Introduction
Ferroptosis, as a novel form of regulated cell
death, has emerged as a promising therapeutic target in cancer
treatment. It not only effectively suppresses tumor growth, but
also reverses cancer cell resistance to conventional therapies
(1). The combined application of
ferroptosis inducers with standard treatments (such as radiotherapy
and immunotherapy) can markedly enhance the antitumor efficacy and
reduce the risk of recurrence (2).
Notably, despite their strong resistance to traditional therapies,
highly aggressive mesenchymal-like cancer cells exhibit exceptional
vulnerability to ferroptosis (3).
Therefore, targeting ferroptosis may provide new therapeutic
opportunities for the treatment of cancer.
Breast cancer is the most common malignant tumor
among women worldwide, with an estimated 2.3 million new cases
diagnosed in 2022 (4). Among these,
TNBC is attracting increasing attention due to its distinct
molecular characteristics and high aggressiveness (3). TNBC is characterized by the absence of
estrogen receptor, progesterone receptor and human epidermal growth
factor receptor 2 (HER2), rendering it insensitive to conventional
endocrine therapy (5). Patients
with TNBC generally have a poorer prognosis, with a 5-year survival
rate of approximately 77%, compared with 93% for hormone
receptor-positive breast cancers (6). Moreover, compared with other breast
cancer subtypes, TNBC exhibits greater susceptibility to
ferroptosis inducers (7).
Ferroptosis is currently being investigated as a potential
therapeutic approach for breast cancer (3,7).
Therefore, exploring the regulatory mechanisms of ferroptosis and
identifying novel therapeutic targets may provide more effective
treatment strategies for TNBC.
Inositol-requiring enzyme 1α (IRE1α) is an
endoplasmic reticulum (ER)-resident protein that serves a crucial
role in the unfolded protein response (UPR) (8). It contains both kinase and
ribonuclease (RNase) domains and is involved in several
pathophysiological processes, including immune responses, cell
death regulation and neurodegenerative diseases (9,10).
Previous research has reported that the RNase activity of IRE1α
enhances the sensitivity of TNBC cells to ferroptosis (11). However, whether the kinase activity
of IRE1α contributes to the regulation of ferroptosis in TNBC
remains unclear.
Therefore, the present study aimed to analyze the
association between IRE1α and breast cancer progression using
existing datasets. Moreover, through pharmacological inhibition and
genetic manipulation, the present study aimed to investigate the
antitumor effects of IRE1α kinase in TNBC cells and explore its
underlying molecular mechanisms of action. The findings of the
present study may deepen the current understanding of the role of
ferroptosis in TNBC and indicate novel therapeutic targets for
future research.
Materials and methods
Reagents and assay kit
Erastin (cat. no. S7242), RAS-selective lethal 3
(RSL3; cat. no. S8155), ML162 (cat. no. S4452), sulfasalazine (SAS;
cat. no. S1576), APY29 (cat. no. S6623), sunitinib (cat. no. S7781)
and IRE1α kinase-inhibiting RNase attenuator (KIRA6; cat. no.
S8658) were purchased from Selleck Chemicals. L-Glutamic acid
(glutamate; cat. no. G8415), L-Cystine (cat. no. C7602),
L-Methionine (cat. no. M5308) and L-Glutamine (cat. no. G8540) were
purchased from Sigma-Aldrich (Merck KGaA).
Lipofectamine® 3000 Transfection Reagent (cat. no.
L3000015), 0.25% Trypsin-EDTA (cat. no. 25200056), Hank's Balanced
Salt Solution (HBSS; cat. no. 14175095), Opti-MEM™ (cat.
no. 11058021), cystine-deficient DMEM (cat. no. 21013024),
BODIPY™ 581/591 C11 (cat. no. D3861), TRIzol®
reagent (cat. no. 15596018) and the SuperScript™ IV
First-Strand Synthesis System Kit (cat. no. 18091050) were
purchased from Thermo Fisher Scientific, Inc. Unless specified, the
rest of the reagents used were purchased from Beyotime
Biotechnology.
Cell line and culture conditions
The MDA-MB-231 (cat. no. SCSP-5043), MDA-MB-468
(cat. no. SCSP-5053), 4T1 (cat. no. SCSP-5056) and HT1080 (cat. no.
TCHu170) cell lines were purchased from The Cell Bank of Type
Culture Collection of The Chinese Academy of Sciences. All cells
were cultured under standard conditions in DMEM supplemented with
10% fetal bovine serum (catalog no. SFBE; NATOCOR,) and maintained
in a humidified incubator at 37°C with 5% CO2. The
medium was replaced with fresh medium every 3 days. Upon reaching
80–90% confluency, the cells were passaged using 0.25% trypsin-EDTA
solution to maintain optimal growth conditions.
Cell viability assay
The aforementioned cell lines (MDA-MB-231,
MDA-MB-468, 4T1 and HT1080) were seeded in 96-well plates (3,000
cells/well). After plating, cells were treated with the indicated
compounds (Erastin, RSL3, ML162, SAS, glutamate, APY29, sunitinib
and KIRA6) at the concentrations detailed in the corresponding
figures. After 24 h of treatment at 37°C, the culture medium was
replaced with a mixture of serum-free medium and Cell Counting
Kit-8 reagent (cat. no. 96992; MilliporeSigma) at a 9:1 ratio,
followed by incubation at 37°C for 2 h. Cell viability was assessed
by measuring the optical density at 450 nm, and the data were
normalized to the control group, as previously described (12).
Assessment of lipid peroxidation with
BODIPY and malondialdehyde (MDA)
Intracellular lipid reactive oxygen species (ROS)
were measured using flow cytometry, as previously described
(13). A total of 100,000
MDA-MB-231 cells were seeded per well in a 6-well plate. After 6 h
of treatment with the indicated compounds (erastin, 2 µM; SAS, 1
mM; glutamate, 20 mM; APY29, 200 nM) at 37°C, cells were harvested,
washed with PBS and incubated with 1 µM BODIPY 581/591 C11 in
serum-free medium for 30 min at 37°C. Cells were then resuspended
in 500 µl HBSS and filtered through a 40-µm cell strainer (BD
Biosciences). Flow cytometry analysis was immediately performed
using an LSRFortessa instrument (BD Biosciences) with a 488 nm
laser. Fluorescence signals from both unoxidized (PE channel) and
oxidized (FITC channel) C11 were monitored. The ratio of the mean
fluorescence intensity of FITC to PE was calculated. Data analysis
was performed using FlowJo v10 software (BD Biosciences).
Cellular MDA levels were assessed using a Lipid
Peroxidation Assay Kit (cat. no. ab233471; Abcam), as previously
described (14). Briefly, cells
were treated with compounds (Erastin, 2 µM; SAS, 1 mM; glutamate,
20 mM; APY29, 200 nM) for 12 h at 37°C. Samples and the MDA color
reagent were added to the wells and incubated for 20 min at room
temperature. Following this, the reaction solution was introduced,
and the mix was incubated for 60 min at room temperature. The
resulting product was subsequently assessed at 695 nm using an
absorbance microplate reader.
Intracellular glutathione (GSH)
assay
MDA-MB-231, MDA-MB-468, and 4T1 cells were treated
with compounds (Erastin, 2 µM; SAS, 1 mM; glutamate, 20 mM; APY29,
200 nM) for 24 h at 37°C and then harvested. GSH was assayed using
the GSH and GSSG Assay Kit (cat. no. S0053; Beyotime
Biotechnology), as previously described (15). Briefly, cells were washed with
ice-cold PBS, harvested and resuspended in three volumes of Protein
Removal Reagent M solution. Cell samples were subjected to two
rapid freeze-thaw cycles by alternating between liquid nitrogen and
a 37°C water bath. The corresponding assay reagents were added to
the appropriate amount of cell sample. After 25 min, absorbance was
measured at 412 nm using a microplate reader. GSH content was then
calculated based on a standard curve.
Cystine restriction treatment
The restricted medium was prepared by supplementing
glutamine-, methionine- and cystine-deficient DMEM with 4 mM
glutamine, 200 µM methionine and 10% fetal bovine serum, as
previously described (16). For
media with specific cystine concentrations, L-cystine was added to
this base restricted medium. Prior to the experiments, cells were
washed three times with ice-cold PBS to remove residual cystine and
then cultured in the cystine-restricted medium. All subsequent
assays, including lipid peroxidation and intracellular GSH
measurement, were performed under a cystine-restricted condition of
50 µM.
Western blotting
MDA-MB-231, MDA-MB-468, and 4T1 cells were harvested
and washed twice with ice cold PBS. Cells were then lysed on ice
using RIPA lysis buffer (cat. no. P0013; Beyotime Biotechnology)
containing the protease inhibitor PMSF (cat. no. ST507; Beyotime
Biotechnology). Protein samples were quantified using a BCA protein
concentration assay kit (cat. no. P0011; Beyotime Biotechnology).
