N-Myc, C-Myc, specialized protein 1, twist-related
protein 1, forkhead box protein M1, E2F1 and sal-like protein 4. By
contrast, Bmi1 is suppressed at the transcriptional level by
Mel-18, Kruppel-like factor 4 and histone deacetylase inhibitors.
The expression of Bmi1 is also regulated by the Notch and Wnt
pathways (120).
In PTEN-null prostate cancer, elevated Bmi1
expression and its role in neoplasia progression are supported by
studies showing that Bmi1 upregulation correlates with PTEN loss
and promotes tumor aggressiveness, while Bmi1 depletion inhibits
progression. Regarding Bmi1 degradation, it is regulated by
βTrCP-mediated ubiquitination: βTrCP recognizes a specific motif in
Bmi1, promoting its proteasomal turnover via the
ubiquitin-proteasome system (121). βTrCP mediates the ubiquitination
of Bmi1 at tyrosine 18 within its N-terminal RING finger domain;
subsequently, the ubiquitinated Bmi1 is shuttled into proteasomes
for degradation (121,122). The degradation of Bmi1 is
increased in MCF10A cells when TrCP is overexpressed, whereas the
opposite occurs in these MCF10A cells with no TrCP overexpression
(122).
In a previous study, compared with original ovarian
carcinomas, metastatic lymph nodes and recurring tumors
significantly overexpressed Bmi1 (123). Furthermore, Bmi1 is highly
expressed in metastatic and recurrent tumors, and was found to be
predictive of both survival and relapse (124). A number of studies have
investigated the role of Bmi1 as a pathway regulator in both stem
cells and cancer cells, validating its oncogenic activation in
various human malignancies Examples include EOC, where Bmi1
overexpression in metastatic/recurrent tumors correlates with
disease progression and platinum resistance (125), NSCLC, where it links EMT to cancer
stem cell properties (126), as
well as lymphomas, prostate cancer, nasopharyngeal carcinoma and
breast cancer, where elevated Bmi1 expression is associated with
tumor aggressiveness and suppressed tumor suppressor genes
(125,126). Upon suppression of Bmi1, cell
proliferation was significantly reduced and a greater proportion of
EOC cells remained in the G1 phase. Previous research
demonstrated that ER-coupled Bmi1 signature activity influences the
status of p16INK4a and cyclin D1, which correlates with tumor
molecular subtypes and biological behavior in breast cancer
(127).
Furthermore, Bmi1 knockout decreased the ability of
EOC cells to migrate and invade; additional studies demonstrated
that Bmi1 is responsible for triggering the EMT, which leads to
tumor invasion and metastasis (128). Additionally, the overexpression of
Bmi1 in EOC cells increased the expression of cyclin D1, CDK4 and
Bcl-2, thereby increasing cell proliferation and inhibiting cell
death (129). By contrast, a
previous study demonstrated that silencing Bmi1 had the opposite
effect on controlling the cell cycle and apoptosis; it was also
shown that Bmi1 knockout increased the susceptibility of EOC cells
to platinum (123). Similarly,
Wang et al (130) reported
that downregulating Bmi1 increased ROS, triggered the DNA damage
repair pathway and ultimately promoted cisplatin-induced apoptosis.
Together, these data provided insight into chemoresistance, an
ongoing and complex problem in the management of EOC.
The Myc gene, which may be located at chromosome
8q24.21, encodes the super-transcription factor Myc (131). Myc oncoproteins (c-Myc, N-Myc and
L-Myc) regulate the transcription of >15% of expressed genes
(132). The PEST-NP Myc, as the
prototypical nuclear transcription factor, dimerizes with MAX to
drive proliferation and repress differentiation via E-box binding
(133). However, Myc requires
cooperative genetic events, such as loss of p53/p16 checkpoints, to
overcome apoptosis/senescence and induce oncogenesis, as
demonstrated in Myc-Ras transgenic models (134). In ovarian cancer, c-Myc
overexpression is detected in 65.9% of epithelial tumors,
correlating with aggressive phenotypes, consistent with its
PEST-mediated instability and oncogenic plasticity.
The wide, unstructured amino-terminal region in Myc
proteins, which is considered critical for transcriptional
activation, contains conserved boxes known as Myc boxes (MBI and
MBII) (135). The PEST sequences,
meanwhile, are responsible for ubiquitination, with two conserved
Myc boxes (MBIII and MBIV) and one NLS for nuclear accumulation
comprising the middle portion of Myc proteins (136). The basic helix-loop-helix leucine
zipper domain is made up of 100 amino acids, which is necessary for
the DNA-protein interactions that start transcription (133,136). Human cancer cells frequently
overexpress the protein Myc; specific examples include EOC, where
c-Myc overexpression correlates with aggressive tumor phenotypes
(133), as well as lymphomas and
prostate cancer, where Myc overexpression has been linked to
malignant transformation processes (133). Protein translation, cell cycle
progression and differentiation, tumor metabolism and ribosome
biogenesis are important downstream effects of Myc. Numerous
biological processes, including cell differentiation, growth,
survival, immune surveillance and apoptosis, are coordinated with
the regulatory process mediated by the downstream effects of Myc
(137). Myc-associated cell cycle
abnormalities occur after SUMOylation, leading to the enzymatic
activation of Myc (135).
The protein expression of c-Myc has previously been
identified in OC and stromal cells via immunohistochemical
techniques (138). In 76% of cases
of early-stage EOC, Skírnisdóttir et al (139) reported positive staining for c-Myc
via the same method. Tumor grade and positivity status are related;
the c-Myc protein was discovered to be overexpressed in 65.9% of
EOC cases compared with that of normal ovary cancer cases,
according to research by Chen et al (140). However, no discernible distinction
in histological subtypes was observed.
However, retrospective clinical data analysis has
indicated that standard histological indicators are more reliable
predictors of tumor behavior than an evaluation of the c-Myc
expression pattern solely by immunostaining. Ning et al
(147) observed that, following
treatment with platinum-based regimens for ovarian carcinomas,
c-Myc upregulation was associated with improved tumor
differentiation, increased p27 levels and decreased Ki-67
expression. Furthermore, Ning et al (147) reported an association between
shorter overall survival and elevated nuclear c-Myc expression in
early-stage OC.
