Endometrial cancer (EC), a malignancy originating
from the uterine endometrium, exhibits a rising global incidence,
with >417,000 new cases and 97,000 deaths reported worldwide in
2022 alone (1). This trend is
largely attributable to the increasing prevalence of risk factors
such as obesity and metabolic syndrome (1–3).
Therapeutically, EC management has been refined by the molecular
classification established by The Cancer Genome Atlas (TCGA), which
categorizes EC into four subtypes [DNA polymerase ε (POLE)
ultramutated, microsatellite instability-high (MSI-H), copy-number
low and copy-number high], each with distinct prognoses and
therapeutic implications (4–6). This
classification provides a critical basis for personalized therapy
(7,8). Surgical intervention remains the
cornerstone of EC management. Chemotherapy is mainly reserved for
cases with advanced or recurrent disease, as well as for patients
with high-risk pathological features post-surgery. Nevertheless,
chemoresistance development presents a major therapeutic challenge
(9), leading to treatment failure
and mortality in >90% of patients with advanced-stage disease. A
notable clinical limitation is the lack of effective prognostic
biomarkers and specific targets to overcome chemoresistance
(10). Dysregulation of regulated
cell death (RCD) mechanisms is a recognized cancer hallmark,
contributing to tumor progression and drug resistance. Emerging
evidence has indicated novel RCD pathways, including autophagy,
ferroptosis and pyroptosis, as potential therapeutic targets to
enhance therapeutic efficacy and reverse chemoresistance in EC
(11–16).
Reactive oxygen species (ROS), primarily generated
via mitochondrial electron transport chain activity (17,18),
NADPH oxidases (19) and exogenous
stressors, exhibit a dual role in cancer initiation, progression,
suppression and therapy (20).
Oxidative stress, defined as an imbalance between ROS production
and elimination, is a critical pathogenic factor in numerous
chronic diseases, including cancer (21), cardiovascular diseases (22,23),
metabolic disorders (24) and
neurodegenerative conditions (such as Alzheimer's and Parkinson's
diseases) (25). Depending on their
levels and cellular context, ROS can promote tumorigenesis by
inducing DNA damage, epithelial-mesenchymal transition (EMT) and
immune evasion, or suppress tumor growth by triggering cell death
pathways such as apoptosis and ferroptosis (20,26).
Ferroptosis is an iron-dependent form of RCD marked
by iron accumulation and extensive lipid peroxidation. Key
morphological characteristics include condensed cellular membranes,
with increased density and reduction, or loss of mitochondrial
cristae (27). Ferroptosis involves
disruption of redox homeostasis, marked by depleted antioxidants
[such as glutathione (GSH) and GSH peroxidase 4 (GPX4)] and
accumulation of oxidants (such as Fe2+ and lipid ROS)
(28). As primary intracellular ROS
sources, mitochondria are intrinsically linked to ferroptosis.
Mitophagy, a selective autophagic process that degrades damaged
mitochondria via ubiquitin (Ub)-dependent or -independent pathways,
serves as a protective mechanism against stress and is crucial for
mitochondrial quality control (29–32).
Mitophagy and ferroptosis, as two distinct forms of RCD, have been
increasingly implicated in EC (11,16,33,34).
Concurrently, oxidative stress, a key regulator of cellular
metabolism, is closely linked to EC progression (35,36).
The triad of mitophagy, ferroptosis and oxidative stress forms a
dynamic equilibrium, collectively contributing to the pathogenesis
of diverse diseases through their interconnected regulatory
networks (37,38). The present review aims to delineate
the role of oxidative stress as a central hub integrating mitophagy
and ferroptosis in EC pathogenesis, offering novel perspectives for
its prevention and treatment.
ROS, including superoxide anions, free radicals and
hydrogen peroxide, are highly reactive molecules generated
primarily through mitochondrial electron transport and NADPH
oxidase activity (16–19,38,39).
Additional sources include peroxisomal metabolism and endoplasmic
reticulum stress (40,41). Oxidative stress occurs due to a
disruption in the balance between ROS generation and clearance,
serving a notable role in the development of cancer (21–23).
Cellular protection against ROS involves a multi-level defense
system, including enzymatic antioxidants (such as superoxide
dismutase, catalase and GSH peroxidase), non-enzymatic scavengers
(such as vitamin C, vitamins E and GSH) and repair systems for
oxidized biomolecules, which collectively maintain redox
homeostasis (39).
Physiological ROS levels regulate essential cellular
processes (including proliferation, differentiation, survival and
apoptosis). However, supraphysiological ROS levels drive
tumorigenesis and progression (42,43).
Excessive endogenous or exogenous ROS levels induce direct DNA
damage and impair DNA repair mechanisms, causing mutations that
inactivate tumor suppressors (such as p53) or activate oncogenes
(such as KRAS) (44–46).
ROS further promote malignancy by activating
proliferative signaling (including the PI3K/AKT/mTOR and MAPK
pathways), and suppressing antitumor immunity via impaired T-cell
differentiation/activation, T-cell death, natural killer cell
dysfunction and M2 macrophage polarization/recruitment (47,48).
ROS facilitate invasion and metastasis through EMT, and induce
EMT-associated cytoskeletal rearrangement (Rho GTPase-dependent),
extracellular matrix degradation (matrix
metalloproteinase-dependent) and angiogenesis [hypoxia-inducible
factor (HIF)-dependent] (49).
Although high ROS levels are crucial in promoting
tumor initiation and progression, excessive ROS levels also serve
an antioncogenic role in cancer (26,50).
Excessively accumulated ROS repress cancer cell growth by
inhibiting cancer cell proliferation through disrupting
nucleotide/ATP synthesis and inducing cell cycle arrest (51). ROS also trigger tumor cell death by
activating endoplasmic reticulum stress-mediated, mitochondrial and
p53-dependent apoptotic pathways, along with the ferroptosis
pathway (26). Therefore, targeting
the regulation of ROS levels (for example, using pro-oxidant agents
or inhibiting antioxidant pathways) has emerged as a novel
therapeutic strategy in cancer treatment, aiming to exploit the
‘dual role’ of oxidative stress to achieve selective elimination of
tumor cells.
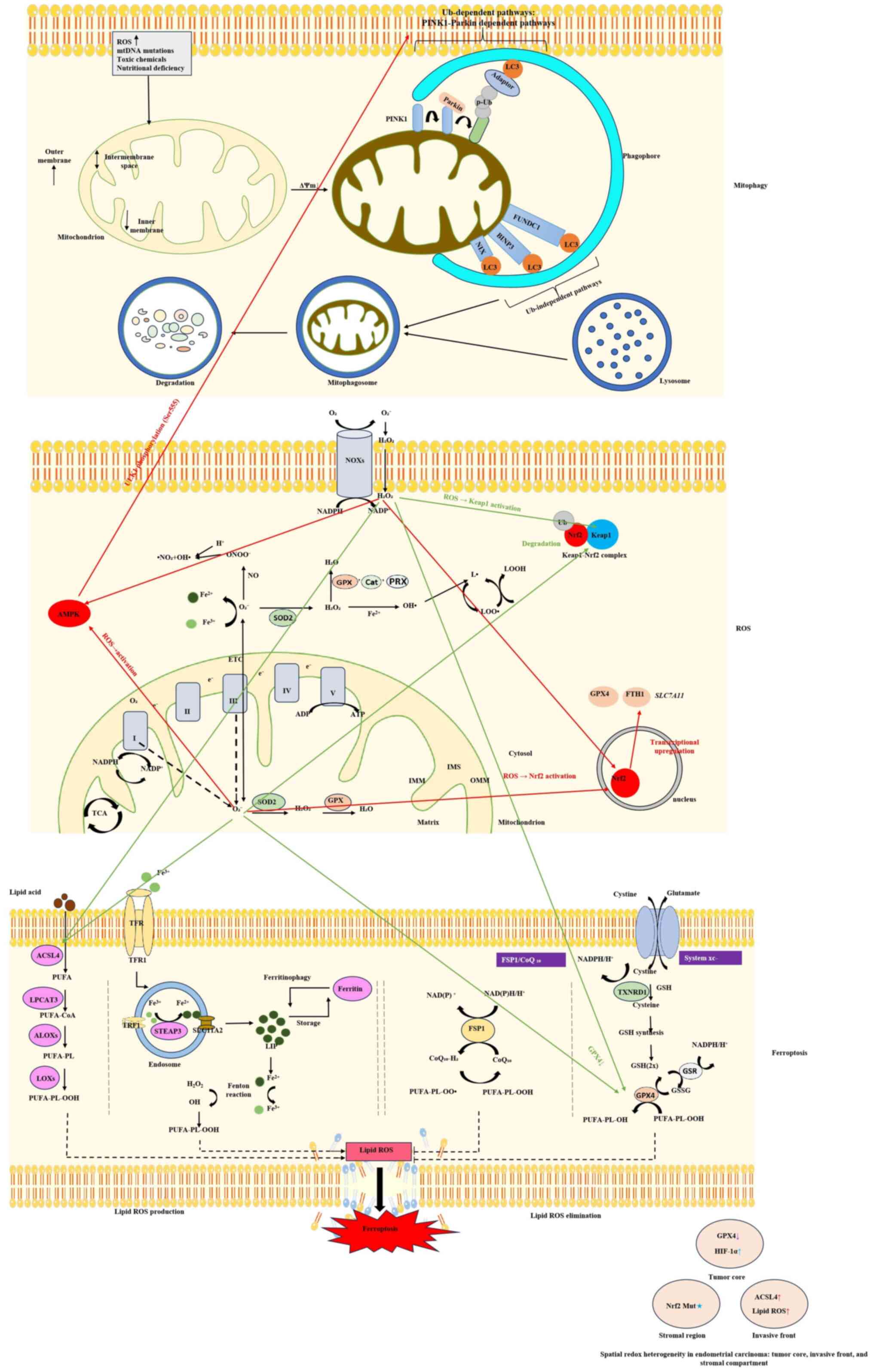
Mitophagy represents a selective type of autophagy
that functions as a protective response to intracellular and
extracellular stress signals (32).