Equal amounts of protein (20 µg per lane) were separated using
4–20% SDS-PAGE and transferred to PVDF membranes using wet
blotting. To reduce nonspecific binding, the membranes were blocked
with 5% skim milk for 1 h at room temperature, followed by
incubation with the corresponding primary antibodies overnight at
4°C with gentle shaking. The following day, the membranes were
washed five times with TBST buffer (containing 0.1% Tween-20) for 5
min each and then incubated with horseradish peroxidase-conjugated
secondary antibodies: Goat Anti-Mouse IgG (1:5,000; cat. no.
ab205719; Abcam) and Goat Anti-Rabbit IgG (1:5,000; cat. no.
ab205718; Abcam) at 30°C for 2 h. After washing three times with
TBST (containing 0.1% Tween-20), the membranes were developed using
an enhanced chemiluminescence detection reagent (cat. no. P10300;
NCM Biotech, China) and images were captured using a ChemiScope
6100 Chemiluminescence Imaging System (CLiNX Science Instruments
Co., Ltd.). Finally, grayscale analysis of target bands was
performed using ImageJ software (v1.53a; National Institutes of
Health). The primary antibodies used in this experiment were as
follows: glutathione peroxidase 4 (GPX4; 1:5,000; cat. no.
ab125066; Abcam), acyl-CoA synthetase long chain family member 4
(ACSL4; 1:5,000; cat. no. ab155282; Abcam), IRE1α (1:1,000; cat.
no. 3294T; Cell Signaling Technology, Inc.), solute carrier family
7 member 11 (SLC7A11; also known as xCT; 1:1,000; cat. no. 12691T;
Cell Signaling Technology, Inc.), and the internal controls β-actin
(1:1,000; cat. no. sc-47778; Santa Cruz Biotechnology, Inc.) and
GAPDH (1:1,000; cat. no. 2118T; Cell Signaling Technology,
Inc.).
Establishment of cell models
overexpressing IRE1α wild-type (WT) or kinase-dead mutants
Human IRE1α WT (cat. no. 20744; Addgene, Inc.) and
human IRE1α kinase-dead mutants (K599A; cat. no. 20745; Addgene,
Inc.) were transfected into MDA-MB-231 cells as previously
described (17,18). Lentiviral particles were produced
using the second-generation packaging plasmids psPAX2 (cat. no.
12260; Addgene, Inc.) and pMD2.G (cat. no. 12259; Addgene, Inc.).
For each transfection, 4 µg of the IRE1α transfer plasmid was
combined with 3 µg of the packaging plasmid (psPAX2) and 1 µg of
the envelope plasmid (pMD2.G). Specifically, the plasmid mix was
diluted in 250 µl Opti-MEM containing 5 µl P3000 reagent, and 10 µl
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) was diluted in 250 µl Opti-MEM. The two solutions
were mixed, incubated at room temperature for 15 min and then added
to the 293T cell (ATCC) culture. Transfected cells were maintained
at 37°C with 5% CO2 for 48–72 h. The lentiviral
supernatant was collected at 24 and 48 h post-transfection, pooled,
filtered (0.45 µm), and concentrated. For transduction, MDA-MB-231
cells were infected at a multiplicity of infection (MOI) of 5 in
the presence of 8 µg/ml polybrene for 24 h. Stable cell lines were
subsequently selected using 2 µg/ml puromycin for 7 days, starting
48 h post-transduction. Following a 5–7 day recovery period,
overexpression was verified by western blotting.
Bioinformatic analysis
Bioinformatics analysis was performed using
web-based bioinformatics tools. ERN1 levels in normal and
breast cancer samples, as well as the profiles of ERN1
levels in different molecular subtypes of breast cancer, were
analyzed using the online University of ALabama at Birmingham
CANcer data analysis Portal (UALCAN) (19) (https://ualcan.path.uab.edu/). The levels of
ERN1 in several malignant tumors were analyzed based on the
web tool Gene Expression Profiling Interactive Analysis 2 (20) (http://gepia2.cancer-pku.cn). The online Kaplan-Meier
Plotter (21) (http://kmplot.com/analysis) was employed to analyze
recurrence-free survival differences between ERN1-high and
-low groups in both overall breast cancer and HER2-positive
subtypes, using the default parameters (probe: 227755_at; cut-off:
median) based on its integrated breast cancer gene expression
dataset. For TNBC-specific analysis, the GSE21653 dataset
(available within the Kaplan-Meier Plotter tool) (22) was selected, applying the
‘auto-select best cut-off’ function.
RNA extraction, cDNA synthesis and
reverse transcription-quantitative PCR (qPCR)
MDA-MB-231, MDA-MB-468 and 4T1 cells were treated
with APY29 (200 nM) for 24 h at 37°C and subsequently harvested.
Total RNA was isolated using TRIzol reagent. cDNA was synthesized
using the SuperScript IV First-Strand Synthesis System with the
following protocol: 25°C for 10 min, 50°C for 10 min, and 80°C for
10 min. qPCR was performed using the SsoAdvanced Universal
SYBR® Green SuperMix (cat. no. 1725270; Bio-Rad
Laboratories, Inc.). The thermocycling conditions were as follows:
initial denaturation at 95°C for 30 sec, followed by 40 cycles of
95°C for 15 sec and 60°C for 30 sec. Gene expression was normalized
to the endogenous reference gene GAPDH using the 2−∆∆Cq
method (23). The results are
expressed as fold changes relative to the control group. Primer
sequences used for qPCR were as follows (24): xCT forward,
5′-TGTGTGGGGTCCTGTCACTA-3′; xCT reverse,
5′-CAGTAGCTGCAGGGCGTATT-3′; GAPDH forward,
5′-ATGGGGAAGGTGAAGGTCG-3′; and GAPDH reverse,
5′-GGGGTCATTGATGGCAACAATA-3′.
Statistical analysis
All experimental data are presented as mean ±
standard error of the mean. Unpaired t-test and one- or two-way
ANOVA with Tukey's post hoc test were performed using GraphPad
Prism 8.0 software (Dotmatics). P<0.05 was considered to
indicate a statistically significant difference. All cell culture
experiments were independently repeated at least three times to
ensure data reproducibility and reliability.
Results
IRE1α is associated with the
progression of breast cancer
Previous research reported that the activation of
IRE1α can promote breast cancer development (25). The present study thus hypothesized
that IRE1α may be pathologically associated with breast cancer and
validated this using existing clinical datasets. Using the online
tool UALCAN, ERN1 (which encodes IRE1α) levels were
analyzed. The results revealed that ERN1 expression was
significantly downregulated in breast cancer tissues compared with
that in normal tissues (Fig. 1A).
Notably, the ERN1 levels were significantly lower in the
more aggressive HER2-positive and TNBC subtypes (26), than in the luminal subtype (Fig. 1B). This suggests that ERN1
may serve a critical role in the pathological process of
HER2-positive and TNBC. Furthermore, pan-cancer analysis further
demonstrated that ERN1 is generally downregulated in
multiple cancer types compared with that in normal tissues
(Fig. S1). Kaplan-Meier survival
analysis also revealed that a low expression of ERN1 was
significantly associated with a worse prognosis of patients with
breast cancer (Fig. 1C). In
patients who were HER2-positive and in those with TNBC with worse
clinical outcomes (27), a lower
expression of ERN1 was also significantly associated a worse
prognosis (Fig. 1D and E).
Collectively, these findings highlight the potential of targeting
IRE1α for breast cancer therapy.