A PEST-NP member currently under investigation is
PCNP, a PEST-containing nuclear protein first identified in the
early 2000s, which consists of 178 amino acids and exhibits
consistent colocalization with the RING finger protein NIRF in the
perinuclear region (152,153). PCNP mRNA has been detected in
several cell types, including normal fibroblasts (WI-38 and TIG-7),
fibrosarcoma (HT-1080) and hepatocellular carcinoma (HepG2) cells
(151). Further evidence, such as
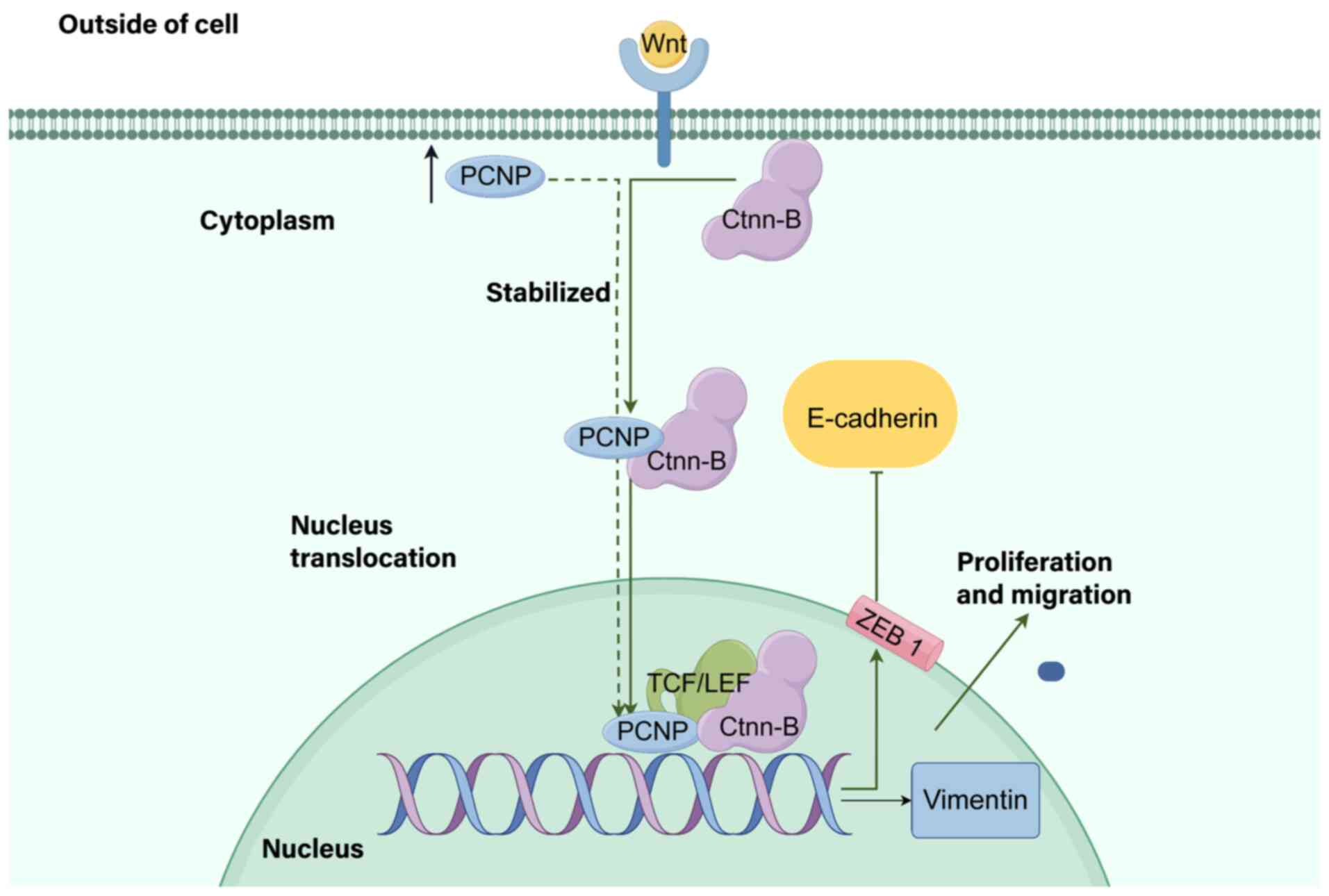
PCNP overexpression in ovarian cancer driving
Wnt/β-catenin-mediated cell proliferation, migration and
epithelial-mesenchymal transition (154), and its interaction with NIRF to
regulate cell cycle-related stability (151). PCNP and NIRF have been
demonstrated to interact with each other both in vitro and
in vivo. The presence of the ubiquitin domain in the
N-terminus and the ring finger catalytic domain in the C-terminus
of NIRF suggests that the PCNP has catalytic activity. The
ubiquitination of PCNP by NIRF is highlighted by the interaction of
PCNP with NIRF in vitro and in vivo, which in turn
controls the stability and regulation of PCNP. The ring finger
proteins MDM2 and p53 share a similar interaction (20,154).
Additionally, NIRF is expressed at a markedly high level in several
malignancies, including EOC, hepatocellular carcinoma and NSCLC
(154,155).
According to previous studies, the MAPK and
PI3K/AKT/mTOR signaling pathways serve pivotal roles together with
PCNP in the proliferation, migration and invasion of OC,
neuroblastoma and lung adenocarcinoma (154,156). Mechanistically, PCNP binds to
β-catenin, accelerates its nuclear translocation and activates
Wnt/β-catenin signaling, suggesting that PCNP is an upstream
regulator of Wnt-mediated OC progression and a potential
therapeutic target (Fig. 3). The
Wnt signaling pathway, which regulates cell proliferation,
apoptosis and EMT, is critical for tumor initiation and
development, and aberrations in this pathway significantly
contribute to OC progression (157–159). Dong et al (154) identified a direct link between
PCNP and the Wnt pathway: PCNP is overexpressed in OC tissues and
cells (compared with that of paracancerous and IOSE80 cells), and
this overexpression promotes OC cell proliferation, migration and
invasion while suppressing apoptosis (validated in SK-OV-3/A2780
cells and xenograft models). High expression levels of PCNP can
reduce apoptosis by increasing the expression levels of the
transcription factor (STAT) activators of phosphor signal
transducers (153). In addition to
TNF-inducible protein 8-like 2, PCNP is also involved in the immune
response in rheumatoid arthritis (160). Although PCNP is known to mediate
caspase activities, upregulating cleaved caspase-3, −8, and −9
in vitro and in vivo, it remains unclear how PCNP
specifically activates these caspases to induce apoptosis (156). The induction of autophagy and
apoptosis in distinct cell lines, however, implies that alternative
nuclear transporters are involved in the dual behavior of the
protein (156). Overall, however,
further research is needed on the nuclear transportation of PCNP
(Fig. 4; Table II and III).
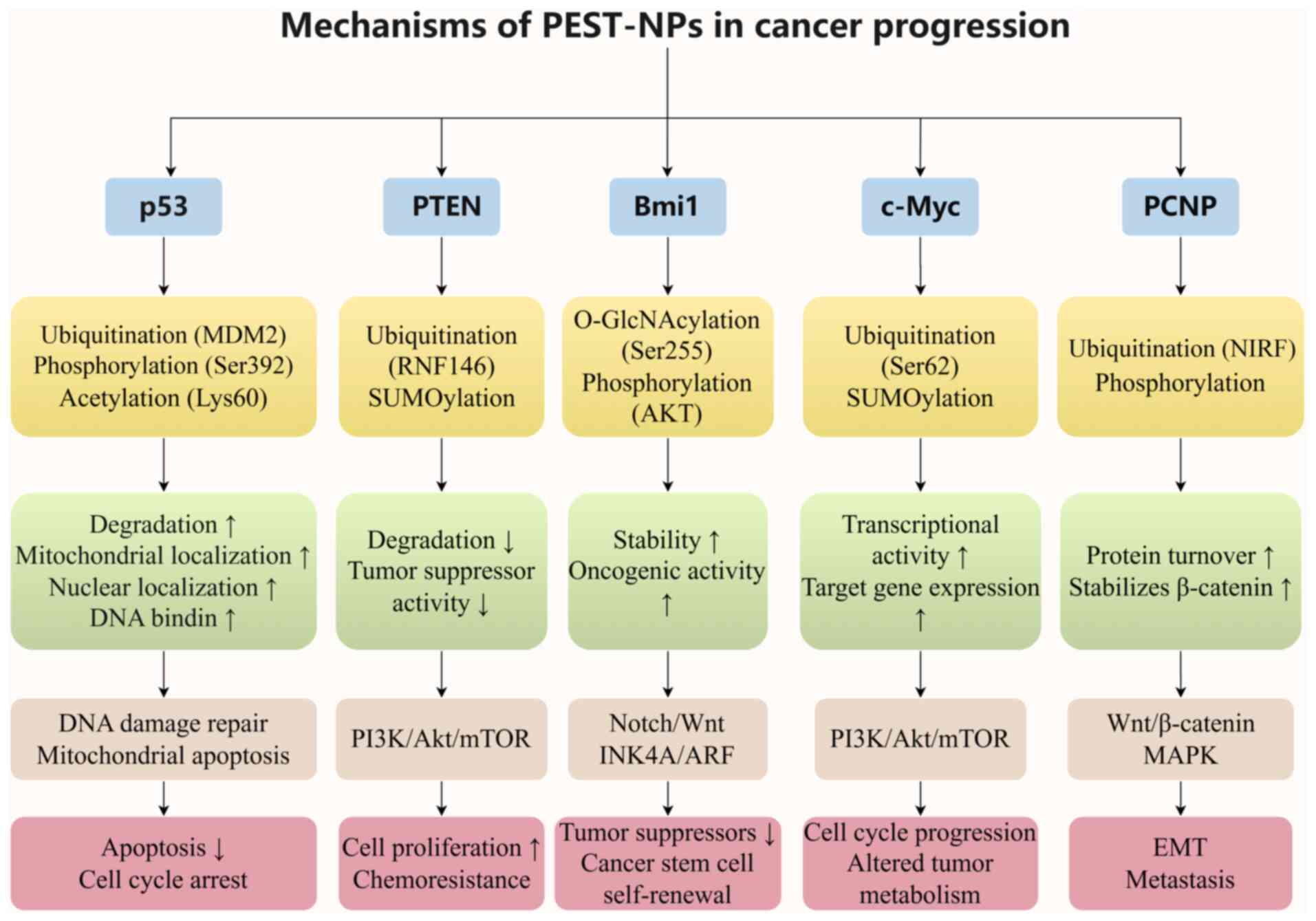
As analyzed in the present review, OC progression
is driven by dysregulated transcription factors and signaling
pathways, with PEST-NPs emerging as key regulators through their
PTMs. PTMs such as phosphorylation, ubiquitination and SUMOylation
control the stability, activation and intracellular localization of
core PEST-NPs, thereby modulating oncogenic pathways such as the
PI3K/Akt/mTOR and Wnt/β-catenin pathways to influence OC growth,
metastasis and chemoresistance.
