Introduction
Colorectal cancer (CRC) is a major cause of
cancer-associated mortality worldwide, In 2023, an estimated 1.97
million new cases of CRC and ~0.93 million deaths were reported
globally (1). The burden of CRC is
increasing in Eastern Asia, the Middle East, and parts of Southeast
Asia, where rapid lifestyle transitions, Westernized diets, and
limited early-screening coverage have contributed to rising
incidence trends (2). In Asia,
~992,000 new CRC cases and ~498,000 related deaths were reported in
2020 (3). Proteomic and genomic
studies have advanced cancer research by revealing molecular
pathways involved in CRC particularly through dysregulated
signaling cascades that drive tumor development and progression,
such as the Wnt/β-catenin, PI3K/AKT signaling, MAPK/ERK pathway,
and alterations in TP53-regulated stress response networks, all of
which play key roles in cell proliferation, survival, and
metastasis (4). Among key molecular
players, claudin-2 (CLDN2), a component of tight junctions,
has emerged as a contributor to malignancy by exhibiting aberrant
expression (5).
The claudin family serves a key role in cell
barriers, differentiation and proliferation. The expression
patterns of claudin knockout (KO) vary throughout malignancies and
organs (6). Thus, claudins have
been proposed as targets for cancer treatment as well as diagnostic
indicators. Previous research has demonstrated an increasing
consensus on the potential value of CLDN2 as a biomarker for
prognostic and therapeutic features in CRC (7–9).
Notably, CLDN2 expression is elevated in inflammatory bowel
disease and colorectal tumors compared with normal tissues
(10).
CLDN2 has diverse biological functions beyond
maintaining epithelial permeability. It contributes to paracellular
water and ion transport, modulates epithelial proliferation and
participates in signaling events associated with oncogenic
transformation (8,11). Elevated CLDN2 levels have
been associated with poor prognosis, advanced tumor stages and
increased metastatic potential in several cancer types, including
CRC (7,12). These findings highlight the
importance of understanding the multifunctional role of
CLDN2 in colorectal carcinogenesis. CLDN2 is the most
distinct member of the claudin family and exhibits a unique
expression pattern because its expression is limited to permeable
epithelial tissues (11). In colon
cancer, CLDN2 appears to function as an oncogene, enhancing
cell proliferation and migration capacity through EGFR-mediated
pathways (12). Notably, the
increasing incidence of CRC has been connected to the presence of
CLDN2 in cellular tight junctions.
Mechanistically, it has been demonstrated that
CLDN2 inhibits the transcription of N-Myc
downstream-regulated gene 1 (NDRG1), a well-established metastasis
suppressor gene. This repression is mediated through the
recruitment of zonula occludens-1 (ZO-1)-associated nucleic
acid binding protein (ZONAB) to the NDRG1 promoter,
where CLDN2 facilitates ZONAB binding and suppresses
NDRG1 transcriptional activity (13).
Elevated CLDN2 expression has been observed
in CRC compared with adjacent normal tissue and is associated with
shorter cancer-specific survival and increased risk of recurrence
in stage II/III CRC receiving adjuvant therapy (14). Although environmental and genetic
variables can affect CRC, such as diet, obesity, and smoking,
understanding the mechanisms by which CLDN2 functions during
CRC development could help identify potential novel treatment
options in the future. High CLDN2 expression patterns
reported shorter survival outcomes, indicating a connection between
high CLDN2 expression and CRC progression (13).
While genomic analyses have identified driver
mutations in CRC, understanding the mechanisms by which specific
proteins such as CLDN2 integrate into oncogenic signaling
networks remains challenging (15–17).
CRISPR/Cas9-based gene editing offers a key tool to dissect
functional roles by enabling precise gene KO models (18). Furthermore, combining CRISPR
approaches with gene expression profiling allows for further
investigation into CRC biology and the identification of potential
novel therapeutic targets in the future (19). Due to the limited mechanistic
understanding of CLDN2 in CRC, the present study aimed to
investigate the effects of CLDN2 deletion on gene expression
patterns associated with migration, invasion and metastasis. A
CRISPR/Cas9-mediated KO approach was used in HCT116 cells to assess
both phenotypic changes and transcriptional alterations, with a
focus on pathways implicated in CRC progression.
Materials and methods
Cell culture
The human CRC cell line HCT116 (cat. no. CCL-247;
American Type Culture Collection) was obtained from Synthego. The
HCT116 cell line is a widely used model of CRC repair deficiency
due to a mutation in the mutL homolog 1 gene. Cells were
authenticated by short tandem repeat profiling, and were confirmed
to be mycoplasma-free prior to use.
Wild-type (Wt) and CLDN2-KO HCT116 cells were
cultured in McCoy's 5A medium (cat. no. 16600082; Thermo Fisher
Scientific, Inc.) supplemented with 10% FBS (cat. no. 10270106;
Thermo Fisher Scientific, Inc.) and 1% penicillin-streptomycin
(cat. no. 15140-122; Thermo Fisher Scientific, Inc.). The cells
were maintained at 37°C in a humidified incubator with 5%
CO2.
Generation of CRISPR/Cas9-mediated
CLDN2-KO cells
CRISPR/Cas9 mediated KO of human CLDN2 (gene
ID, 9075) in HCT116 cells was produced by Synthego. Guide RNA
design and off-target prediction were performed using the Synthego
CRISPR Design Tool (v1.2; Synthego). Ribonucleoprotein complexes
consisting of Cas9 protein and a synthetic, chemically modified
single-guide RNA (sgRNA) targeting exon 2 of human CLDN2
(sgRNA sequence, 5′-GGUGCUAUAGAUGUCACACU-3′) were electroporated
into HCT116 cells. The sgRNA cut site was chrX:106,928,419.
Following electroporation (20),
single-cell clones were generated by fluorescence-activated cell
sorting into 96- or 384-well plates and expanded (21). The assessment of editing efficiency
was performed 48 h after electroporation. The process involved
extracting genomic DNA (gDNA) from a subset of cells, followed by
PCR amplification and sequencing using the Sanger sequencing
method. Sequencing was performed in the forward direction using a
20-nt primer, generating 700–900 nt reads. Sanger sequencing
services were provided by Synthego. PCR primers were as follows:
Forward, 5′-CAGCCTGAAGACAAGGGAGC-3′; and reverse,
5′-TGTCTTTGGCTCGGGATTCC-3′. The sequencing primer used was as
follows: 5′-CAGCCTGAAGACAAGGGAGC-3′. The chromatograms obtained
were analyzed using the Synthego Inference of CRISPR edits (ICE;
version 2.0 (Synthego) (22).
For insertion-deletion (indel), the indel
examination was performed using the Illumina sequencing platform
(Illumina, Inc.), according to the manufacturer's protocol
(23). A total of 20 ng gDNA was
utilized as a template for amplifying the region surrounding the
sgRNA target site, using the specified primers. To generate
monoclonal cell populations, cell pools with altered CLDN2
gene (CLDN2-KO) were distributed at a density of 1 cell/well
using a single cell printer onto either 96- or 384-well plates
(24). Every 3 days, all wells were
recorded to verify the growth of a clone originating from a single
cell. The PCR-Sanger-ICE genotyping technique was used to screen
and identify clonal populations.
Wound healing assay
The migratory capacity of Wt and CLDN2-KO
cells was assessed using a wound healing assay (25). Briefly, 3×105 cells/well
were seeded into 6-well plates and cultured until they reached 90%
confluence. After which, a sterile pipette tip was used to scratch
across the center of each well, creating a defined wound area.
