Introduction
Rhabdomyosarcoma (RMS) is a non-epithelial malignant
tumor that differentiates into immature skeletal muscle with
rhabdomyoblastic differentiation (1). The 2020 WHO classification of tumors
of soft tissue and bones has introduced new subtypes of spindle
cell/sclerosing RMS (SS-RMS). SS-RMS exhibit significant clinical
and genetic heterogeneity. Based on molecular characteristics,
SS-RMS can be further subdivided into i) congenital/infantile
spindle cell RMS, which contains gene fusions involving
vestigial-like family member 2, nuclear receptor coactivator 1/2
(NCOA1/2) and serum response factor (2,3); ii)
ss-RMS with myogenic differentiation 1 mutations (4); and iii) intraosseous RMS with EWS RNA
binding protein 1/FUS RNA binding protein-transcription factor
cellular promoter 2 (EWSR1/FUS-TFCP2) fusions (collectively
referred to as FET-TFCP2 fusion RMS) or Meis homeobox 1-NCOA2
fusions (5).
RMS with TFCP2 rearrangements (TFCP2-RMS) were first
described in 2018 (6). The tumor
consists of spindle and epithelioid cells, predominantly occurs in
the craniofacial bones and most commonly affects the mandible.
TFCP2-RMS exhibits positive expression of myogenic markers,
including desmin, myogenin and myoblast determination protein 1
(MyoD1), positive expression of epithelial markers, such as pan-CK
and epithelial membrane antigen (EMA), and anaplastic lymphoma
kinase (ALK) upregulation (5). The
tumor has a predilection for the craniofacial bones, particularly
the jaws of young adults, and is often associated with a rapid
clinical course and poor prognosis (7).
To the best of our knowledge, to date, a total of
108 cases have been reported, mainly affecting the craniofacial
bones, with only 1 case occurring in the rib (8). The present study reports an additional
case of TFCP2-RMS occurring in the rib and reviews the relevant
literature. The aim of the present study is to enhance the
understanding of this rare subtype of RMS by summarizing its
clinicopathological features, immunophenotype, molecular
alterations and prognosis.
Case report
Case presentation
A 29-year-old male was admitted to the General
Hospital of Southern Theater Command (Guangzhou, China) in June
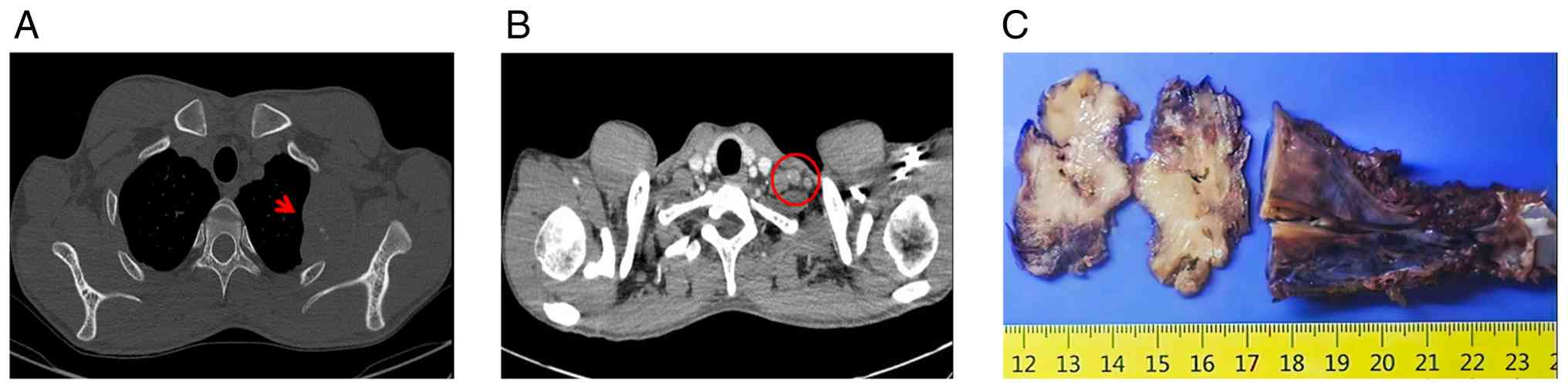
2023 with a 3-month history of an asymptomatic rib mass. A computed
tomography scan demonstrated a destructive lesion arising from the
second rib bone with a surrounding soft-tissue mass, measuring
~51×40 mm in diameter. The mass showed mild enhancement after
contrast, along with left cervical lymphadenopathy (Fig. 1A and B). The patient underwent
surgical resection of the tumor in the left second rib and a left
cervical lymph node dissection.
Gross examination revealed that the tumor exhibited
a destructive growth pattern, with erosion of the rib and absence
of normal bone tissue. The tumor appeared solid, grayish-white and
firm in consistency (Fig. 1C).
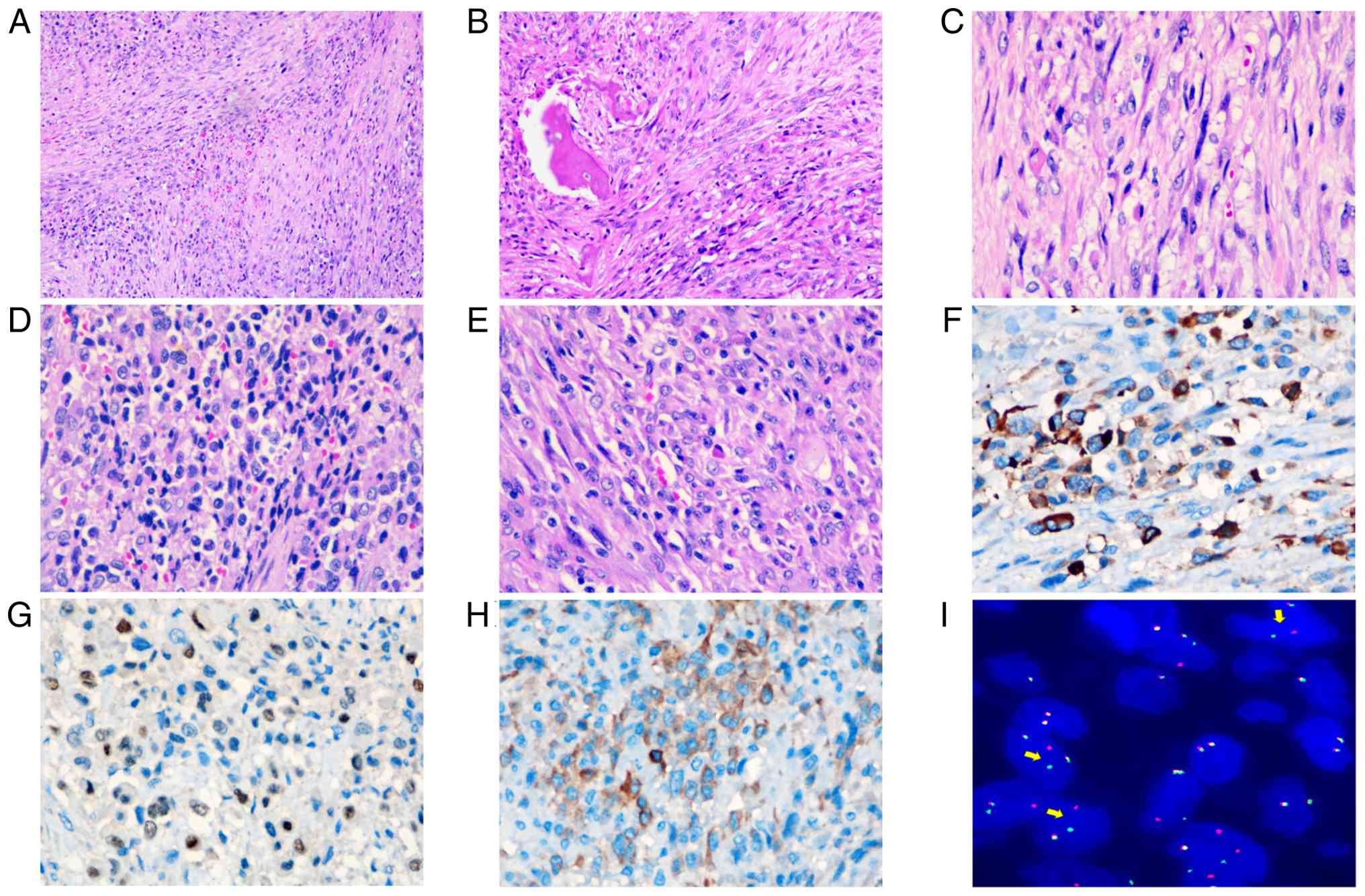
Microscopically, the neoplasm exhibited a biphasic population of
spindled and epithelioid tumor cells. The spindled component was
arranged in intersecting fascicles with focal, ill-defined
storiform architecture and displayed permeative infiltration of
osseous trabeculae (Fig. 2A and B).