Mitophagy maintains mitochondrial and cellular homeostasis by
eliminating dysfunctional mitochondria, including depolarized,
damaged or superfluous organelles, through lysosomal degradation.
The core molecular mechanisms can be divided into Ub-dependent
pathways and Ub-independent pathways (31). The PTEN-induced kinase 1
(PINK1)/Parkin pathway represents the most well-characterized
Ub-dependent mechanism. Upon mitochondrial depolarization or
damage, PINK1 stabilizes on the outer mitochondrial membrane (OMM),
and phosphorylates Ub and the E3 Ub ligase Parkin, leading to the
ubiquitination of OMM proteins. Autophagy receptors such as
p62/sequestosome 1 (SQSTM1) and optineurin then recognize
ubiquitinated substrates and link them to microtubule-associated
proteins 1A/1B light chain 3B (LC3)-positive autophagosomal
membranes for engulfment (31).
Alternatively, Ub-independent pathways utilize OMM-resident
receptors, including BCL2/adenovirus E1B 19 kDa protein-interacting
protein 3 (BNIP3), BNIP3L (also known as NIX) and FUN14
domain-containing protein 1 (FUNDC1), which directly interact with
LC3/GABA type A receptor-associated protein via LC3-interacting
regions to initiate mitophagy. These coordinated mechanisms ensure
the precise elimination of dysfunctional mitochondria, and their
dysregulation is broadly implicated in various pathological states,
including cancer (30,52,53).
Mitophagy is widely reported to exhibit aberrant
activity levels in various cancer types compared with under normal
physiological conditions (54,55).
Current research has revealed its dual role in cancer biology,
demonstrating both tumor-promoting and tumor-suppressive functions
depending on the cellular context (56,57).
In its pro-tumorigenic capacity, mitophagy is known to enable
cancer cell survival under stress and facilitates malignant
progression by clearing dysfunctional mitochondria (58). Under hypoxia, mitophagy mediated by
BNIP3 and NIX is known to enhance tumor aggressiveness by lowering
mitochondrial ROS levels, stabilizing HIF-1α, promoting glycolytic
shift and inducing EMT, which collectively increase invasiveness
and metastatic potential (59).
Chemotherapy- or radiotherapy-induced mitochondrial damage can
activate the PINK1/Parkin pathway, which ubiquitinates damaged
mitochondria to block cytochrome c release, thereby potentially
enabling cancer cells to evade apoptosis and develop therapeutic
resistance (60). During
metastasis, FUNDC1-dependent mitophagy is suggested to optimize
energy metabolism to fuel ATP production, supporting cancer cell
migration and invasion (61).
Additionally, mitophagy may suppress NLR family pyrin domain
containing 3 (NLRP3) inflammasome activation by removing
mitochondrial damage-associated molecular patterns [mitochondrial
DNA (mtDNA) and cardiolipin], thereby reducing secretion of the
pro-inflammatory cytokine IL-1β and impairing antitumor immunity to
promote immune evasion (62).
Conversely, mitophagy also exerts tumor-suppressive
effects, as supported by multiple lines of evidence (59,63).
In precancerous or early-stage tumors, mitophagy impedes malignant
transformation by eliminating mitochondria harboring oncogenic
damage. For example, mtDNA mutations, which drive early
carcinogenesis, are selectively cleared via NIX/BNIP3-dependent
mitophagy, thereby mitigating ROS-induced nuclear genomic
instability and delaying tumor initiation (64). The tumor suppressor p53 has been
shown to enhance mitophagy by transcriptionally activating DNA
damage-regulated autophagy modulator 1, facilitating the removal of
dysfunctional mitochondria and inducing apoptosis in premalignant
cells, as validated in colorectal precancerous models (65,66).
Furthermore, excessive mitochondrial damage can trigger ‘autosis’,
a form of autophagic cell death, via mitochondrial membrane
potential collapse and ATP depletion, irreversibly eliminating
premalignant cells. In inflammation-associated carcinogenesis,
mitophagy is proposed to block chronic inflammation-driven
tumorigenesis by clearing mitochondrial damage-associated molecular
patterns (such as cardiolipin and mtDNA), thereby inhibiting NLRP3
inflammasome activation and IL-1β secretion (67). Critically, the tumor-suppressive
effects of mitophagy are context- and stage-dependent: Activation
of mitophagy in PTEN-deficient or KRAS-mutant precancerous models
reduces hyperproliferative lesions, whereas the same intervention
in advanced tumors may paradoxically promote malignancy due to
metabolic rewiring and survival adaptation. This spatiotemporal
duality underscores the necessity of stage-specific targeting of
mitophagy pathways in cancer therapy (68,69).
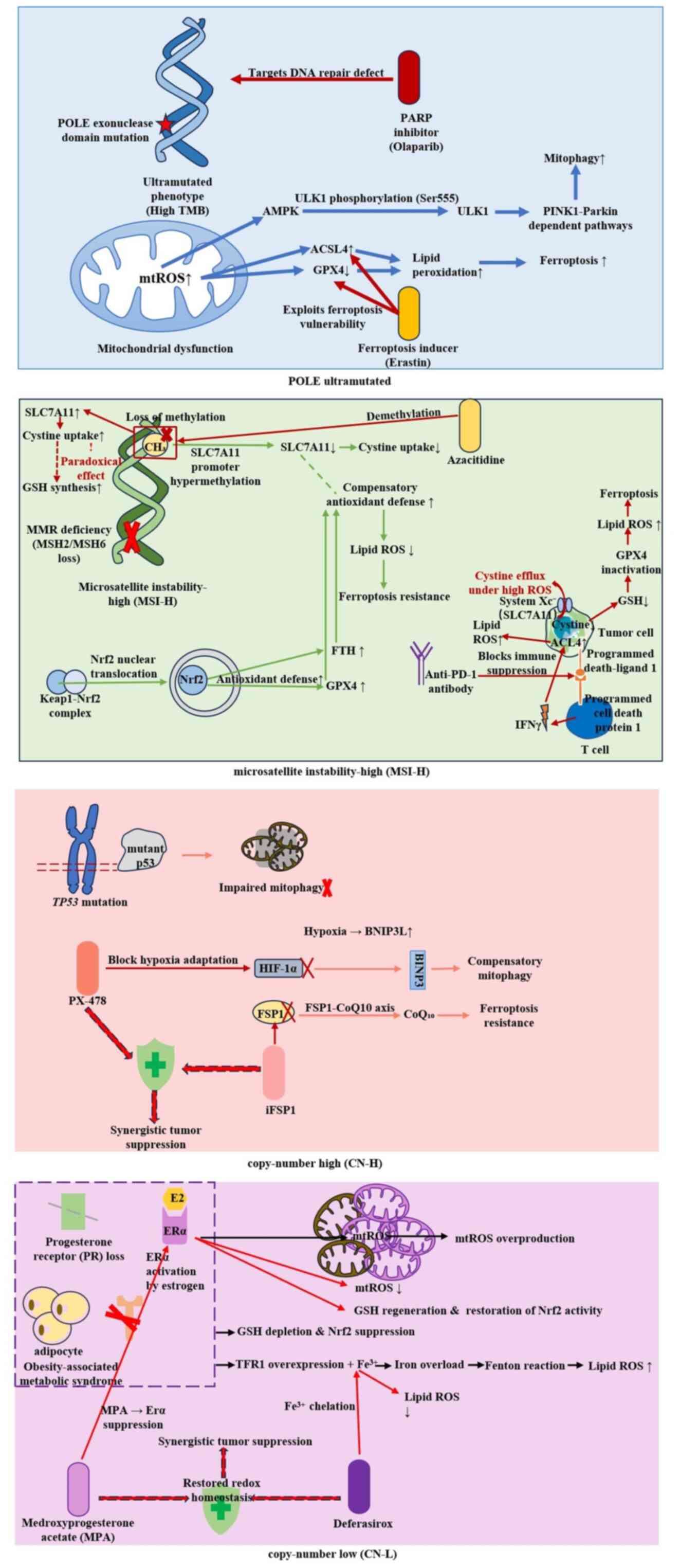
Emerging evidence highlights stage- and molecular
context-specific regulation of mitophagy in EC, although research
in this field remains sparse (70).
