Introduction
Colorectal cancer (CRC) is the third most common
malignant tumor and the second leading cause of cancer-related
death worldwide, ranking first in mortality among malignancies of
the digestive system (1–4). The total number of mortalities from
colon and rectal cancer is predicted to increase by 60.0 and 71.5%,
respectively, when comparing between 2013 and the projection for
2035 (colon cancer: 158,816 vs. 254,165 cases; rectal cancer:
72,649 vs. 124,614 cases) (5). In
China, the 5-year survival rate for patients with metastatic CRC is
limited to 10–25% (6,7). Early-stage CRC can be treated with
conventional modalities, such as surgical resection, chemotherapy
and radiotherapy; these approaches usually yield limited efficacy
and are associated with high recurrence rates (8), with >29% of CRC cases experiencing
recurrence ≥5 years following primary surgical resection, while
merely one-third of CRC cases are diagnosed at the localized stage
(9,10) Recently, immune checkpoint inhibitors
(ICIs), including anti-programmed cell death protein-1
(PD-1)/programmed death-ligand 1 (PD-L1) antibodies, have improved
patient survival with several solid tumors, such as melanoma and
lung cancer, by modulating the immune system to induce cytotoxic
CD8+ T cell-mediated tumor cell death (3,4,11,12).
Despite the promise of immunotherapy as a potential treatment for
CRC, its clinical success has been limited by factors such as
aberrant signaling pathway activation leading to immune evasion,
impaired antigen presentation and accumulation of immunosuppressive
cells (5,6,13,14).
This challenge is particularly evident in microsatellite-stable
CRC, where a low tumor mutation burden and a paucity of neoantigens
render immunotherapy less effective (7,15).
Therefore, novel combinatorial treatment strategies are urgently
needed to enhance the immune response in patients with CRC.
The glucagon-like peptide-1 receptor (GLP-1R) is
primarily expressed on pancreatic β-cells, serves a key role in
regulating blood glucose levels and metabolism through its specific
interaction with GLP-1 (8,16). Initially, the research focused on
treating diabetes and cardiovascular diseases (9,10,17,18);
however, GLP-1R agonist therapies indirectly alleviate hepatic
inflammation and fibrosis, offering benefits for the management of
metabolic liver diseases (11,19).
Recent studies have highlighted the potential of targeting GLP-1R
beyond conventional metabolic endpoints, such as gastrointestinal
stress attenuation and pain modulation (20–22).
Notably, emerging evidence indicates that GLP-1R activation
modulates various innate immune cells, including macrophages and
innate-like T lymphocytes (23–25)
Furthermore, several T cell subsets express functional GLP-1R in
animal models and human participants, underscoring its notable role
in immune regulation (26). In
preclinical murine CRC models, GLP-1R has been shown to be a
negative co-stimulatory molecule that dampens T-cell function and
attenuates graft immune responses, whereas GLP-1R antagonists
provoke robust antitumor immunity. Collectively, these findings
suggest that the immunomodulatory properties of GLP-1R have
potential for the development of innovative cancer immunotherapy
strategies in the future.
Due to the potential of GLP-1R antagonists to
modulate T-cell regulation and the evidence that enhancing T-cell
infiltration can effectively transform ‘cold’ tumors into ‘hot’
tumors (27–29), it is plausible that combining GLP-1R
antagonists with ICIs may synergistically modulate T-cell-mediated
immune sensitivity to improve tumor treatment outcomes. However,
current investigations into GLP-1R in patients with cancer have
predominantly focused on its role in mitigating cardiovascular
complications or their side effects (30,31).
For example, a retrospective cohort study indicated that GLP-1RAs
may help alleviate cardiac dysfunction caused by anticancer
treatments, such as chemotherapy or radiotherapy (32). Common side effects of GLP-1RAs,
including semaglutide, involve gastrointestinal reactions such as
nausea and vomiting. Further research suggested that simultaneous
activation of the glucose-dependent insulinotropic polypeptide
receptor may mitigate these adverse effects through an anti-emetic
mechanism (33). The potential
antitumor benefits of combining GLP-1R antagonism with immune
checkpoint inhibition in CRC remain to be explored.
In the present study, the effects of the GLP-1R
antagonist Exendin 9–39 (Exe-9) on T-cell function, CD8+
T cell-mediated tumor cell killing and the synergistic antitumor
efficacy of its combination with ICIs were systematically evaluated
via in vitro co-culture systems and in vivo CRC mouse
tumor models. Furthermore, the relationship between GLP-1R
expression levels and immunotherapy response was analyzed in
samples of patients with CRC to assess its potential as a
predictive clinical outcomes biomarker. Therefore, the present
study aimed to provide experimental evidence for the combined
application of GLP-1R antagonists and ICIs in CRC, offering novel
strategies for optimizing combination therapies for this disease in
the future.
Materials and methods
Ethics statement
All experimental procedures involving animals and
human tissues were approved by the Ethics Committee of Guangzhou
Development District Hospital (approval no. F2024-030; Guangzhou,
China).