The epithelioid cells were arranged in small clusters, nests or
cords, with eosinophilic cytoplasm and rhabdoid features, vesicular
nuclei and prominent nucleoli (Fig. 2C
and D). The tumor cells demonstrated marked cytological atypia
and frequent mitotic activity. The tumor showed marked
collagenous/sclerotic and occasionally myxoid stroma. The tumor
cells had metastasized to the left cervical lymph nodes (Fig. 2E). Immunohistochemical analysis
revealed that the tumor cells exhibited focal positivity for CK
(Fig. 2F), myogenin (Fig. 2G), ALK (Fig. 2H) and S-100 (Fig. S1A). Diffuse positivity was observed
for α-smooth muscle actin (SMA), myogenic differentiation 1
(MyoD1), desmin and vimentin (Fig.
S1B-E). The tumor cells were negative for SRY-box transcription
factor 10 (SOX-10), cyclin-dependent kinase 4 (CDK4), mouse double
minute 2 (MDM2), special AT-rich sequence binding protein 2
(SATB2), friend leukaemia virus integration 1 gene (Fli-1) and
cluster of differentiation 34 (CD34) proteins (Fig. S1F-K). Additionally, the expression
of trimethylated histone H3 at lysine 27 (H3K27me3), Brahma-related
gene 1 (BRG-1) and integrase interactor-1 (INI-1) retained intact
expression (Fig. S1L-N). The Ki-67
proliferation index was approximately 30% (Fig. S1O). Next generation sequencing
(NGS) revealed TFCP-FUS gene fusion and deletion of CDKN2A and
CDKN2B. Fluorescence in situ hybridization (FISH) using a
break-apart probe showed a translocation of TFCP2 (Fig. 2I). The patient did not receive
postoperative adjuvant radiotherapy/chemotherapy due to: i)
TFCP2-rearranged rhabdomyosarcoma being an extremely rare, highly
aggressive subtype with no recognized standard adjuvant regimen and
limited response to conventional chemotherapy; and ii) rapid
postoperative deterioration of the patient's general condition
precluding treatment tolerance, following full family
communication. The patient succumbed 4 months after surgery.
Materials and methods
Hematoxylin-eosin (H&E) staining
and Immunohistochemistry (IHC)
Tissue samples for conventional microscopy were
processed as follows: Fixation in 10% neutral buffered formalin at
37°C for 12 h, followed by sequential routine dehydration, clearing
and paraffin embedding. Following section preparation at a
thickness of 4 µm, the sections were subjected to H&E staining.
Briefly, after deparaffinization in xylene and rehydration through
a graded ethanol series, the sections were stained with hematoxylin
at room temperature for 3–8 min, differentiated in 1% acid alcohol
at room temperature for several seconds, and then blued in running
tap water or a weak alkaline solution at room temperature.
Subsequently, the sections were counterstained with eosin Y
solution at room temperature for 1–3 min. Finally, the sections
were dehydrated through an ascending alcohol series, cleared in
xylene and mounted with a resinous medium. The stained sections
were used for subsequent microscopic observation with an Olympus
BX43 upright biological light microscope (Olympus Corporation). The
formalin-fixed paraffin-embedded (FFPE) tissue sections were
deparaffinized and rehydrated using xylene and graded alcohols.
Following antigen retrieval, sections were blocked with 10% normal
goat serum (cat. no. X090710; Agilent Technologies, Inc.) at room
temperature for 30 min to reduce non-specific binding. The sections
were incubated overnight at 4°C with one of the following primary
antibodies. Staining was performed with ready-to-use antibodies,
including Pan-CK (cat. no. RTU-AE1/AE3-601-QH; QuanhHui
International Trading Co., Ltd.), MyoD1 (cat. no. IHC630-7;
GenomeMe Lab, Inc.), Ki67 (cat. no. ZM-0166; Beijing Zhongshan
Jinqiao Biotechnology Co., Ltd.), SMA (cat. no. ZM-0003; Beijing
Zhongshan Jinqiao Biotechnology Co., Ltd.), vimentin (cat. no.
RTU-Vim-U9-QH; QUANHUI), SOX-10 (cat. no. ZA-0624; Beijing
Zhongshan Jinqiao Biotechnology Co., Ltd.), CDK4 (cat. no. ZA-0614;
Beijing Zhongshan Jinqiao Biotechnology Co., Ltd.), MDM2 (cat. no.
ZM-0425; Beijing Zhongshan Jinqiao Biotechnology Co., Ltd.), SATB2
(cat. no. ZM-0163; Beijing Zhongshan Jinqiao Biotechnology Co.,
Ltd.), Fli-1 (cat. no. RTU-FLI-1-QH; QUANHUI), CD34 (cat. no.
ZA-0550; Beijing Zhongshan Jinqiao Biotechnology Co., Ltd.),
H3K27me3 (cat. no. RMA-0843; Fuzhou Maixin Biotech Co., Ltd.),
BRG-1 (cat. no. ZA-0673; Beijing Zhongshan Jinqiao Biotechnology
Co., Ltd.) and INI-1 (cat. no. ZA-0696; Beijing Zhongshan Jinqiao
Biotechnology Co., Ltd.) on the Dako Link48 platform (Agilent
Technologies, Inc.). External positive control tissues were
provided for each slide to validate staining specificity.
Subsequently, the sections were incubated with the secondary
antibody using the ready-to-use Dako Real Envision Detection System
(HRP-labeled polymer; cat. no. K5007; Agilent Technologies, Inc.)
at room temperature for 30 min, and were stained using the Dako
Real Envision kit (Agilent Technologies, Inc.). The sections were
then observed using an Olympus BX43 upright biological light
microscope.
Fluorescence in situ hybridization
(FISH)
FISH was performed on 4-µm FFPE sections with a
TFCP2 (12q13) break-apart probe (Guangzhou Amoytop Medical
Technology Co., Ltd.) according to the manufacturer's instructions.
Briefly, the procedure was as follows: 4 µm FFPE tissue sections
were baked at 65°C for 12–16 h, deparaffinized in xylene and
rehydrated through graded alcohols. Subsequently, the sections were
boiled at 100°C for 25 min, digested with pepsin for 10 min and
then dehydrated to dryness via graded alcohols. Under dark
conditions, 10 µl of hybridization solution containing the probe
was added to the sample area, followed by cover slipping and
sealing. The sections were denatured at 85°C for 5 min and then
hybridized at 37°C for 10–18 h. After hybridization, the sealing
gel was removed, and the sections were washed with 0.1% NP-40/2X
SSC washing buffer at 37°C to eliminate non-specific binding.
Following graded alcohol dehydration and air-drying, DAPI
counterstain was applied, and the sections were incubated in the
dark for a short period before observation under an Olympus BX53
fluorescence microscope (Olympus Corporation). Normal interphase
nuclei exhibited two red-green fused signals, whereas the presence
of one red, one green and one fused signal was indicative of TFCP2
gene breakage.
DNA and RNA NGS analysis
NGS experiments were commissioned to Hybribio
Biotech Co., Ltd., for execution. For sample processing, genomic
DNA (gDNA) samples were prepared using the Haipu HP FFPE Tissue
gDNA Extraction Kit (magnetic bead-based; catalog no. HP036;
HaploX; Shenzhen HaploS Biotechnology Co., Ltd.). In the sample
quality assessment stage, the integrity of processed samples was
evaluated using an Agilent 4200 Bioanalyzer (Agilent Technologies,
Inc.), while sample concentrations were quantified via quantitative
polymerase chain reaction (qPCR). Sequencing was performed on the
Illumina NovaSeq 6000 platform (Illumina Inc.), employing a
paired-end 150 bp read length; the corresponding sequencing kit
used was the NovaSeq 6000 S4 Reagent Kit v1.5 (300 cycles; catalog
no. 20046933; Illumina, Inc.). The final library was loaded at a
concentration of 1.8 nM, as quantified by qPCR. Differential gene
expression analysis was performed using Cuffdiff within the
Cufflinks package (version 2.2.1; http://cole-trapnell-lab.github.io/cufflinks/). Genes
with differential expression defined as q<0.05 and
|log2(fold change)|>0.8 were further analyzed and
validated by qPCR.