Dysregulation of the PTEN/PI3K/AKT pathway, observed in >80% of
EC cases, impairs PINK1/Parkin-mediated mitophagy, resulting in the
accumulation of dysfunctional mitochondria, increased ROS levels,
heightened genomic instability and estrogen-driven proliferation
(71). However, evidence has
revealed that PTEN-deficient EC cells activate an alternative
mitophagy pathway mediated by the autophagy and beclin 1 regulator
1-autophagy-related protein 5 complex, which bypasses Unc-51 like
autophagy activating kinase 1 (ULK1) inhibition and maintains
mitochondrial quality control under mTORC1 hyperactivation
(13). In type I EC, hyperactivated
mTORC1 phosphorylates ULK1 at Ser757, disrupting the
ULK1-AMP-activated protein kinase (AMPK) complex essential for
mitophagy initiation and promoting tumor cell survival (34). Notably, advanced TP53-mutant ECs
exhibit hypoxia-driven compensatory upregulation of
BNIP3L/NIX-mediated mitophagy, which is transcriptionally activated
by HIF-1α under hypoxic conditions to sustain mitochondrial
metabolism and chemoresistance, thereby contributing to the poor
prognosis of p53-aberrant tumors (33). This dual regulatory role,
tumor-suppressive in early stages and pro-tumorigenic in advanced
disease, underscores the need for stage-specific therapeutic
strategies. Restoring PINK1/Parkin signaling in PTEN-deficient
models may improve mitochondrial quality control, while targeting
BNIP3L/NIX in TP53-mutant tumors could enhance chemosensitivity.
However, the molecular mechanisms underlying these
context-dependent effects require further elucidation.
In conclusion, mitophagy serves as both a ‘metabolic
accomplice’ and a ‘genomic custodian’ in EC, with its dualistic
role underscoring the therapeutic conundrum in targeting
mitochondrial quality control. The transition from
tumor-suppressive to pro-survival functions is intricately linked
to disease stage, molecular subtypes (such as POLE-mutated and
TP53-aberrant) and metabolic rewiring driven by PTEN/PI3K
dysregulation or obesity-associated stress (33). Future endeavors must prioritize
stage-adaptive strategies, activating mitophagy to enforce genomic
fidelity in precancerous lesions while inhibiting
FUNDC1/PINK1/Parkin-driven pathways to disrupt metabolic plasticity
in advanced tumors. Integrating multi-omics profiling (such as
mitophagic flux mapping and spatial metabolomics) with clinical
staging will unlock context-specific vulnerabilities, enabling the
design of precision therapies that convert mitophagy from a foe to
an ally in the battle against EC, as illustrated in Fig. 1.
Ferroptosis is an iron-dependent RCD pathway that is
driven by lipid peroxidation, originating from redox imbalance,
dysregulated lipid metabolism and disrupted iron homeostasis
(72); however, cellular adaptation
to these stressors hinges on dynamic modifications of core
regulatory pathways, which paradoxically form the molecular basis
of ferroptosis resistance (29).
The GPX4/GSH axis is a central defense system: System
Xc− [composed of solute carrier family 7 member 11
(SLC7A11) and solute carrier family 3 member 2] imports cystine for
GSH synthesis and GPX4 utilizes GSH to reduce lipid peroxides.
Resistance often arises from GPX4 or System Xc−
upregulation (73). Alternatively,
the ferroptosis suppressor protein 1 (FSP1)-coenzyme Q10 (CoQ10)
axis has been identified as a key GPX4-independent pathway wherein
FSP1 reduces ubiquinone to ubiquinol, which quenches lipid radicals
(28). Acyl-CoA synthetase
long-chain family member 4 (ACSL4) is a well-established promoter
of ferroptosis that enriches membranes with polyunsaturated fatty
acids; its suppression diminishes ferroptosis sensitivity (74). Iron metabolism serves a dual role:
Iron overload amplifies lipid peroxidation, while iron storage
proteins [such as ferritin heavy chain 1 (FTH1)] buffer toxicity.
The nuclear factor erythroid 2-related factor 2 (Nrf2) pathway is a
recognized suppressor of ferroptosis, primarily by activating
antioxidant genes (such as GPX4 and FTH1) (29,75).
p53 exerts context-dependent effects, which may include inhibiting
SLC7A11 or activating spermidine/spermine N1-acetyltransferase 1
(65–67).
Ferroptosis exhibits a dynamically regulated dual
role in EC progression: Its suppressed state drives tumor invasion,
metastasis and therapy resistance through multifaceted molecular
mechanisms, while ferroptosis-inducing strategies have the
potential to reverse drug resistance and inhibit metastasis
(16,76–78).
The core regulatory hubs GPX4 and Nrf2 are upregulated in EC and
are functionally synergistic. GPX4 maintains membrane stability by
reducing toxic lipid peroxides (such as phosphatidylethanolamine
hydroperoxide), whereas Nrf2 enhances antioxidant defense via
transcriptional activation of SLC7A11 (cystine/glutamate
antiporter) and FTH1 (iron storage protein) (79–81). A
multivariate analysis of the TCGA-uterine corpus endometrial
carcinoma cohort revealed that GPX4/Nrf2 co-upregulation was
independently associated with lymph node metastasis risk [odds
ratio (OR), 3.21; 95% confidence interval (CI), 1.38–7.45; P=0.007]
and International Federation of Gynecology and Obstetrics III/IV
staging (OR, 2.94; P=0.012) (82,83).
EC cells evade ferroptosis through epigenetic reprogramming,
including the microRNA (miR)-424-5p-mediated destabilization of
ACSL4 mRNA via 3′untranslated region binding (validated by
dual-luciferase assays) and hypermethylation of the ACSL4 promoter
CpG island (methylation β-value >0.6; P<0.001), collectively
suppressing pro-ferroptotic lipid peroxidation (83). Iron metabolism dysregulation
exacerbates malignant phenotypes: FTH1 upregulation reduces the
labile iron pool by enhancing iron storage, thereby inhibiting
Fenton reaction-driven lipid peroxidation, while divalent metal
transporter 1 mediates lysosomal iron efflux to amplify ferroptosis
sensitivity in metastatic niches. Concurrently, transferrin
receptor 1 (TFR1)-mediated iron overload activates pro-angiogenic
factors (such as VEGF) via the mitochondrial ROS/HIF-1α axis
(84). Clinical translational
studies have highlighted the therapeutic potential of combining
erastin with cisplatin: Cisplatin-resistant EC cells (Ishikawa-CR
line) acquire ferroptosis resistance via GPX4/FSP1 dual-pathway
upregulation, whereas erastin/cisplatin co-treatment induces
caspase-3-independent cell death (4.2-fold apoptosis increase;
P<0.001) (11,85). Metformin suppresses SLC7A11
transcription via the AMPK/p53 axis (chromatin
immunoprecipitation-quantitative PCR-validated), reducing
peritoneal metastases by 68% in patient-derived xenograft (PDX)
models (P=0.002) (86,87). Interim analysis of the NCT04817332
phase II trial demonstrated improved objective response rates with
erastin/carboplatin-paclitaxel combination therapy in recurrent EC
(51 vs. 32% for monotherapy; P=0.039), albeit with increased
ferroptosis-related toxicity (grade 3 anemia, 28 vs. 12%) (88). Parallel preclinical investigations
provided complementary insights: To further elucidate the cellular
diversity underlying treatment responses, single-cell RNA
sequencing of patient-derived EC samples revealed significant
heterogeneity in ferroptosis-related gene expression profiles.
Meanwhile, leveraging nanodelivery systems to encapsulate
ferroptosis inducers has been established as a promising strategy
in preclinical models to enhance tumor selectivity and reduce
systemic toxicity (89). These
findings systematically elucidate the pivotal role of ferroptosis
dysregulation in EC progression and provide novel directions for
developing redox metabolism-targeted precision therapies (16).
ROS serve as key signaling molecules that
dynamically coordinate the balance between mitophagy and
ferroptosis. Mitochondrial ROS promote PINK1/Parkin-mediated
mitophagy, which clears damaged mitochondria and limits lipid
peroxidation (31,52). Conversely, endoplasmic
reticulum-derived ROS activate the inositol-requiring enzyme
1α/c-Jun N-terminal kinase pathway, upregulating ACSL4 to enhance
membrane polyunsaturated fatty acid incorporation and ferroptosis
sensitivity (90). Lipid
peroxidation products [such as phospholipid hydroperoxides (PLOOH)]
further amplify mitochondrial damage, establishing a feedforward
loop (91). This interplay exhibits
concentration dependence in EC: Physiological ROS levels promote
cytoprotective mitophagy via AMPK-ULK1 signaling to suppress early
malignant transformation (92),
whereas chemotherapy (such as cisplatin)-induced ROS trigger
ferroptosis-dominated cell death by depleting GSH and inhibiting
GPX4 (93). A previous study has
revealed that estrogen receptor α (ERα) activation in EC cells
simultaneously upregulates mitochondrial ROS production and
autophagy-related genes (such as BNIP3), driving dynamic imbalances
between mitophagy and ferroptosis to fuel tumor progression
(94).
Mitophagy and ferroptosis are interconnected through
shared molecular nodes, forming a bidirectional regulatory network
(95). On the one hand, mitophagy
suppresses ferroptosis by degrading ACSL4, a key enzyme that
catalyzes polyunsaturated fatty acid-phospholipid synthesis, and
removing lipid peroxidation precursors (96). On the other hand,
ferroptosis-derived lipid peroxides (such as PLOOH) activate
PINK1/Parkin or NIX/BNIP3L-dependent mitophagy via mitochondrial
membrane oxidation (97). Emerging
evidence demonstrates that ACSL4, a ferroptosis-specific regulator,
induces mitochondrial Ca2+ overload, leading to membrane
depolarization and subsequent mitophagy initiation (98). Additionally, labile iron released
during ferroptosis exacerbates mitochondrial oxidative damage
through Fenton reactions, creating an ‘iron-ROS-mitophagy’ cycle.