All animal experiments were approved by the
Institutional Animal Care and Use Committee of Guangzhou
Development District Hospital (Guangzhou, China). Mice were housed
in a specific pathogen-free facility, maintained in a controlled
environment (22±2°C, 50 ± 10% humidity, 12 h light/dark cycle) and
provided ad libitum access to food and water. All efforts
were made to minimize animal suffering. For human studies, this
research utilized a retrospective design. Peripheral blood samples
were retrospectively collected from healthy donors, and matched
tumor tissue samples were obtained from patients with CRC,
respectively, following the provision of written informed consent
in accordance with the Declaration of Helsinki. The study protocol,
including the retrospective use of these samples, was approved by
the Institutional Review Board of the Guangzhou Development
District Hospital (Guangzhou, China).
Clinical sample collection and
grouping
Peripheral blood samples were collected from healthy
donors via venipuncture and drawn into sterile
ethylenediaminetetraacetic acid-coated tubes on ice. Clinical tumor
specimens were collected from 11 patients with colon cancer who
received ICI therapy at Guangzhou Development District Hospital
(Guangzhou, China) between July 2022 and December 2024. Patients
were enrolled based on the following criteria: i) Histologically
confirmed diagnosis of colon adenocarcinoma; ii) scheduled to
undergo or having undergone first-line ICI therapy (either as
monotherapy or in combination); iii) availability of pre-treatment
tumor tissue samples and complete clinical follow-up records; iv)
aged ≥18 years. Key exclusion criteria included: i) Prior history
of other active malignancies ≤5 years; ii) severe concurrent
autoimmune diseases or active infections; iii) receipt of any other
form of anticancer therapy (such as chemotherapy or radiotherapy)
≤4 weeks prior to sample collection; v) incomplete clinical or
pathological data. Surgical resection or core needle biopsy was
used to obtain tumor tissues, which were immediately placed in
ice-cold RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.).
Based on the Response Evaluation Criteria for Solid Tumors
(34), tumor responses were
evaluated using contrast-enhanced computed tomography. Patients
whose target lesions exhibited either complete response (CR) or
partial response (PR) were categorized as responders (n=5), whereas
those with stable disease (SD) or progressive disease (PD) were
classified as non-responders (n=6). CR was defined as the
disappearance of all target lesions and PR as a ≥30% reduction in
their total diameter. SD was characterized by insufficient
shrinkage to qualify as a partial response and PD was defined as a
minimum 20% increase in the target lesions. The clinical cohort
consisted of 7 men and 4 women, with a median age of 62 years
(range, 48–71 years).
Cell maintenance and treatment
The CRC cell lines MC38 (cat. no. iCell-m032; Mirror
Point (Shanghai) Cell Technology Co., Ltd) and HT-29 (cat. no.
iCell-h078; Mirror Point (Shanghai) Cell Technology Co., Ltd) were
maintained according to the manufacturer's protocols. They were
cultured with activated T cells (specifically, in
vitro-activated human peripheral blood-derived T cells and
isolated murine CD8+ T cells from OT-1 mice, as detailed
in the ‘T-cell separation’ section) at 37°C in a 5% CO2
incubator. MC38 cells were genetically modified to overexpress
SIINFEKL via a lentiviral vector system (cat. no. MC38-OVA; iCell
Biotech). Cells were cultured in DMEM (Gibco; Thermo Fisher
Scientific, Inc.), supplemented with 10% fetal bovine serum (FBS;
Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin, 100
µg/ml streptomycin and maintained in a humidified incubator at 37°C
with 5% CO2. For the treatment experiments, cells were
either cultured individually or co-cultured with activated T cells
at a ratio of 3:1 for 48 h in the presence of Exe-9 (GLP-1R
antagonist; cat. no. E7269; MilliporeSigma) at concentrations of 10
and 50 nM, whereas parallel cultures received phosphate-buffered
saline (PBS) as the vehicle control.
T-cell separation
Human peripheral blood mononuclear cells were
isolated from whole blood of healthy donors via density gradient
centrifugation using Lymphoprep (cat. no. 10970; STEMCELL
Technologies). Specifically, blood was collected in BD CPT tubes
(cat. no. 362782; BD Biosciences) containing Ficoll™ density
gradient medium (Ρ=1.077 g/cm3) and sodium citrate.
Tubes were incubated at room temperature for 20 min to stabilize
the temperature, then centrifuged at 1,650 × g for 20 min at 20°C
with the brake on. This formed a density barrier separating PBMCs
and serum. The PBMC fraction was gently mixed with the serum by
inversion and transferred to 15 ml tubes for further processing.
and subsequently cultured in CTS™ AIIM V™ SFM medium (cat. no.
A3021002; Gibco; Thermo Fisher Scientific, Inc.) supplemented with
ImmunoCult™ Human CD3/CD28/CD2 T Cell Activation Reagent (cat. no.