Literature review
Since the first report of TFCP2-RMS in 2018, 109
cases have been reported (including the current study) (Tables I and II) (5–37). A
review of the literature showed that the tumors mostly affect young
adults but may also occur in elderly patients and infants. The
median age of diagnosis is 33 years (range, 7–86 years). There is
slight female predominance, with a female to male of 1.53:1
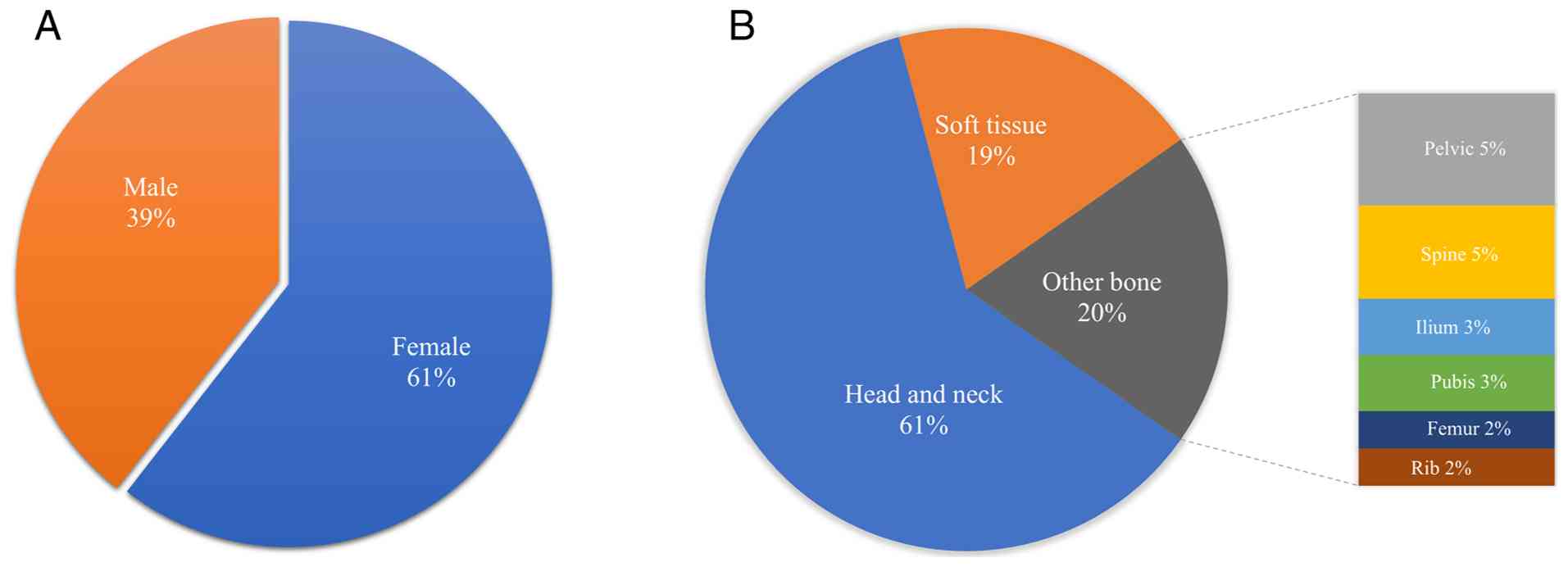
(66:43). The majority of cases (n=92) have an intraosseous
component. A total of 66 cases (60.6%) have been located in
craniofacial regions, most commonly in the mandible (n=22), while
other less common sites (20.2%; n=22) include the pelvic bones,
spine, ilium, pubis, femurs and ribs. The remaining 19.3% (n=21)
affect the soft tissues, such as the skin (11,15,25),
bladder (13,36), chest wall (6), abdominal wall (13,33),
peritoneum (7), mediastinum
(24) and ileum (8) (Fig.
3).
 | Table I.Molecular genetic features. |
Table I.
Molecular genetic features.
| First author,
year | Age, years | Sex | Location | Myogenin | MyoD1 | Desmin | CK | ALK | EMA | Molecular | ALK gene | (Refs.) |
|---|
| Ishiyama et
al, 2023 | 58 | M | Cutaneous
(scalp) | + | F+ | F+ | + | + | NA | F::T | Fusion | (1) |
| Si et al,
2025 | 39 | F | Mandible | - | F+ | - | D+ | D+ | NA | T | NA | (9) |
| Le Loarer et
al, | 16 | F | Sphenoid bone | F+ | + | D+ | + | + | - | F::T | 50% + | (7) |
| 2020 | 26 | F | Sacrum | F+ | + | + | + | + | - | F::T | 50% + |
|
|
| 38 | F | Peritoneum | F+ | + | + | + | + | F+ | E::T | 50% + |
|
|
| 32 | M | Hard palate and
upper lip | - | + | D+ | + | - | - | E::T | WT |
|
|
| 20 | M |
Orbito-temporosphenoid | F+ | + | D+ | + | - | - | F::T | <5% + |
|
|
| 86 | M | Inguinal | F+ | + | + | F+ | + | - | E::T | 100% + |
|
|
| 18 | F | Femur | + | F+ | D+ | + | + | - | E::T | 100% + |
|
|
| 17 | F | Cervico-occipital
junction | - | + | + | NA | + | F+ | F::T | 100% + |
|
|
| 31 | M | Occipital bone | F+ | D+ | D+ | + | + | - | F::T | 100% + |
|
|
| 32 | M | Mandible | F+ | D+ | + | + | + | - | F::T | 70% |
|
|
| 58 | F | Mandible | F+ | + | D+ | + | + | - | F::T | 70% |
|
|
| 12 | F | Mandible | + | + | D+ | + | + | F+ | F::T | 50% |
|
|
| 11 | F | Maxilla | F+ | + | D+ | W+ | W+ | F+ | E::T | <5% + |
|
|
| 25 | M | Mandible | - | + | - | + | + | - | E::T | 80% |
|
| Panferova et
al, 2022 | 16 | F | Mandible | F+ | NA | + | W+ | F+ | NA | E::T | NA | (10) |
| Duan et
al, | 54 | F | Lower back,
skin | S+ | + | + | S+ | + | + | F::T,T::A | Fusion | (11) |
| 2023 | 28 | M | Maxilla | - | + | + | + | + | NA | E::T | NA |
|
| Chen et
al, | 31 | F | Maxilla | - | + | P+ | P+ | F+ | NA | E::T | NA | (12) |
| 2022 | 49 | F | Maxilla | - | + | + | - | F+ | NA | E::T | NA |
|
| Li et al,
2024 | 6-36, | F (n=9), | Head and neck | 3/14 b + | 14/14+ | 14/14+ | 8/14+ | 9/14+ | 5/14+ | F::T | 14*NA | (13) |
|
| mid22 | M (n=5) | region (n=9), |
|
|
|
|
|
| (n=8), |
|
|
|
| (n=14) |
| pelvis (n=2),
bladder |
|
|
|
|
|
| E::T |
|
|
|
|
|
| (n=1), pubic
bone |
|
|
|
|
|
| (n=6) |
|
|
|
|
|
| (n=1), abdominal
wall, humerus and pubic bone (n=1) |
|
|
|
|
|
|
|
|
|
| Carrillo- | 26 | F | Mandible | - | + | NA | + | P+ | NA | F::T | WT | (14) |
| Ng et al,
2023 | 59 | F | Mandible | - | + | +a | + | + | - | F::T | WT |
|
| Demirkesen et
al, 2023 | 35 | F | Scapular-skin | W+ | + | + | + | + | - | F::T | Fusion | (15) |
| Csizmok et
al, 2024 | 31 | M | Mandible | NA | NA | F+ | + | F+ | - | F::T | Deletion | (32) |
| Haug et al,
2023 | 55 | F | Thoracic
vertebrae | - | + | + | + | + | NA | E::T | Upregulation | (17) |
| Chrisinger et
al, | 20 | F | Pelvic | S+ | + | + | NA | + | NA | F::T | NA | (18) |
| 2020 | 20-30 | F | Frontal bone | - | + | - | + | P+ | NA | E::T | NA |
|
| Li et al,
2023 | 30 | F | Abdominal wall | + | + | + | + | + | - | E::T | WT | (33) |
| Agaram et
al, 2019 | 27 | M | Other bone | + | + | + | + | + | NA | F::T | NA | (5) |
| Koutlas et
al, 2021 | 15 | M | Mandible | F+ | + | P+ | + | - | NA | E::T | NA | (19) |
| Dehner et
al, | 21 | F | Skull | + | + | + | + | + | NA | F::T | NA | (8) |
| 2023 | 30 | F | Maxilla | + | NA | + | + | NA | NA | F::T | NA |
|
|
| 60 | M | Right shoulder | - | + | + | + | + | NA | E::T | NA |
|
|
| 18 | M | Left pubic
ramus | + | + | + | + | + | NA | F::T | NA |
|
|
| 43 | F | Maxilla | + | + | + | + | + | NA | F::T | NA |
|
|
| 31 | F | Posterior iliac
crest | - | + | + | - | + | NA | E::T | NA |
|
|
| 41 | F | Pelvic | - | + | + | NA | + | NA | E::T | NA |
|
|
| 32 | F | L5 vertebra | - | + | + | NA | + | NA | F::T | NA |
|
|
| 26 | F | Left iliac
bone | + | + | + | - | + | NA | F::T | NA |
|
|
| 48 | F | Pelvic | NA | NA | + | NA | NA | NA | F::T | NA |
|
|
| 43 | M | Fourth rib | + | + | + | - | + | NA | F::T | NA |
|
|
| 13 | M | Maxilla | + | + | + | - | NA | NA | F::T | NA |
|
|
| 27 | F | Maxilla | + | + | + | + | + | NA | F::T | NA |
|
|
| 44 | M | Ileum | NA | NA | NA | NA | NA | NA | F::T | NA |
|
|
| 70 | F | Mandible | + | + | + | - | - | NA | E::T | NA |
|
|
| 62 | F | T7 vertebra | - | + | + | NA | + | NA | F::T | NA |
|
| Xu et al,
2021 | 22 | M | Mandible | F+ | + | F+ | + | + | NA | F::T | WT | (20) |
|
| 27 | F | Skull | F+ | + | + | + | + | NA | E::T | NA |
|
|
| 20 | F | Maxilla | + | NA | F+ | + | + | NA | E::T | NA |
|
|
| 29 | M | Skull | + | + | + | + | NA | NA | E::T | WT |
|
|
| 33 | F | Maxilla | + | + | F+ | + | + | NA | E::T | NA |
|
|
| 18 | M | Skull | - | NA | - | + | + | NA | F::T | WT |
|
|
| 40 | F | Neck superficial
soft tissue | + | NA | + | + | + | NA | F::T | Deletion |
|
|
| 43 | F | Mandible | R + | + | + | + | + | NA | F::T | NA |
|
|
| 34 | M | Mandible | NA | P+ | + | - | - | NA | F::T | Deletion |
|
|
| 16 | M | Mandible | F+ | F+ | F+ | + | + | NA | F::T | Deletion |
|
| Zhu et al,
2019 | 74c | F | Maxilla | F+ | P+ | + | - | + | - | F::T | WT | (16) |
| Silva | 17 | F | Maxilla | S + | + | + | NA | NA | NA | F::T | NA | (21) |
| Cunha et al,
2022 |
|
|
|
|
|
|
|
|
|
|
|
|
| Valerio et