Detached cells were removed by washing with 500 µl PBS and fresh
culture medium was added. Subsequently, cell migration into the
scratched area was monitored and captured at 0 and 24 h using a
Nikon E 600 phase-contrast microscope. Images of the wound area
were analyzed using ImageJ software (version 2.9.0/1.53t; National
Institutes of Health) (26). Wound
closure percentage was calculated as
[(At=0-At=∆t)/At=0] ×100, where
At=0 is the scratch area at 0 h and At=∆t is
the scratch area at 24 h.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
RNA was isolated in accordance with the
manufacturer's protocol using an RNA extraction kit (cat. no.
R1200; Beijing Solarbio Science & Technology Co., Ltd.).
Subsequently, cDNA was synthesized using random nonamer primers and
the First-Strand Synthesis System (MilliporeSigma; Merck KGaA).
EvaGreen fluorescence-based RT-qPCR was performed using reagents
purchased from Applied Biological Materials, Inc. RT-qPCR reactions
were performed following standard protocols as previously described
(27). The RT-qPCR primers used in
the present study are listed in Table
I. The gene expression levels in the samples were investigated
using RT-qPCR and the cDNA synthesis and PCR procedures were
optimized to ensure high-quality results.
 | Table I.Primers for RT-qPCR used in the
present study. |
Table I.
Primers for RT-qPCR used in the
present study.
| Gene symbol | Oligo sequence
(5′-3′) |
|---|
| AF-6
(AFDN) | F:
AGTCGGTTGTGAAAGGAGGTGC |
|
| R:
TCCTGAGAGAGTCCAACCAGAC |
| APC | F:
AGGCTGCATGAGAGCACTTGTG |
|
| R:
CACACTTCCAACTTCTCGCAACG |
| Bax | F:
TCAGGATGCGTCCACCAAGAAG |
|
| R:
TGTGTCCACGGCGGCAATCATC |
| Bcl-2 | F:
ATCGCCCTGTGGATGACTGAGT |
|
| R:
GCCAGGAGAAATCAAACAGAGGC |
| Bcl-6 | F:
CATGCAGAGATGTGCCTCCACA |
|
| R:
TCAGAGAAGCGGCAGTCACACT |
| Caspase
3 | F:
GGAAGCGAATCAATGGACTCTGG |
|
| R:
GCATCGACATCTGTACCAGACC |
| CDK4 | F:
CCATCAGCACAGTTCGTGAGGT |
|
| R:
TCAGTTCGGGATGTGGCACAGA |
| CLDN2 | F:
GTGACAGCAGTTGGCTTCTCCA |
|
| R:
GGAGATTGCACTGGATGTCACC |
| CLDN14 | F:
CCAAGACCACCTTTGCCATCCT |
|
| R:
AGTTCTGCACCACGTCGTTGGT |
| CLDN23 | F:
CCTGGTGCGACGAGCGTTGTC |
|
| R:
GTCGCTGTAGTACTTGATGGCG |
| IL-6 | F:
AGACAGCCACTCACCTCTTCAG |
|
| R:
TTCTGCCAGTGCCTCTTTGCTG |
| MMP7 | F:
TCGGAGGAGATGCTCACTTCGA |
|
| R:
GGATCAGAGGAATGTCCCATACC |
| NDRG1 | F:
ATCACCCAGCACTTTGCCGTCT |
|
| R:
GACTCCAGGAAGCATTTCAGCC |
| p53 | F:
CCTCAGCATCTTATCCGAGTGG |
|
| R:
TGGATGGTGGTACAGTCAGAGC |
| PTMS | F:
AGAAACTGCCGAGGATGGAGAG |
|
| R:
TGCCGTTTGGGATCCGCTTCAT |
| TCN1 | F:
CAGTGTGATGGAGAAAGCCCAG |
|
| R:
CCACTCAGAAGTTCCCAGTAGG |
| VDR | F:
CGCATCATTGCCATACTGCTGG |
|
| R:
CCACCATCATTCACACGAACTGG |
| ZO-1
(TJP1) | F:
GTCCAGAATCTCGGAAAAGTGCC |
|
| R:
CTTTCAGCGCACCATACCAACC |
| ZONAB
(YBX3) | F:
TGGTCCAAACCAGCCGTCTGTT |
|
| R:
GTTCTCAGTTGGTGCTTCACCTG |
| GAPDH
(RG) | F:
GTCTCCTCTGACTTCAACAGCG |
|
| R:
ACCACCCTGTTGCTGTAGCCAA |
Relative and normalized fold
expression values calculation
The gene expression patterns of several target genes
were examined in samples from both Wt and CLDN2-KO cells
using RT-qPCR. Relative expression levels were calculated using the
comparative Cq method (ΔΔCq), normalized to GAPDH as the reference
gene (28). For every target gene,
the Cq values that were derived from the amplification curves were
used to compute ΔCq, copy number, ΔΔCq and fold change. ΔCq was
calculated using the following formula: ΔCq=Cq(target gene;
same sample)-Cq(control gene; same sample). Copy
number was calculated using the following formula: Copy number=100
× 2−ΔCq. ΔΔCq was calculated using the following
formula: ΔΔCq=ΔCq(same gene; target
sample)-ΔCq(same gene; control sample). Fold
change was calculated using the following formula: Fold
change=2−ΔΔCq.
The housekeeping gene GAPDH was used as an
internal control for normalization. The expression levels of
GAPDH were relatively stable across all samples. Data are
represented as the mean of three independent experiments.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism software (version 8.0; Dotmatics). Data are presented as the
mean ± SEM, with at least three independent biological replicates.
Normality was assessed using the Shapiro-Wilk test. Statistical
significance was assessed using Welch's unpaired t-test for ΔCq
values Comparisons between two groups (Wt vs. CLDN2-KO) were
made using an unpaired two-tailed Student's t-test if data were
normally distributed or a Mann-Whitney U test for non-parametric
comparisons. P<0.05 was considered to indicate a statistically
significant difference.
Results
High efficiency of CRISPR/Cas9 editing
of the CLDN2 gene
The Synthego ICE CRISPR Analysis tool was employed
to assess the effectiveness of CRISPR CLDN2 gene KO
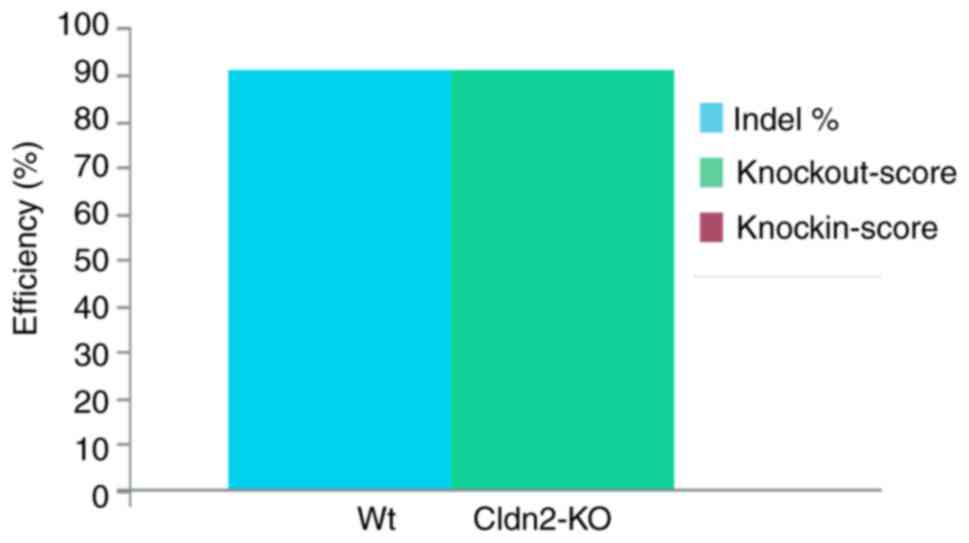
(Fig. 1). The ICE value of 91%
indicated a high degree of editing efficiency, suggesting that a
notable proportion of the cells within the edited population
harbored the intended CLDN2 gene KO. Particularly, 91% of
cells contained the indel of the CLDN2 gene after
CRISPR/Cas9 editing.