10970; STEMCELL Technologies) and recombinant 1,000 U/ml human
interleukin (IL)-2 (cat. no. 11848-HNAY1; Sino Biological, Inc.)
for 7 days to obtain activated human T cells. CD8+ T
cells were isolated from the spleens of OT-1 mice (cat. no. 003831;
THE JACKSON LABORATORY), aged 6–8 weeks old with a body weight of
18–25 g using the EasySep™ Mouse CD8+ T Cell Isolation
Kit (STEMCELL Technologies) in accordance with the manufacturer's
instructions. Collected cells were cultured briefly in complete T
cell medium comprising RPMI-1640 (Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 10% FBS, 20 mM HEPES, 1 mM sodium pyruvate,
0.05 mM 2-mercaptoethanol, 2 mM L-glutamine and 50 U/ml
streptomycin/penicillin, with recombinant mouse IL-2 (cat. no.
CK24; Novoprotein Scientific, Inc.) and incubated for 5 days.
Cell viability assay
After treatment, the adherent cancer cells were
washed thrice with PBS, fixed with 100% methanol at room
temperature for 10 min and stained with a 0.5% crystal violet
solution at room temperature for 30 min. After staining, the wells
were rinsed with PBS to remove excess dye and subsequently
solubilized by adding 10% acetic acid for 10 min. Optical density
(OD) was measured at 570 nm using a BioTek Epoch 2 microplate
reader (BioTek Instruments; Agilent Technologies, Inc.) to
quantitatively determine cell viability.
Cytokine quantification
To quantify interferon (IFN)-γ and tissue necrotic
factor (TNF)-α levels, the supernatants of the colon cancer cell
culture were collected after a 48 h incubation period at 37°C and
centrifuged at 13,000 × g for 5 min at 4°C. The supernatants were
analyzed using the Mouse IFN-γ enzyme-linked immunosorbent assay
(ELISA) Kit (cat. no. PI508; Beyotime Biotechnology), Mouse TNF α
ELISA Kit (cat. no. ab285327; Abcam), Human IFN-γ ELISA Kit (cat.
no. PI521; Beyotime Biotechnology) and human TNF α ELISA Kit (cat.
no. ab181421; Abcam), in strict accordance with the manufacturer's
instructions. Briefly, 100 µl each standard and sample was
dispensed into pre-coated wells of a 96-well microplate and
incubated for 2 h at room temperature. After washing with 0.05%
Tween-20 in PBS, 100 µl horseradish peroxidase (HRP)-conjugated
detection antibody (provided within the respective ELISA kits was
added.
The antibody in the aforementioned IFN-γ reagent kit
was used directly (utilizing the pre-prepared formulation provided
with the kit, which required no additional dilution), while the
antibody in the TNF-α reagent kits was used at a 1:100 dilution;
both antibody application protocols followed the specifications in
the respective manufacturers' instructions; cat. no. 80220; Alpha
Diagnostic Intl. Inc.) was added and the plate was incubated for an
additional 1 h at room temperature. After a series of washes, 100
µl tetramethylbenzidine substrate solution (MilliporeSigma) was
added and the enzymatic reaction proceeded in the dark for 20 min.
The reaction was terminated by the addition of 50 µl 2 M sulfuric
acid and the absorbance was measured at 450 nm. Cytokine
concentrations were determined by generating standard curves using
recombinant cytokine standards provided in the kits.
Animal experiment
Male BALB/c mice (6–8 weeks old; 22–25 g) were
purchased from Beijing Vital River Laboratories Animal Technology
Co., Ltd. and subcutaneously injected with 1×106 MC38
cells (day 0) into the left flank. When tumor volumes reached ~150
mm3, the mice were randomized into treatment groups
(n=10). To assess the antitumor efficacy of Exe-9, mice were
randomly allocated to either the Exe-9 group (n=10) or the control
group (n=10). To evaluate the synergistic effect of Exe-9 combined
with Anti-PD-1, mice were randomly assigned to four groups: Control
(n=10), Exe-9 (n=10), Anti-PD-1 (n=10) and Exe-9 + Anti-PD-1
(n=10). Mice in the Exe-9 group received intratumoral injections of
Exe-9 at a concentration of 25 nmol/kg in saline on days 10, 12, 14
and 16. Mice in the Anti-PD-1 group were administered 100 µg of
anti-PD-1 antibody (cat. no. BE0146; Bio X Cell, Lebanon) via
intraperitoneal injection on days 10, 13, 16 and 19. The control
mice received equivalent volumes of sterile saline or isotype
immunoglobulin G (IgG; cat. no. 1054; Bio X Cell), as appropriate.
To further elucidate whether the antitumor effects of Exe-9 were
mediated by the modulation of tumor immunity, an additional
experiment was conducted in male BALB/c nude mice (Beijing Vital
River Laboratories Animal Technology Co., Ltd.) using the same
group size (n=10 per group) following the same injection schedule
as the Exe-9 group.