al, 2023 | 19 | F | Mandible | - | + | F+ | + | D+ | - | F::T | Deletion | (22) |
| Watson et
al, | 27 | F | Chest wall | + | + | + | NA | NA | NA | E::T | NA | (6) |
| 2018 | 27 | F | Pelvic | + | + | + | NA | NA | NA | F::T | NA |
|
|
| 27 | F | Sphenoid bone | + | + | + | NA | NA | NA | F::T | NA |
|
| Chen et al,
2024 | 40 | F | Mandible | - | + | + | + | + | NA | E::T | NA | (23) |
| Schöpf et
al, | 17 | F | Iliac bone | NA | NA | NA | NA | NA | NA | F::T | WT | (24) |
| 2024 | 60 | M | Mediastinum | NA | NA | NA | NA | NA | NA | E::T | WT |
|
|
| 9 | F | Mandible | NA | NA | NA | NA | NA | NA | E::T | WT |
|
|
| 48 | M | Maxillar | NA | NA | NA | NA | NA | NA | F::T | WT |
|
|
| 35 | M | Occipital/nuchal
soft tissue | + | + | + | + | W+ | NA | F::T | Deletion |
|
|
| 49 | F | Mandible | NA | NA | NA | NA | NA | NA | F::T | WT |
|
|
| 25 | M | Shoulder soft
tissue | NA | NA | NA | NA | NA | NA | E::T | Deletion |
|
|
| 14 | F | Maxillar | NA | NA | NA | NA | NA | NA | F::T | Deletion |
|
|
| 15 | F | Temporal/sphenoid
bone | NA | NA | NA | NA | NA | NA | F::T | WT |
|
|
| 38 | M | Maxillar | NA | NA | NA | NA | NA | NA | F::T | Deletion |
|
|
| 40 | F | Occipital/nuchal
soft tissue | W+ | F+ | F+ | NA | + | NA | F::T | WT |
|
|
| 58 | M | Ethmoidal
cells/frontal sinus | NA | NA | NA | NA | NA | NA | E::T | Deletion |
|
| Fang et al,
2024 | 13 | M | Mandible | + | + | + | F+ | + | - | F::T | NA | (34) |
| Machado et
al, 2025 | 49 | F | Cutaneous (left
lower back) | F+ | + | + | + | + | + | F::T | WT | (25) |
|
| 55 | M | Cutaneous (right
flank) | NA | + | F+ | + | + | NA | F::T | WT |
|
|
| 67 | M | Cutaneous (thoracic
skin) | F+ | + | F+ | + | + | - | F::T | Upregulation |
|
|
| 25 | M | Cutaneous
(shoulder/back) | + | + | + | + | + | - | E::T | Upregulation,
rearrangement |
|
| Flaitz, 2022 | 15 | F | Mandible | R + | D+ | R+ | NA | + | NA | F::T | NA | (26) |
| Plotzke et
al, 2024 | 18 | M | Femur | - | + | F+ | + | + | NA | E::T | Upregulation | (35) |
| Zhong et al,
2024 | 26 | M | Mandible | - | S+ | + | + | + | NA | F::T | NA | (27) |
| Gallagher et
al, | 58 | M | Maxilla | + | + | + | + | + | NA | T | NA | (28) |
| 2023 | 22 | M | Maxilla | NA | + | + | + | + | NA | T | NA |
|
|
| 43 | F | Zygomatic
region | - | + | + | + | + | NA | NA | NA |
|
| Tagami et
al, 2019 | 70 | F | Lumbar
vertebra | F+ | + | F+ | + | + | NA | F::T | NA | (29) |
| Dashti et
al, 2018 | 72 | M | Mandible | + | + | + | + | + | NA | F::T | Upregulation | (30) |
| Bradova et
al, 2024 | 65 | M | Supraumbilical area
soft tissues | Few+ | D+ | NA | F+ | D+ | + | F::T | NA | (31) |
|
| 7 | F | Mandible | NA | D+ | Few+ | F+ | D+ | NA | E::T,2*V::A | NA |
|
|
| 51 | F | Maxilla | NA | D+ | NA | W+ | D+ | NA | F::T | NA |
|
| Ma et al,
2023 | 8 | F | Bladder | - | + | + | - | F+ | NA | E::T | Deletion | (36) |
| Brunac et
al, 2020 | 16 | F | Craniovertebral
junction | + | + | + | - | + | NA | F::T | NA | (37) |
| Present study | 29 | M | Lumps in the
ribs | Few+ | + | + | F+ | F+ | + | F::T | NA |
|
| Table II.Clinicopathological features. |
Table II.
Clinicopathological features.
| First author,
year | Age, years | Sex | Location | Cytological | Evolution | Treatment | Outcome, time in
months | (Refs.) |
|---|
| Ishiyama et
al, 2023 | 58 | M | Cutaneous
(scalp) | s, ro | Local recur, met to
lymph node | ST | AWD, 7 | (1) |
| Si et al,
2025 | 39 | F | Mandible | s, e | No | ST, CHT, RT | AWOD, 3 | (9) |
| Le Loarer et
al, 2020 | 16 | F | Sphenoid bone | s, e | Pro, local recur
and met femur bone | ST, CHT | DOD, 15 | (7) |
|
| 26 | F | Sacrum | e | Local pro | CHT | DOD, 4 |
|
|
| 38 | F | Peritoneum | s, e | Pro and
lymphangitic cardinomatosis | CHT | DOD, 2 |
|
|
| 32 | M | Hard palate and
upper lip | s, e | Local pro | CHT | DOD, 8 |
|
|
| 20 | M |
Orbito-temporo-sphenoid | s, e | Local pro | CHT | DOD, 6 |
|
|
| 86 | M | Inguinal | s, e | Local recur and
pro | ST | DOD, 6 |
|
|
| 18 | F | Femur | s, e | Local pro | CHT | DOD, 8 |
|
|
| 17 | F | Cervico-occipital
junction | ro | Stable with ALK
inhibitor | CHT, RT,
Crizotinib, alectinib | AWD, 15 |
|
|
| 31 | M | Occipital bone | s, e | Local recur, lung
met | ST | DOD, 6 |
|
|
| 32 | M | Mandible | s | Local recur,
mandible, lung, soft tissue met | ST, CHT | AWD, 14 |
|
|
| 58 | F | Mandible | s, e | No | ST, CHT, RT | ANED, 21 |
|
|
| 12 | F | Mandible | s, e | No | ST, CHT, RT | ANED, 21 |
|
|
| 11 | F | Maxilla | e | Local pro | CHT, RT | DOD, NA |
|
|
| 25 | M | Mandible | e | No | ST, CHT | ANED, 20 |
|
| Panferova et
al, 2022 | 16 | F | Mandible | s | Local recur, lymph
node met | ST, CHT, RT | DOD, 11.5 | (10) |
| Duan et al,
2023 | 54 | F | Lower back,
skin | s, e | Lung met | ST, Crizotinib,
alectinib | AWOD, 11 | (11) |
|
| 28 | M | Maxilla | s, ro, e | NA | ST, RT | NA, 3 |
|
| Chen et al,
2022 | 31 | F | Maxilla | s | Lymph node | ST | AWD, 5 | (12) |
|
| 49 | F | Maxilla | s, e | Recur | ST, RT,
apatinib | AWD, 32 |
|
| Li et al,
2024 | 6-36, | F (n=9), | Head and neck
region | s, e(n=14) | met or
recur(n=12), | ST, CHT | DOD(n=7), | (13) |
|
| mid2 | M (n=5) | (n=9), pelvis
(n=2), |
| no
(n=1),NA(n=1) | (n=8);ST, | AWD(n=5), |
|
|
| (n=14) |
| bladder (n=1),
pubic bone |
|
| CHT, RT | AWOD(n=1), |
|
|
|
|
| (n=1), abdominal
wall, |
|
| (n=5;CHT | NA(n=1), |
|
|
|
|
| humerus and
pubic |
|
| (n=1) anti- | 5-37 |
|
|
|
|
| bone (n=1) |
|
| ALK c |
|
|
| Carrillo-Ng et
al, | 26 | F | Mandible | s | Recur | ST, RT | AWD, 60 | (14) |
| 2023 | 59 | F | Mandible | s | Recur | ST | AWD, 22 |
|
| Demirkesen et
al, 2023 | 35 | F | Scapular-skin | s, e#(R) | Local recur, met to
lymph node | ST, RT | AWD, 9 | (15) |
| Csizmok et
al, 2024 | 31 | M | Mandible | s, e | Local recur, met to
lung met alectinib | ST, CHT, RT, | DOD, 12 | (32) |
| Haug et al,
2023 | 55 | F | Thoracic
vertebrae | s, e | Recur | CHT, RT | DOD, 6 | (17) |
| Chrisinger et
al, 2020 | 20 | F | Pelvic | s, e, ro, rha | Met (lung, gluteal
soft tissue nodules, liver, osseous) | CHT, RT | DOD, 11 | (18) |
|
| 20-30 | F | Frontal bone | s, e | Recur in