Notably, PCNP and Bmi1, as key PEST-NPs, may hold
translational potential for OC therapy based on their characterized
roles. The direct interaction of PCNP with β-catenin accelerates
nuclear translocation and activates Wnt/β-catenin signaling, a
pathway critical for OC proliferation, migration and invasion.
Targeting PCNP could therefore involve strategies to block its
binding to β-catenin or inhibit its phosphorylation, which
modulates its oncogenic activity in regulating the MAPK and
PI3K/Akt/mTOR pathways. Given that PCNP is overexpressed in OC
tissues and cells compared with that of their normal counterparts,
such interventions may specifically disrupt tumor-promoting
signaling without affecting normal cells. The ability of Bmi1 to be
overexpressed in metastatic and recurrent OC supports its utility
as a therapeutic target. Inhibiting Bmi1 could reduce its
stabilization via O-GlcNAcylation or block its phosphorylation by
AKT, thereby reversing its oncogenic effects on cell cycle
progression and stemness. This may sensitize OC cells to
platinum-based therapies, as Bmi1 silencing has been shown to
increase platinum susceptibility in preclinical models.
These insights deepen the current understanding of
OC pathogenesis by linking PEST motif-mediated protein dynamics to
tumor behavior, thus underscoring PEST-NPs as valuable diagnostic
biomarkers and therapeutic targets. To advance clinical
translation, future research should focus on elucidating the
intricate interactions of PCNP with cancer-related genes and
pathways, validating PEST-NP-targeted strategies in preclinical
models and exploring PEST-NP expression/PTM profiles as predictors
of treatment response, which will serve to bridge the gap between
laboratory research and clinical applications.
Not applicable.
The present work was supported by the National Natural Science
Foundation of China (grant nos. 31902287 and 81670988), Cultivation
Project for Innovation Team in Teachers' Teaching Proficiency by
Zhengzhou Health College (grant no. 2024jxcxtd01).
Not applicable.
HL, ZLJ, YL, YHZ, MUA and ZDL conceived the study
and drafted the manuscript. MBK, SK and UAKS prepared the figures,
participated in manuscript content revision and table legend
editing. YZ and XYJ provided funding and supervision, reviewed the
manuscript framework, revised key sections and ensured academic
consistency. ZLJ and YL participated in the extensive review,
revision, and figure/table production of the review. Data
authentication is not applicable. All the authors read and approved
the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Menon U, Griffin M and Gentry-Maharaj A:
Ovarian cancer screening-current status, future directions. Gynecol
Oncol. 132:490–495. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gupta KK, Gupta VK and Naumann RW: Ovarian
cancer: Screening and future directions. Int J Gynecol Cancer.
29:195–200. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang MH, Zhang HH, Du XH, Gao J, Li C,
Shi HR and Li SZ: UCHL3 promotes ovarian cancer progression by
stabilizing TRAF2 to activate the NF-κB pathway. Oncogene.
39:322–333. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hubackova M, Vaclavikova R, Ehrlichova M,
Mrhalova M, Kodet R, Kubackova K, Vrána D, Gut I and Soucek P:
Association of superoxide dismutases and NAD(P)H quinone
oxidoreductases with prognosis of patients with breast carcinomas.
Int J Cancer. 130:338–348. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ke Y, Chen X, Su Y, Chen C, Lei S, Xia L,
Wei D, Zhang H, Dong C, Liu X and Yin F: Low expression of SLC7A11
confers drug resistance and worse survival in ovarian cancer via
inhibition of cell autophagy as a competing endogenous RNA. Front
Oncol. 11:7449402021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li D, Hong X, Zhao F, Ci X and Zhang S:
Targeting Nrf2 may reverse the drug resistance in ovarian cancer.
Cancer Cell Int. 21:1162021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gao Y, Liu X, Li T, Wei L, Yang A, Lu Y,
Zhang J, Li L, Wang S and Yin F: Cross-validation of genes
potentially associated with overall survival and drug resistance in
ovarian cancer. Oncol Rep. 37:3084–3092. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hydbring P, Castell A and Larsson LG: MYC
modulation around the CDK2/p27/SKP2 axis. Genes (Basel). 8:1742017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ammirante M, Kuraishy AI, Shalapour S,
Strasner A, Ramirez-Sanchez C, Zhang W, Shabaik A and Karin M: An
IKKα-E2F1-BMI1 cascade activated by infiltrating B cells controls
prostate regeneration and tumor recurrence. Genes Dev.
27:1435–1440. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Park JY, Wang PY, Matsumoto T, Sung HJ, Ma
W, Choi JW, Anderson SA, Leary SC, Balaban RS, Kang JG and Hwang
PM: p53 improves aerobic exercise capacity and augments skeletal
muscle mitochondrial DNA content. Circ Res. 105:705–712. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ho SR, Mahanic CS, Lee YJ and Lin WC:
RNF144A, an E3 ubiquitin ligase for DNA-PKcs, promotes apoptosis
during DNA damage. Proc Natl Acad Sci USA. 111:E2646–E2655. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bakhanashvili M, Grinberg S, Bonda E,
Simon AJ, Moshitch-Moshkovitz S and Rahav G: p53 in mitochondria
enhances the accuracy of DNA synthesis. Cell Death Differ.
15:1865–1874. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nithipongvanitch R, Ittarat W, Velez JM,
Zhao R, St Clair DK and Oberley TD: Evidence for p53 as guardian of
the cardiomyocyte mitochondrial genome following acute adriamycin
treatment. J Histochem Cytochem. 55:629–639. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nie M, Moser BA, Nakamura TM and Boddy MN:
SUMO-targeted ubiquitin ligase activity can either suppress or
promote genome instability, depending on the nature of the DNA
lesion. PLoS Genet. 13:e10067762017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou ZD, Chan CH, Xiao ZC and Tan EK: Ring
finger protein 146/Iduna is a poly(ADP-ribose) polymer binding and
PARsylation dependent E3 ubiquitin ligase. Cell Adh Migr.