Prior to the invasive procedures, the mice were
anesthetized via inhalation of isoflurane (3–4% for induction and
1.5–2% for maintenance in oxygen). Mice demonstrating severe
distress signs at the end of the experiment were humanely
euthanized via CO2 inhalation, followed by cervical
dislocation.
Tumor length and width were recorded at the same
intervals as the injections using digital calipers to generate
tumor growth curves. According to institutional ethical guidelines,
the maximum tumor volume permitted was 1,500 mm3 and the
maximum diameter did not exceed 20 mm in any dimension. In the
present study, the largest tumor measured was ~1,200 mm3
in volume and 17 mm in diameter. The mice were euthanized on day
20; tumor tissue and spleen were collected for further assays.
Euthanasia was performed by gradual CO2 inhalation at a
displacement rate of 30–40% volume per min of the chamber, followed
by cervical dislocation to ensure mortality. The tumors were
harvested and weighed to assess their anticancer efficacy every 2
days starting from day 10.
Immunohistochemistry (IHC)
Excised mouse or patient tumor tissues were fixed in
10% neutral-buffered formalin for 24 h at room temperature and
processed for paraffin embedding. Paraffin blocks were sectioned at
5 µm thickness and mounted onto slides. Sections were
deparaffinized in xylene and rehydrated using a graded ethanol
series (100, 95 and 70%) before rinsing in distilled water. After
heating in a 10 mM citrate buffer (pH 6.0) at 92°C for 15 min,
slides were incubated in 3% hydrogen peroxide for 10 min at room
temperature. For the detection of the intracellular/membrane
protein GLP-1R, sections were permeabilized with 0.1% Triton X-100
in PBS for 10 min at room temperature. Blocking was performed with
5% normal goat serum (cat. no. 5425; Cell Signaling Technology,
Inc.) for 30 min at room temperature and the sections were
incubated overnight at 4°C with a primary anti-CD8 (cat. no.
PA007381.r2b; Syd Labs) or anti-GLP-1R antibody (cat. no. EPR21819;
Abcam) diluted 1:100 in blocking solution. After three washes in
PBS, a biotinylated goat anti-mouse IgG secondary antibody (1:200;
cat. no. ab6789; Abcam), was applied for 1 h at room temperature.
Immunoreactivity was visualized using the VECTASTAIN®
Elite ABC HRP Kit (Vector Laboratories, Inc.) with
diaminobenzidine, following the manufacturer's protocol.
Subsequently, the sections were counterstained with hematoxylin
(MilliporeSigma) at a concentration of 1% for 2 min at room
temperature, dehydrated using graded ethanol and xylene solutions
and cover slipped. The stained slides were examined and imaged
under an Olympus BX53 light/fluorescence microscope (Olympus
Corporation). Quantitative analysis of IHC staining was performed
using the Alpathwell Pathology Image Analysis System (version 3.5;
Servicebio Technology Co., Ltd.).
GLP-1R staining was semi-quantified using the
H-score method (35). For each
case, five tumor fields (×400 magnification) were randomly selected
within the tumor area. The percentage of tumor cells demonstrating
weak (1+), moderate (2+) or strong (3+) staining intensity was
recorded (P1-P3; total, 0–100%) and the H-score was calculated as
(1×P1 + 2×P2 + 3×P3), yielding a range of 0–300. Two
board-certified pathologists independently scored all slides in a
blinded manner. In cases of initial disagreement, the two
pathologists first re-examined the corresponding slides together to
reach a consensus. If a consensus could not be achieved, a third
senior pathologist was consulted to make the final
determination.
Enzyme-linked immunosorbent spot
(ELISpot) assay
Spleens harvested from Exp-9 treated mice or the
naive control were mechanically dissociated and filtered through a
70 µm cell strainer to obtain a cell suspension. Cells were then
plated at a density of 1×105 cells per well in ELISpot
plates pre-coated with anti-IFN-γ capture antibody, as provided in
the Mouse IFN-γ ELISpot Kit (cat. no. 3321-4APT-2; Mabtech). For
restimulation, cells were incubated with either irradiated MC38
cells or the major histocompatibility complex (MHC)-I restricted
AH1 peptide derived from MC38 cells (MilliporeSigma) at a final
concentration of 1 µg/ml. After being cultured for 20 h at 37°C,
the plates were washed with PBS and incubated with a biotinylated
anti-IFN-γ detection antibody for 2 h at room temperature. After
subsequent washing steps, an HRP conjugate was added and the
reaction was developed using a 3-amino-9-ethylcarbazole substrate
solution (cat. no. SK-4200; Vector Laboratories, Inc.) at room
temperature in the dark for 15 min. The enzymatic reaction was
stopped by rinsing the plates with distilled water and the spots
were counted using an automated ELISpot reader (ELR08; Cellular
Technology Limited).