acetabulum, iliac bone, lung | ST, CHT, RT | DOD, 17 |
|
| Li et al,
2023 | 30 | F | Abdominal wall | s, e | Recur | ST, TCM | AWD, 24 | (33) |
| Agaram et
al, 2019 | 27 | M | Other bone | s | NA | NA | NA | (5) |
| Koutlas et
al, 2021 | 15 | M | Mandible | s, e | Met (lymph node,
bone) | ST, CHT | AWD, 7 | (19) |
| Dehner et
al, 2023 | 21 | F | Skull | s, e | Local recur | RT | AWD, 2 | (8) |
|
| 30 | F | Maxilla | s, e | Local recur | ST, CHT, RT | DOD, 24 |
|
|
| 60 | M | Right shoulder | s, e | Met to lymph
node | ST | NA |
|
|
| 18 | M | Left pubic
ramus | s, e | Met (lymph nodes,
bone, bilateral lung, skull) | CHT | AWD, 3 |
|
|
| 43 | F | Maxilla | s, e, rha | Met to bone | CHT | AWD, 24 |
|
|
| 31 | F | Posterior iliac
crest | s, scl | Met to bone | Palliative
care | DOD, 1 |
|
|
| 41 | F | Pelvic | s, scl | Met to bone,
lung | CHT, RT | AWD, 8 |
|
|
| 32 | F | L5 vertebra | s, scl | Met to bone,
retroperitoneum, soft tissue | ST, CHT | DOD, 15.5 |
|
|
| 26 | F | Left iliac
bone | s, e | Met (lung, bone,
thyroid, adrenal, liver, soft tissue) | CHT, RT | DOD, 11 |
|
|
| 48 | F | Pelvic | s, scl | Met to lung, bone,
liver | CHT | DOD, 5 |
|
|
| 43 | M | Fourth rib | s, e | Met to soft tissue,
bone | CHT, RT | DOD, 5 |
|
|
| 13 | M | Maxilla | s, e | NA | CHT, RT | NA |
|
|
| 27 | F | Maxilla | s, e | NA | NA | NA |
|
|
| 44 | M | Ileum | s, e, rha | NA | CHT | AWD, 1 |
|
|
| 70 | F | Mandible | s, e | NA | NA | NA |
|
|
| 62 | F | T7 vertebra | s, e | No | ST, CHT | DOD, 34 |
|
| Xu et al,
2021 | 22 | M | Mandible | s, e | Met to lymph
node | CHT, RT | NA | (20) |
|
| 27 | F | Skull | s, e | Met to bone | NA | AWD, 1 |
|
|
| 20 | F | Maxilla | s, e | Met to bone | NA | NA |
|
|
| 29 | M | Skull | s, e | Met to lung | NA | AWD, 2 |
|
|
| 33 | F | Maxilla | s, e | NA | ST | NED, 108 |
|
|
| 18 | M | Skull | s, e | NA | NA | NA |
|
|
| 40 | F | Neck superficial
soft tissue | s, ro, e | NA | NA | NA |
|
|
| 43 | F | Mandible | s, e | NA | CHT, RT | NA |
|
|
| 34 | M | Mandible | s, e, rha | No | CHT, RT | AWD, 10 |
|
|
| 16 | M | Mandible | s, e, rha | Recur, met (bone,
lung, lymph node) | CHT, RT | DOD, 20 |
|
| Zhu et al,
2019; | 74d | F | Maxilla | s, e | Recur, met to lymph
node | NA | DOD, 21 | (16) |
| Silva Cunha et
al, 2022 | 17 | F | Maxilla | s, e | Recur | ST, CHT | DOD, 9 | (21) |
| Valerio et
al, 2023 | 19 | F | Mandible | s, e | Recur | ST, CHT, RT,
alectinib, Lorlatinib | DOD, 11 | (22) |
| Watson et
al, 2018 | 27 | F | Chest wall | e | NA | NA | DOD, 5 | (6) |
|
| 27 | F | Pelvic | e | NA | NA | DOD, 5 |
|
|
| 27 | F | Sphenoid bone | e | NA | NA | DOD, 5 |
|
| Chen et al,
2024 | 40 | F | Mandible | s, e | No | ST, RT | AWOD, 6 | (23) |
| Schöpf et
al, 2024 | 17 | F | Iliac bone | NA | Met (bone, pleura,
lung) | ST, CHT, RT,
alectinib, lorlatinib | DOD, 34 | (24) |
|
| 60 | M | Mediastinum | s | met to lung | ST, RT,
crizotinib | DOD, 20 |
|
|
| 9 | F | Mandible | NA | Local pro, met
(lung, lymph nodes) | CHT | DOD, 25 |
|
|
| 48 | M | Maxillar | s | Met (lung, lymph
nodes, bone) | ST, CHT,
crizotinib | DOD, 16 |
|
|
| 35 | M | Occipital/nuchal
soft tissue | s | Recur, met (lung,
lymph nodes, malignant pleural effusion) | ST, ceritinib | DOD, 42 |
|
|
| 49 | F | Mandible | s, e, rha | Met to lymph
nodes | ST, CHT, RT | DOD, 36 |
|
|
| 25 | M | Shoulder soft
tissue | e | Met (lymph nodes,
lung, bone) | ST, RT | AWD, 19 |
|
|
| 14 | F | Maxillar | NA | Local pro | CHT | DOD, 14 |
|
|
| 15 | F | Temporal/sphenoid
bone | s, e, rha | Local pro | CHT, RT | DOD, 9 |
|
|
| 38 | M | Maxillar | s, e, rha | Local relapse,
NA | ST, CHT, RT,
crizotinib | DOD, 48 |
|
|
| 40 | F | Occipital/nuchal
soft tissue | s | Local relapse,
NA | ST | DOD, 42 |
|
|
| 58 | M | Ethmoidal
cells/frontal sinus | s | Local pro | CHT,
crizotinib | DOD, 33 |
|
| Fang et al,
2024 | 13 | M | Mandible | s | Met (vertebra and
chest) | ST, CHT | DOD, 6 | (34) |
| Machado et
al, 2025 | 49 | F | Cutaneous (left
lower back) | s, e | No | NA | NED, 4 | (25) |
|
| 55 | M | Cutaneous (riht
flank) | e | No | ST, RT | NED, 49 |
|
|
| 67 | M | Cutaneous (thoracic
skin) | s, e | Local recur | ST, crizotinib | NED, 50 |
|
|
| 25 | M |
Cutaneous(shoulder/back) | e, rha | Lymph node, met
(lung and bone) | ST, RT | DOD, 19 |
|
| Flaitz, 2022 | 15 | F | Mandible | s, e | NA | ST, CHT, RT | NA | (26) |
| Plotzke et
al, 2024 | 18 | M | Femur | s, e | Recur, met to bone
chest | ST, CHT | DOD, 25 | (35) |
| Zhong et al,
2024 | 26 | M | Mandible | s, e | Local recur | ST | DOD, 6 | (27) |
| Gallagher et
al, 2023 | 58 | M | Maxilla | s, e | NA | CHT | DOD, 3 | (28) |
|
| 22 | M | Maxilla | s, e | NA | NA | NA |
|
|
| 43 | F | Zygomatic
region | s, e | NA | NA | NA |
|
| Tagami et
al, 2019 | 70 | F | Lumbar
vertebra | s, ro, rha | No | CHT, RT | AWD, 6 | (29) |
| Dashti et
al, 2018 | 72 | M | Mandible | s, e | No | ST | AWOD, 2 | (30) |
| Bradova et
al, 2024 | 65 | M | Supraumbilical area
soft tissues | S | No | ST | ANED, 38 | (31) |
|
| 7 | F | Mandible | s, e | No | RT and CHT,
crizotinib | AWD, 8 |
|
|
| 51 | F | Maxilla | s, e | No | ST | AWD, 48 |
|
| Ma et al,
2023 | 8 | F | Bladder | s | Local recur, met to
lung | ST, CHT | DOD, 14 | (36) |
| Brunac et
al, 2020 | 16 | F | Craniovertebral
junction | ro | Recur, stable with
ALK inhibitor | ST, CHT, RT,
crizotinib, alectinib, lorlatinib | AWD, 19 | (37) |
| Present study | 29 | M | Lumps in the
ribs | s, e, rha | Pro | ST | DOD, 4 |
|
Histologically, the tumors are composed of a mixture
of different cellular phenotypes, including spindle cells,
epithelioid cells, round cells and rhabdomyoblasts. Among the 109
cases reported in the literature (including the present case),
except for 3 cases without morphological description, the most
frequent pattern described is a combined spindle and epithelioid
morphology (64 cases), followed by heterogeneous mixtures of two to
four cell types (18 cases). Pure phenotypes include spindle cell
(14 cases), epithelioid (8 cases) and round cell (2 cases).