In EC, PTEN loss-induced dysregulation of the PI3K/AKT/mTOR pathway
disrupts redox homeostasis via dual mechanisms: Impairing ULK1
phosphorylation to block protective mitophagy and downregulating
GPX4 to heighten ferroptosis susceptibility (33). Combined targeting of both pathways
(for example, using mitophagy inducer urolithin A + ferroptosis
activator RSL3) synergistically inhibits tumor growth in
preclinical models, highlighting the therapeutic potential of
modulating the ROS-mitophagy-ferroptosis axis (16,71),
as illustrated in Fig. 2.
EC is characterized by alterations in redox
homeostasis that is driven by hormonal imbalances and somatic
mutations. Estrogen dominance, a hallmark of type I EC, activates
ERα-mediated mitochondrial biogenesis, amplifying superoxide
generation while epigenetically suppressing Nrf2-dependent
antioxidant defenses through promoter hypermethylation of NFE2 like
bZIP transcription factor 2 (NFE2L2) (99). Conversely, progesterone exerts
protective effects by binding to the progesterone receptor-B
isoform, upregulating thioredoxin reductase and GSH synthetase
through cAMP-response element binding protein phosphorylation, a
mechanism disrupted in obesity-associated progesterone resistance
(100). PTEN loss (occurring in
40–80% of cases) exacerbates this imbalance via dual pathways: i)
Hyperactivation of PI3K/AKT/mTORC1 inhibits ULK1-driven mitophagy,
allowing the accumulation of ROS-generated damaged mitochondria;
and ii) transcriptional repression of the FSP1/CoQ10 axis through
yes-associated protein and transcriptional coactivator with
PDZ-binding motif nuclear translocation, sensitizing cells to
ferroptosis (101,102). Single-cell multi-omics has
identified PTEN-null CD44-positive aldehyde dehydrogenase 1 family
member A1-high cancer stem cells (CSCs) that evade ferroptosis via
aldehyde detoxification and CoQ10 recycling, establishing a
redox-buffered niche resistant to oxidative stress (103).
Redox adaptations exhibit temporal evolution during
disease progression: i) Premalignant lesions exhibit compensatory
upregulation of mitophagic machinery (elevated Parkin RBR E3
ubiquitin-protein ligase and BNIP3 levels) with concomitant
suppression of ferroptosis drivers (low ACSL4 expression); ii)
early invasive tumors activate the FSP1/CoQ10 antioxidant system
and suppress mitophagy through mTORC1-mediated transcription factor
EB phosphorylation, resulting in lipid peroxide accumulation; and
iii) advanced tumors undergo metabolic rewiring (glutamine-driven
oxidative phosphorylation and HIF-1α signaling) to enforce
mitophagy dependency and ferroptosis resistance (104–106). These stage-specific adaptations
inform rational therapeutic targeting: mTOR inhibition with
rapamycin restores protective mitophagy in premalignant states,
while FSP1 inhibitors combined with ferroptosis inducers (such as
imidazole ketone erastin) overcome therapy resistance in advanced
disease.
Dynamic regulation of the Nrf2/Kelch-like
ECH-associated protein 1 (Keap1) axis and EC-specific
dysregulation
The Nrf2/Keap1 pathway serves as a central hub for
oxidative stress responses, integrating antioxidant defenses,
mitophagy and ferroptosis to maintain cellular homeostasis. Under
physiological conditions, Keap1-mediated ubiquitination and
degradation of Nrf2 restrict its activity, while oxidative stress
triggers Nrf2 nuclear translocation, activating target genes such
as heme oxygenase 1 (which degrades pro-ferroptotic labile iron)
and SQSTM1/p62 (which promotes clearance of damaged mitochondria
via selective autophagy) (107).
In EC, KEAP1 loss-of-function mutations or NFE2L2 (encoding Nrf2)
amplifications lead to constitutive Nrf2 activation, driving
p62-dependent mitophagy and suppressing ferroptosis to favor tumor
survival. Estrogen enhances NFE2L2 transcription via ERα binding to
its promoter, while progesterone upregulates Keap1 expression
through PR-B, establishing hormone-dependent oscillations in Nrf2
activity (100,108). Furthermore, Nrf2-hyperactive
tumors reprogram metabolism by upregulating glutaminase and the
cystine transporter xCT/SLC7A11, sustaining mitochondrial function
and ferroptosis resistance, a mechanism particularly prominent in
PTEN-deficient subtypes (13).
Fe-S cluster biogenesis acts as a critical node
linking mitochondrial integrity and ferroptosis sensitivity.
Deficiencies in mitochondrial Fe-S assembly systems [such as
iron-sulfur cluster assembly enzyme (ISCU) and frataxin] disrupt
electron transport chain function, causing superoxide accumulation
and PINK1/Parkin-mediated mitophagy, while cytosolic labile iron
pool expansion exacerbates lipid peroxidation via Fenton reactions,
increasing ferroptosis susceptibility (109). In EC, ISCU promoter
hypermethylation and miR-214 upregulation synergistically impair
Fe-S cluster synthesis, whereas cancer-associated fibroblasts
rescue redox homeostasis by delivering Fe-S complementation factors
(such as cysteine desulfurase) via exosomes (110,111). Therapeutic Fe-S cluster donors
(such as Fe4S4) restore mitochondrial
function while inducing ferroptosis, exhibiting synergistic
efficacy in PTEN-deficient organoid models and offering a novel
strategy to overcome therapy resistance (103).
The spatiotemporal dynamics of oxidative stress
critically influence therapeutic responses in EC. Chemotherapy
(e.g., cisplatin) induces biphasic ROS generation (112): Acute-phase mitochondrial ROS
activate AMPK-ULK1-mediated protective mitophagy, promoting cell
survival, while chronic-phase ROS accumulation triggers ferroptosis
via GSH depletion and GPX4 inactivation. The circadian regulators
BMAL1 and CLOCK rhythmically control the expression of autophagy
genes like LC3B. This regulation drives a peak in autophagic
activity at night, which helps to counteract the oxidative damage
that accumulates during the day (113). Chronotherapy strategies leveraging
these rhythms [e.g., administering ferroptosis inducers (such as
erastin) during active circadian phases] synergize with endogenous
ROS fluctuations, enhancing treatment efficacy (114,115).
Spatial heterogeneity further shapes oxidative
stress regulation: Hypoxic tumor cores suppress ferroptosis via
HIF-1α-mediated downregulation of ACSL4 and TFR1, while
upregulating BNIP3-driven mitophagy to maintain CSC stemness. By
contrast, high-ROS microenvironments at invasive fronts activate
NF-κB/STAT3 signaling to promote EMT and FSP1/CoQ10-dependent
ferroptosis resistance (112,116). CSCs remodel stromal niches via
exosomal transfer of antioxidants (such as glutathione
S-transferase P1) and iron chelators, creating localized ‘oxidative
sanctuaries’ (117).
Nanotherapeutic strategies exploiting this heterogeneity, such as
ROS-responsive carriers delivering autophagy inhibitors to cores
and ferroptosis inducers to invasive fronts, exhibit potent
antitumor effects in preclinical models (118).
In summary, the spatiotemporal regulation of
oxidative stress defines distinct therapeutic vulnerabilities,
enabling chronotherapeutic and microenvironment-targeted
interventions.
The oxidative stress network offers novel biomarkers
and therapeutic strategies (119).
Combined evaluation of mitophagy (for example, mtDNA copy number)
and ferroptosis markers (for example, lipid peroxidation products)
enables real-time monitoring of tumor redox status (120). CD8+ T cell-derived IFNγ
enhances ferroptosis susceptibility via STAT1/interferon regulatory
factor 1/ACSL4 signaling, suggesting synergies between immune
checkpoint inhibitors and ferroptosis inducers (121). Advanced imaging technologies (such
as nanoprobe-based iron tracking) integrated with artificial
intelligence (AI) algorithms accurately predict ferroptosis
sensitivity (122).
Spatiotemporally optimized combinations, such as
sequential autophagy inhibition followed by ferroptosis induction,
overcome compensatory resistance (123,124). ROS-responsive nanocarriers achieve
precise drug release by exploiting microenvironmental features
(such as low pH and high GSH levels), effectively targeting both
hypoxic cores and invasive fronts. Additionally, targeting
PTEN-null vulnerabilities with mTOR and glutaminase inhibitors
synergistically blocks mitophagy and induces ferroptosis, achieving
remarkable responses in patient-derived models (101).
These advances highlight the clinical potential of
targeting oxidative stress dynamics for precision therapy in
EC.
Key challenges remain in modeling oxidative stress
dynamics. Current organoid systems lack spatiotemporal resolution
for tumor-microenvironment crosstalk (for example, immune cell
interactions and hormone gradients) (125). Integrating single-cell
transcriptomics with spatial metabolomics is essential to resolve
redox heterogeneity and niche-specific adaptations (126,127). Mechanistic gaps persist regarding
the subcellular localization of ferroptosis regulators (such as
FSP1) and the crosstalk between Nrf2 and circadian pathways
(96–99). Compensatory activation, such as GPX4
inhibition upregulating FSP1/CoQ10, requires sequential targeting
strategies tailored to tumor heterogeneity (128–131). AI-driven drug discovery (for
example, using deep generative models) predicts synergistic
combinations (such as mTOR inhibitors with ferroptosis inducers),
validated in PDX models (132).