Reverse transcription
(RT)-quantitative PCR
Total RNA was extracted from freshly frozen tissue
samples using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.), following the manufacturer's instructions. RNA
purity and concentration was assessed using a NanoDrop™ 2000
spectrophotometer (Thermo Fisher Scientific, Inc.). Subsequently, 1
µg total RNA was reverse transcribed into cDNA using the
PrimeScript™ RT reagent Kit (Takara Bio, Inc.). The quantitative
real-time PCR was performed on an ABI 7500 Real-Time PCR System
(Applied Biosystems, Inc.; Thermo Fisher Scientific, Inc.) using
SYBR® Premix Ex Taq II (Takara Bio, Inc.) The PCR
protocol comprised three stages: Initial denaturation at 95°C for
30 sec to activate the DNA polymerase and denature templates; 40
cycles of amplification, each consisting of denaturation at 95°C
for 5 sec followed by a combined annealing/extension at 60°C for 30
sec (the temperature of this step was optimized based on the primer
Tm); and a melting curve analysis (95°C for 15 sec; 60°C for 1 min;
then gradual heating to 95°C) to confirm amplicon specificity and
exclude nonspecific products. The primers used to detect GLP-1R and
GAPDH for normalization are listed in Table SI. PCR conditions were set as: 95°C
for 30 sec, followed by 40 cycles of 95°C for 5 sec and 60°C for 30
sec. Relative gene expression was calculated using the
2−ΔΔCq method (36). All
primers were designed for human genes and validated by National
Center for Biotechnology Information Basic Local Alignment Search
Tool (National Institutes of Health).
Western blotting analysis
Tissue samples from patients with CRC were
homogenized and lysed using radioimmunoprecipitation assay buffer
supplemented with 1% protease inhibitor cocktail and 1% phosphatase
inhibitor cocktail (MilliporeSigma) on ice for 30 min. Lysates were
clarified via centrifugation at 12,000 × g for 15 min at 4°C and
protein concentrations were determined using a BCA protein assay
kit (Thermo Fisher Scientific, Inc.). Equal amounts of proteins (30
µg per lane) were resolved on 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis gels and transferred
onto polyvinylidene difluoride membranes (MilliporeSigma) using a
Bio-Rad Trans-Blot Turbo system. The membranes were blocked with 5%
(w/v) skim milk in Tris-buffered saline containing 0.1% Tween-20
(TBST) for 1 h at room temperature. Membranes were incubated
overnight at 4°C with primary antibodies targeting GLP-1R (1:500).
After washing, the membranes were incubated with HRP-conjugated
secondary IgG (1:2,000, Beyotime Biotechnology) for 1 h at room
temperature. Protein bands were visualized using an enhanced
chemiluminescence detection kit (Thermo Fisher Scientific, Inc.)
and imaged using the ChemiDoc XRS+ Imaging System (Bio-Rad
Laboratories, Inc.). Densitometric band intensity analysis was
performed using the ImageJ 2.0 software (National Institutes of
Health), with GAPDH used for normalization. The antibodies used for
western blotting analysis are listed in Table SII.
Statistical analysis
All data were analyzed using the GraphPad Prism
software (version 9.0; Dotmatics). Continuous variables were
expressed as mean ± SEM and the Shapiro-Wilk test was employed to
assess data normality. For normally distributed data, one-way ANOVA
followed by Tukey's honest significant difference post hoc test was
used for group comparisons, whereas for non-normally distributed
data, the Kruskal-Wallis test with Dunn's multiple comparison test
was applied. Pearson's correlation analysis was performed to
evaluate the correlation between tumor volume and the number of
IFN-γ-secreting cells, as well as between GLP-1R expression and the
immunotherapy response. P<0.05 was considered to indicate a
statistically significant difference.