Immunohistochemically, TFCP2-RMS invariably exhibits
dual myogenic and epithelial differentiation. The myogenic
compartment shows uniform, robust expression: MyoD1 (91/91; 100%),
desmin (91/95; 95.8%) and myogenin (57/91; 62.6%). Epithelial
commitment is reflected in diffuse, strong staining for cytokeratin
(70/86; 81.4%) and EMA (13/41; 31.7%). ALK is expressed in nearly
all cases (81/90; 90.0%).
Among 109 initially enrolled cases, molecular
genetic testing was successfully performed on 108 cases (1 excluded
due to insufficient tissue). FUS::TFCP2 fusions were identified in
67 cases (62.0%), including 1 case of dual fusion (FUS::TFCP2 and
TIMP3::ALK), while EWSR1::TFCP2 fusions were detected in 38 cases
(35.2%), including 1 case of triple fusion (EWSR1 exon5::TFCP2
exon2, VAX2 exon2::ALK exon2 and VAX2 intron2::ALK exon2). FISH
revealed TFCP2 rearrangements with unknown partners in 3 cases
(2.8%). Among 49 cases analyzed by RNA sequencing and FISH, ALK
alterations included: No alterations (19/49;38.8%), focal
upregulation (16/49; 32.7%), partial deletions (12/49; 24.5%) and
ALK gene fusions (2/49; 4.1%).
Treatment for TFCP2-RMS primarily involves surgery,
frequently supplemented with chemotherapy and/or radiotherapy.
After excluding 15 undocumented cases from the literature review,
94 patients received documented treatments: 1 underwent radiation
therapy, 1 received palliative care for multiple metastases, 14 had
chemotherapy, 17 underwent surgery and 61 received surgery with
radiotherapy and/or chemotherapy. Among these 94 patients, 19 were
treated with ALK inhibitors) (7,11–13,22,24,25,31,32,37).
Due to limited treatment duration and follow-up, the prognosis was
unknown for 8 of these ALK-treated patients. Of the remaining 11, a
subgroup of 6 experienced temporary remission before eventual
disease progression, while 5 maintained stable disease while on ALK
inhibitors. Follow-up information was available for 93 patients.
Among the 93 patients followed up for a mean time of 34 months
(median, 15 months; range, 0–108 months), 55.9% (n=52) died of the
disease within a mean time of 18 months (median, 14 months; range,
1–48 months), whereas 30.1% (n=28) remained alive with disease
after a mean follow-up time of 17.6 months (median, 14.5 months;
range, 1–60 months), while 14.0% (n=13) were disease-free with a
mean follow-up time of 27 months (median, 21 months; range, 2–108
months).
Univariate comparisons were used to statistically
evaluated clinical parameters as potential predictors of overall
survival (OS) in patients, using Kaplan-Meier curves and log-rank
tests (Table III). The median
survival time was 21 months (Fig.
S2). Among the clinical parameters of sex, age, ALK status,
treatment, recurrence and tumor location, patients <30 years
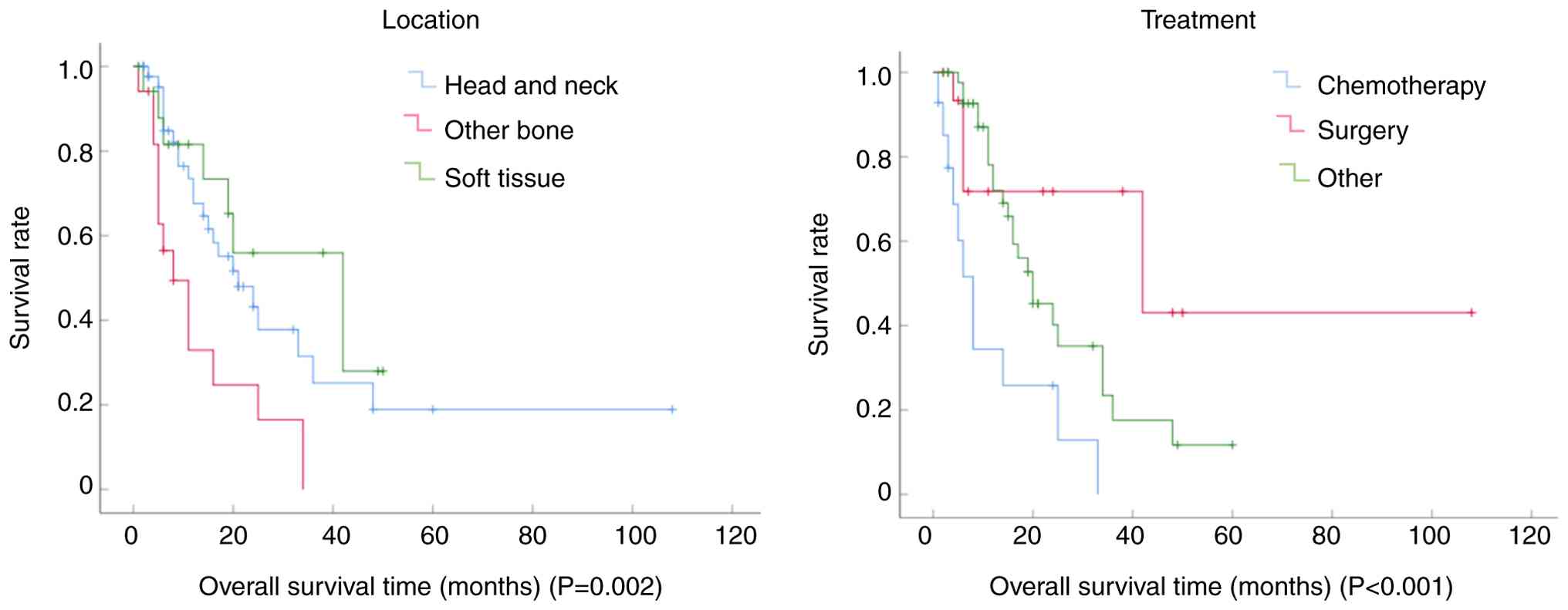
showed the worst prognosis. Tumor location significantly impacted
survival, with bone tumors arising outside the head and neck region
associated with a worse prognosis. By contrast, soft-tissue tumors
demonstrated no statistically significant survival difference
compared with head and neck tumors (Fig. 4A). Treatment with standard
chemotherapy adversely affected OS compared with surgery alone or
combined modality therapy (surgery with radiotherapy and/or
chemotherapy), which was potentially attributable to more advanced
clinical stages at diagnosis (Fig.
4B). Patients experiencing disease recurrence also had a
significantly poorer prognosis. However, no statistically
significant associations were observed for sex or ALK status levels
(Table III).
| Table III.Overall survival of selected
prognostically relevant parameters. |
Table III.
Overall survival of selected
prognostically relevant parameters.
| Concomitant
variable | Standard error (95%
confidence interval) | P-value |
|---|
| Sex | 0.423
(20.170–21.830) | 0.106 |
| Age | 2.929
(14.258–25.742) | 0.011a |
| ALK | 0.423
(20.170–21.830) | 0.066 |
| Treatment | 3.704
(121.741–27.259) |
<0.001a |
| Recurrence | 4.166
(11.835–28.165) | 0.001a |
| Location | 2.929
(14.258–25.742) | 0.002a |
Discussion
TFCP2-RMS is an extremely rare tumor, with only 108
cases having been reported to date, to the best of our knowledge
(6). The present study aims to
broaden our knowledge about the biological behavior, histological
characteristics, treatment and prognosis of these rare tumors.
Despite diverse histological patterns, TFCP2-RMS
typically displays features of mesenchymal malignant neoplasms,
including infiltrative growth, marked cellular pleomorphism, high
mitotic activity and necrosis. Rhabdomyoblasts are either absent or
only focally present. Immunohistochemistry is required to confirm a
RMS diagnosis. Notably, desmin, MyoD1 and myogenin are positively
expressed in TFCP2-RMS. Additionally, CK and ALK positivity are
critical for diagnosing TFCP2-RMS (6,13).