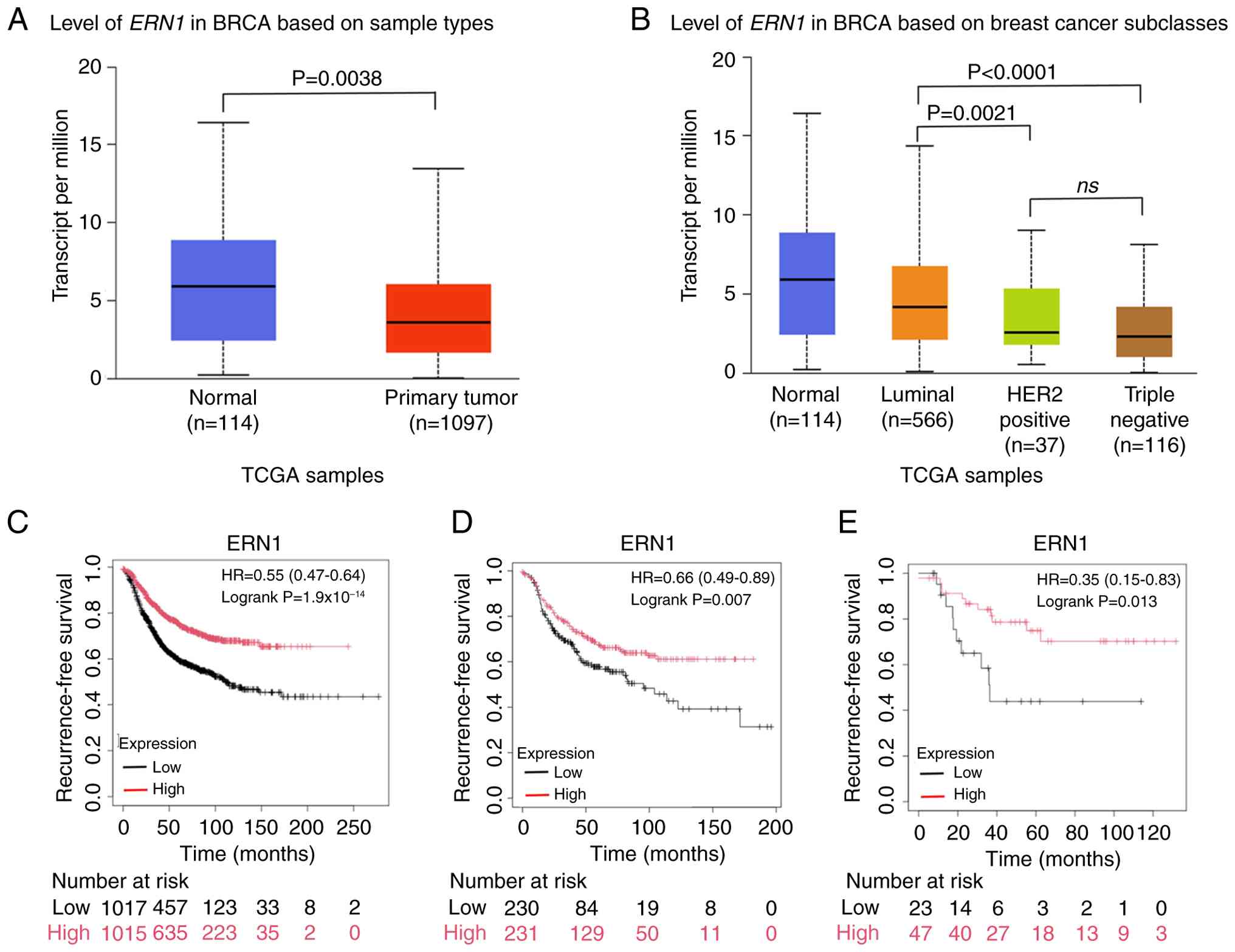 | Figure 1.IRE1α is associated with BRCA
progression. (A) UALCAN analysis of the level of ERN1 in
BRCA. (B) UALCAN analysis of the level of ERN1 in BRCA based
on BRCA subclasses. The Kaplan-Meier Plotter was used to analyze
the outcomes patients with BRCA, and the differences in
recurrence-free survival were compared between groups stratified by
ERN1 status: (C) Overall BRCA, (D) HER2-positive breast
cancer subset and (E) triple negative BRCA subset. IRE1α,
inositol-requiring enzyme 1α; UALCAN, University of ALabama at
Birmingham CANcer data analysis Portal; ERN1, endoplasmic
reticulum to nucleus signaling 1; BRCA, breast cancer; TCGA, The
Cancer Genome Atlas; HER2, human epidermal growth factor receptor
2; HR, hazard ratio; ns, not significant. |
IRE1α kinase pharmacological
inhibition confers ferroptosis resistance in TNBC cells
Ferroptosis has emerged as a promising therapeutic
target in breast cancer (3), and
TNBC exhibits a heightened susceptibility to ferroptosis compared
with other molecular subtypes (7).
Furthermore, IRE1α possesses both a kinase domain and an RNase
domain (8), and whilst previous
research has established the involvement of its RNase in modulating
ferroptosis sensitivity (11), the
potential role of its kinase in ferroptosis regulation remains
unexplored. In the present study, to investigate the association
between IRE1α kinase and ferroptosis in TNBC, MDA-MB-231 cells were
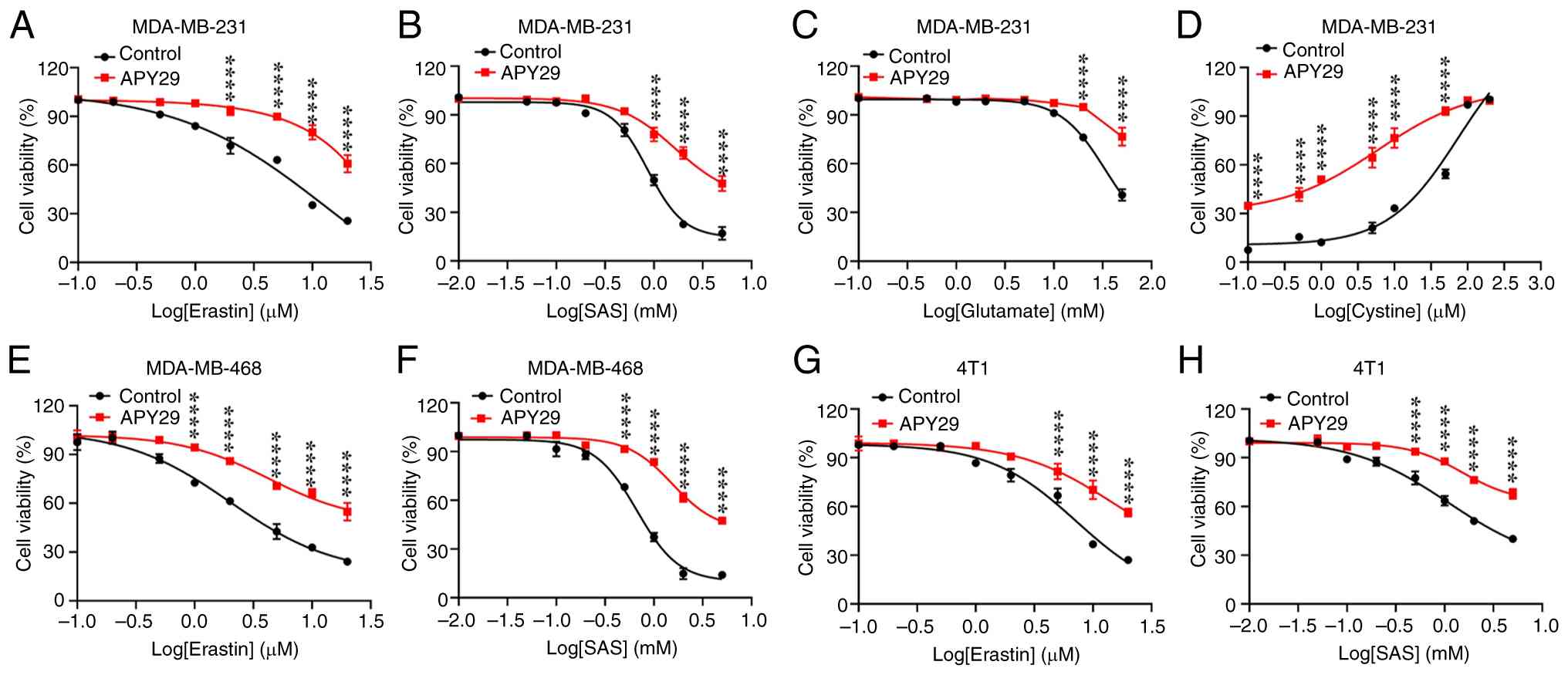
treated with the type I kinase inhibitor APY29 (28) and a dose-effect curve was determined
(Fig. S2A). Following subsequent
experiments at a non-toxic concentration of APY29 (0.2 µM), it was
observed that treatment with APY29 significantly enhanced the
resistance of MDA-MB-231 cells to multiple ferroptosis-inducing
agents, including erastin, SAS, glutamate and cystine restriction
(Fig. 2A-D; P<0.0001 vs. Control
at key concentrations). In contrast to the dose-dependent decrease
in viability observed with erastin, SAS, and glutamate (which
inhibit cystine uptake), cell viability increased with higher
cystine concentration in the deprivation assay (Fig. 2D), as cystine itself is a critical
substrate for the cellular antioxidant defense against ferroptosis.
Notably, these inducers all trigger ferroptosis by directly or
indirectly inhibiting system Xc−, the cystine/glutamate
antiporter critical for antioxidant defense (29). This protective effect was further
confirmed in MDA-MB-468 and 4T1 TNBC cells, as well as in HT1080
fibrosarcoma cells (Figs. 2E-H,
S2B-D and S3A-D; P<0.0001 vs. Control at key
concentrations). Notably, APY29 did not exert a significant effect
on ferroptosis induced by the GPX4 inhibitors RSL3 or ML162
(Fig. S3E-H; P>0.05 vs.