Future studies must integrate computational biology
and high-resolution omics to map the oxidative stress landscape,
enabling personalized therapeutic strategies.
The establishment of oxidative stress as a nexus
between mitophagy and ferroptosis unveils a novel therapeutic
landscape in EC (95,120,133). However, clinical translation is
impeded by defined knowledge gaps and practical challenges that
must be prioritized in future research.
A primary unresolved question is the spatiotemporal
dynamics of this interplay within the tumor microenvironment (TME).
The precise thresholds of oxidative stress that dictate a switch
from pro-survival mitophagy to lethal ferroptosis across different
molecular subtypes and disease stages remain unmapped (120). This context-dependent duality
necessitates a deeper understanding of how metabolic and hormonal
cues influence this balance. Concurrently, the immunological
consequences of targeting this axis constitute a critical knowledge
gap. It is not known whether ferroptosis induction or mitophagy
modulation can enhance antitumor immunity by, for example,
triggering immunogenic cell death or reversing immunosuppressive
niches, which would create a rationale for combinations with
immunotherapy.
The compelling preclinical evidence stands in stark
contrast to the current clinical trial landscape, highlighting a
significant translation gap. To definitively assess the clinical
translation of these mechanisms, a systematic search of the
ClinicalTrials.gov registry (https://clinicaltrials.gov/) was conducted on
September 10, 2025. The search strategy utilized keywords including
‘endometrial cancer’, ‘ferroptosis’, ‘mitophagy’ and related terms.
Crucially, the following exclusion criteria were applied: i) Trials
not focused on EC; ii) non-interventional studies; and iii) trials
where direct targeting of mitophagy or induction of ferroptosis was
not an explicit primary strategy. It is important to note that this
search constituted a systematic search of trial registries and not
a systematic review; as such, a formal quality assessment of the
identified trials or a synthesis of data from existing publications
was not performed, which is a limitation of this methodological
approach.
This rigorous search confirmed a complete absence of
clinical trials specifically designed to directly target mitophagy
or induce ferroptosis as a primary therapeutic strategy in EC.
However, the search identified several interventional trials
investigating agents with indirect mechanistic links to these
pathways: i) mTOR inhibitors: Active and completed trials
investigating everolimus (such as NCT01797523 and NCT00870337) are
relevant due to the established role of mTOR signaling in
regulating the mitophagic flux and cellular stress responses; and
ii) AMPK activators: Multiple trials involving metformin (such as
NCT04576104, NCT07145827 and NCT01205672) are of interest based on
preclinical evidence suggesting its potential to modulate AMPK
activity and sensitize cells to ferroptosis.
The absence of direct-targeting trials underscores
the nascent stage of translating these concepts into clinical
applications. Conversely, the prevalence of trials involving mTOR
inhibitors and AMPK activators suggests a growing clinical
recognition of the importance of targeting broader metabolic and
stress-response pathways in EC, which may indirectly influence
mitophagy and ferroptosis (134,135).
Translation of these insights faces significant
barriers. The lack of robust, dynamic biomarkers (such as
circulating lipid peroxidation products) beyond static molecular
classification severely hinders patient stratification.
Furthermore, managing systemic toxicity, particularly of potent
ferroptosis inducers, is a paramount concern. Overcoming this
requires the development of advanced nano-theranostic platforms
engineered for EC-specific targeting [e.g., against folate receptor
α (FRα)] and TME-responsive drug release (e.g., triggered by high
ROS levels) to maximize efficacy and minimize off-target effects.
The inherent compensatory activation of parallel pathways (such as
upregulation of the FSP1/CoQ10 axis upon GPX4 inhibition) further
necessitates the rational design of combination therapies.
In summary, the present review consolidates evidence
that oxidative stress functions as a critical mechanistic nexus
coordinating mitophagy and ferroptosis in EC. The deregulation of
this axis presents a compelling therapeutic vulnerability.
The future of targeting this nexus lies in precision
medicine strategies that directly address the aforementioned
challenges, with tailored approaches for both mitophagy and
ferroptosis: i) Biomarker-guided stratification: Future efforts
must integrate functional redox biomarkers with molecular subtyping
to identify patient populations most likely to benefit, such as
those with PTEN-deficient tumors exhibiting heightened ferroptosis
sensitivity. For mitophagy, this necessitates stage-aware
strategies, such as activating it to enforce genomic fidelity in
precancerous/early-stage lesions (such as PTEN-deficient) while
inhibiting its pro-survival function in advanced or
therapy-resistant tumors (such as TP53-mutant or BNIP3L/NIX-high).
ii) Intelligent therapeutic platforms: The development of
stimuli-responsive nanocarriers is indispensable to overcome the
dual challenges of drug delivery efficiency and systemic toxicity,
enabling spatially controlled therapy within the TME for
ferroptosis inducers. Complementary efforts should develop
mitochondrially-targeted delivery systems for mitophagy modulators
(such as PINK1 activators or FUNDC1 inhibitors) to achieve
organelle-specific precision. iii) Rational combination therapies:
Overcoming adaptive resistance necessitates ‘two-pronged’
strategies, such as co-inhibiting parallel defense pathways (for
example, GPX4 and FSP1) or combining ferroptosis inducers with
immunotherapy in appropriate subtypes (such as MSI-H) to exploit
immunogenic cell death. Similarly, mitophagy inhibition can be
rationally combined with standard chemo-/radiotherapy to prevent
treatment resistance or with ferroptosis inducers to
synergistically disrupt mitochondrial metabolism and redox
homeostasis.
By addressing the knowledge gaps and embracing these
precision approaches, the transformative potential of targeting the
oxidative stress-mitophagy-ferroptosis axis can be realized, paving
the way for novel and effective management strategies in EC.
Not applicable.
This work was supported by the Medical and Health Science and
Technology Development Program Project of Shandong Province (grant
no. 2019WS335).
Not applicable.
FL designed the study and critically revised the
manuscript. QY wrote the initial draft of the manuscript. LR
performed analysis and interpretation of the underlying data to
draw the figures and authored and revised the figure legends and
the corresponding part of the Methods section. FR participated in
the revision of the manuscript. All authors read and approved the
final version of the manuscript. Data authentication is not
applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Bray F, Laversanne M, Sung H, ME JF,
Siegel RL, Soerjomataram I and DVM AJ: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J. 74:229–263. PubMed/NCBI
|
|
2
|
Mili N, Paschou SA, Goulis DG, Dimopoulos
MA, Lambrinoudaki I and Psaltopoulou T: Obesity, metabolic
syndrome, and cancer: Pathophysiological and therapeutic
associations. Endocrine. 74:478–497. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Henley SJ, Ward E, Scott S, Ma J, Anderson
RN, Firth AU, Thomas CC, Islami F, Weir HK, Lewis DR, et al: Annual
report to the nation on the status of cancer, Part 1: National
cancer statistics. Cancer. 126:2225–2249. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Avgerinos KI, Spyrou N, Mantzoros CS and
Dalamaga M: Obesity and cancer risk: Emerging biological mechanisms
and perspectives. Metabolism. 92:121–135. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Karpel HC, Slomovitz B, Coleman RL and
Pothuri B: Treatment options for molecular subtypes of endometrial
cancer in 2023. Curr Opin Obstet Gynecol. 35:270–278. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Alexa M, Hasenburg A and Battista MJ: The
TCGA molecular classification of endometrial cancer and its
possible impact on adjuvant treatment decisions. Cancers.
13:14782021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vermij L, Smit V, Nout R and Bosse T:
Incorporation of molecular characteristics into endometrial cancer
management. Histopathology. 76:52–63. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Galant N, Krawczyk P, Monist M, Obara A,
Gajek Ł, Grenda A, Nicoś M, Kalinka E and Milanowski J: Molecular
classification of endometrial cancer and its impact on therapy
selection. Int J Mol Sci. 25:58932024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brasseur K, Gévry N and Asselin E:
Chemoresistance and targeted therapies in ovarian and endometrial
cancers. Oncotarget. 8:4008–4042. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wilson EM, Eskander RN and Binder PS:
Recent therapeutic advances in gynecologic oncology: A review.
Cancers. 16:7702024. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Žalytė E: Ferroptosis, metabolic rewiring,
and endometrial cancer. Int J Mol Sci. 25:752023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Al Mamun A, Geng P, Wang S and Shao C:
Role of pyroptosis in endometrial cancer and its therapeutic
regulation. J Inflamm Res. 17:7037–7056. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Devis-Jauregui L, Eritja N, Davis ML,
Matias-Guiu X and Llobet-Navàs D: Autophagy in the physiological
endometrium and cancer. Autophagy. 17:1077–1095. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fukuda T and Wada-Hiraike O: The Two-faced
role of autophagy in endometrial cancer. Front Cell Dev Biol.
10:8394162022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nuñez-Olvera SI, Gallardo-Rincón D,
Puente-Rivera J, Salinas-Vera YM, Marchat LA, Morales-Villegas R
and López-Camarillo C: Autophagy machinery as a promising
therapeutic target in endometrial cancer. Front Oncol. 9:13262019.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu J, Zhang L, Wu S and Liu Z:
Ferroptosis: Opportunities and challenges in treating endometrial
cancer. Front Mol Biosci. 9:9298322022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brand MD: The sites and topology of
mitochondrial superoxide production. Exp Gerontol. 45:466–472.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
West AP, Shadel GS and Ghosh S:
Mitochondria in innate immune responses. Nat Rev Immunol.