NGS-detected homozygous loss of CDKN2A/CDKN2B
simultaneously disables the two key cell-cycle brakes,
retinoblastoma-associated protein and tumor protein p53 (p53), in
FUS-TFCP2 RMS (38). This dual
disruption may explain the rapid progression of the tumor despite
only moderate Ki-67 labeling, providing both a biomarker of
aggressive behavior and a rationale for CDK4/6 or p53
pathway-directed therapies.
The TFCP2 gene, also known as late SV40 factor,
encodes a transcription factor CP2 (also known as late SV40
factor), and is located on human chromosome 12q13.12 (12). TFCP2 plays a critical role in DNA
synthesis, cell survival and anti-apoptosis by regulating cell
cycle-related genes and modulating apoptosis-related genes (such as
Bcl-2 family members). Aberrant TFCP2 expression or rearrangement
has been implicated in diverse cancer types, including
hepatocellular, pancreatic, renal, thyroid, oral, breast, cervical
and colorectal cancer, where its upregulation or gene fusion drives
tumor progression and correlates with a poor prognosis (39,40).
For TFCP2-RMS, TFCP2 rearrangements generate fusion proteins that
further augment tumor cell proliferation and aggressiveness, which
may underlie the high aggressiveness and poor prognosis of this
subtype.
TFCP2-RMS, a bone-predominant sarcoma, should be
differentiated from metastatic sarcomatoid carcinoma, mesenchymal
chondrosarcoma, osteosarcoma, dedifferentiated chondrosarcoma and
epithelioid sarcoma-like hemangioendothelioma. For soft-tissue
tumors, the main differential diagnoses include epithelioid
hemangioendothelioma, pseudomyogenic hemangioendothelioma, pleural
malignant mesothelioma, malignant peripheral nerve sheath tumor,
inflammatory myofibroblastic tumor, EWSR1-POZ/BTB and AT hook
containing zinc finger 1 fusion-associated spindle and round cell
sarcomas, and embryonal rhabdomyosarcoma (12).
The standard treatment regimen comprises surgery
combined with chemoradiotherapy. The present survival analysis
demonstrated that surgical intervention alone or in conjunction
with radiotherapy resulted in significantly improved overall
survival compared with chemotherapy alone, which may offer critical
guidance for clinical decision-making. Despite a median overall
survival time of 21 months reported in general, the present case
presented with lymph node metastasis at diagnosis, which precluded
chemotherapy, and the survival time was only 4 months. We
hypothesize that tumors originating in the ribs may exhibit
particularly aggressive biological behavior, a finding consistent
with the observation that primary bone tumors at other sites are
associated with shorter survival times than those in the head and
neck. Notably, another rib case in the literature also demonstrated
a limited survival time (5 months) despite receiving
chemoradiotherapy without surgical resection (8).
In conclusion, TFCP2-RMS represents a rare and
highly aggressive subtype of RMS; it predominantly involves the
craniofacial bones in young males, with the present case being only
the second reported example of rib origin. The unusual rib location
and diffuse CK expression pose significant diagnostic challenges.
Heightened recognition of its key clinicopathological features,
predominance in young adults, epithelioid-spindled morphology,
expression of rhabdomyoblastic markers and frequent ALK positivity,
is therefore critical for accurate diagnosis. Molecular detection
of TFCP2 translocation further confirms the diagnosis. Notably,
bone tumors arising outside the head and neck, recurrence, standard
chemotherapy use and age <30 years adversely impact overall
survival time.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by the Natural Science Foundation of
Guangdong Province of China (grant no. 2023A1515012384) and
Guangdong Provincial Medical Science and Technology Research Fund
(grant no. A2023190).
Availability of data and materials
The sequencing data generated in the present study
are available in the NCBI SRA under accession numbers SRP650875
(BioProject) and SRR36280525 (Run). The respective URLs are:
https://trace.ncbi.nlm.nih.gov/Traces/?view=study&acc=SRP65087
and https://www.ncbi.nlm.nih.gov/sra/SRR36280525.
Additional data are available from the corresponding author upon
request.
Authors' contributions
DLY, MW and WW conceived and designed the study. DLY
and MW wrote the manuscript. JWZ, WPZ and JZ acquired MRI and CT
images, and performed immunohistochemical analysis. GNY and DLY
analyzed and interpreted the results. All authors have read and
approved the final manuscript. DLY, MW and WW confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
This study involving humans was approved by the
Ethics Committee of General Hospital of Southern Theater Command,
People's Liberation Army of China (Guangzhou, China; approval no.
NZLLKZ2025021). The study was conducted in accordance with the
local legislation and institutional requirements. The participants
provided their written informed consent to participate in this
study.
Patient consent for publication
Written informed consent was obtained from the
patient for the case information and images to be published in the
present case report.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ishiyama T, Kato I, Ito J, Matsumura M,
Saito K, Kawabata Y, Kato S, Takeyama M and Fujii S:
Rhabdomyosarcoma with FUS::TFCP2 fusion in the scalp: A rare case
report depicting round and spindle cell morphology. Int J Surg
Pathol. 31:805–812. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Alaggio R, Zhang L, Sung YS, Huang SC,
Chen CL, Bisogno G, Zin A, Agaram NP, LaQuaglia MP, Wexler LH and
Antonescu CR: A molecular study of pediatric spindle and sclerosing
rhabdomyosarcoma: Identification of novel and recurrent
VGLL2-related fusions in infantile cases. Am J Surg Pathol.
40:224–235. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Karanian M, Pissaloux D, Gomez-Brouchet A,
Chevenet C, Le Loarer F, Fernandez C, Minard V, Corradini N, Castex
MP, Duc-Gallet A, et al: SRF-FOXO1 and SRF-NCOA1 fusion genes
delineate a distinctive subset of well-differentiated
rhabdomyosarcoma. Am J Surg Pathol. 44:607–616. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Agaram NP, LaQuaglia MP, Alaggio R, Zhang
L, Fujisawa Y, Ladanyi M, Wexler LH and Antonescu CR: MYOD1-mutant
spindle cell and sclerosing rhabdomyosarcoma: An aggressive subtype
irrespective of age. A reappraisal for molecular classification and
risk stratification. Mod Pathol. 32:27–36. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Agaram NP, Zhang L, Sung YS, Cavalcanti
MS, Torrence D, Wexler L, Francis G, Sommerville S, Swanson D,
Dickson BC, et al: Expanding the spectrum of intraosseous
rhabdomyosarcoma: Correlation between 2 distinct gene fusions and
phenotype. Am J Surg Pathol. 43:695–702. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Watson S, Perrin V, Guillemot D, Reynaud
S, Coindre JM, Karanian M, Guinebretiere JM, Freneaux P, Le Loarer
F, Bouvet M, et al: Transcriptomic definition of molecular
subgroups of small round cell sarcomas. J Pathol. 245:29–40. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Le Loarer F, Cleven AHG, Bouvier C, Castex
MP, Romagosa C, Moreau A, Salas S, Bonhomme B, Gomez-Brouchet A,
Laurent C, et al: A subset of epithelioid and spindle cell
rhabdomyosarcomas is associated with TFCP2 fusions and common ALK
upregulation. Mod Pathol. 33:404–419. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dehner CA, Broski SM, Meis JM, Murugan P,
Chrisinger JSA, Sosa C, Petersen M, Halling KC, Gupta S and Folpe
AL: Fusion-driven spindle cell rhabdomyosarcomas of bone and soft
tissue: A clinicopathologic and molecular genetic study of 25
cases. Mod Pathol. 36:1002712023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Si C, Wang Y and Zhu J: A rare case report
of intraosseous spindle and epithelioid rhabdomyosarcoma with TFCP2
rearrangement: A pathological diagnostic conundrum and literature
review. Int J Surg Pathol. 33:125–130. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Panferova A, Sinichenkova KY, Abu Jabal M,
Usman N, Sharlai A, Roshchin V, Konovalov D and Druy A: EWSR1-TFCP2
in an adolescent represents an extremely rare and aggressive form
of intraosseous spindle cell rhabdomyosarcomas. Cold Spring Harb
Mol Case Stud. 8:a0062092022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Duan FL, Yang H, Gong X, Zuo Z, Qin S, Ji
J, Zhou C, Dai J, Guo P and Liu Y: Clinicopathological features of
rhabdomyosarcoma with novel FET::TFCP2 and TIMP3::ALK fusion:
Report of two cases and literature review. Histopathology.