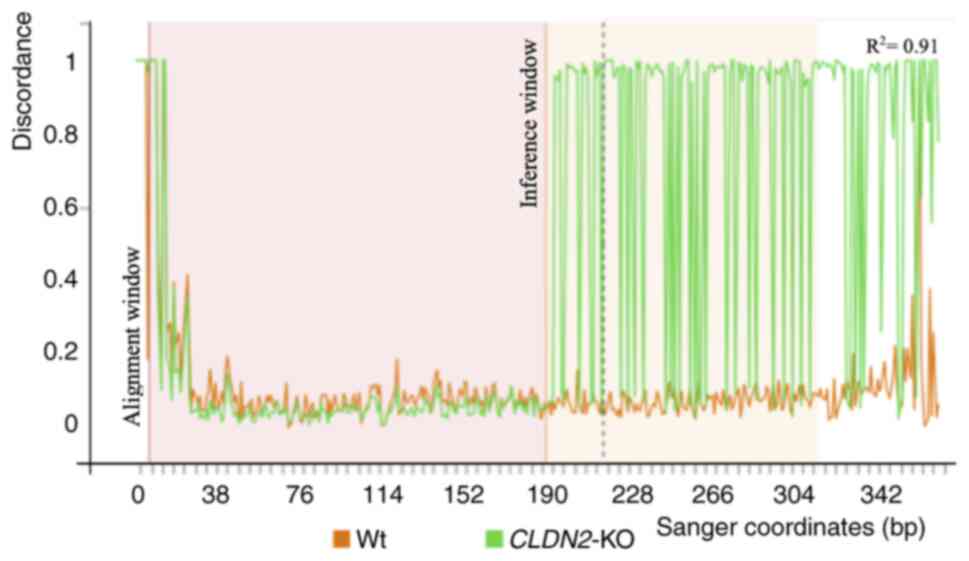
Fig. 2 presents the
Sanger sequencing discordance plot, comparing sequencing traces
from Wt and CLDN2-KO cells around the CRISPR cut site. Prior
to the cut site, the Wt and CLDN2-KO traces overlapped. At
the target site, a sharp increase in sequence discordance was
observed in CLDN2-KO cells, evidenced by the divergence
between Wt (green) and CLDN2-KO (orange) signal lines. This
pattern is characteristic of successful genome editing.
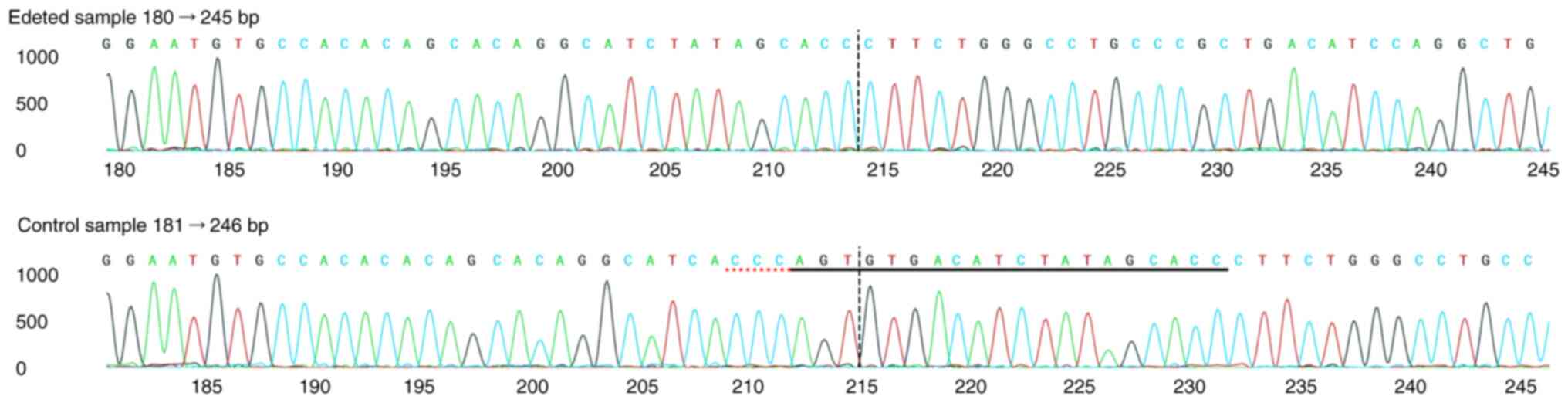
Furthermore, Fig. 3 displays the
Sanger sequence alignment surrounding the sgRNA target site, with
the sgRNA sequence underlined in black and the protospacer adjacent
motif underlined in red. A vertical line marks the cut site, where
sequence differences between Wt and CLDN2-KO cells become
evident. These results confirmed that the CLDN2 gene was
effectively knocked out in HCT116 cells with high efficiency.
Consistent with the sequencing results, qPCR analysis demonstrated
a significant reduction in CLDN2 expression in
CLDN2-KO cells relative to Wt cells (Fig. S1).
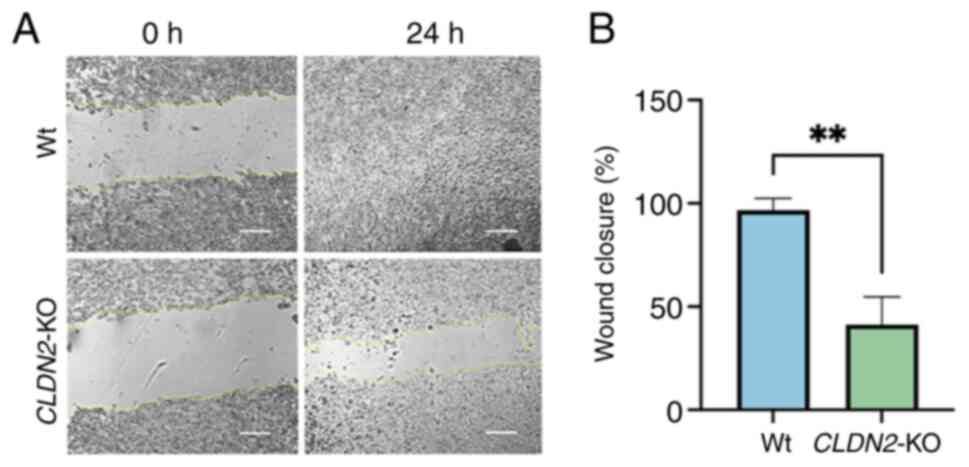
CLDN2-KO impairs HCT116 cell
migration
At 24 h, Wt cells exhibited near-complete wound
closure (~96%), while CLDN2-KO cells achieved only ~41%
closure. Quantification of wound healing (Fig. 4B) demonstrated a significant
reduction in migratory capacity in CLDN2-KO cells compared
with Wt controls (P=0.0027; unpaired t-test; n=3 independent
experiments). These findings indicated that CLDN2 is key for
the efficient migration of CRC cells.