Control). These results suggest that IRE1α kinase modulates the
susceptibility to ferroptosis through the regulation of system
Xc− rather than through GPX4.
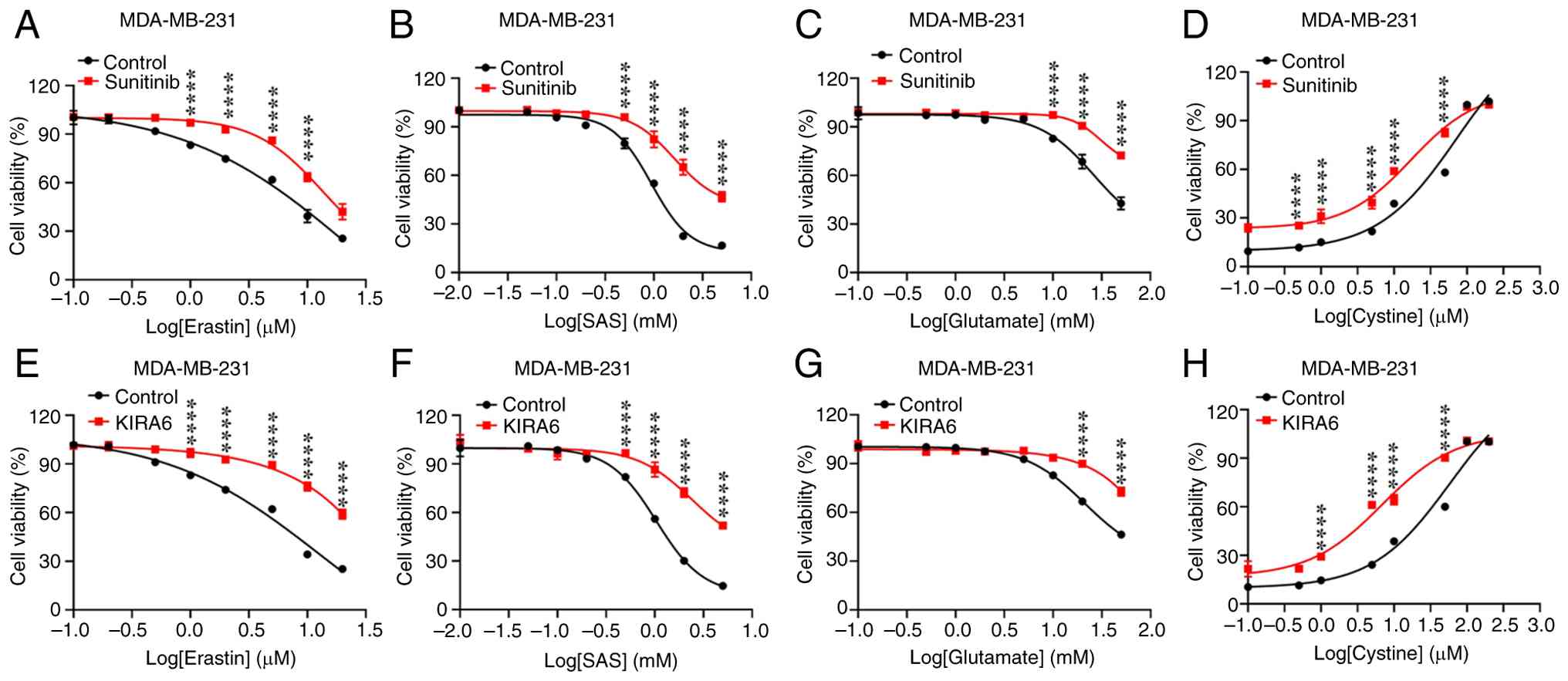
The present study further selected two known IRE1α
modulators, sunitinib [a type I inhibitor that suppresses IRE1α
kinase activity without affecting its RNase function (30)] and KIRA6 [an ATP-competitive IRE1α
kinase inhibiting RNase attenuator (31)], for subsequent investigations.
First, the cytotoxicity of these compounds was evaluated in
MDA-MB-231 cells (Fig. S2E and F).
Subsequent experiments were then performed at non-toxic
concentrations (sunitinib at 0.2 µM; KIRA6 at 0.5 µM). The results
revealed that sunitinib significantly suppressed the ferroptosis
induced by erastin, SAS, glutamate or cystine restriction (Fig. 3A-D; P<0.0001 vs. Control at key
concentrations). Similarly, KIRA6 exerted marked cytoprotective
effects (Fig. 3E-H; P<0.0001 vs.
Control at key concentrations). These findings suggest that
targeting the IRE1α kinase signaling pathway may represent an
effective strategy for modulating ferroptosis.
Overexpression of IRE1α-WT enhances
ferroptosis
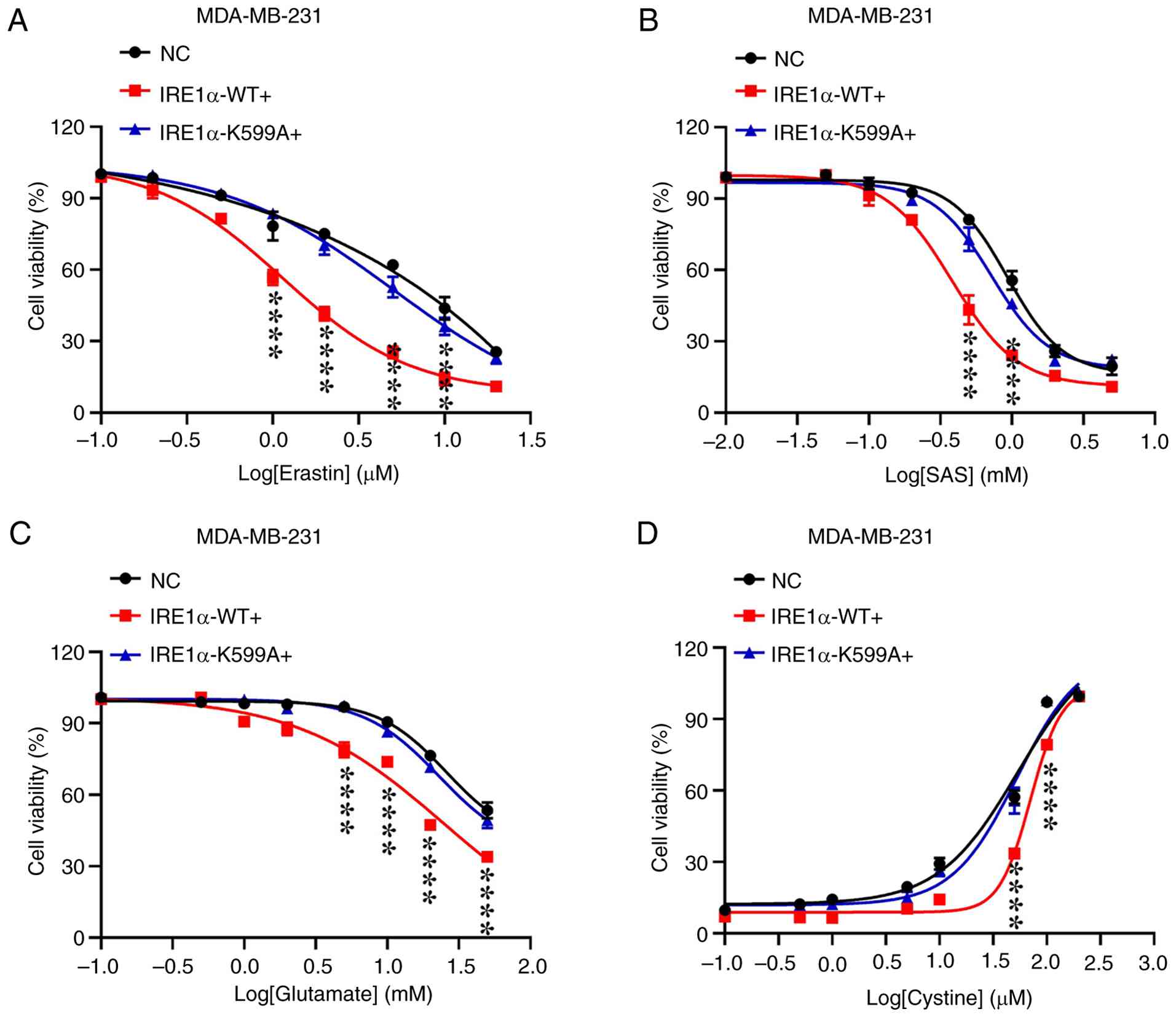
To further investigate the role of IRE1α kinase in
ferroptosis in MDA-MB-231, the present study established stable
cell lines overexpressing either IRE1α-WT or IRE1α-K599A, which
abolishes the kinase function of IRE1α (Fig. S4). Subsequent drug resistance
assays demonstrated that, compared with the control cells, the
overexpression of IRE1α-WT significantly enhanced cellular
sensitivity to ferroptosis induced by several stimuli, including
erastin, SAS, glutamate and cystine restriction. By contrast, the
overexpression of the kinase-dead mutant (IRE1α-K599A) did not
exert a significant effect on ferroptosis sensitivity (Fig. 4; P<0.0001 for IRE1α-WT vs. NC,
P>0.05 for IRE1α-K599A vs. NC at key concentrations). Taken
together, these findings demonstrate that the overexpression of
IRE1α-WT, but not the kinase-dead mutant increased the sensitivity
of TNBC cells to ferroptosis.