5:463–471. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Freed-Pastor WA and Prives C: Mutant p53:
One name, many proteins. Genes Dev. 26:1268–2686. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kang HC, Lee YI, Shin JH, Andrabi SA, Chi
Z, Gagné JP, Lee Y, Ko HS, Lee BD, Poirier GG, et al: Iduna is a
poly(ADP-ribose) (PAR)-dependent E3 ubiquitin ligase that regulates
DNA damage. Proc Natl Acad Sci USA. 108:14103–14108. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu L, Zhang H, Bergholz J, Sun S and Xiao
ZX: MDM2/MDMX: Master negative regulators for p53 and RB. Mol Cell
Oncol. 3:e11066352016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tuna M, Ju Z, Yoshihara K, Amos CI, Tanyi
JL and Mills GB: Clinical relevance of TP53 hotspot mutations in
high-grade serous ovarian cancers. Br J Cancer. 122:405–412. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Afzal A, Sarfraz M, Li GL, Ji SP, Duan SF,
Khan NH, Wu DD and Ji XY: Taking a holistic view of PEST-containing
nuclear protein (PCNP) in cancer biology. Cancer Med. 8:6335–6343.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rogers S, Wells R and Rechsteiner M: Amino
acid sequences common to rapidly degraded proteins: The PEST
hypothesis. Science. 234:364–368. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Roth AF, Sullivan DM and Davis NG: A large
PEST-like sequence directs the ubiquitination, endocytosis, and
vacuolar degradation of the yeast a-factor receptor. J Cell Biol.
142:949–961. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhuang X, Northup JK and Ray K: Large
putative PEST-like sequence motif at the carboxyl tail of human
calcium receptor directs lysosomal degradation and regulates cell
surface receptor level. J Biol Chem. 287:4165–4176. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sekhar KR and Freeman ML: PEST sequences
in proteins involved in cyclic nucleotide signalling pathways. J
Recept Signal Transduct Res. 18:113–132. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bellini E, Pavesi G, Barbiero I, Bergo A,
Chandola C, Nawaz MS, Rusconi L, Stefanelli G, Strollo M, Valente
MM, et al: MeCP2 post-translational modifications: A mechanism to
control its involvement in synaptic plasticity and homeostasis?
Front Cell Neurosci. 8:2362014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bereshchenko OR, Gu W and Dalla-Favera R:
Acetylation inactivates the transcriptional repressor BCL6. Nat
Genet. 32:606–613. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao JF, Shyue SK and Lee TS: Excess
nitric oxide activates TRPV1-Ca(2+)-calpain signaling and promotes
PEST-dependent degradation of liver X receptor α. Int J Biol Sci.
12:18–29. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu R, Hong J, Xu X, Feng Q, Zhang D, Gu
Y, Shi J, Zhao S, Liu W, Wang X, et al: Gut microbiome and serum
metabolome alterations in obesity and after weight-loss
intervention. Nat Med. 23:859–868. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pan S and Chen R: Pathological implication
of protein post-translational modifications in cancer. Mol Aspects
Med. 86:1010972022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Afzal A, Sarfraz M, Wu Z, Wang G and Sun
J: Integrated scientific data bases review on asulacrine and
associated toxicity. Crit Rev Oncol Hematol. 104:78–86. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lang V, Aillet F, Da Silva-Ferrada E,
Xolalpa W, Zabaleta L, Rivas C and Rodriguez MS: Analysis of PTEN
ubiquitylation and SUMOylation using molecular traps. Methods.
77–78. 112–118. 2015.
|
|
33
|
Li N, Zhang Y, Han X, Liang K, Wang J,
Feng L, Wang W, Songyang Z, Lin C, Yang L, et al: Poly-ADP
ribosylation of PTEN by tankyrases promotes PTEN degradation and
tumor growth. Genes Dev. 29:157–170. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shumway SD, Maki M and Miyamoto S: The
PEST domain of IkappaBalpha is necessary and sufficient for in
vitro degradation by mu-calpain. J Biol Chem. 274:30874–30881.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sarfraz M, Afzal A, Khattak S, Saddozai
UAK, Li HM, Zhang QQ, Madni A, Haleem KS, Duan SF, Wu DD, et al:
Multifaceted behavior of PEST sequence enriched nuclear proteins in
cancer biology and role in gene therapy. J Cell Physiol.
236:1658–1676. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chipuk JE, Bouchier-Hayes L, Kuwana T,
Newmeyer DD and Green DR: PUMA couples the nuclear and cytoplasmic
proapoptotic function of p53. Science. 309:1732–1735. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lisachev PD, Pustylnyak VO and Shtark MB:
Mdm2-dependent regulation of p53 expression during long-term
potentiation. Bull Exp Biol Med. 158:333–335. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Geyer RK, Yu ZK and Maki CG: The MDM2
RING-finger domain is required to promote p53 nuclear export. Nat
Cell Biol. 2:569–573. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lu W, Pochampally R, Chen L, Traidej M,
Wang Y and Chen J: Nuclear exclusion of p53 in a subset of tumors
requires MDM2 function. Oncogene. 19:232–240. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang DY, Hong Y, Chen YG, Dong PZ, Liu SY,
Gao YR, Lu D, Li HM, Li T, Guo JC, et al: PEST-containing nuclear
protein regulates cell proliferation, migration, and invasion in
lung adenocarcinoma. Oncogenesis. 8:222019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ye J, Zhong L, Xiong L, Li J, Yu L, Dan W,
Yuan Z, Yao J, Zhong P, Liu J, et al: Nuclear import of NLS-RARα is
mediated by importin α/β. Cell Signal. 69:1095672020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Panagiotopoulos AA, Polioudaki C, Ntallis
SG, Dellis D, Notas G, Panagiotidis CA, Theodoropoulos PA, Castanas
E and Kampa M: The sequence [EKRKI(E/R)(K/L/R/S/T)] is a nuclear
localization signal for importin 7 binding (NLS7). Biochim Biophys
Acta Gen Subj. 1865:1298512021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zheng Y, Yu K, Lin JF, Liang Z, Zhang Q,
Li J, Wu QN, He CY, Lin M, Zhao Q, et al: Deep learning prioritizes
cancer mutations that alter protein nucleocytoplasmic shuttling to
drive tumorigenesis. Nat Commun. 16:25112025. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lang YD and Jou YS: PSPC1 is a new
contextual determinant of aberrant subcellular translocation of
oncogenes in tumor progression. J Biomed Sci. 28:572021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Rubbi CP and Milner J: Disruption of the
nucleolus mediates stabilization of p53 in response to DNA damage
and other stresses. EMBO J. 22:6068–6077. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang X, Blaskovich MA, Forinash KD and
Sebti SM: Withacnistin inhibits recruitment of STAT3 and STAT5 to
growth factor and cytokine receptors and induces regression of
breast tumours. Br J Cancer. 111:894–902. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Baugh EH, Ke H, Levine AJ, Bonneau RA and
Chan CS: Why are there hotspot mutations in the TP53 gene in human
cancers? Cell Death Differ. 25:154–160. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kastan MB and Berkovich E: p53: A
two-faced cancer gene. Nat Cell Biol. 9:489–491. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Levine AJ and Oren M: The first 30 years
of p53: Growing ever more complex. Nat Rev Cancer. 9:749–758. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wojnarowicz PM, Oros KK, Quinn MC, Arcand
SL, Gambaro K, Madore J, Birch AH, de Ladurantaye M, Rahimi K,
Provencher DM, et al: The genomic landscape of TP53 and p53
annotated high grade ovarian serous carcinomas from a defined
founder population associated with patient outcome. PLoS One.
7:e454842012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Jayson GC, Kohn EC, Kitchener HC and
Ledermann JA: Ovarian cancer. Lancet. 384:1376–1388. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Matulonis UA, Sood AK, Fallowfield L,
Howitt BE, Sehouli J and Karlan BY: Ovarian cancer. Nat Rev Dis
Primers. 2:160612016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Joerger AC and Fersht AR: The tumor
suppressor p53: From structures to drug discovery. Cold Spring Harb
Perspect Biol. 2:a0009192010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhu G, Pan C, Bei JX, Li B, Liang C, Xu Y
and Fu X: Mutant p53 in cancer progression and targeted therapies.