Results
GLP-1R antagonist potentiates
T-cell-mediated cytotoxicity against colon cancer cells
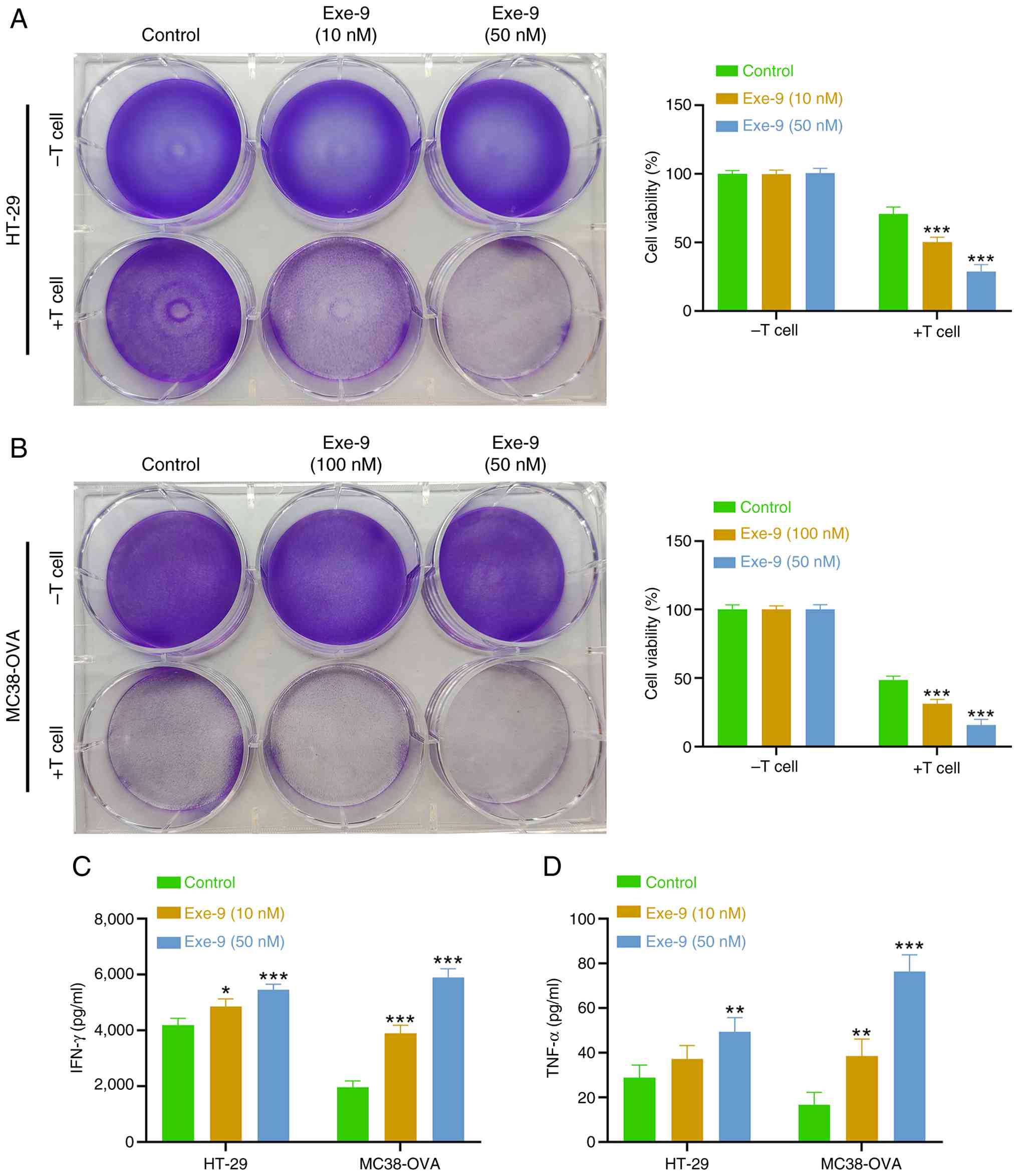
The colon cancer cell lines HT-29 and MC38-OVA were
cultured either individually or co-cultured with activated T cells
for 48 h in the presence of the GLP-1R antagonist Exe-9 at 10 or 50
nM or PBS as the vehicle control. The viability of HT-29 (Fig. 1A) and MC38-OVA (Fig. 1B) cells was assessed. In the absence
of T cells, Exe-9 treatment did not significantly alter cancer cell
viability, as no statistically significant differences were
detected between the PBS- and Exe-9-treated groups. By contrast,
when co-cultured with activated T cells, a significant
dose-dependent reduction in cell viability was observed, indicating
enhanced T-cell-mediated cytotoxicity in the presence of Exe-9. In
parallel, ELISA measurements of the culture supernatants revealed
that levels of IFN-γ (Fig. 1C) and
TNF-α (Fig. 1D) increased following
Exe-9 treatment compared with the control, with a dose-dependent
effect evident particularly in the MC38-OVA cultures. Collectively,
these results suggest that Exe-9 enhances the killing efficiency of
CD8+ T cells against colon cancer cells, potentially by
modulating the cytokine milieu that governs T-cell-mediated
cytotoxicity.