IRE1α kinase inhibition reduces lipid
peroxidation
As lipid peroxidation is a critical process in the
execution of ferroptosis (3), the
present study examined the effects of IRE1α kinase inhibition on
MDA [an end product of lipid peroxidation (32)] and lipid ROS levels. The findings
demonstrated that treatment with APY29 significantly inhibited the
accumulation of MDA (Fig. 5A-D) and
lipid ROS (Fig. 5E-H) induced by
erastin, SAS, glutamate or cystine restriction (compared with the
respective inducer-only groups), whereas IRE1α kinase inhibition
with APY29 did not affect the baseline levels of MDA or lipid ROS
(compared with the control group) (Fig.
5).
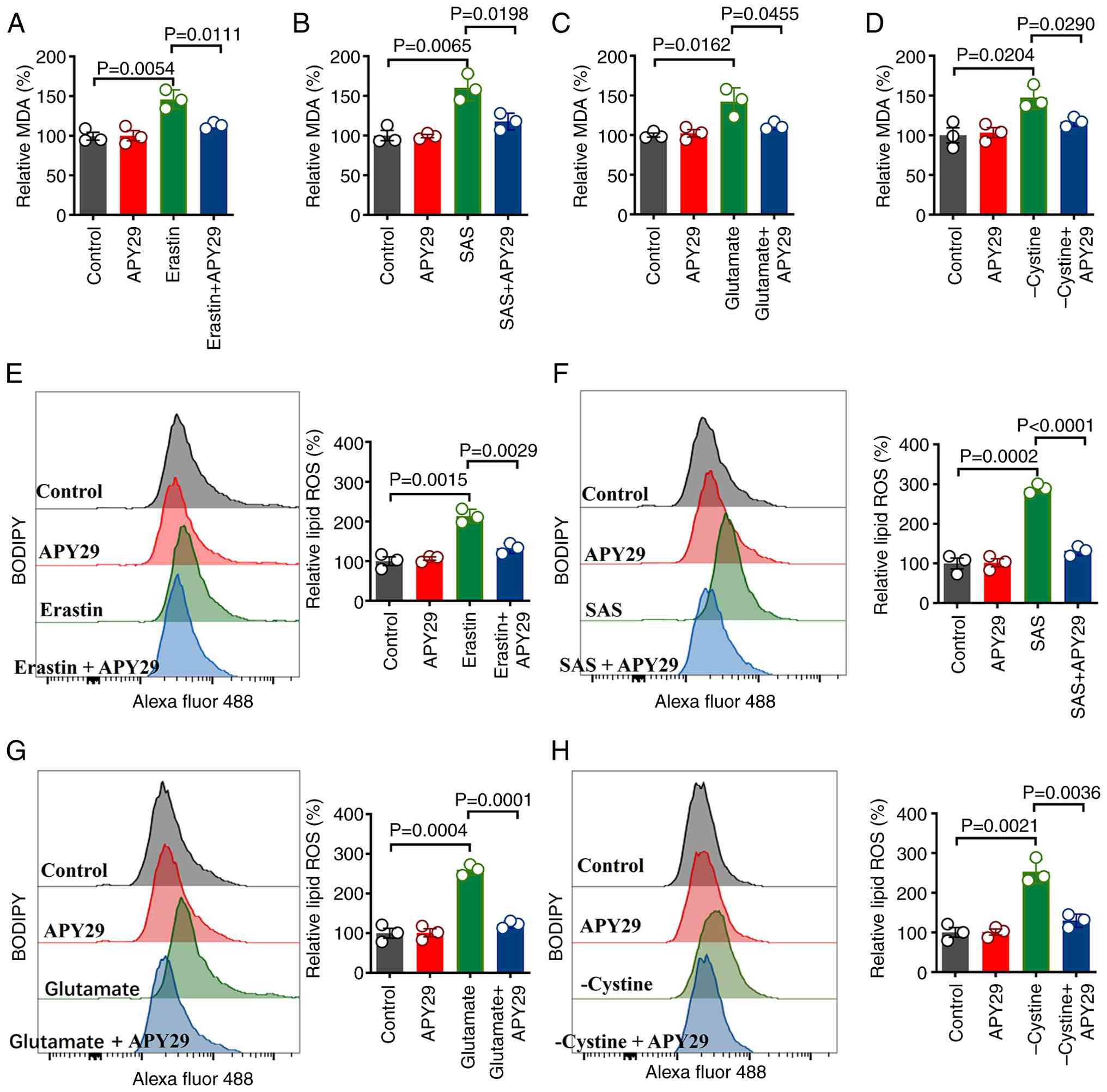 | Figure 5.Pharmacological inhibition of IRE1α
kinase attenuates lipid peroxidation. MDA levels were measured in
MDA-MB-231 cells following 12-h treatment with (A) erastin (5 µM),
(B) SAS (1 mM), (C) glutamate (20 mM) or (D) cystine-restricted
medium, in the presence or absence of APY29 (0.2 µM). Lipid ROS
levels were measured in MDA-MB-231 cells following 6-h treatment
with (E) erastin (5 µM), (F) SAS (1 mM), (G) glutamate (20 mM) or
(H) cystine-restricted medium, in the presence or absence of APY29
(0.2 µM), with corresponding statistical histograms. Data are
presented as mean ± standard error of the mean (n=3). IRE1α,
inositol-requiring enzyme 1α; MDA, malondialdehyde; ROS, reactive
oxygen species; SAS, sulfasalazine. |
Further investigations revealed that under
ferroptosis-inducing conditions, the overexpression of IRE1α-WT
significantly increased the accumulation of MDA (Fig. 6A-D) and lipid ROS (Fig. 6E-H) compared with that in the
control cells and in cells expressing the kinase-dead mutant. Taken
together, these results indicate that the inhibition of IRE1α
kinase alleviates lipid peroxidation, thereby suppressing
ferroptosis.