Front Oncol. 10:5951872020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Cole AJ, Dwight T, Gill AJ, Dickson KA,
Zhu Y, Clarkson A, Gard GB, Maidens J, Valmadre S, Clifton-Bligh R
and Marsh DJ: Assessing mutant p53 in primary high-grade serous
ovarian cancer using immunohistochemistry and massively parallel
sequencing. Sci Rep. 6:261912016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Flemming A: Cancer: Mutant p53 rescued by
aggregation inhibitor. Nat Rev Drug Discov. 15:852016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Duffy MJ, Synnott NC, O'Grady S and Crown
J: Targeting p53 for the treatment of cancer. Semin Cancer Biol.
79:58–67. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Okal A, Cornillie S, Matissek SJ, Matissek
KJ, Cheatham TE III and Lim CS: Re-engineered p53 chimera with
enhanced homo-oligomerization that maintains tumor suppressor
activity. Mol Pharm. 11:2442–2452. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Okal A, Matissek KJ, Matissek SJ, Price R,
Salama ME, Janát-Amsbury MM and Lim CS: Re-engineered p53 activates
apoptosis in vivo and causes primary tumor regression in a dominant
negative breast cancer xenograft model. Gene Ther. 21:903–912.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Brady CA and Attardi LD: p53 at a glance.
J Cell Sci. 123:2527–2532. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wei H, Qu L, Dai S, Li Y, Wang H, Feng Y,
Chen X, Jiang L, Guo M, Li J, et al: Structural insight into the
molecular mechanism of p53-mediated mitochondrial apoptosis. Nat
Commun. 12:22802021. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Leu JI, Dumont P, Hafey M, Murphy ME and
George DL: Mitochondrial p53 activates Bak and causes disruption of
a Bak-Mcl1 complex. Nat Cell Biol. 6:443–450. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Heyne K, Schmitt K, Mueller D, Armbruester
V, Mestres P and Roemer K: Resistance of mitochondrial p53 to
dominant inhibition. Mol Cancer. 7:542008. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Chen S, Cavazza E, Barlier C, Salleron J,
Filhine-Tresarrieu P, Gavoilles C, Merlin JL and Harlé A: Beside
P53 and PTEN: Identification of molecular alterations of the
RAS/MAPK and PI3K/AKT signaling pathways in high-grade serous
ovarian carcinomas to determine potential novel therapeutic
targets. Oncol Lett. 12:3264–3272. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Toledo F and Wahl GM: MDM2 and MDM4: p53
regulators as targets in anticancer therapy. Int J Biochem Cell
Biol. 39:1476–1482. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Astanehe A, Arenillas D, Wasserman WW,
Leung PC, Dunn SE, Davies BR, Mills GB and Auersperg N: Mechanisms
underlying p53 regulation of PIK3CA transcription in ovarian
surface epithelium and in ovarian cancer. J Cell Sci. 121:664–674.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hu L, Zaloudek C, Mills GB, Gray J and
Jaffe RB: In vivo and in vitro ovarian carcinoma growth inhibition
by a phosphatidylinositol 3-kinase inhibitor (LY294002). Clin
Cancer Res. 6:880–886. 2000.PubMed/NCBI
|
|
68
|
Raab M, Kostova I, Peña-Llopis S, Fietz D,
Kressin M, Aberoumandi SM, Ullrich E, Becker S, Sanhaji M and
Strebhardt K: Rescue of p53 functions by in vitro-transcribed mRNA
impedes the growth of high-grade serous ovarian cancer. Cancer
Commun (Lond). 44:101–126. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Faramarzi L, Dadashpour M, Sadeghzadeh H,
Mahdavi M and Zarghami N: Enhanced anti-proliferative and
pro-apoptotic effects of metformin encapsulated PLGA-PEG
nanoparticles on SKOV3 human ovarian carcinoma cells. Artif Cells
Nanomed Biotechnol. 47:737–746. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Guo X, Fang Z, Zhang M, Yang D, Wang S and
Liu K: A Co-delivery system of curcumin and p53 for enhancing the
sensitivity of drug-resistant ovarian cancer cells to cisplatin.
Molecules. 25:26212020. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Huang X, Cao Z, Qian J, Ding T, Wu Y,
Zhang H, Zhong S, Wang X, Ren X, Zhang W, et al: Nanoreceptors
promote mutant p53 protein degradation by mimicking selective
autophagy receptors. Nat Nanotechnol. 19:545–553. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zhang XF and Gurunathan S: Combination of
salinomycin and silver nanoparticles enhances apoptosis and
autophagy in human ovarian cancer cells: An effective anticancer
therapy. Int J Nanomedicine. 11:3655–3675. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wang J, Qu C, Shao X, Song G, Sun J, Shi
D, Jia R, An H and Wang H: Carrier-free nanoprodrug for p53-mutated
tumor therapy via concurrent delivery of zinc-manganese dual ions
and ROS. Bioact Mater. 20:404–417. 2022.PubMed/NCBI
|
|
74
|
Lee JM and Johnson JA: An important role
of Nrf2-ARE pathway in the cellular defense mechanism. J Biochem
Mol Biol. 37:139–143. 2004.PubMed/NCBI
|
|
75
|
Castrogiovanni C, Waterschoot B, De Backer
O and Dumont P: Serine 392 phosphorylation modulates p53
mitochondrial translocation and transcription-independent
apoptosis. Cell Death Differ. 25:190–203. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Ai G, Dachineni R, Kumar DR, Marimuthu S,
Alfonso LF and Bhat GJ: Aspirin acetylates wild type and mutant p53
in colon cancer cells: Identification of aspirin acetylated sites
on recombinant p53. Tumour Biol. 37:6007–6016. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Oreskes N: Beyond the ivory tower. The
scientific consensus on climate change. Science. 306:16862004.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Haddadi N, Lin Y, Travis G, Simpson AM,
Nassif NT and McGowan EM: PTEN/PTENP1: ‘Regulating the regulator of
RTK-dependent PI3K/Akt signalling’, new targets for cancer therapy.
Mol Cancer. 17:372018. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Jiang C, Song Y, Rorive S, Allard J, Tika
E, Zahedi Z, Dubois C, Salmon I, Sifrim A and Blanpain C: Innate
immunity and the NF-κB pathway control prostate stem cell
plasticity, reprogramming and tumor initiation. Nat Cancer.
6:1537–1558. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Ren G, Chen J, Pu Y, Yang EJ, Tao S, Mou
PK, Chen LJ, Zhu W, Chan KL, Luo G, et al: BET inhibition induces
synthetic lethality in PTEN deficient colorectal cancers via dual
action on p21CIP1/WAF1. Int J Biol Sci. 20:1978–1991.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Georgescu MM, Kirsch KH, Akagi T, Shishido
T and Hanafusa H: The tumor-suppressor activity of PTEN is
regulated by its carboxyl-terminal region. Proc Natl Acad Sci USA.
96:10182–10187. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Meyer RD, Srinivasan S, Singh AJ, Mahoney
JE, Gharahassanlou KR and Rahimi N: PEST motif serine and tyrosine
phosphorylation controls vascular endothelial growth factor
receptor 2 stability and downregulation. Mol Cell Biol.
31:2010–2025. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Trotman LC, Wang X, Alimonti A, Chen Z,
Teruya-Feldstein J, Yang H, Pavletich NP, Carver BS, Cordon-Cardo
C, Erdjument-Bromage H, et al: Ubiquitination regulates PTEN
nuclear import and tumor suppression. Cell. 128:141–156. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Hu X, Xu X, Zeng X, Jin R, Wang S, Jiang
H, Tang Y, Chen G, Wei J, Chen T and Chen Q: Gut microbiota
dysbiosis promotes the development of epithelial ovarian cancer via
regulating Hedgehog signaling pathway. Gut Microbes.
15:22210932023. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Willner J, Wurz K, Allison KH, Galic V,
Garcia RL, Goff BA and Swisher EM: Alternate molecular genetic
pathways in ovarian carcinomas of common histological types. Hum
Pathol. 38:607–613. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Karlsson T, Krakstad C, Tangen IL, Hoivik
EA, Pollock PM, Salvesen HB and Lewis AE: Endometrial cancer cells
exhibit high expression of p110β and its selective inhibition
induces variable responses on PI3K signaling, cell survival and
proliferation. Oncotarget. 8:3881–3894. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Singer G, Oldt R III, Cohen Y, Wang BG,
Sidransky D, Kurman RJ and Shih IeM: Mutations in BRAF and KRAS
characterize the development of low-grade ovarian serous carcinoma.