GLP-1R antagonist enhances
T-cell-mediated antitumor efficacy in mice
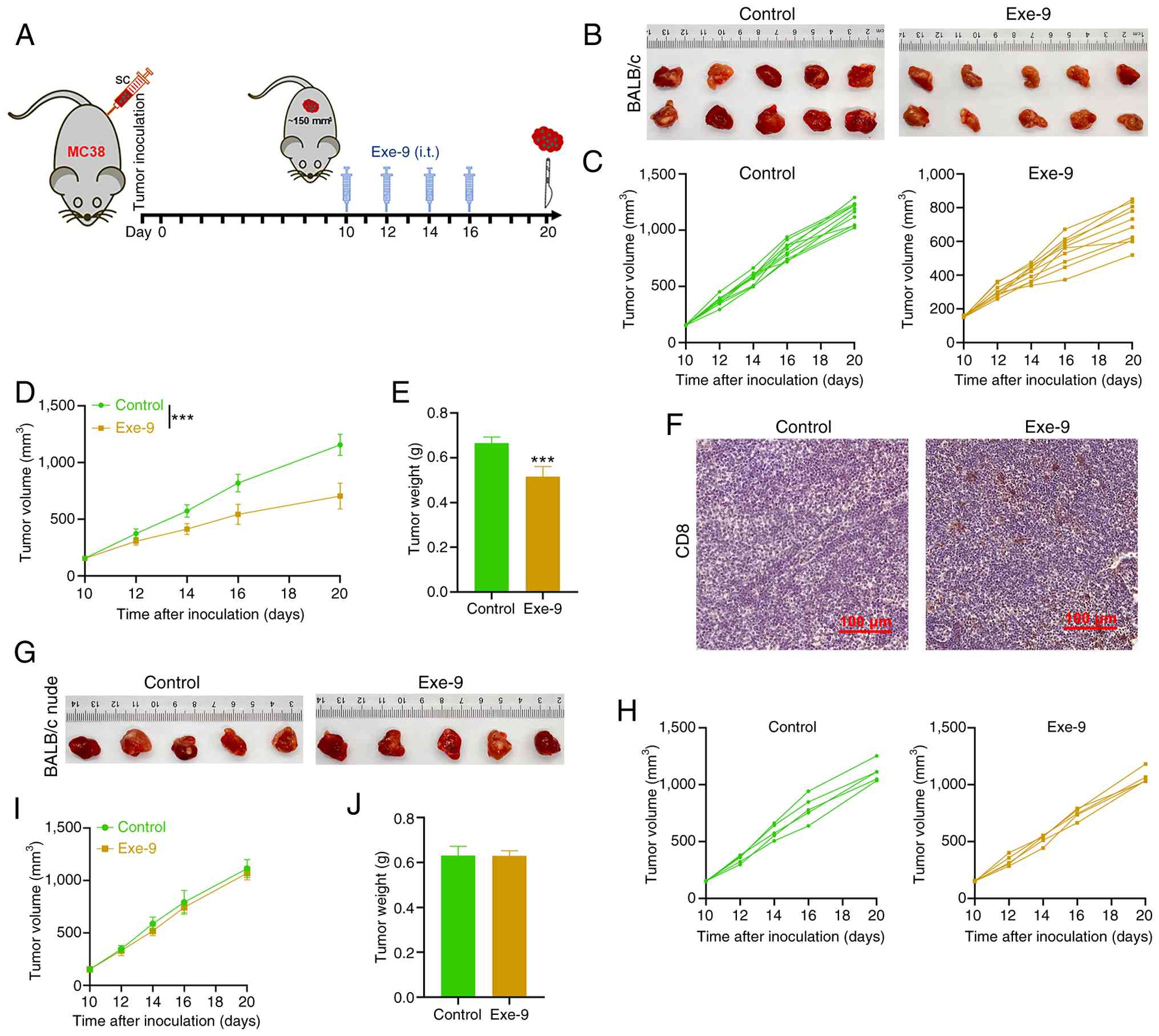
To evaluate the effects of the GLP-1R antagonist on
tumor growth, male mice were subcutaneously injected with MC38
tumor cells. When tumors reached ~150 mm3 on day 10,
Exe-9 was administered intratumorally at days 10, 12, 14 and 16,
while control mice received equivalent volumes of saline. Tumor
tissues were collected on day 20 for analysis (Fig. 2A). Notably, the tumors in
Exe-9-treated mice were markedly smaller compared with those in the
control group (Fig. 2B). The tumor
volume measurements recorded at each treatment point demonstrated a
continuous increase in both groups. However, the Exe-9 group
exhibited a markedly lower growth rate (Fig. 2C) compared with the control group.
At the endpoint, tumor volume (Fig.
2D) and weight (Fig. 2E) in the
control group were significantly greater compared with those in the
Exe-9-treated group. IHC analysis revealed that tumors from
Exe-9-treated mice displayed markedly higher CD8+ T-cell
infiltration compared with those from the control group (Fig. 2F), suggesting enhanced antitumor
immunity. To investigate the role of T cells in this response, the
experiment was replicated using nude mice lacking functional T
cells. In this model, no significant differences in tumor size were
observed between the Exe-9 and saline-treated groups (Fig. 2G); however, the tumor volume
continued to increase in both groups throughout the treatment
period (Fig. 2H and I). Tumor
weights collected on day 20 were also not significantly different
between the two groups (Fig. 2J).
These findings suggested that the antitumor effects of Exe-9 depend
on the presence of functional T cells, potentially through the
enhancement of CD8+ T-cell-mediated immunity.