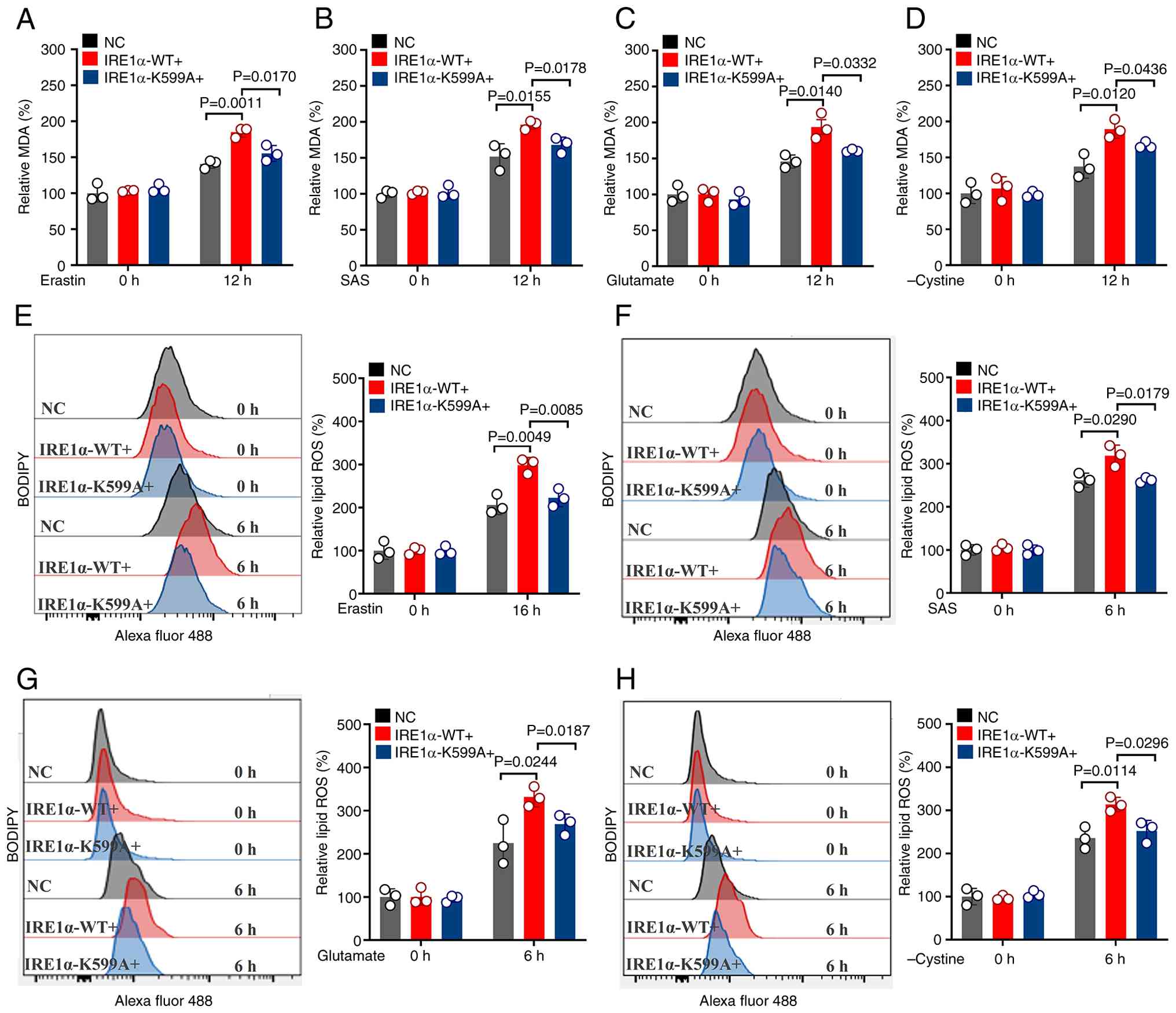 | Figure 6.Upregulation of IRE1α kinase
increases lipid peroxidation. MDA levels in MDA-MB-231 cells
overexpressing empty vector, wild-type IRE1α or kinase-dead IRE1α
mutant following 12-h treatment with (A) erastin (5 µM), (B) SAS (1
mM), (C) glutamate (20 mM) or (D) cystine-restricted medium. Lipid
ROS levels in MDA-MB-231 cells overexpressing empty vector,
wild-type IRE1α or kinase-dead IRE1α mutant following 6-h treatment
with (E) erastin (5 µM), (F) SAS (1 mM), (G) glutamate (20 mM) or
(H) cystine-restricted medium, with corresponding statistical
histograms. Data are presented as mean ± standard error of the mean
(n=3). IRE1α, inositol-requiring enzyme 1α; MDA, malondialdehyde;
ROS, reactive oxygen species; NC, negative control; WT, wildtype;
SAS, sulfasalazine. |
Inhibiting IRE1α kinase prevents GSH
depletion in ferroptosis
xCT is a key component of system Xc− and
serves a crucial role in regulating intracellular redox homeostasis
and determining cellular sensitivity to ferroptosis (1). In the present study, the results
revealed that the inhibition of IRE1α kinase activity significantly
alleviated ferroptosis induced by system Xc− suppression
(Fig. 2). Specifically, Fig. 2 demonstrates that co-treatment with
APY29 markedly improved cell viability compared with cells treated
with erastin, SAS, glutamate, or under cystine restriction alone.
Thus, it was hypothesized that IRE1α kinase may regulate
ferroptosis through xCT. In comparison with controls, treatment of
MDA-MB-231 cells with the IRE1α kinase inhibitor, APY29,
significantly increased xCT mRNA levels and protein expression
(Figs. 7A and S5A). This effect was also observed in
MDA-MB-468 (Figs. S5B and S6A) and 4T1 cells (Figs. S5C and S6B). However, the expression of GPX4 and
ACSL4, two key ferroptosis regulatory genes (1), remained unaltered (Fig. 7B and C).
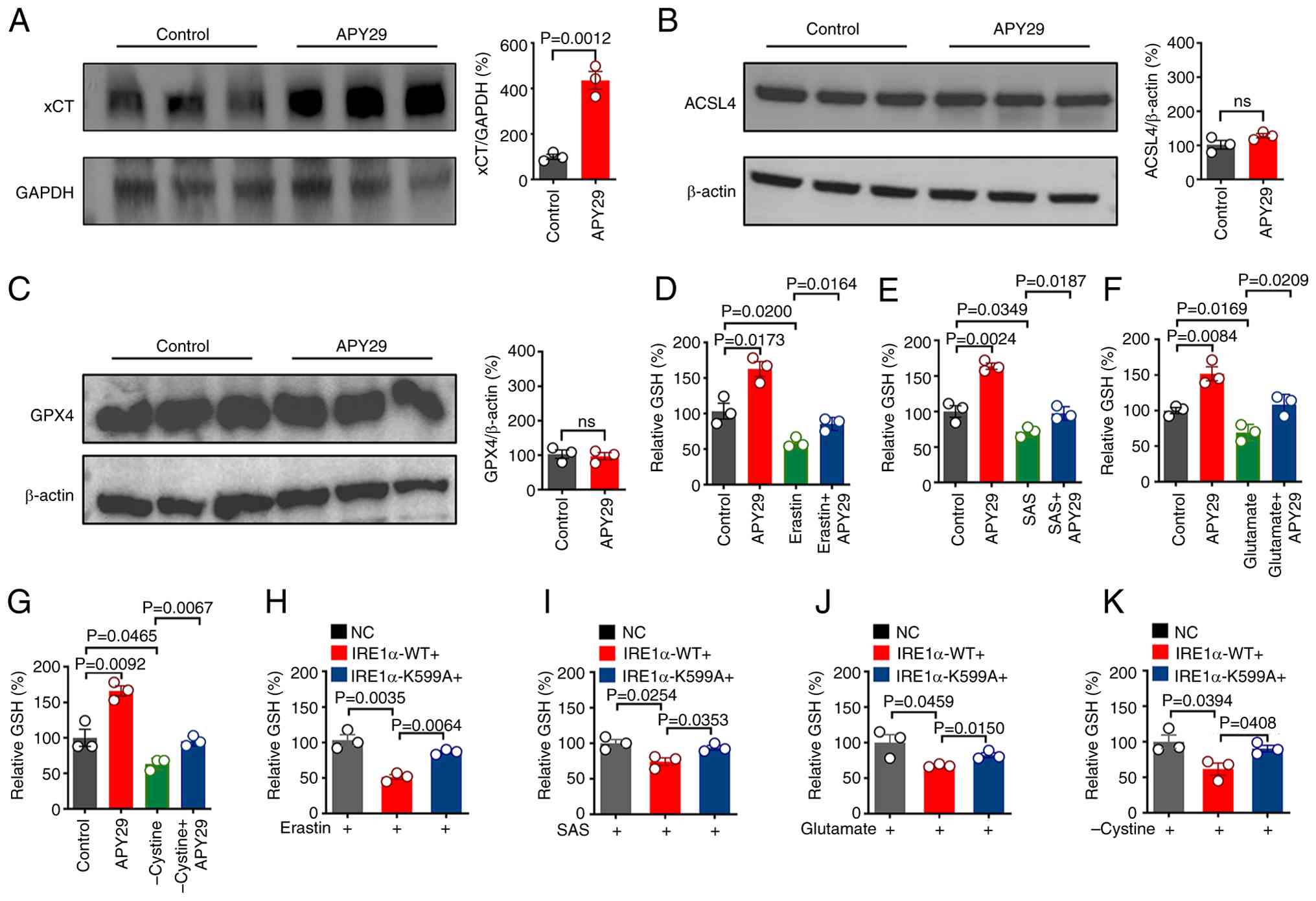 | Figure 7.IRE1α regulates GSH and participates
in ferroptosis. MDA-MB-231 cells were treated with APY29 (0.2 µM)
for 24 h, and the protein expression of (A) xCT, (B) ACSL4 and (C)
GPX4 was analyzed using western blotting. GSH levels were measured
in MDA-MB-231 cells following 12-h treatment with (D) erastin (5
µM), (E) SAS (1 mM), (F) glutamate (20 mM) or (G)
cystine-restricted medium, in the presence or absence of APY29 (0.2
µM). Cellular GSH levels were measured in MDA-MB-231 cells
overexpressing empty vector, wild-type IRE1α or the kinase-dead
IRE1α mutant following 12-h treatment with (H) erastin (5 µM), (I)
SAS (1 mM), (J) glutamate (20 mM) or (K) cystine-restricted medium.
Data are presented as mean ± standard error of the mean (n=3).
IRE1α, inositol-requiring enzyme 1α; GSH, glutathione; xCT, solute
carrier family 7 member 11; ACSL4, acyl-CoA synthetase long chain
family member 4; GPX4, glutathione peroxidase 4; NC, negative
control; WT, wildtype; ns, not significant; SAS, sulfasalazine. |
The system Xc− inhibits ferroptosis by
maintaining GSH synthesis (33).