Gene expression profiles in Wt and
CLDN2-KO samples
To investigate the transcriptional effects of
CLDN2 deletion, the expression levels of multiple genes
implicated in invasion and metastasis was analyzed using RT-qPCR.
Fig. 5 displays the copy numbers of
target genes in Wt and CLDN2-KO cells. Analysis of copy
number variations for all genes consistently demonstrated a
significant decrease in copy number in CLDN2-KO samples
compared with the Wt cells (all P<0.05). The observed patterns
indicated that CLDN2-KO has a notable impact on its
interacting partners, causing their downregulation.
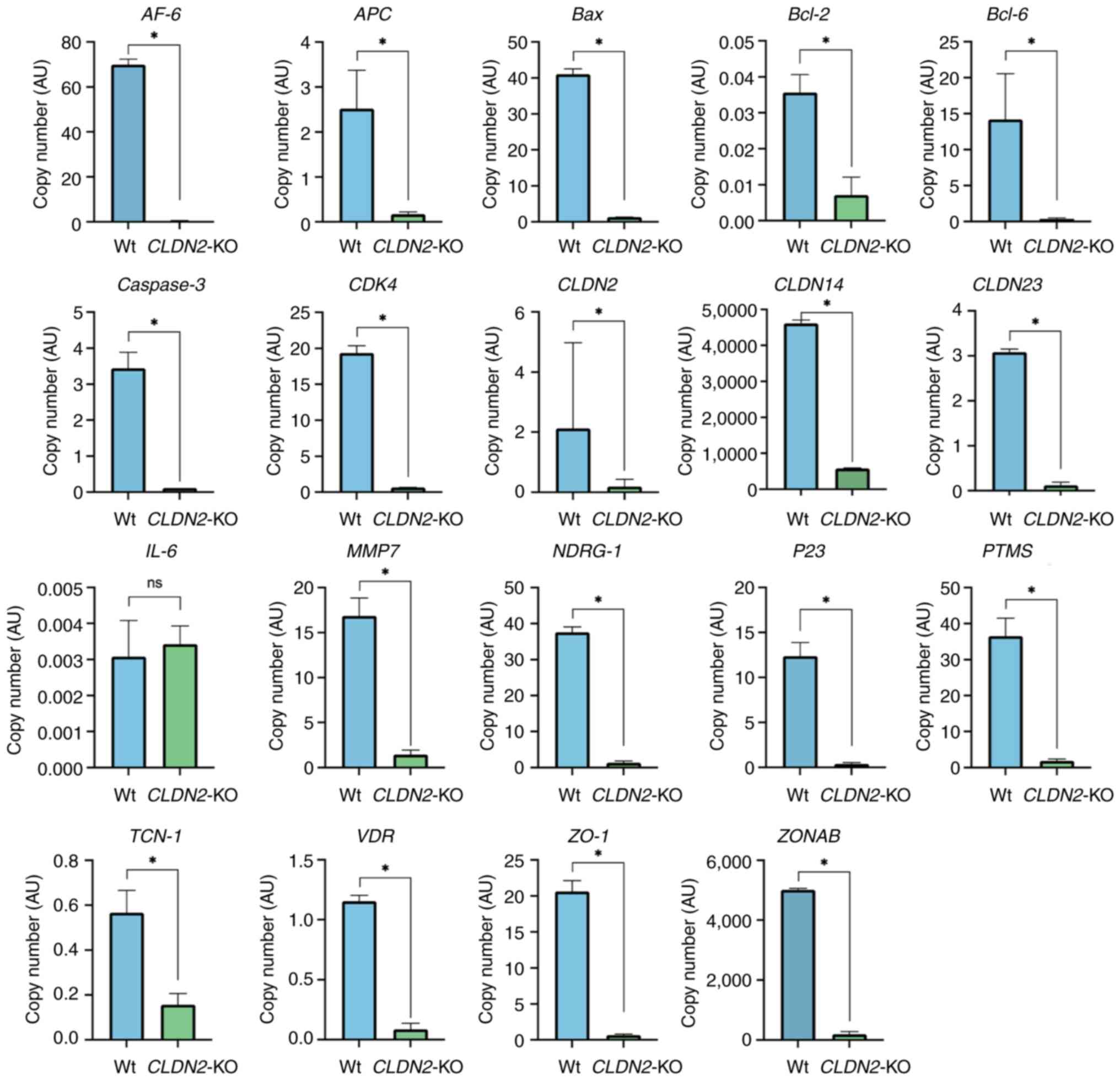 | Figure 5.Gene expression analysis of invasion-
and metastasis-related genes in CLDN2-KO vs. Wt HCT116
cells. Reverse transcription-quantitative PCR exhibited significant
downregulation of multiple target genes, including ZONAB, NDRG1,
CLDN14, CLDN23, Bcl-2, p53 and Bcl-6. Gene expression
levels were normalized to GAPDH. Data are represented as mean ± SEM
(n=3). Statistical comparisons were made using unpaired two-tailed
t-tests. *P<0.05. ns, not significant; Wt, wild-type;
CLDN2-KO, claudin-2 knock out; ZO-1, zonula occludens-1;
VDR, vitamin D receptor; ZONAB, ZO-1-associated
nucleic acid binding protein; NDRG1, N-Myc downstream-regulated
gene 1; APC, adenomatous polyposis coli; AF-6/AFDN, Afadin;
TJP1, tight junction protein 1; YBX3, Y-box binding protein 3;
PTMS, parathymosin; TCN-1, transcobalamin 1. |
Fig. 6 depicts the
fold-change values of several genes in both Wt and CLDN2-KO
samples. Of all the genes that were evaluated, IL-6 was the
least downregulated in CLDN2-KO samples, with a fold-change
of 0.718 and maintains a relatively stable expression pattern. By
contrast, AF-6, which encodes Afadin, exhibited the most
marked downregulation, with a fold-change of 0.008. These results
suggested that CLDN2 loss disrupts a network of
pro-metastatic gene expression programs that may contribute to
impaired cellular migration and reduced metastatic potential.