82:478–484. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen XY, Chen G, Zhu Q, Zhu WF, He C and
Huang RF: Clinicopathological features of rhabdomyosarcoma with
TFCP2 fusions. Zhonghua Bing Li Xue Za Zhi. 51:545–547. 2022.(In
Chinese). PubMed/NCBI
|
|
13
|
Li HL, Mo CH, Xie L, Wu YX, Zeng M and Mao
RJ: Clinicopathological study of epithelioid and spindle cell
rhabdomysarcoma with EWSR1/FUS-TFCP2 fusion. Zhonghua Bing Li Xue
Za Zhi. 53:58–63. 2024.(In Chinese). PubMed/NCBI
|
|
14
|
Carrillo-Ng H, Liang Y, Chang S, Afkhami
M, Gernon T, Bell D and Arias-Stella JA: Complete mimicry:
Rhabdomyosarcoma with FUS::TFCP2 fusion masquerading as
carcinoma-diagnostic challenge and report of two cases. Genes
Chromosomes Cancer. 62:430–436. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Demirkesen C, Danyeli AE, Yildiz P,
Ertekin SS, Yilmaz B, Karahan SI and Bahrami A: Cutaneous
rhabdomyosarcoma with FUS::TFCP2 fusion: A case report emphasizing
early detection. J Cutan Pathol. 50:1059–1064. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhu G, Benayed R, Ho C, Mullaney K,
Sukhadia P, Rios K, Berry R, Rubin BP, Nafa K, Wang L, et al:
Diagnosis of known sarcoma fusions and novel fusion partners by
targeted RNA sequencing with identification of a recurrent
ACTB-FOSB fusion in pseudomyogenic hemangioendothelioma. Mod
Pathol. 32:609–620. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Haug L, Doll J, Appenzeller S, Kunzmann V,
Rosenwald A, Maurus K and Gerhard-Hartmann E: Epithelioid and
spindle cell rhabdomyosarcoma with EWSR1::TFCP2 fusion mimicking
metastatic lung cancer: A case report and literature review. Pathol
Res Pract. 249:1547792023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chrisinger JSA, Wehrli B, Dickson BC,
Fasih S, Hirbe AC, Shultz DB, Zadeh G, Gupta AA and Demicco EG:
Epithelioid and spindle cell rhabdomyosarcoma with FUS-TFCP2 or
EWSR1-TFCP2 fusion: Report of two cases. Virchows Arch.
477:725–732. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Koutlas IG, Olson DR and Rawwas J:
FET(EWSR1)-TFCP2 Rhabdomyosarcoma: An additional example of this
aggressive variant with predilection for the gnathic bones. Head
Neck Pathol. 15:374–380. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu B, Suurmeijer AJH, Agaram NP, Zhang L
and Antonescu CR: Head and neck rhabdomyosarcoma with TFCP2 fusions
and ALK overexpression: A clinicopathological and molecular
analysis of 11 cases. Histopathology. 79:347–357. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Silva Cunha JL, Cavalcante IL, da Silva
Barros CC, Alves PM, Nonaka CFW, Albuquerque AFM, de Almeida OP, de
Andrade BAB and Cavalcante RB: Intraosseous rhabdomyosarcoma of the
maxilla with TFCP2 fusion: A rare aggressive subtype with
predilection for the gnathic bones. Oral Oncol. 130:1058762022.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Valerio E, Furtado Costa JL, Perez Fraile
NM, Credidio CH, Taveira Garcia MR, Neto CS and Costa FD:
Intraosseous spindle Cell/Epithelioid rhabdomyosarcoma with TFCP2
rearrangement: A recent recognized subtype with partial response to
alectinib. Int J Surg Pathol. 31:861–865. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen F, Wang J, Sun Y and Zhang J:
Mandibular rhabdomyosarcoma with TFCP2 rearrangement and osteogenic
differentiation: A case misdiagnosed as fibrous dysplasia or
low-grade central osteosarcoma. Oral Surg Oral Med Oral Pathol Oral
Radiol. 137:e143–e149. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schöpf J, Uhrig S, Heilig CE, Lee KS,
Walther T, Carazzato A, Dobberkau AM, Weichenhan D, Plass C,
Hartmann M, et al: Multi-omic and functional analysis for
classification and treatment of sarcomas with FUS-TFCP2 or
EWSR1-TFCP2 fusions. Nat Commun. 15:512024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Machado I, Wardelmann E, Zhao M, Song J,
Wang Y, Braun SA, Catasus L, Ferre M, Leoveanu I, Westhoff J, et
al: Primary cutaneous rhabdomyosarcoma with EWSR1/FUS::TFCP2
fusion: Four new cases with distinctive morphology,
immunophenotypic, and genetic profile. Virchows Arch.
486:1187–1198. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Flaitz C and Hicks J: Primary Intraosseous
Rhabdomyosarcoma: Rare Subtype Involving Mandible with Unique
Translocation. Oral Surgery Oral Med Oral Pathol Oral Radiol.
133:1572022. View Article : Google Scholar
|
|
27
|
Zhong P, Wei S, Xiao H and Zeng Y:
Rhabdomyosarcoma with FUS::TFCP2 fusion in the mandible: A
rare aggressive subtype, but can be misdiagnosed as ossifying
fibroma. Int J Surg Pathol. 32:758–766. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gallagher KPD, Roza A, Tager E, Mariz B,
Soares CD, Rocha AC, Abrahao AC, Romanach MJ, Carlos R, Hunter KD,
et al: Rhabdomyosarcoma with TFCP2 rearrangement or typical
Co-expression of AE1/AE3 and ALK: Report of three new cases in the
head and neck region and literature review. Head Neck Pathol.
17:546–561. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tagami Y, Sugita S, Kubo T, Iesato N,
Emori M, Takada K, Tsujiwaki M, Segawa K, Sugawara T, Kikuchi T and
Hasegawa T: Spindle cell rhabdomyosarcoma in a lumbar vertebra with
FUS-TFCP2 fusion. Pathol Res Pract. 215:1523992019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dashti NK, Wehrs RN, Thomas BC, Nair A,
Davila J, Buckner JC, Martinez AP, Sukov WR, Halling KC, Howe BM
and Folpe AL: Spindle cell rhabdomyosarcoma of bone with FUS-TFCP2
fusion: Confirmation of a very recently described rhabdomyosarcoma
subtype. Histopathology. 73:514–520. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bradova M, Mosaieby E, Michal M, Vanecek
T, Ing SK, Grossmann P, Koshyk O, Kinkor Z, Laciok S, Nemcova A, et
al: Spindle cell rhabdomyosarcomas: With TFCP2 rearrangements, and
novel EWSR1::ZBTB41 and PLOD2::RBM6 gene fusions. A study of five
cases and review of the literature. Histopathology. 84:776–793.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Csizmok V, Grisdale CJ, Williamson LM, Lim
HJ, Lee L, Renouf DJ, Jones SJM, Marra MA, Laskin J and Smrke A:
Diagnostic and therapeutic implications of a FUS::TFCP2 fusion and
ALK activation in a metastatic rhabdomyosarcoma. Genes Chromosomes
Cancer. 63:e232592024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li Y, Li D, Wang J and Tang J: Epithelioid
and spindle rhabdomyosarcoma with TFCP2 rearrangement in abdominal
wall: A distinctive entity with poor prognosis. Diagn Pathol.
18:412023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fang Z, Duan C, Wang S, Fu L, Yang P, Yu
T, Deel MD, Lau LMS, Ma X, Ni X and Su Y: Pediatric spindle
cell/sclerosing rhabdomyosarcoma with FUS-TFCP2 fusion: A case
report and literature review. Transl Pediatr. 13:178–191. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Plotzke JM, Rabah R, Robinson DR, Edmonds
A, Bloom DA, Mody R and Heider A: Primary intraosseous spindle cell
rhabdomyosarcoma: A case report in an unusual location. Pediatr Dev
Pathol. 27:597–602. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ma Y, Feng J, Ding D, Zhao J and Tian F:
TFCP2-rearranged epithelioid and spindle cell rhabdomyosarcoma in
the bladder: A rare case in an 8-year-old female child. Pediatr
Blood Cancer. 70:e299352023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Brunac AC, Laprie A, Castex MP, Laurent C,
Le Loarer F, Karanian M, Le Guellec S, Guillemot D, Pierron G and
Gomez-Brouchet A: The combination of radiotherapy and ALK
inhibitors is effective in the treatment of intraosseous
rhabdomyosarcoma with FUS-TFCP2 fusion transcript. Pediatr Blood
Cancer. 67:e281852020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ginn MP, Denu RA, Ingram DR, Wani KM,
Lazar AJ, Harrison DJ, Nakazawa MS, Conley AP, Patel S and
Livingston JA: TFCP2 Fusion-Positive Rhabdomyosarcomas: A Report of
10 cases and a review of the literature. Cancers (Basel).
17:14412025. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kotarba G, Krzywinska E, Grabowska AI,
Taracha A and Wilanowski T: TFCP2/TFCP2L1/UBP1 transcription
factors in cancer. Cancer Lett. 420:72–79. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hsu WH, LaBella KA, Lin Y, Xu P, Lee R,
Hsieh CE, Yang L, Zhou A, Blecher JM, Wu CJ, et al: Oncogenic KRAS
drives lipofibrogenesis to promote angiogenesis and colon cancer
progression. Cancer Discov. 13:2652–2673. 2023. View Article : Google Scholar : PubMed/NCBI
|