Further experiments revealed that treatment with APY29 increased
baseline GSH levels in MDA-MB-231 cells compared with the untreated
control and effectively reversed GSH depletion induced by erastin,
SAS, glutamate and cystine restriction compared with the respective
inducer-only groups (Fig. 7D-G).
This effect was also observed in MDA-MB-468 and 4T1 cells (Fig. S6C-F). Moreover, under these
ferroptosis-inducing conditions, the overexpression of wild-type
IRE1α significantly reduced GSH levels compared with that in both
the control and kinase-dead mutant overexpression groups (Fig. 7H-K). Full original western blot
images from Figs. 7 and S6 are presented in Fig. S7. Collectively, the aforementioned
results indicate that the inhibition of IRE1α kinase activity
attenuates GSH depletion, thereby enhancing cellular resistance to
ferroptosis.
Discussion
The present study, through the analysis of existing
datasets, demonstrated that ERN1 levels in patients with
breast cancer were significantly lower than those in normal
samples. Moreover, a low expression of ERN1 was associated
with a poor prognosis of patients with breast cancer. Its low level
also predicted a poor prognosis of patients with HER2-positive
breast cancer and TNBC. Mechanistically, the inhibition of IRE1α
kinase activity upregulated xCT expression and enhanced GSH
production, thereby suppressing ferroptosis in TNBC cells. These
findings suggest that IRE1α kinase is a potential therapeutic
target for TNBC. Notably, similar effects observed in HT1080 cells
indicate that this regulatory mechanism may extend to broader
cancer therapies.
IRE1α serves multifaceted roles in several types of
cancer by modulating the tumor microenvironment, antitumor immunity
and chemoresistance (9,34). IRE1α regulates tumor progression
through distinct functional domains; small-molecule drugs targeting
IRE1α are currently under development as potential anticancer
therapeutics (35,36). STF-083010, is a compound
specifically targeting the catalytic core of the IRE1α RNase domain
that inhibits RNase activity without altering kinase function or
the oligomerization state (28).
Similarly, 4µ8C selectively inhibits the IRE1α RNase domain,
specifically targeting and suppressing regulated IRE1α-dependent
decay (RIDD), whilst preserving the kinase activity of IRE1α
(37). Previous studies have
reported that both these selective IRE1α RNase inhibitors can
effectively inhibit the ferroptosis process (11,38).
Furthermore, the present study demonstrated that the IRE1α kinase
inhibitors, APY29 and sunitinib [which selectively inhibit IRE1α
kinase activity without affecting RNase function (30,39)],
effectively rescued the ferroptosis of TNBC cells induced by the
inhibitory system Xc−. This suggests that IRE1α kinase
activity is as crucial as its RNase activity in the regulation of
ferroptosis. Additionally, KIRA6, a dual inhibitor targeting both
the kinase and RNase domains (31),
also exhibits ferroptosis-suppressing effects. In summary, the
pharmacological modulation of IRE1α provides a promising
therapeutic strategy for the treatment of disease by regulating the
ferroptosis pathway.
The UPR is a cellular stress response triggered by
the accumulation of misfolded proteins in the ER (40). A total of three major pathways
mediate the UPR: Protein kinase R-like ER kinase (PERK), activating
transcription factor 6 (ATF6) and IRE1α (41). Emerging evidence indicates the
crucial role of UPR in regulating the ferroptosis of tumor cells:
It has been demonstrated that the loss of PERK function promotes
ferroptosis by downregulating xCT in colorectal cancer (42). Moreover, ATF6 exists in two isoforms
(α and β), with ATF6α being the predominant form (43). In prostate cancer, ATF6α deficiency
has been reported to enhance ferroptotic cell death (44). Additionally, IRE1α contains two
functional domains: A kinase domain and an RNase domain (45). Its RNase activity enhances cellular
susceptibility to ferroptosis by downregulating xCT expression
through the RIDD pathway (11).
However, the role of IRE1α kinase in the regulation of ferroptosis
remains unclear. The present study demonstrated that the inhibition
of IRE1α kinase activity upregulated xCT expression and promoted
GSH synthesis, thereby rescuing the ferroptosis of TNBC cells
induced by the inhibitory system Xc-. Notably, although
both the kinase and RNase domains can modulate ferroptosis through
xCT, these data suggest they may function through independent
pathways, as the inhibition of either domain alone is sufficient to
confer ferroptosis resistance. To explain this kinase-mediated
effect, the present study proposes a model wherein its
pharmacological inhibition mimics the functional consequence of
IRE1α kinase domain cysteine sulfenylation, which is a
redox-sensing modification known to repress its canonical UPR
function and activate the NRF2 pathway (46). Therefore, we hypothesize that the
observed transcriptional upregulation of SLC7A11 is likely mediated
through NRF2 activation. Overall, the findings of the present study
unveil a novel role of IRE1α kinase in ferroptosis and provide a
more comprehensive understanding of the function of IRE1α in the
ferroptosis pathway.
However, the therapeutic implications of the
findings of the present study require careful consideration. The
data demonstrate that inhibiting IRE1α kinase activity protects
TNBC cells from ferroptosis. This key observation argues against
the use of IRE1α kinase inhibitors as a monotherapy, as it would
promote tumor survival. Instead, the present work suggests more
rational strategies. Firstly, tumors with low IRE1α expression may
be intrinsically resistant to ferroptosis inducers, a factor that
should be considered in patient stratification. Secondly, a novel
combinatorial approach could involve developing IRE1α kinase
activators to hypersensitize tumors to ferroptosis inducers,
thereby leveraging this axis for therapeutic benefit.
Limitations of the present study should be noted.
The findings are primarily based on in vitro experiments
using a limited set of TNBC cell lines. Future in vivo
studies and validation in clinical samples are required to confirm
the translational relevance of the IRE1α kinase-xCT-GSH axis.
In conclusion, the inhibition of IRE1α kinase
activity increases xCT expression and promotes GSH synthesis, a
mechanism that serves a critical role in regulating the ferroptosis
of TNBC MDA-MB-231 cells. These findings support the IRE1α
kinase-xCT-GSH axis as a potential therapeutic strategy for the
treatment of TNBC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present work was supported by the Startup Research Fund from
Sichuan Urban Vocational College.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SX raised funds and contributed to the conception
and design of the study. SX and SQY performed the experiments,
analyzed the data and wrote the manuscript. SX and SQY confirm
authenticity of all the raw data. Both authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zhang C, Liu X, Jin S, Chen Y and Guo R:
Ferroptosis in cancer therapy: A novel approach to reversing drug
resistance. Mol Cancer. 21:472022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Diao J, Jia Y, Dai E, Liu J, Kang R, Tang
D, Han L, Zhong Y and Meng L: Ferroptotic therapy in cancer:
Benefits, side effects, and risks. Mol Cancer. 23:892024.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xu S, Tuo QZ, Meng J, Wu XL, Li CL and Lei
P: Thrombin induces ferroptosis in triple-negative breast cancer
through the cPLA2α/ACSL4 signaling pathway. Transl Oncol.
39:1018172024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kim J, Harper A, McCormack V, Sung H,
Houssami N, Morgan E, Mutebi M, Garvey G, Soerjomataram I and
Fidler-Benaoudia MM: Global patterns and trends in breast cancer
incidence and mortality across 185 countries. Nat Med.
31:1154–1162. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xu T, Ma D, Chen S, Tang R, Yang J, Meng
C, Feng Y, Liu L, Wang J, Luo H and Yu K: High GPER expression in
triple-negative breast cancer is linked to pro-metastatic pathways
and predicts poor patient outcomes. NPJ Breast Cancer. 8:1002022.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hsu JY, Chang CJ and Cheng JS: Survival,
treatment regimens and medical costs of women newly diagnosed with
metastatic triple-negative breast cancer. Sci Rep. 12:7292022.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li J, He D, Li S, Xiao J and Zhu Z:
Ferroptosis: The emerging player in remodeling triple-negative
breast cancer. Front Immunol. 14:12840572023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Raymundo DP, Doultsinos D, Guillory X,
Carlesso A, Eriksson LA and Chevet E: Pharmacological targeting of
IRE1 in cancer. Trends Cancer. 6:1018–1030. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Unal B, Kuzu OF, Jin Y, Osorio D, Kildal
W, Pradhan M, Kung SHY, Oo HZ, Daugaard M, Vendelbo M, et al:
Targeting IRE1alpha reprograms the tumor microenvironment and
enhances anti-tumor immunity in prostate cancer. Nat Commun.
15:88952024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shi M, Chai Y, Zhang J and Chen X:
Endoplasmic reticulum Stress-associated neuronal death and innate
immune response in neurological diseases. Front Immunol.