11:389–402. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lambeth JD and Neish AS: Nox enzymes and
new thinking on reactive oxygen: A Double-edged sword revisited.
Annu Rev Pathol Mech Dis. 9:119–145. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Y, Qi H, Liu Y, Duan C, Liu X, Xia T,
Chen D, Piao HL and Liu HX: The double-edged roles of ROS in cancer
prevention and therapy. Theranostics. 11:4839–4857. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Perillo B, Di Donato M, Pezone A, Di Zazzo
E, Giovannelli P, Galasso G, Castoria G and Migliaccio A: ROS in
cancer therapy: the bright side of the moon. Exp Mol Med.
52:192–203. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
van der Pol A, van Gilst WH, Voors AA and
van der Meer P: Treating oxidative stress in heart failure: Past,
present and future. Eur J Heart Fail. 21:425–435. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Senoner T and Dichtl W: Oxidative stress
in cardiovascular diseases: Still a therapeutic target? Nutrients.
11:20902019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rezzani R and Franco C: Liver, oxidative
stress and metabolic syndromes. Nutrients. 13:3012021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Teleanu DM, Niculescu AG, Lungu II, Radu
CI, Vladâcenco O, Roza E, Costăchescu B, Grumezescu AM and Teleanu
RI: An overview of oxidative stress, neuroinflammation, and
neurodegenerative diseases. Int J Mol Sci. 23:59382022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang R, Chen H, Liang J, Li Y, Yang J,
Luo C, Tang Y, Ding Y, Liu X, Yuan Q, et al: Dual role of reactive
oxygen species and their application in cancer therapy. J Cancer.
12:5543–5561. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An Iron-dependent form of Non-apoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tang D, Chen X, Kang R and Kroemer G:
Ferroptosis: Molecular mechanisms and health implications. Cell
Res. 31:107–125. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao
N, Sun B and Wang G: Ferroptosis: Past, present and future. Cell
Death Dis. 11:882020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu Y, Shen J and Ran Z: Emerging views of
mitophagy in immunity and autoimmune diseases. Autophagy. 16:3–17.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lu Y, Li Z, Zhang S, Zhang T, Liu Y and
Zhang L: Cellular mitophagy: Mechanism, roles in diseases and small
molecule pharmacological regulation. Theranostics. 13:736–766.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Onishi M, Yamano K, Sato M, Matsuda N and
Okamoto K: Molecular mechanisms and physiological functions of
mitophagy. EMBO J. 40:e1047052021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Czegle I, Huang C, Soria PG, Purkiss DW,
Shields A and Wappler-Guzzetta EA: The role of genetic mutations in
mitochondrial-driven cancer growth in selected tumors: Breast and
gynecological malignancies. Life. 13:9962023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Musicco C, Cormio G, Pesce V, Loizzi V,
Cicinelli E, Resta L, Ranieri G and Cormio A: Mitochondrial
dysfunctions in type I endometrial carcinoma: Exploring their role
in oncogenesis and tumor progression. Int J Mol Sci. 19:20762018.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yıldırım E, Türkler C, Görkem Ü, Şimşek
ÖY, Yılmaz E and Aladağ H: The relationship between oxidative
stress markers and endometrial hyperplasia: A case-control study.
Turk J Obstet Gynecol. 18:298–303. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bukato K, Kostrzewa T, Gammazza AM,
Gorska-Ponikowska M and Sawicki S: Endogenous estrogen metabolites
as oxidative stress mediators and endometrial cancer biomarkers.
Cell Commun Signal. 22:2052024. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gao Y, Sun W, Wang J, Zhao D, Tian H, Qiu
Y, Ji S, Wang S, Fu Q, Zhang F, et al: Oxidative stress induces
ferroptosis in tendon stem cells by regulating mitophagy through
cGAS-STING pathway. Int Immunopharmacol. 138:1126522024. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Granata S, Votrico V, Spadaccino F,
Catalano V, Netti GS, Ranieri E, Stallone G and Zaza G: Oxidative
Stress and Ischemia/reperfusion injury in kidney transplantation:
Focus on ferroptosis, mitophagy and new antioxidants. Antioxidants
(Basel). 11:7692022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jomova K, Alomar SY, Alwasel SH,
Nepovimova E, Kuca K and Valko M: Several lines of antioxidant
defense against oxidative stress: antioxidant enzymes,
nanomaterials with multiple enzyme-mimicking activities, and
low-molecular-weight antioxidants. Arch Toxicol. 98:1323–1367.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial reactive oxygen species (ROS) and ROS-Induced ROS
Release. Physiol Rev. 94:909–950. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Phaniendra A, Jestadi DB and Periyasamy L:
Free Radicals: Properties, sources, targets, and their implication
in various diseases. Indian J Clin Biochem. 30:11–26. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Canli Ö, Nicolas AM, Gupta J, Finkelmeier
F, Goncharova O, Pesic M, Neumann T, Horst D, Löwer M, Sahin U and
Greten FR: Myeloid Cell-derived reactive oxygen species induce
epithelial mutagenesis. Cancer Cell. 32:869–883.e5. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kumari S, Badana AK G MM, G S and Malla R:
Reactive oxygen species: A key constituent in cancer survival.
Biomark Insights. 13:11772719187553912018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Iskandar K, Foo J, Liew AQX, Zhu H, Raman
D, Hirpara JL, Leong YY, Babak MV, Kirsanova AA and Armand AS: A
novel MTORC2-AKT-ROS axis triggers mitofission and
mitophagy-associated execution of colorectal cancer cells upon
drug-induced activation of mutant KRAS. Autophagy. 20:1418–1441.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Srinivas US, Tan BWQ, Vellayappan BA and
Jeyasekharan AD: ROS and the DNA damage response in cancer. Redox
Biol. 25:1010842018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu B, Chen Y and St Clair DK: ROS and
p53: Versatile partnership. Free Radic Biol Med. 44:1529–1535.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang Y, Choksi S, Chen K, Pobezinskaya Y,
Linnoila I and Liu ZG: ROS play a critical role in the
differentiation of alternatively activated macrophages and the
occurrence of Tumor-associated macrophages. Cell Res. 23:898–914.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhong LM, Liu ZG, Zhou X, Song SH, Weng
GY, Wen Y, Liu FB, Cao DL and Liu YF: Expansion of PMN-myeloid
derived suppressor cells and their clinical relevance in patients
with oral squamous cell carcinoma. Oral Oncol. 95:157–163. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jiang J, Wang K, Chen Y, Chen H, Nice EC
and Huang C: Redox regulation in tumor cell epithelial-mesenchymal
transition: molecular basis and therapeutic strategy. Signal
Transduct Target Ther. 2:170362017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Snezhkina AV, Kudryavtseva AV, Kardymon
OL, Savvateeva MV, Melnikova NV, Krasnov GS and Dmitriev AA: ROS
generation and antioxidant defense systems in normal and malignant
cells. Oxid Med Cell Longev. 2019:61758042019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Dey P, Baddour J, Muller F, Wu CC, Wang H,
Liao WT, Lan Z, Chen A, Gutschner T, Kang Y, et al: Genomic
deletion of malic enzyme 2 confers collateral lethality in
pancreatic cancer. Nature. 542:119–123. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang S, Long H, Hou L, Feng B, Ma Z, Wu Y,
Zeng Y, Cai J, Zhang DW and Zhao G: The mitophagy pathway and its
implications in human diseases. Signal Transduct Target Ther.
8:3042023. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Orvedahl A, Sumpter R, Xiao G, Ng A, Zou
Z, Tang Y, Narimatsu M, Gilpin C, Sun Q and Roth M: Image-based
Genome-Wide siRNA screen identifies selective autophagy factors.
Nature. 480:113–117. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Vara-Perez M, Felipe-Abrio B and Agostinis
P: Mitophagy in cancer: A tale of adaptation. Cells. 8:4932019.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ferro F, Servais S, Besson P, Roger S,
Dumas JF and Brisson L: Autophagy and mitophagy in cancer metabolic
remodelling. Semin Cell Dev Biol. 98:129–138. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Peoples JN, Saraf A, Ghazal N, Pham TT and
Kwong JQ: Mitochondrial dysfunction and oxidative stress in heart
disease. Exp Mol Med. 51:1622019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Scheibye-Knudsen M, Fang EF, Croteau DL,
Wilson DM and Bohr VA: Protecting the mitochondrial powerhouse.
Trends Cell Biol. 25:158–170. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wu W, Xu H, Wang Z, Mao Y, Yuan L, Luo W,
Cui Z, Cui T, Wang XL and Shen YH: PINK1-Parkin-Mediated mitophagy
protects mitochondrial integrity and prevents metabolic
stress-induced endothelial injury. PLoS One. 10:e01324992015.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Song C, Pan S, Zhang J, Li N and Geng Q:
Mitophagy: A novel perspective for insighting into cancer and
cancer treatment. Cell Prolif. 55:e133272022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Chen Z, Liu L, Cheng Q, Li Y, Wu H, Zhang
W, Zhang W, Wang Y, Sehgal SA, Siraj S, et al: Mitochondrial E3
ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO
Rep. 18:495–509. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Li J, Agarwal E, Bertolini I, Seo JH,
Caino MC, Ghosh JC, Kossenkov AV, Liu Q, Tang HY, Goldman AR, et
al: The mitophagy effector FUNDC1 controls mitochondrial
reprogramming and cellular plasticity in cancer cells. Sci Signal.