J Natl Cancer Inst. 95:484–486. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Dinulescu DM, Ince TA, Quade BJ, Shafer
SA, Crowley D and Jacks T: Role of K-ras and Pten in the
development of mouse models of endometriosis and endometrioid
ovarian cancer. Nat Med. 11:63–70. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Zhao X, Lai H, Li G, Qin Y, Chen R, Labrie
M, Stommel JM, Mills GB, Ma D, Gao Q and Fang Y: Rictor
orchestrates β-catenin/FOXO balance by maintaining redox
homeostasis during development of ovarian cancer. Oncogene.
44:1820–1832. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Tamura T, Nagai S, Masuda K, Imaeda K,
Sugihara E, Yamasaki J, Kawaida M, Otsuki Y, Suina K, Nobusue H, et
al: mTOR-mediated p62/SQSTM1 stabilization confers a robust
survival mechanism for ovarian cancer. Cancer Lett. 616:2175652025.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Trillsch F, Czogalla B, Mahner S, Loidl V,
Reuss A, du Bois A, Sehouli J, Raspagliesi F, Meier W, Cibula D, et
al: Risk factors for anastomotic leakage and its impact on survival
outcomes in radical multivisceral surgery for advanced ovarian
cancer: An AGO-OVAR.OP3/LION exploratory analysis. Int J Surg.
111:2914–2922. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Russo A, Czarnecki AA, Dean M, Modi DA,
Lantvit DD, Hardy L, Baligod S, Davis DA, Wei JJ and Burdette JE:
PTEN loss in the fallopian tube induces hyperplasia and ovarian
tumor formation. Oncogene. 37:1976–1990. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Coscia F, Watters KM, Curtis M, Eckert MA,
Chiang CY, Tyanova S, Montag A, Lastra RR, Lengyel E and Mann M:
Integrative proteomic profiling of ovarian cancer cell lines
reveals precursor cell associated proteins and functional status.
Nat Commun. 7:126452016. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Labidi-Galy SI, Papp E, Hallberg D,
Niknafs N, Adleff V, Noe M, Bhattacharya R, Novak M, Jones S,
Phallen J, et al: High grade serous ovarian carcinomas originate in
the fallopian tube. Nat Commun. 8:10932017. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Roh MH, Yassin Y, Miron A, Mehra KK,
Mehrad M, Monte NM, Mutter GL, Nucci MR, Ning G, Mckeon FD, et al:
High-grade fimbrial-ovarian carcinomas are unified by altered p53,
PTEN and PAX2 expression. Mod Pathol. 23:1316–1324. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Dean M, Jin V, Bergsten TM, Austin JR,
Lantvit DD, Russo A and Burdette JE: Loss of PTEN in fallopian tube
epithelium results in multicellular tumor spheroid formation and
metastasis to the ovary. Cancers (Basel). 11:8842019. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Zhang L, Ma T, Brozick J, Babalola K,
Budiu R, Tseng G and Vlad AM: Effects of Kras activation and Pten
deletion alone or in combination on MUC1 biology and
epithelial-to-mesenchymal transition in ovarian cancer. Oncogene.
35:5010–5020. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Alkema MJ, Wiegant J, Raap AK, Berns A and
van Lohuizen M: Characterization and chromosomal localization of
the human proto-oncogene BMI-1. Hum Mol Genet. 2:1597–1603. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Calao M, Sekyere EO, Cui HJ, Cheung BB,
Thomas WD, Keating J, Chen JB, Raif A, Jankowski K, Davies NP, et
al: Direct effects of Bmi1 on p53 protein stability inactivates
oncoprotein stress responses in embryonal cancer precursor cells at
tumor initiation. Oncogene. 32:3616–3626. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Su WJ, Fang JS, Cheng F, Liu C, Zhou F and
Zhang J: RNF2/Ring1b negatively regulates p53 expression in
selective cancer cell types to promote tumor development. Proc Natl
Acad Sci USA. 110:1720–1725. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Ginjala V, Nacerddine K, Kulkarni A, Oza
J, Hill SJ, Yao M, Citterio E, van Lohuizen M and Ganesan S: BMI1
is recruited to DNA breaks and contributes to DNA damage-induced
H2A ubiquitination and repair. Mol Cell Biol. 31:1972–1982. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Itahana K, Zou Y, Itahana Y, Martinez JL,
Beausejour C, Jacobs JJ, Van Lohuizen M, Band V, Campisi J and
Dimri GP: Control of the replicative life span of human fibroblasts
by p16 and the polycomb protein Bmi-1. Mol Cell Biol. 23:389–401.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Yadav AK, Sahasrabuddhe AA, Dimri M, Bommi
PV, Sainger R and Dimri GP: Deletion analysis of BMI1 oncoprotein
identifies its negative regulatory domain. Mol Cancer. 9:1582010.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Jacobs JJ, Scheijen B, Voncken JW, Kieboom
K, Berns A and van Lohuizen M: Bmi-1 collaborates with c-Myc in
tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF.
Genes Dev. 13:2678–2690. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Song LB, Li J, Liao WT, Feng Y, Yu CP, Hu
LJ, Kong QL, Xu LH, Zhang X, Liu WL, et al: The polycomb group
protein Bmi-1 represses the tumor suppressor PTEN and induces
epithelial-mesenchymal transition in human nasopharyngeal
epithelial cells. J Clin Invest. 119:3626–3636. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Bhattacharyya J, Mihara K, Ohtsubo M,
Yasunaga S, Takei Y, Yanagihara K, Sakai A, Hoshi M, Takihara Y and
Kimura A: Overexpression of BMI-1 correlates with drug resistance
in B-cell lymphoma cells through the stabilization of survivin
expression. Cancer Sci. 103:34–41. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Fan C, He L, Kapoor A, Gillis A, Rybak AP,
Cutz JC and Tang D: Bmi1 promotes prostate tumorigenesis via
inhibiting p16(INK4A) and p14(ARF) expression. Biochim Biophys
Acta. 1782:642–648. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Vonlanthen S, Heighway J, Altermatt HJ,
Gugger M, Kappeler A, Borner MM, van Lohuizen M and Betticher DC:
The bmi-1 oncoprotein is differentially expressed in non-small cell
lung cancer and correlates with INK4A-ARF locus expression. Br J
Cancer. 84:1372–1376. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Song LB, Zeng MS, Liao WT, Zhang L, Mo HY,
Liu WL, Shao JY, Wu QL, Li MZ, Xia YF, et al: Bmi-1 is a novel
molecular marker of nasopharyngeal carcinoma progression and
immortalizes primary human nasopharyngeal epithelial cells. Cancer
Res. 66:6225–6232. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Fasano CA, Dimos JT, Ivanova NB, Lowry N,
Lemischka IR and Temple S: shRNA knockdown of Bmi-1 reveals a
critical role for p21-Rb pathway in NSC self-renewal during
development. Cell Stem Cell. 1:87–99. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Mao L, Ding J, Perdue A, Yang L, Zha Y,
Ren M, Huang S, Cui H and Ding HF: Cyclin E1 is a common target of
BMI1 and MYCN and a prognostic marker for neuroblastoma
progression. Oncogene. 31:3785–3795. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Dimri GP, Martinez JL, Jacobs JJ, Keblusek
P, Itahana K, Van Lohuizen M, Campisi J, Wazer DE and Band V: The
Bmi-1 oncogene induces telomerase activity and immortalizes human
mammary epithelial cells. Cancer Res. 62:4736–4745. 2002.PubMed/NCBI
|
|
113
|
Ismail IH, Gagné JP, Caron MC, McDonald D,
Xu Z, Masson JY, Poirier GG and Hendzel MJ: CBX4-mediated SUMO
modification regulates BMI1 recruitment at sites of DNA damage.