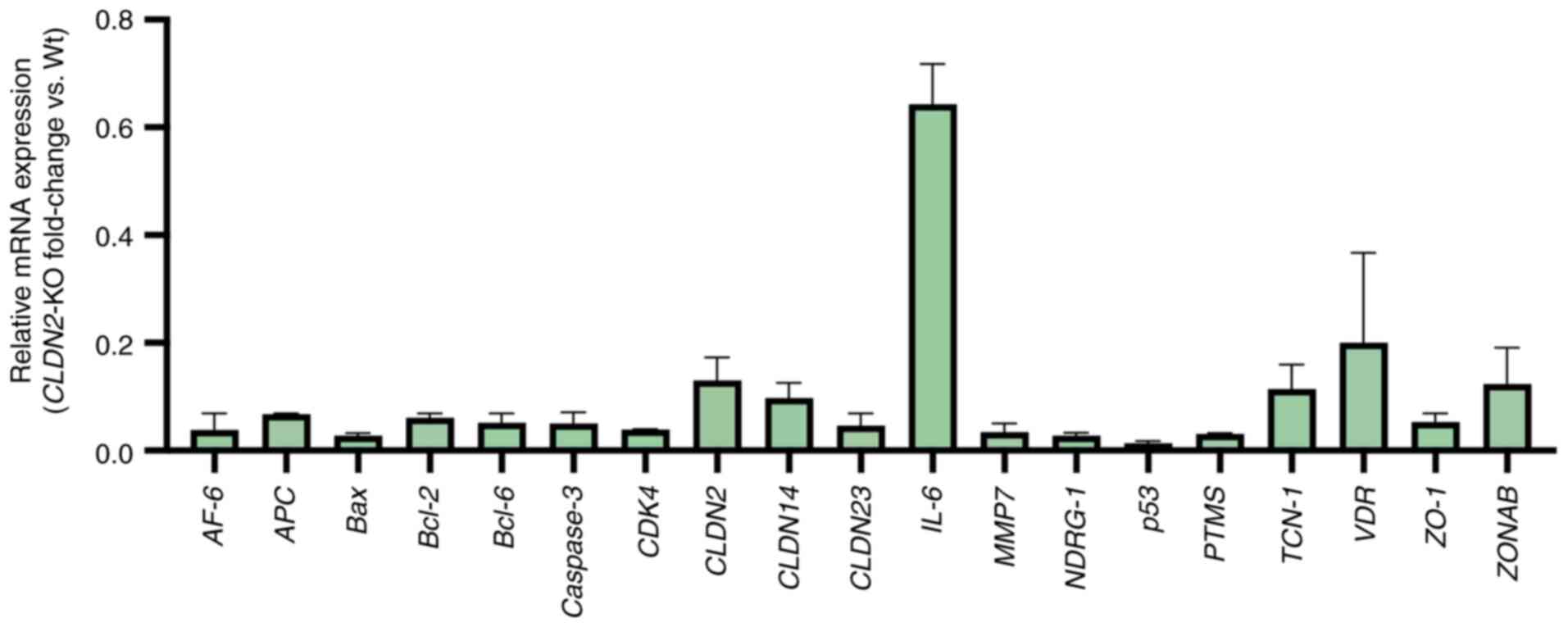 | Figure 6.Relative expression levels of
metastasis-associated genes in Wt and CLDN2-KO HCT116 cells.
Gene expression was quantified using reverse
transcription-quantitative PCR. Each bar represents the mean ± SEM
of three independent experiments. Values were calculated using the
2−ΔΔCq method and normalized to GAPDH. Expression in Wt
cells was set to 1.0 and knock out values were expressed relative
to this baseline. Each bar corresponds to a specific gene and the
height of the bars indicates the magnitude of the fold-change
observed in the CLDN2-KO samples compared with the Wt
samples. The highest bars on the figure represent the gene with the
lowest degree of expression variation. IL-6 exhibited the
lowest degree of downregulation (fold-change, 0.718), whereas AF-6
demonstrated the most pronounced reduction (fold-change, 0.008).
Wt, wild type; ZO-1, zonula occludens-1; VDR, vitamin
D receptor; ZONAB, ZO-1-associated nucleic acid binding
protein; NDRG1, N-Myc downstream-regulated gene 1; APC, adenomatous
polyposis coli; AF-6, Afadin; PTMS, parathymosin; TCN-1,
transcobalamin 1; CLDN2-KO, claudin-2 knock out; AU,
arbitrary Units. |
Discussion
Claudins are key components of tight junctions,
serving key roles in the maintenance of epithelial integrity and
regulating paracellular permeability (8). Previous studies have suggested that
claudins are involved in the development of malignancies. However,
the specific process has not yet been fully elucidated (9,29,30).
In the present study, the role of CLDN2 in
CRC progression was investigated. Using CRISPR/Cas9-mediated
CLDN2-KO in HCT116 cells, it was demonstrated that the loss
of CLDN2 leads to significant downregulation of genes
associated with invasion, metastasis and cell motility. The
findings of the present study aligned with previous studies
suggesting that CLDN2 promotes tumorigenicity and metastasis
in CRC (8–11,26,31).
Particularly, it was observed that CLDN2-KO resulted in
significantly reduced expression levels of ZONAB and NDRG1, two
factors which regulate epithelial proliferation and metastasis
suppression, respectively.
ZONAB is a Y-box transcription factor that interacts
with the tight junction protein ZO-1. When retained at tight
junctions, its transcriptional activity is restricted, whereas
nuclear translocation of ZONAB promotes the expression levels of
genes involved in proliferation and epithelial-mesenchymal
transition (32,33). By contrast, NDRG1 functions as a
metastasis suppressor. It inhibits invasion and migration,
modulates epithelial differentiation and negatively regulates
oncogenic pathways such as PI3K/AKT and Wnt/β-catenin
(34,35). Downregulation of NDRG1 is
associated with poor prognosis in CRC (13). Thus, CLDN2-driven suppression
of NDRG1 through ZONAB activity provides a mechanistic
association between tight junction dysregulation and enhanced tumor
progression. These results support the hypothesis that CLDN2
may modulate the CLDN2/ZO-1/ZONAB signaling axis, thereby
influencing transcriptional programs key for cancer cell migration
and invasion (36). Previous
studies suggested that the abundance of CLDN2 increases with
cell confluence and the maturation of tight junctions and that
ZO-1 (and ZO-2) help stabilize CLDN2 by
reducing its turnover. Knockdown of ZO-1 leads to notable
loss of CLDN2 abundance and promoter activity (37). In the present study, the
downregulation of ZO-1 in CLDN2-KO cells indicated
that downstream signaling is disrupted in the absence of
CLDN2.
In addition to tight junction-related proteins,
CLDN2-KO affected the expression levels of other
metastasis-associated genes. Downregulation of CLDN14 and
CLDN23, tight junction components previously associated with
CRC progression (38,39), was observed. Furthermore, reduced
expression level of AF-6 (encoding Afadin), a scaffolding
protein involved in cell-cell adhesion and migration, was also
notable. Previous research has demonstrated that CLDN2
physically interacts with Afadin via its PDZ-binding motif and this
complex contributes to metastatic behavior in breast cancer models.
In addition, loss of Afadin impairs colony formation and metastasis
(40,41). This provides a precedent to consider
a CLDN2-Afadin axis in CRC.
Furthermore, the data in the present study revealed
a decreased expression level of CDK4, a cyclin-dependent kinase key
for cell cycle progression. Since ZONAB (regulated by CLDN2)
can associate with and influence CDK4 nuclear activity, reduced
CDK4 expression may reflect the disruption of
CLDN2/ZO-1/ZONAB signaling (32,33,42).
Collectively, these transcriptional changes highlight the broad
regulatory influence of CLDN2 on multiple pathways driving
CRC metastasis.
Notably, several key oncogenic and tumor suppressor
genes were also modulated by CLDN2 deletion. The
proto-oncogene c-Myc, a master regulator of proliferation
and metabolism, was significantly downregulated. Reduced
c-Myc expression may contribute to the suppression of
migratory and invasive phenotypes in CLDN2-deficient cells,
consistent with its role in CRC aggressiveness (43).
An inverse association between p53 and
CLDN2 has been reported in mouse colon tissue during dextran
sulfate sodium-induced colitis (44), where reduced p53 expression
was accompanied by increased CLDN2 levels, suggesting that
loss of p53-mediated regulation may contribute to
CLDN2 upregulation in inflamed and neoplastic intestinal
epithelium (45). Furthermore, a
previous study suggested a negative regulatory complex involving
p53 and hepatocyte nuclear factor 4 α that may influence
CLDN2 expression (44).