13:eaaz82402020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Poole LP and Macleod KF: Mitophagy in
tumorigenesis and metastasis. Cell Mol Life Sci. 78:3817–3851.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Akabane S, Matsuzaki K, Yamashita S, Arai
K, Okatsu K, Kanki T, Matsuda N and Oka T: Constitutive activation
of PINK1 protein leads to proteasome-mediated and Non-apoptotic
cell death independently of mitochondrial autophagy. J Biol Chem.
291:16162–16174. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Song C, Pan S, Zhang J, Li N and Geng Q:
Mitophagy: A novel perspective for insighting into cancer and
cancer treatment. Cell Prolif. 55:e133272022. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Liu K, Shi Y, Guo XH, Ouyang YB, Wang SS,
Liu DJ, Wang AN, Li N and Chen DX: Phosphorylated AKT inhibits the
apoptosis induced by DRAM-mediated mitophagy in hepatocellular
carcinoma by preventing the translocation of DRAM to mitochondria.
Cell Death Dis. 5:e10782014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Yin K, Lee J, Liu Z, Kim H, Martin DR, Wu
D, Liu M and Xue X: Mitophagy protein PINK1 suppresses colon tumor
growth by metabolic reprogramming via p53 activation and reducing
acetyl-CoA production. Cell Death Differ. 28:2421–2435. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Chen L, Mao LS, Xue JY, Jian YH, Deng ZW,
Mazhar M, Zou Y, Liu P, Chen MT, Luo G and Liu MN: Myocardial
ischemia-reperfusion injury: The balance mechanism between
mitophagy and NLRP3 inflammasome. Life Sci. 355:1229982024.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ikeda H, Kawase K, Nishi T, Watanabe T,
Takenaga K, Inozume T, Ishino T, Aki S, Lin J, Kawashima S, et al:
Immune evasion through mitochondrial transfer in the tumour
microenvironment. Nature. 638:225–236. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wang SF, Tseng LM and Lee HC: Role of
mitochondrial alterations in human cancer progression and cancer
immunity. J Biomed Sci. 30:612023. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Sun R, Zhou X, Wang T, Liu Y, Wei L, Qiu
Z, Qiu C and Jiang J: Novel insights into tumorigenesis and
prognosis of endometrial cancer through systematic investigation
and validation on mitophagy-related signature. Hum Cell.
36:1548–1563. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Zhao MM, Wang B, Huang WX, Zhang L, Peng R
and Wang C: Verteporfin suppressed mitophagy via PINK1/parkin
pathway in endometrial cancer. Am J Cancer Res. 14:1935–1946. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology, and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zhang C, Liu X, Jin S, Chen Y and Guo R:
Ferroptosis in cancer therapy: A novel approach to reversing drug
resistance. Mol Cancer. 21:472022. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Chen X, Li J, Kang R, Klionsky DJ and Tang
D: Ferroptosis: Machinery and regulation. Autophagy. 17:2054–2081.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Pandrangi SL, Chittineedi P, Chikati R,
Lingareddy JR, Nagoor M and Ponnada SK: Role of dietary iron
revisited: In metabolism, ferroptosis and pathophysiology of
cancer. Am J Cancer Res. 12:974–985. 2022.PubMed/NCBI
|
|
76
|
Gao W, Wang X, Zhou Y, Wang X and Yu Y:
Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor
immunotherapy. Signal Transduct Target Ther. 7:1962022. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Dai E, Chen X, Linkermann A, Jiang X, Kang
R, Kagan VE, Bayir H, Yang WS, Garcia-Saez AJ, Ioannou MS, et al: A
guideline on the molecular ecosystem regulating ferroptosis. Nat
Cell Biol. 26:1447–1547. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Mou Y, Wang J, Wu J, He D, Zhang C, Duan C
and Li B: Ferroptosis, a new form of cell death: opportunities and
challenges in cancer. J Hematol Oncol. 12:342019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zhang M, Zhang T, Song C, Qu J, Gu Y, Liu
S, Li H, Xiao W, Kong L, Sun Y and Lv W: Guizhi Fuling Capsule
ameliorates endometrial hyperplasia through promoting
p62-Keap1-NRF2-mediated ferroptosis. J Ethnopharmacol.
274:1140642021. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
López-Janeiro Á, Ruz-Caracuel I,
Ramón-Patino JL, De Los Ríos V, Villalba Esparza M, Berjón A,
Yébenes L, Hernández A, Masetto I, Kadioglu E, et al: Proteomic
analysis of Low-grade, early-stage endometrial carcinoma reveals
new dysregulated pathways associated with cell death and cell
signaling. Cancers (Basel). 13:7942021. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Tang S and Chen L: The recent advancements
of ferroptosis of gynecological cancer. Cancer Cell Int.
24:3512024. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Huang JY and Yu HN: The role of the Nrf2
pathway in inhibiting ferroptosis in kidney disease and its future
prospects. Pathol Res Pract. 272:1560842025. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Doll S, Proneth B, Tyurina YY, Panzilius
E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A,
et al: Acsl4 dictates ferroptosis sensitivity by shaping cellular
lipid composition. Nat Chem Biol. 13:91–98. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Rosenblum SL: Inflammation, dysregulated
iron metabolism, and cardiovascular disease. Front Aging.
4:11241782023. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Wei S, Yu Z, Shi R, An L, Zhang Q, Zhang
Q, Zhang T, Zhang J and Wang H: GPX4 suppresses ferroptosis to
promote malignant progression of endometrial carcinoma via
transcriptional activation by ELK1. BMC Cancer. 22:8812022.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Hirayama T, Nagata Y, Nishida M, Matsuo M,
Kobayashi S, Yoneda A, Kanetaka K, Udono H and Eguchi S: Metformin
prevents peritoneal dissemination via Immune-suppressive cells in
the tumor microenvironment. Anticancer Res. 39:4699–4709. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Zhao Y, Wang Y, Zhang X, Han S and Yang B:
Metformin-induced RBMS3 expression enhances ferroptosis and
suppresses ovarian cancer progression. Reprod Biol. 25:1009682025.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
van den Heerik ASVM, Horeweg N, de Boer
SM, Bosse T and Creutzberg CL: Adjuvant therapy for endometrial
cancer in the era of molecular classification: Radiotherapy,
chemoradiation and novel targets for therapy. Int J Gynecol Cancer.
31:594–604. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Liu Q, Zhao Y, Zhou H and Chen C:
Ferroptosis: Challenges and opportunities for nanomaterials in
cancer therapy. Regen Biomater. 10:rbad0042023. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Sotomayor-Flores C, Rivera-Mejías P,
Vásquez-Trincado C, López-Crisosto C, Morales PE, Pennanen C,
Polakovicova I, Aliaga-Tobar V, García L, Roa JC, et al:
Angiotensin-(1–9) prevents cardiomyocyte hypertrophy by controlling
mitochondrial dynamics via miR-129-3p/PKIA pathway. Cell Death
Differ. 27:2586–2604. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Yang WS, SriRamaratnam R, Welsch ME,
Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji
AF, Clish CB, et al: Regulation of ferroptotic cancer cell death by
GPX4. Cell. 156:317–33. 20141 View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Egan DF, Shackelford DB, Mihaylova MM,
Gelino SR, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor
R, et al: Phosphorylation of ULK1 (hATG1) by AMP-activated protein
kinase connects energy sensing to mitophagy. Science. 331:456–461.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Zhao L, Zhou X, Xie F and Zhang L, Yan H,
Huang J, Zhang C, Zhou F, Chen J and Zhang L: Ferroptosis in cancer
and cancer immunotherapy. Cancer Commun. 42:88–116. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Cormio A, Musicco C, Gasparre G, Cormio G,
Pesce V, Sardanelli AM and Gadaleta MN: Increase in proteins
involved in mitochondrial fission, mitophagy, proteolysis and
antioxidant response in type I endometrial cancer as an adaptive
response to respiratory complex I deficiency. Biochem Biophys Res
Commun. 491:85–90. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Liu C, Li Z, Li B, Liu W, Zhang S, Qiu K
and Zhu W: Relationship between ferroptosis and mitophagy in
cardiac ischemia reperfusion injury: A mini-review. PeerJ.
11:e149522023. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Bi Y, Liu S, Qin X, Abudureyimu M, Wang L,
Zou R, Ajoolabady A, Zhang W, Peng H, Ren J and Zhang Y: FUNDC1
interacts with GPx4 to govern hepatic ferroptosis and fibrotic
injury through a mitophagy-dependent manner. J Adv Res. 55:45–60.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Wilhelm LP, Zapata-Muñoz J, Villarejo-Zori
B, Pellegrin S, Freire CM, Toye AM, Boya P and Ganley IG:
BNIP3L/NIX regulates both mitophagy and pexophagy. EMBO J.