Nucleic Acids Res. 40:5497–5510. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Voncken JW, Niessen H, Neufeld B,
Rennefahrt U, Dahlmans V, Kubben N, Holzer B, Ludwig S and Rapp UR:
MAPKAP kinase 3pK phosphorylates and regulates chromatin
association of the polycomb group protein Bmi1. J Biol Chem.
280:5178–5187. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Bruggeman SW, Hulsman D, Tanger E, Buckle
T, Blom M, Zevenhoven J, van Tellingen O and van Lohuizen M: Bmi1
controls tumor development in an Ink4a/Arf-independent manner in a
mouse model for glioma. Cancer Cell. 12:328–341. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Xu CR, Lee S, Ho C, Bommi P, Huang SA,
Cheung ST, Dimri GP and Chen X: Bmi1 functions as an oncogene
independent of Ink4A/Arf repression in hepatic carcinogenesis. Mol
Cancer Res. 7:1937–1945. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Liu Y, Liu F, Yu H, Zhao X, Sashida G,
Deblasio A, Harr M, She QB, Chen Z, Lin HK, et al: Akt
phosphorylates the transcriptional repressor bmi1 to block its
effects on the tumor-suppressing ink4a-arf locus. Sci Signal.
5:ra772012. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Cui H, Hu B, Li T, Ma J, Alam G, Gunning
WT and Ding HF: Bmi-1 is essential for the tumorigenicity of
neuroblastoma cells. Am J Pathol. 170:1370–1378. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Sahasrabuddhe AA, Dimri M, Bommi PV and
Dimri GP: βTrCP regulates BMI1 protein turnover via ubiquitination
and degradation. Cell Cycle. 10:1322–1330. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
López-Arribillaga E, Rodilla V,
Pellegrinet L, Guiu J, Iglesias M, Roman AC, Gutarra S, González S,
Muñoz-Cánoves P, Fernández-Salguero P, et al: Bmi1 regulates murine
intestinal stem cell proliferation and self-renewal downstream of
Notch. Development. 142:41–50. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Goel HL, Chang C, Pursell B, Leav I, Lyle
S, Xi HS, Hsieh CC, Adisetiyo H, Roy-Burman P, Coleman IM, et al:
VEGF/neuropilin-2 regulation of Bmi-1 and consequent repression of
IGF-IR define a novel mechanism of aggressive prostate cancer.
Cancer Discov. 2:906–921. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Zhang J and Sarge KD: Identification of a
polymorphism in the RING finger of human Bmi-1 that causes its
degradation by the ubiquitin-proteasome system. FEBS Lett.
583:960–964. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Zhao Q, Qian Q, Cao D, Yang J, Gui T and
Shen K: Role of BMI1 in epithelial ovarian cancer: Investigated via
the CRISPR/Cas9 system and RNA sequencing. J Ovarian Res.
11:312018. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Gui T, Bai H, Zeng J, Zhong Z, Cao D, Cui
Q, Chen J, Yang J and Shen K: Tumor heterogeneity in the recurrence
of epithelial ovarian cancer demonstrated by polycomb group
proteins. Onco Targets Ther. 7:1705–1716. 2014.PubMed/NCBI
|
|
125
|
Bhattacharya R, Nicoloso M, Arvizo R, Wang
E, Cortez A, Rossi S, Calin GA and Mukherjee P: MiR-15a and MiR-16
control Bmi-1 expression in ovarian cancer. Cancer Res.
69:9090–9095. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Koren A, Rijavec M, Kern I, Sodja E,
Korosec P and Cufer T: BMI1, ALDH1A1, and CD133 transcripts connect
epithelial-mesenchymal transition to cancer stem cells in lung
carcinoma. Stem Cells Int. 2016:97143152016. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Wang H, Liu H, Li X, Zhao J, Zhang H, Mao
J, Zou Y, Zhang H, Zhang S, Hou W, et al: Estrogen receptor
α-coupled Bmi1 regulation pathway in breast cancer and its clinical
implications. BMC Cancer. 14:1222014. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Qiao B, Chen Z, Hu F, Tao Q and Lam AK:
BMI-1 activation is crucial in hTERT-induced epithelial-mesenchymal
transition of oral epithelial cells. Exp Mol Pathol. 95:57–61.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Kim BR, Kwon Y and Rho SB: BMI-1 interacts
with sMEK1 and inactivates sMEK1-induced apoptotic cell death.
Oncol Rep. 37:579–586. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Wang E, Bhattacharyya S, Szabolcs A,
Rodriguez-Aguayo C, Jennings NB, Lopez-Berestein G, Mukherjee P,
Sood AK and Bhattacharya R: Enhancing chemotherapy response with
Bmi-1 silencing in ovarian cancer. PLoS One. 6:e179182011.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Chen H, Liu H and Qing G: Targeting
oncogenic Myc as a strategy for cancer treatment. Signal Transduct
Target Ther. 3:52018. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Dang CV, O'Donnell KA, Zeller KI, Nguyen
T, Osthus RC and Li F: The c-Myc target gene network. Semin Cancer
Biol. 16:253–264. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Adhikary S and Eilers M: Transcriptional
regulation and transformation by Myc proteins. Nat Rev Mol Cell
Biol. 6:635–645. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Alexander WS, Adams JM and Cory S:
Oncogene cooperation in lymphocyte transformation: Malignant
conversion of E mu-myc transgenic pre-B cells in vitro is enhanced
by v-H-ras or v-raf but not v-abl. Mol Cell Biol. 9:67–73. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Kessler JD, Kahle KT, Sun T, Meerbrey KL,
Schlabach MR, Schmitt EM, Skinner SO, Xu Q, Li MZ, Hartman ZC, et
al: A SUMOylation-dependent transcriptional subprogram is required
for Myc-driven tumorigenesis. Science. 335:348–353. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Conacci-Sorrell M, McFerrin L and Eisenman
RN: An overview of MYC and its interactome. Cold Spring Harb
Perspect Med. 4:a0143572014. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Ward PS and Thompson CB: Signaling in
control of cell growth and metabolism. Cold Spring Harb Perspect
Biol. 4:a0067832012. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Souza JL, Martins-Cardoso K, Guimarães IS,
de Melo AC, Lopes AH, Monteiro RQ and Almeida VH: Interplay between
EGFR and the platelet-activating factor/PAF receptor signaling axis
mediates aggressive behavior of cervical cancer. Front Oncol.
10:5572802020. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Skírnisdóttir IA, Sorbe B, Lindborg K and
Seidal T: Prognostic impact of p53, p27, and C-MYC on
clinicopathological features and outcome in early-stage (FIGO I–II)
epithelial ovarian cancer. Int J Gynecol Cancer. 21:236–244. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Chen CH, Shen J, Lee WJ and Chow SN:
Overexpression of cyclin D1 and c-Myc gene products in human
primary epithelial ovarian cancer. Int J Gynecol Cancer.
15:878–883. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Plisiecka-Hałasa J, Karpińska G, Szymańska
T, Ziółkowska I, Madry R, Timorek A, Debniak J, Ułańska M, Jedryka
M, Chudecka-Głaz A, et al: P21WAF1, P27KIP1, TP53 and C-MYC
analysis in 204 ovarian carcinomas treated with platinum-based
regimens. Ann Oncol. 14:1078–1085. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
van Dam PA, Vergote IB, Lowe DG, Watson
JV, van Damme P, van der Auwera JC and Shepherd JH: Expression of
c-erbB-2, c-myc, and c-ras oncoproteins, insulin-like growth factor
receptor I, and epidermal growth factor receptor in ovarian
carcinoma. J Clin Pathol. 47:914–919. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Watson JV, Curling OM, Munn CF and Hudson
CN: Oncogene expression in ovarian cancer: A pilot study of c-myc
oncoprotein in serous papillary ovarian cancer. Gynecol Oncol.