However, in the present study, downregulation of p53 in the
KO condition, indicated by a fold-change of 0.02, suggests that
CLDN2 may have a regulatory role in its own expression. The
reduced copy number further supports the notion of downregulation
of p53 in the CLDN2-KO cells.
A previous study provided evidence for the
qualitative and quantitative expression levels of Bcl-6
involvement in human CRC progression, and demonstrated that
Bcl-6 appears to be involved in tumor development, from the
earliest stage of carcinogenesis (46). In the present study, in the KO
condition, Bcl-6 exhibited a downregulation, indicating a
possible regulatory function for CLDN2 in its expression.
Although direct evidence of a CLDN2-Bcl-6 axis is limited,
to the best of our knowledge, perturbations in tight junction
integrity and nuclear signaling have been associated with
transcriptional repressor modulation in cancer (47,48),
suggesting that CLDN2 loss may indirectly influence
Bcl-6 expression.
Furthermore, the tumor suppressor gene adenomatous
polyposis coli (APC), a key component of the Wnt/β-catenin
signaling pathway, was markedly reduced. APC dysfunction is a
hallmark of CRC initiation and progression (49,50).
Its downregulation following CLDN2-KO highlights the complex
interplay between tight junction integrity and oncogenic pathways.
Loss of APC may also reflect feedback from junctional disruption on
Wnt signaling regulation, further implicating CLDN2 in the
modulation of oncogenic cascades. Activation of the Wnt/β-catenin
signaling pathway has been documented in a notable proportion of
gastric cancer cases. For example, nuclear β-catenin accumulation,
a hallmark of Wnt pathway activation, has been reported in ~1/3 of
gastric adenocarcinomas (51).
Recent studies estimated that 30–50% of gastric tumor specimens
exhibit hyperactivation of this pathway (52,53).
In addition, activation of Wnt/β-catenin signaling has been
reported to increase the expression levels of MMP7 at both
the mRNA and protein levels in triple-negative breast cancer,
providing a mechanistic association between Wnt signaling and
invasive phenotypes (54,55). These findings further support the
role of Wnt pathway activation in promoting cancer progression
across multiple tumor types.
Furthermore, the vitamin D receptor (VDR), which
regulates proliferation, differentiation and epithelial barrier
function, exhibited decreased expression. Multiple studies have
indicated that VDR can directly bind and regulate
CLDN2 transcription in intestinal tissues, and that
VDR dysregulation has been implicated in colorectal
tumorigenesis (56,57). This supports a bidirectional
regulatory association between VDR and CLDN2
expression, and suggests that CLDN2 loss may contribute to
impaired VDR signaling in CRC.
Notably, elevated CLDN2 expression has been
associated with poor prognosis in CRC, liver cancer and other
gastrointestinal malignancies (7,12,58).
The findings of the present study supported this clinical
association by demonstrating that Cldn2 loss impairs migratory
potential and suppresses pro-metastatic gene expression. Thus,
targeting CLDN2 may represent a promising therapeutic
strategy to inhibit CRC metastasis in the future, although careful
evaluation of potential side effects on normal epithelial function
is warranted in future studies.
It is key to acknowledge the limitations of the
present study. While the present study results demonstrated a
strong association between CLDN2 loss and altered gene
expression, mechanistic experiments, such as rescue assays or
pathway-specific inhibition, were not performed to directly
establish causality. The findings were also based on in
vitro analyses in a single CRC cell line; validation using
in vivo models or additional cell lines in future studies
would strengthen these conclusions. Another limitation is that
CLDN2 protein loss was not validated by western blotting,
although the CRISPR editing efficiency was demonstrated by qPCR,
ICE analysis and Sanger sequencing. In addition, cell migration was
assessed only by the wound healing assay. While this method
reflects overall migratory capacity, it does not capture
chemotactic or invasive behavior. Incorporating approaches such as
Transwell migration and invasion assays would provide complementary
evidence and these are planned for future research. Lastly, further
studies are warranted to dissect the downstream mechanisms by which
CLDN2 modulates signaling pathways. Rescue experiments
restoring CLDN2 expression, pathway-specific analyses (such
as PI3K/AKT, Wnt/β-catenin and yes-associated
protein/transcriptional co-activator with PDZ-binding motif) and
investigation of the interaction partners of CLDN2, such as
ZONAB and Afadin, will be key to elucidate its precise role in CRC
progression in the future.
In conclusion, the present study demonstrated that
CLDN2 serves a key role in regulating metastasis-associated
gene networks in CRC. Disruption of CLDN2 in HCT116 cells
significantly impaired migration and was accompanied by the
downregulation of key invasion- and metastasis-related genes,
including ZONAB, NDRG1, AF-6, CLDN14, CLDN23, CDK4,
Bcl−6 and APC. These transcriptional changes
highlight the broad regulatory influence of CLDN2 on
pathways governing cell adhesion, proliferation and Wnt/β-catenin
signaling. While additional mechanistic studies, such as rescue
assays and pathway-specific analyses, are warranted to further
dissect these interactions, the present study findings provide
notable genetic and phenotypic evidence that CLDN2
contributes to CRC progression. By directly associating
CLDN2 deletion with the suppression of specific
pro-metastatic genes, the present study suggests that targeting
CLDN2 may represent a promising therapeutic strategy to
potentially inhibit CRC metastasis in the future.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by the Deanship of Scientific
Research (DSR) at King Abdulaziz University, Jeddah, Saudi Arabia
(grant no. G: 48-665-1443).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
RA conceived and designed the study. MA designed
the study. RA and MA performed experiments, analyzed and
interpreted the data and wrote the manuscript. RA and MA confirm
the authenticity of all the raw data. Both authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
During the preparation of this work, artificial
intelligence tools were used to improve the readability and
language of the manuscript or to generate images, and subsequently,
the authors revised and edited the content produced by the
artificial intelligence tools as necessary, taking full
responsibility for the ultimate content of the present
manuscript.
References
|
1
|
Siegel RL, Wagle NS, Cercek A, Smith RA
and Jemal A: Colorectal cancer statistics, 2023. CA Cancer J Clin.
73:233–254. 2023.
|
|
2
|
Xi Y and Xu P: Global colorectal cancer
burden in 2020 and projections to 2040. Transl Oncol.
14:1011742021. View Article : Google Scholar
|
|
3
|
Mousavi SE, Ilaghi M, Hamidi Rad R and
Nejadghaderi SA: Epidemiology and socioeconomic correlates of
colorectal cancer in Asia in 2020 and its projection to 2040. Sci
Rep. 15:266392025. View Article : Google Scholar
|
|
4
|
Sarkar S: Proteogenomic approaches to
understand gene mutations and protein structural alterations in
colon cancer. Physiologia. 3:11–29. 2023. View Article : Google Scholar
|
|
5
|
Tabariès S, Dupuy F, Dong Z, Monast A,
Annis MG, Spicer J, Ferri LE, Omeroglu A, Basik M, Amir E, et al:
Claudin-2 promotes breast cancer liver metastasis by facilitating
tumor cell interactions with hepatocytes. Mol Cell Biol.