12:7945802021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jiang D, Guo Y, Wang T, Wang L, Yan Y, Xia
L, Bam R, Yang Z, Lee H, Iwawaki T, et al: IRE1α determines
ferroptosis sensitivity through regulation of glutathione
synthesis. Nat Commun. 15:41142024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zheng X, Wei J, Li W, Li X, Wang W, Guo J
and Fu Z: PRDX2 removal inhibits the cell cycle and autophagy in
colorectal cancer cells. Aging (Albany NY). 12:16390–16409. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiao L, Li X, Luo Y, Wei J, Ding X, Xiong
H, Liu X and Lei P: Iron metabolism mediates microglia
susceptibility in ferroptosis. Front Cell Neurosci. 16:9950842022.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang X, Chen X, Zhou W, Men H, Bao T, Sun
Y, Wang Q, Tan Y, Keller BB, Tong Q, et al: Ferroptosis is
essential for diabetic cardiomyopathy and is prevented by
sulforaphane via AMPK/NRF2 pathways. Acta Pharm Sin B. 12:708–722.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang H, Zhang S, Li Y, Liu Z, Mi L, Cai
Y, Wang X, Chen L, Ran H, Xiao D, et al: Suppression of
mitochondrial ROS by prohibitin drives glioblastoma progression and
therapeutic resistance. Nat Commun. 12:37202021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang X, Wang Z, Samovich SN, Kapralov AA,
Amoscato AA, Tyurin VA, Dar HH, Li Z, Duan S, Kon N, et al:
PHLDA2-mediated phosphatidic acid peroxidation triggers a distinct
ferroptotic response during tumor suppression. Cell Metab.
36:762–777.e9. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen L, Zhao X, Sheng R, Lazarovici P and
Zheng W: Artemisinin alleviates astrocyte overactivation and
neuroinflammation by modulating the IRE1/NF-κB signaling pathway in
in vitro and in vivo Alzheimer's disease models. Free Radic Biol
Med. 229:96–110. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sanese P, Fasano C, Buscemi G, Bottino C,
Corbetta S, Fabini E, Silvestri V, Valentini V, Disciglio V, Forte
G, et al: Targeting SMYD3 to sensitize homologous
Recombination-proficient tumors to PARP-Mediated synthetic
lethality. iScience. 23:1016042020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chandrashekar DS, Karthikeyan SK, Korla
PK, Patel H, Shovon AR, Athar M, Netto GJ, Qin ZS, Kumar S, Manne
U, et al: UALCAN: An update to the integrated cancer data analysis
platform. Neoplasia. 25:18–27. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gyorffy B, Lanczky A, Eklund AC, Denkert
C, Budczies J, Li Q and Szallasi Z: An online survival analysis
tool to rapidly assess the effect of 22,277 genes on breast cancer
prognosis using microarray data of 1,809 patients. Breast Cancer
Res Treat. 123:725–731. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sabatier R, Finetti P, Adelaide J, Guille
A, Borg JP, Chaffanet M, Lane L, Birnbaum D and Bertucci F:
Down-regulation of ECRG4, a candidate tumor suppressor gene, in
human breast cancer. PLoS One. 6:e276562011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang W, Sun Y, Bai L, Zhi L, Yang Y, Zhao
Q, Chen C, Qi Y, Gao W, He W, et al: RBMS1 regulates lung cancer
ferroptosis through translational control of SLC7A11. J Clin
Invest. 131:e1520672021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang X, Wang Q, Wang H, Cai G, An Y, Liu
P, Zhou H, Chen HW, Ji S, Ye J, et al: Small protein ERSP encoded
by LINC02870 promotes triple negative breast cancer progression via
IRE1α/XBP1s activation. Cell Death Differ. 32:1014–1025. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Alanko J, Tanner M, Vanninen R, Auvinen A
and Isola J: Triple-negative and HER2-positive breast cancers found
by mammography screening show excellent prognosis. Breast Cancer
Res Treat. 187:267–274. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hoeferlin LA, C EC and Park MA: Challenges
in the treatment of triple negative and HER2-Overexpressing breast
cancer. J Surg Sci. 1:3–7. 2013.PubMed/NCBI
|
|
28
|
Park H, Shin DH, Sim JR, Aum S and Lee MG:
IRE1α kinase-mediated unconventional protein secretion rescues
misfolded CFTR and pendrin. Sci Adv. 6:eaax99142020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jiang Y and Sun M: SLC7A11: The Achilles
heel of tumor? Front Immunol. 15:14388072024. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Makhov P, Naito S, Haifler M, Kutikov A,
Boumber Y, Uzzo RG and Kolenko VM: The convergent roles of NF-κB
and ER stress in sunitinib-mediated expression of pro-tumorigenic
cytokines and refractory phenotype in renal cell carcinoma. Cell
Death Dis. 9:3742018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tang X, Teder T, Samuelsson B and
Haeggstrom JZ: The IRE1α Inhibitor KIRA6 blocks leukotriene
biosynthesis in human phagocytes. Front Pharmacol. 13:8062402022.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gawel S, Wardas M, Niedworok E and Wardas
P: Malondialdehyde (MDA) as a lipid peroxidation marker. Wiad Lek.
57:453–455. 2004.(In Polish). PubMed/NCBI
|
|
33
|
Koppula P, Zhuang L and Gan B: Cystine
transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient
dependency, and cancer therapy. Protein Cell. 12:599–620. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xu L, Peng F, Luo Q, Ding Y, Yuan F, Zheng
L, He W, Zhang SS, Fu X, Liu J, et al: IRE1α silences dsRNA to
prevent taxane-induced pyroptosis in triple-negative breast cancer.
Cell. 187:7248–7266.e34. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Harnoss JM, Le Thomas A, Shemorry A,
Marsters SA, Lawrence DA, Lu M, Chen YA, Qing J, Totpal K, Kan D,
et al: Disruption of IRE1α through its kinase domain attenuates
multiple myeloma. Proc Natl Acad Sci USA. 116:16420–16429. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Abbasi S, Rivand H, Eshaghi F, Moosavi MA,
Amanpour S, McDermott MF and Rahmati M: Inhibition of IRE1 RNase
activity modulates tumor cell progression and enhances the response
to chemotherapy in colorectal cancer. Med Oncol. 40:2472023.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cross BC, Bond PJ, Sadowski PG, Jha BK,
Zak J, Goodman JM, Silverman RH, Neubert TA, Baxendale IR, Ron D,
et al: The molecular basis for selective inhibition of
unconventional mRNA splicing by an IRE1-binding small molecule.
Proc Natl Acad Sci USA. 109:E869–E878. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chan KY, Yu Y, Kong Y, Cheng L, Yao R, Yin
Chair PS, Wang P, Wang R, Sun WY, He RR, et al: GPX4-dependent
ferroptosis sensitivity is a fitness trade-off for cell
enlargement. iScience. 28:1123632025. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li C, Tan YP, Gao D, Su R, Xu K, Liu SC,
Li XF, Lu YH, Yi LT, Wang G, et al: IRE1alpha RNase activity is
critical for early embryo development by degrading maternal
transcripts. Nucleic Acids Res. 53:gkaf5202025. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Read A and Schroder M: The unfolded
protein response: An overview. Biology (Basel).
10:3842021.PubMed/NCBI
|
|
41
|
Kim P: Understanding the unfolded protein
response (UPR) pathway: Insights into neuropsychiatric disorders
and therapeutic potentials. Biomol Ther (Seoul). 32:183–191. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Saini KK, Chaturvedi P, Sinha A, Singh MP,
Khan MA, Verma A, Nengroo MA, Satrusal SR, Meena S, Singh A, et al:
Loss of PERK function promotes ferroptosis by downregulating
SLC7A11 (System Xc−) in colorectal cancer. Redox Biol.
65:1028332023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Oka OB, van Lith M, Rudolf J, Tungkum W,
Pringle MA and Bulleid NJ: ERp18 regulates activation of ATF6alpha
during unfolded protein response. EMBO J. 38:e1009902019.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhao R, Lv Y, Feng T, Zhang R, Ge L, Pan
J, Han B, Song G and Wang L: ATF6α promotes prostate cancer
progression by enhancing PLA2G4A-mediated arachidonic acid
metabolism and protecting tumor cells against ferroptosis.
Prostate. 82:617–629. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ghosh R, Wang L, Wang ES, Perera BG,
Igbaria A, Morita S, Prado K, Thamsen M, Caswell D, Macias H, et
al: Allosteric inhibition of the IRE1α RNase preserves cell
viability and function during endoplasmic reticulum stress. Cell.
158:534–548. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hourihan JM, Moronetti Mazzeo LE,
Fernandez-Cardenas LP and Blackwell TK: Cysteine sulfenylation
directs IRE-1 to activate the SKN-1/Nrf2 antioxidant response. Mol
Cell. 63:553–566. 2016. View Article : Google Scholar : PubMed/NCBI
|