28:137–150. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Sasano H, Nagura H and Silverberg SG:
Immunolocalization of c-myc oncoprotein in mucinous and serous
adenocarcinomas of the ovary. Hum Pathol. 23:491–495. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Li XS, Sun J and He XL: Expression of
c-myc and mutation of the KRAS gene in patients with ovarian
mucinous tumors. Genet Mol Res. 14:10752–10759. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Polacarz SV, Hey NA, Stephenson TJ and
Hill AS: C-myc oncogene product P62c-myc in ovarian mucinous
neoplasms: Immunohistochemical study correlated with malignancy. J
Clin Pathol. 42:148–152. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Ning YX, Luo X, Xu M, Feng X and Wang J:
Let-7d increases ovarian cancer cell sensitivity to a genistein
analog by targeting c-Myc. Oncotarget. 8:74836–74845. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Curling M, Stenning S, Hudson CN and
Watson JV: Multivariate analyses of DNA index, p62c-myc, and
clinicopathological status of patients with ovarian cancer. J Clin
Pathol. 51:455–461. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Jung M, Russell AJ, Kennedy C, Gifford AJ;
Australian Ovarian Cancer Study Group, ; Mallitt KA, Sivarajasingam
S, Bowtell DD, DeFazio A, Haber M, et al: Clinical importance of
Myc family oncogene aberrations in epithelial ovarian cancer. JNCI
Cancer Spectr. 2:pky0472018. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Yamamoto A, Kurata M, Yamamoto K, Nogawa
D, Inoue M, Ishibashi S, Ikeda M, Miyasaka N and Kitagawa M: High
amplification of PVT1 and MYC predict favorable prognosis in early
ovarian carcinoma. Pathol Res Pract. 216:1531752020. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Mori T, Li Y, Hata H, Ono K and Kochi H:
NIRF, a novel RING finger protein, is involved in cell-cycle
regulation. Biochem Biophys Res Commun. 296:530–536. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Khan NH, Chen HJ, Fan Y, Surfaraz M,
Ahammad MF, Qin YZ, Shahid M, Virk R, Jiang E, Wu DD and Ji XY:
Biology of PEST-containing nuclear protein: A potential molecular
target for cancer research. Front Oncol. 12:7845972022. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Xu T, Wu K, Shi J, Ji L, Song X, Tao G,
Zheng S, Zhang L and Jiang B: LINC00858 promotes colon cancer
progression through activation of STAT3/5 signaling by recruiting
transcription factor RAD21 to upregulate PCNP. Cell Death Discov.
8:2282022. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Dong P, Fu H, Chen L, Zhang S, Zhang X, Li
H, Wu D and Ji X: PCNP promotes ovarian cancer progression by
accelerating β-catenin nuclear accumulation and triggering EMT
transition. J Cell Mol Med. 24:8221–8235. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Alhosin M, Sharif T, Mousli M,
Etienne-Selloum N, Fuhrmann G, Schini-Kerth VB and Bronner C:
Down-regulation of UHRF1, associated with re-expression of tumor
suppressor genes, is a common feature of natural compounds
exhibiting anti-cancer properties. J Exp Clin Cancer Res.
30:412011. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Wu DD, Gao YR, Li T, Wang DY, Lu D, Liu
SY, Hong Y, Ning HB, Liu JP, Shang J, et al: PEST-containing
nuclear protein mediates the proliferation, migration, and invasion
of human neuroblastoma cells through MAPK and PI3K/AKT/mTOR
signaling pathways. BMC Cancer. 18:4992018. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Clevers H: Wnt/beta-catenin signaling in
development and disease. Cell. 127:469–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Ghahhari NM and Babashah S: Interplay
between microRNAs and WNT/β-catenin signalling pathway regulates
epithelial-mesenchymal transition in cancer. Eur J Cancer.
51:1638–1649. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Arend RC, Londoño-Joshi AI, Straughn JM Jr
and Buchsbaum DJ: The Wnt/β-catenin pathway in ovarian cancer: A
review. Gynecol Oncol. 131:772–779. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Shi-Bai Z, Rui-Min L, Ying-Chuan S, Jie Z,
Chao J, Can-Hua Y, Xi C and Wen-Wei Q: TIPE2 expression is
increased in peripheral blood mononuclear cells from patients with
rheumatoid arthritis. Oncotarget. 8:87472–87479. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Rivlin N, Brosh R, Oren M and Rotter V:
Mutations in the p53 tumor suppressor gene: important milestones at
the various steps of tumorigenesis. Genes Cancer. 2:466–474. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Momand J, Zambetti GP, Olson DC, George D
and Levine AJ: The mdm-2 oncogene product forms a complex with the
p53 protein and inhibits p53-mediated transactivation. Cell.
69:1237–1245. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Lin J, Chen J, Elenbaas B and Levine AJ:
Several hydrophobic amino acids in the p53 amino-terminal domain
are required for transcriptional activation, binding to mdm-2 and
the adenovirus 5 E1B 55-kD protein. Genes Dev. 8:1235–1346. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Stramucci L, Pranteda A and Bossi G:
Insights of crosstalk between p53 protein and the MKK3/MKK6/p38
MAPK signaling pathway in cancer. Cancers (Basel). 10:1312018.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Uhlen M, Zhang C, Lee S, Sjöstedt E,
Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, et
al: A pathology atlas of the human cancer transcriptome. Science.
357:eaan25072017. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Thul PJ, Åkesson L, Wiking M, Mahdessian
D, Geladaki A, Ait Blal H, Alm T, Asplund A, Björk L, Breckels LM,
et al: A subcellular map of the human proteome. Science.
356:eaal33212017. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Cheaib B, Auguste A and Leary A: The
PI3K/Akt/mTOR pathway in ovarian cancer: Therapeutic opportunities
and challenges. Chin J Cancer. 34:4–16. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Hopkins BD, Hodakoski C, Barrows D, Mense
SM and Parsons RE: PTEN function: The long and the short of it.
Trends Biochem Sci. 39:183–190. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Mabuchi S, Kuroda H, Takahashi R and
Sasano T: The PI3K/AKT/mTOR pathway as a therapeutic target in
ovarian cancer. Gynecol Oncol. 137:173–179. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Siddique HR, Parray A, Tarapore RS, Wang
L, Mukhtar H, Karnes RJ, Deng Y, Konety BR and Saleem M: BMI1
polycomb group protein acts as a master switch for growth and death
of tumor cells: Regulates TCF4-transcriptional factor-induced BCL2
signaling. PLoS One. 8:e606642013. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Wang J, Guo Y, Chu H, Guan Y, Bi J and
Wang B: Multiple functions of the RNA-binding protein HuR in cancer
progression, treatment responses and prognosis. Int J Mol Sci.
14:10015–10041. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Cao L, Bombard J, Cintron K, Sheedy J,
Weetall ML and Davis TW: BMI1 as a novel target for drug discovery
in cancer. J Cell Biochem. 112:2729–2741. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Glinsky GV, Berezovska O and Glinskii AB:
Microarray analysis identifies a death-from-cancer signature
predicting therapy failure in patients with multiple types of
cancer. J Clin Invest. 115:1503–1521. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Gray F, Cho HJ, Shukla S, He S, Harris A,
Boytsov B, Jaremko Ł, Jaremko M, Demeler B, Lawlor ER, et al: BMI1
regulates PRC1 architecture and activity through homo- and
hetero-oligomerization. Nat Commun. 7:133432016. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Luo W, Chen J, Li L, Ren X, Cheng T, Lu S,
Lawal RA, Nie Q, Zhang X and Hanotte O: c-Myc inhibits myoblast
differentiation and promotes myoblast proliferation and muscle
fibre hypertrophy by regulating the expression of its target genes,
miRNAs and lincRNAs. Cell Death Differ. 26:426–442. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Rechsteiner M and Rogers SW: PEST
sequences and regulation by proteolysis. Trends Biochem Sci.
21:267–271. 1996. View Article : Google Scholar : PubMed/NCBI
|