32:2979–2991. 2012. View Article : Google Scholar
|
|
6
|
Cherradi S, Ayrolles-Torro A, Vezzo-Vié N,
Gueguinou N, Denis V, Combes E, Boissière F, Busson M,
Canterel-Thouennon L, Mollevi C, et al: Antibody targeting of
claudin-1 as a potential colorectal cancer therapy. J Exp Clin
Cancer Res. 36:892017. View Article : Google Scholar
|
|
7
|
Tabariès S, Annis MG, Lazaris A, Petrillo
SK, Huxham J, Abdellatif A, Palmieri V, Chabot J, Johnson RM, Van
Laere S, et al: Claudin-2 promotes colorectal cancer liver
metastasis and is a biomarker of the replacement type growth
pattern. Commun Biol. 4:6572021. View Article : Google Scholar
|
|
8
|
Venugopal S, Anwer S and Szászi K:
Claudin-2: Roles beyond permeability functions. Int J Mol Sci.
20:56552019. View Article : Google Scholar
|
|
9
|
Alghamdi RA and Al-Zahrani MH:
Identification of key claudin genes associated with survival
prognosis and diagnosis in colon cancer through integrated
bioinformatic analysis. Front Genet. 14:12218152023. View Article : Google Scholar
|
|
10
|
Weber CR, Nalle SC, Tretiakova M, Rubin DT
and Turner JR: Claudin-1 and claudin-2 expression is elevated in
inflammatory bowel disease and may contribute to early neoplastic
transformation. Lab Invest. 88:1110–120. 2008. View Article : Google Scholar
|
|
11
|
Rosenthal R, Milatz S, Krug SM, Oelrich B,
Schulzke JD, Amasheh S, Günzel D and Fromm M: Claudin-2, a
component of the tight junction, forms a paracellular water
channel. J Cell Sci. 123:1913–1921. 2010. View Article : Google Scholar
|
|
12
|
Dhawan P, Ahmad R, Chaturvedi R, Smith JJ,
Midha R, Mittal MK, Krishnan M, Chen X, Eschrich S, Yeatman TJ, et
al: Claudin-2 expression increases tumorigenicity of colon cancer
cells: Role of epidermal growth factor receptor activation.
Oncogene. 30:3234–3247. 2011. View Article : Google Scholar
|
|
13
|
Wei M, Zhang Y, Yang X, Ma P, Li Y, Wu Y,
Chen X, Deng X, Yang T, Mao X, et al: Claudin-2 promotes colorectal
cancer growth and metastasis by suppressing NDRG1 transcription.
Clin Transl Med. 11:e6672021. View Article : Google Scholar
|
|
14
|
Al-Zahrani M, Lary S, Assidi M, Hakamy S,
Dallol A, Alahwal M, Al-Maghrabi J, Sibiany A, Abuzenadah A,
Al-Qahtani M and Buhmeida A: Evaluation of prognostic potential of
CD44, Claudin-2 and EpCAM expression patterns in colorectal
carcinoma. Adv Environmental Biol. 10:147–156. 2016.
|
|
15
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou S, Diaz LA Jr and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 2013. View Article : Google Scholar
|
|
16
|
Kim D and Cho KH: Hidden patterns of gene
expression provide prognostic insight for colorectal cancer. Cancer
Gene Ther. 30:11–21. 2022. View Article : Google Scholar
|
|
17
|
Cancer Genome Atlas Network, .
Comprehensive molecular characterization of human colon and rectal
cancer. Nature. 487:330–337. 2012. View Article : Google Scholar
|
|
18
|
Zhao Z, Li C, Tong F, Deng J, Huang G and
Sang Y: Review of applications of CRISPR-Cas9 gene-editing
technology in cancer research. Biol Proced Online. 23:142021.
View Article : Google Scholar
|
|
19
|
Meng H, Nan M, Li Y, Ding Y, Yin Y and
Zhang M: Application of CRISPR-Cas9 gene editing technology in
basic research, diagnosis and treatment of colon cancer. Front
Endocrinol (Lausanne). 14:11484122023. View Article : Google Scholar
|
|
20
|
Neumann E, Schaefer-Ridder M, Wang Y and
Hofschneider P: Gene transfer into mouse lyoma cells by
electroporation in high electric fields. EMBO J. 1:841–845. 1982.
View Article : Google Scholar
|
|
21
|
Enzmann BL and Wronski A: Synthego's
engineered cells allow scientists to ‘Cut Out’ CRISPR optimization
[SPONSORED]. CRISPR J. 1:255–257. 2018. View Article : Google Scholar
|
|
22
|
Roginsky J: Analyzing CRISPR editing
results: Synthego developed a tool called ICE to be more efficient
than other methods. Genet Eng Biotechnol News. 38 (Suppl):S24–S26.
2018. View Article : Google Scholar
|
|
23
|
Bentley DR, Balasubramanian S, Swerdlow
HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL,
Bignell HR, et al: Accurate whole human genome sequencing using
reversible terminator chemistry. Nature. 456:53–59. 2008.
View Article : Google Scholar
|
|
24
|
Gross A, Schoendube J, Zimmermann S, Steeb
M, Zengerle R and Koltay P: Technologies for single-cell isolation.
Int J Mol Sci. 16:16897–16919. 2015. View Article : Google Scholar
|
|
25
|
Liang CC, Park AY and Guan JL: In vitro
scratch assay: A convenient and inexpensive method for analysis of
cell migration in vitro. Nat Protoc. 2:329–333. 2007. View Article : Google Scholar
|
|
26
|
Schneider CA, Rasband WS and Eliceiri KW:
NIH Image to ImageJ: 25 years of image analysis. Nat Methods.
9:671–675. 2012. View Article : Google Scholar
|
|
27
|
Bustin SA, Benes V, Garson JA, Hellemans
J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL,
et al: The MIQE guidelines: Minimum information for publication of
quantitative real-time PCR experiments. Clin Chem. 55:611–622.
2009. View Article : Google Scholar
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)). Method. 25:402–408. 2001. View Article : Google Scholar
|
|
29
|
Zhu L, Han J, Li L, Wang Y, Li Y and Zhang
S: Claudin family participates in the pathogenesis of inflammatory
bowel diseases and Colitis-associated colorectal cancer. Front
Immunol. 10:14412019. View Article : Google Scholar
|
|
30
|
Al-Zahrani MH, Assidi M, Pushparaj PN,
Al-Maghrabi J, Zari A, Abusanad A, Buhmeida A and Abu-Elmagd M:
Expression pattern, prognostic value and potential microRNA
silencing of FZD8 in breast cancer. Oncol Lett. 26:4772023.
View Article : Google Scholar
|
|
31
|
Ahmad R, Kumar B, Thapa I, Tamang RL,
Yadav SK, Washington MK, Talmon GA, Yu AS, Bastola DK, Dhawan P and
Singh AB: Claudin-2 protects against colitis-associated cancer by
promoting colitis-associated mucosal healing. J Clin Invest.
133:e1707712023. View Article : Google Scholar
|
|
32
|
Sourisseau T, Georgiadis A, Tsapara A, Ali
RR, Pestell R, Matter K and Balda MS: Regulation of PCNA and Cyclin
D1 expression and epithelial morphogenesis by the ZO-1-regulated
transcription factor ZONAB/DbpA. Mol Cell Biol. 26:2387–2398. 2006.
View Article : Google Scholar
|
|
33
|
Balda MS: The tight junction protein ZO-1
and an interacting transcription factor regulate ErbB-2 expression.