41:e1111152022. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Yamashita SI, Sugiura Y, Matsuoka Y, Maeda
R, Inoue K, Furukawa K, Fukuda T, Chan DC and Kanki T: Mitophagy
mediated by BNIP3 and NIX protects against ferroptosis by
downregulating mitochondrial reactive oxygen species. Cell Death
Differ. 31:651–661. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Yang CH, Almomen A, Wee YS, Jarboe EA,
Peterson CM and Janát-Amsbury MM: An estrogen-induced endometrial
hyperplasia mouse model recapitulating human disease progression
and genetic aberrations. Cancer Med. 4:1039–1050. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Fan R, Wang Y, Wang Y, Wei L and Zheng W:
Mechanism of progestin resistance in endometrial precancer/cancer
through Nrf2-survivin pathway. Am J Transl Res. 9:1483–1491.
2017.PubMed/NCBI
|
|
101
|
Yi J, Zhu J, Wu J, Thompson CB and Jiang
X: Oncogenic activation of PI3K-AKT-mTOR signaling suppresses
ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci USA.
117:31189–31197. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Álvarez-Garcia V, Tawil Y, Wise HM and
Leslie NR: Mechanisms of PTEN loss in cancer: It's all about
diversity. Semin Cancer Biol. 59:66–79. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Nero C, Ciccarone F, Pietragalla A and
Scambia G: PTEN and gynecological cancers. Cancers. 11:14582019.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Wang P, Zhang XP, Liu F and Wang W:
Progressive Deactivation of Hydroxylases Controls Hypoxia-Inducible
Factor-1α-Coordinated Cellular Adaptation to Graded Hypoxia.
Research. 8:06512025. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Wang P, Dai X, Jiang W, Li Y and Wei W:
RBR E3 ubiquitin ligases in tumorigenesis. Semin Cancer Biol.
67:131–144. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Zhang HL, Hu BX, Li ZL, Du T, Shan JL, Ye
ZP, Peng XD, Li X, Huang Y, Zhu XY, et al: PKCβII phosphorylates
ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nature
Cell Biol. 24:88–98. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Jaramillo MC and Zhang DD: The emerging
role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev.
27:2179–2191. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Yang B, Hu M, Fu Y, Sun D, Zheng W, Liao
H, Zhang Z and Chen X: LASS2 mediates Nrf2-driven progestin
resistance in endometrial cancer. Am J Transl Res. 13:1280–1289.
2021.PubMed/NCBI
|
|
109
|
Read AD, Bentley RET, Archer SL and
Dunham-Snary KJ: Mitochondrial iron-sulfur clusters: Structure,
function, and an emerging role in vascular biology. Redox Biol.
47:1021642021. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Luo J, Zhang X, Liang Z, Zhuang W, Jiang
M, Ma M, Peng S, Huang S, Qiao G and Chen Q: ISCU-p53 axis
orchestrates macrophage polarization to dictate immunotherapy
response in esophageal squamous cell carcinoma. Cell Death Dis.
16:4622025. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Yang H, Yao X, Liu Y, Shen X, Li M and Luo
Z: Ferroptosis Nanomedicine: Clinical Challenges and Opportunities
for Modulating Tumor Metabolic and Immunological Landscape. ACS
Nano. 17:15328–15353. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Glorieux C, Liu S, Trachootham D and Huang
P: Targeting ROS in cancer: Rationale and strategies. Nature
Reviews Drug Discovery. 23:583–606. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
McKee CA, Polino AJ, King MW and Musiek
ES: Circadian clock protein BMAL1 broadly influences autophagy and
endolysosomal function in astrocytes. Proc Natl Acad Sci USA.
120:e22205511202025. View Article : Google Scholar
|
|
114
|
Makovec T: Cisplatin and beyond: Molecular
mechanisms of action and drug resistance development in cancer
chemotherapy. Radiol Oncol. 53:148–1458. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Mauro-Lizcano M, Sotgia F and Lisanti MP:
Mitophagy and cancer: Role of BNIP3/BNIP3L as energetic drivers of
stemness features, ATP production, proliferation, and cell
migration. Aging (Albany NY). 16:9334–9349. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Li K, Xu K, He Y, Yang Y, Tan M, Mao Y,
Zou Y, Feng Q, Luo Z and Cai K: Oxygen Self-generating nanoreactor
mediated ferroptosis activation and immunotherapy in
Triple-negative breast cancer. ACS Nano. 17:4667–4687. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Xu L, Fang Q, Miao Y, Xu M, Wang Y, Sun L
and Jia X: The role of CCR2 in prognosis of patients with
endometrial cancer and tumor microenvironment remodeling.
Bioengineered. 12:3467–3484. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Nizami ZN, Aburawi HE, Semlali A, Muhammad
K and Iratni R: Oxidative stress inducers in cancer therapy:
Preclinical and clinical evidence. Antioxidants. 12:11592023.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Li J, Jia Y Chen, Ding Y Xuan, Bai J, Cao
F and Li F: The crosstalk between ferroptosis and mitochondrial
dynamic regulatory networks. Int J Biol Sci. 19:2756–2771. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
CD8+ T cells and fatty acids orchestrate
tumor ferroptosis and immunity via ACSL4-PMC [Internet], . [cited
2025 Sept 10]. Available from. https://pmc.ncbi.nlm.nih.gov/articles/PMC9007863/
|
|
122
|
Huo K, Yang Y, Yang T, Zhang W and Shao J:
Identification of drug targets and agents associated with
ferroptosis-related osteoporosis through integrated network
pharmacology and molecular docking technology. Curr Pharm Des.
30:1103–1114. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Hu G, Yuan Z and Wang J: Autophagy
inhibition and ferroptosis activation during atherosclerosis:
Hypoxia-inducible factor 1α inhibitor PX-478 alleviates
atherosclerosis by inducing autophagy and suppressing ferroptosis
in macrophages. Biomed Pharmacother. 161:1143332023. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Bhatt V, Lan T, Wang W, Kong J, Lopes EC,
Wang J, Khayati K, Raju A, Rangel M, Lopez E, et al: Inhibition of
autophagy and MEK promotes ferroptosis in Lkb1-deficient
Kras-driven lung tumors. Cell Death Dis. 14:612023. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Crouigneau R, Li YF, Auxillos J,
Goncalves-Alves E, Marie R, Sandelin A and Pedersen SF: Mimicking
and analyzing the tumor microenvironment. Cell Rep Methods.
4:1008662024. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Early JO, Menon D, Wyse CA,
Cervantes-Silva MP, Zaslona Z, Carroll RG, Palsson-McDermott EM,
Angiari S, Ryan DG, Corcoran SE, et al: Circadian clock protein
BMAL1 regulates IL-1β in macrophages via NRF2. Proc Natl Acad Sci
USA. 115:E8460–E8468. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Wang Z, Ge S, Liao T, Yuan M, Qian W, Chen
Q, Liang W, Cheng X, Zhou Q, Ju Z, et al: Integrative single-cell
metabolomics and phenotypic profiling reveals metabolic
heterogeneity of cellular oxidation and senescence. Nat Commun.
16:27402025. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Doll S, Freitas FP, Shah R, Aldrovandi M,
da Silva MC, Ingold I, Grocin AG, da Silva TNX, Panzilius E, Scheel
CH, et al: FSP1 is a glutathione-independent ferroptosis
suppressor. Nature. 575:693–698. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Bersuker K, Hendricks J, Li Z, Magtanong
L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al:
The CoQ oxidoreductase FSP1 acts in parallel to GPX4 to inhibit
ferroptosis. Nature. 575:688–692. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Pekovic-Vaughan V, Gibbs J, Yoshitane H,
Yang N, Pathiranage D, Guo B, Sagami A, Taguchi K, Bechtold D,
Loudon A, et al: The circadian clock regulates rhythmic activation
of the NRF2/glutathione-mediated antioxidant defense pathway to
modulate pulmonary fibrosis. Genes Dev. 28:548–560. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee
H, Koppula P, Wu S, Zhuang L, Fang B, et al: DHODH-mediated
ferroptosis defense is a targetable vulnerability in cancer.
Nature. 593:586–590. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Oh M, Jang SY, Lee JY, Kim JW, Jung Y, Kim
J, Seo J, Han TS, Jang E, Son HY, et al: The lipoprotein-associated
phospholipase A2 inhibitor Darapladib sensitises cancer cells to
ferroptosis by remodelling lipid metabolism. Nat Commun.
14:57282023. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Błachnio-Zabielska AU, Sadowska P,
Zdrodowski M, Laudański P and Szamatowicz J and Szamatowicz J: The
Interplay between Oxidative Stress and Sphingolipid Metabolism in
Endometrial Cancer. Int J Mol Sci. 25:102432024. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Hsin IL, Shen HP, Chang HY, Ko JL and Wang
PH: Suppression of PI3K/Akt/mTOR/c-Myc/mtp53 Positive Feedback Loop
Induces Cell Cycle Arrest by Dual PI3K/mTOR Inhibitor PQR309 in
Endometrial Cancer Cell Lines. Cells. 10:29162021. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Ruiz-Mitjana A, Vidal-Sabanés M, Navaridas
R, Perramon-Güell A, Yeramian A, Nicholson-Sabaté N, Egea J,
Encinas M, Matias-Guiu X and Dolcet X: Metformin exhibits
antineoplastic effects on Pten-deficient endometrial cancer by
interfering with TGF-β and p38/ERK MAPK signalling. Biomed
Pharmacother. 168:1158172023. View Article : Google Scholar : PubMed/NCBI
|