EMBO J. 19:2024–2033. 2000. View Article : Google Scholar
|
|
34
|
Kovacevic Z and Richardson DR: The
metastasis suppressor, Ndrg-1: A new ally in the fight against
cancer. Carcinogenesis. 27:2355–2366. 2006. View Article : Google Scholar
|
|
35
|
Sun J, Zhang D, Bae DH, Sahni S, Jansson
P, Zheng Y, Zhao Q, Yue F, Zheng M, Kovacevic Z and Richardson DR:
Metastasis suppressor, NDRG1, mediates its activity through
signaling pathways and molecular motors. Carcinogenesis.
34:1943–1954. 2013. View Article : Google Scholar
|
|
36
|
Tao D, Guan B, Li H and Zhou C: Expression
patterns of claudins in cancer. Heliyon. 9:e213382023. View Article : Google Scholar
|
|
37
|
Amoozadeh Y, Anwer S, Dan Q, Venugopal S,
Shi Y, Branchard E, Liedtke E, Ailenberg M, Rotstein OD, Kapus A
and Szászi K: Cell confluence regulates claudin-2 expression:
Possible role for ZO-1 and Rac. Am J Physiol Physiol.
314:C366–C378. 2018. View Article : Google Scholar
|
|
38
|
Yang L and Zhang W, Li M, Dam J, Huang K,
Wang Y, Qiu Z, Sun T, Chen P, Zhang Z and Zhang W: Evaluation of
the prognostic relevance of differential claudin gene expression
highlights Claudin-4 as Being Suppressed by TGFβ1 inhibitor in
colorectal cancer. Front Genet. 13:7830162022. View Article : Google Scholar
|
|
39
|
Chen W, Zhu XN, Wang J, Wang YP and Yang
JL: The expression and biological function of claudin-23 in
colorectal cancer. Sichuan Da Xue Xue Bao Yi Xue Ban. 49:331–336.
2018.(In Chinese).
|
|
40
|
Tabariès S, McNulty A, Ouellet V, Annis
MG, Dessureault M, Vinette M, Hachem Y, Lavoie B, Omeroglu A, Simon
HG, et al: Afadin cooperates with Claudin-2 to promote breast
cancer metastasis. Genes Dev. 33:180–193. 2019. View Article : Google Scholar
|
|
41
|
Zhang X, Wang H, Li Q and Li T: CLDN2
inhibits the metastasis of osteosarcoma cells via down-regulating
the afadin/ERK signaling pathway. Cancer Cell Int. 18:1602018.
View Article : Google Scholar
|
|
42
|
González-Mariscal L, Miranda J,
Gallego-Gutiérrez H, Cano-Cortina M and Amaya E: Relationship
between apical junction proteins, gene expression and cancer.
Biochim Biophys Acta Biomembr. 1862:1832782020. View Article : Google Scholar
|
|
43
|
Dang CV: MYC on the path to cancer. Cell.
149:22–35. 2012. View Article : Google Scholar
|
|
44
|
Hirota C, Takashina Y, Ikumi N, Ishizuka
N, Hayashi H, Tabuchi Y, Yoshino Y, Matsunaga T and Ikari A:
Inverse regulation of claudin-2 and −7 expression by p53 and
hepatocyte nuclear factor 4α in colonic MCE301 cells. Tissue
Barriers. 9:18604092021. View Article : Google Scholar
|
|
45
|
Kim HY, Jeon H, Bae CH, Lee Y, Kim H and
Kim S: Rumex japonicus Houtt. alleviates dextran sulfate
sodium-induced colitis by protecting tight junctions in mice.
Integr Med Res. 9:1003982020. View Article : Google Scholar
|
|
46
|
Sena P, Mariani F, Benincasa M, De Leon
MP, Di Gregorio C, Mancini S, Cavani F, Smargiassi A, Palumbo C and
Roncucci L: Morphological and quantitative analysis of BCL6
expression in human colorectal carcinogenesis. Oncol Rep.
31:103–110. 2014. View Article : Google Scholar
|
|
47
|
Li J: Context-dependent roles of claudins
in tumorigenesis. Front Oncol. 11:6767812021. View Article : Google Scholar
|
|
48
|
Nehme Z, Roehlen N, Dhawan P and Baumert
TF: Tight junction protein signaling and cancer biology. Cells.
12:2432023. View Article : Google Scholar
|
|
49
|
Miwa N, Furuse M, Tsukita S, Niikawa N,
Nakamura Y and Furukawa Y: Involvement of claudin-1 in the
beta-catenin/Tcf signaling pathway and its frequent upregulation in
human colorectal cancers. Oncol Res. 12:469–476. 2001. View Article : Google Scholar
|
|
50
|
Mankertz J, Hillenbrand B, Tavalali S,
Huber O, Fromm M and Schulzke JD: Functional crosstalk between Wnt
signaling and Cdx-related transcriptional activation in the
regulation of the claudin-2 promoter activity. Biochem Biophys Res
Commun. 314:1001–1007. 2004. View Article : Google Scholar
|
|
51
|
Clements WM, Wang J, Sarnaik A, Kim OJ,
MacDonald J, Fenoglio-Preiser C, Groden J and Lowy AM: Beta-Catenin
mutation is a frequent cause of Wnt pathway activation in gastric
cancer. Cancer Res. 62:3503–3506. 2002.
|
|
52
|
Chiurillo MA: Role of the Wnt/β-catenin
pathway in gastric cancer: An in-depth literature review. World J
Exp Med. 5:84–102. 2015. View Article : Google Scholar
|
|
53
|
Jang JH, Jung J, Kang HG, Kim W, Kim WJ,
Lee H, Cho JY, Hong R, Kim JW, Chung JY, et al: Kindlin-1 promotes
gastric cancer cell motility through the Wnt/β-catenin signaling
pathway. Sci Rep. 15:24812025. View Article : Google Scholar
|
|
54
|
Zhang N, Wei P, Gong A, Chiu WT, Lee HT,
Colman H, Huang H, Xue J, Liu M, Wang Y, et al: FoxM1 promotes
β-catenin nuclear localization and controls Wnt target-gene
expression and glioma tumorigenesis. Cancer Cell. 20:427–442. 2011.
View Article : Google Scholar
|
|
55
|
Dey N, Young B, Abramovitz M, Bouzyk M,
Barwick B, De P and Leyland-Jones B: Differential activation of
Wnt-β-catenin pathway in triple negative breast cancer increases
MMP7 in a PTEN dependent manner. PLoS One. 8:e774252013. View Article : Google Scholar
|
|
56
|
Zhang YG, Wu S, Lu R, Zhou D, Zhou J,
Carmeliet G, Petrof E, Claud EC and Sun J: Tight junction CLDN2
gene is a direct target of the Vitamin D receptor. Sci Rep.
5:106422015. View Article : Google Scholar
|
|
57
|
Pereira F, Fernández-Barral A, Larriba MJ,
Barbáchano A and González-Sancho JM: From molecular basis to
clinical insights: A challenging future for the vitamin D endocrine
system in colorectal cancer. FEBS J. 291:2485–2518. 2024.
View Article : Google Scholar
|
|
58
|
Mariadason JM, Nicholas C, L'Italien KE,
Zhuang M, Smartt HJM, Heerdt BG, Yang W, Corner GA, Wilson AJ,
Klampfer L, et al: Gene expression profiling of intestinal
epithelial cell maturation along the crypt-villus axis.
Gastroenterology. 128:1081–1088. 2005. View Article : Google Scholar
|