GLP-1R antagonist elicits
tumor-specific CD8+ T-cell replication
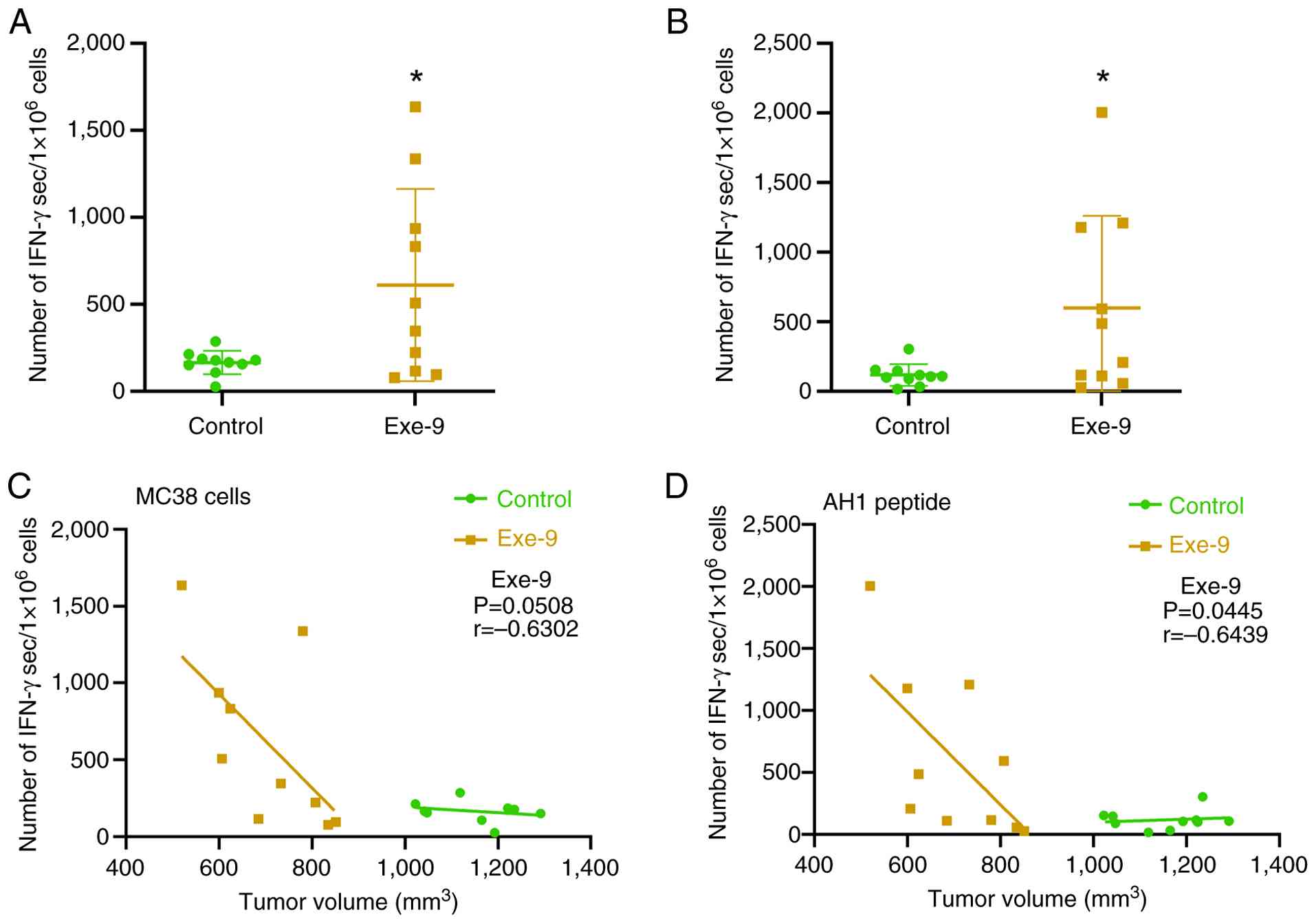
To investigate the effect of the GLP-1R antagonist
on tumor-specific CD8+ T-cell responses, splenocytes
were collected from mice and restimulated for 20 h with either MC38
tumor cells or MHC-I-restricted AH1 peptide. The number of
IFN-γ-secreting cells was quantified using ELISpot assays.
Exe-9-treated mice exhibited significantly higher numbers of
IFN-γ-secreting cells compared with the control mice under MC38
(Fig. 3A) and AH1 (Fig. 3B) restimulation conditions.
Correlation analysis between tumor volume and IFN-γ-secreting cells
revealed distinct patterns. In the Exe-9-treated group, a negative
correlation trend was observed between them following MC38
restimulation, although the trend was not statistically significant
(r=−0.6302; P=0.0508; Fig. 3C).
However, a significant negative correlation was identified when the
AH1 peptide was restimulated (r=−0.6439; P=0.0445; Fig. 3D), suggesting that Exe-9 treatment
enhanced tumor-specific CD8+ T-cell activation. By
contrast, splenocytes of mice from the control group consistently
exhibited low numbers of IFN-γ-secreting cells, regardless of tumor
volume or type of restimulation antigen (Fig. 3C and D). These results revealed that
Exe-9 promotes the generation of tumor-specific CD8+ T
cells capable of robust IFN-γ production. These results further
suggest that the enhanced antitumor response induced by Exe-9 is
likely mediated by an increase in antigen-specific CD8+
T-cell activity.
Combination of GLP-1R antagonist and
ICIs synergistically suppresses colon cancer growth in vivo
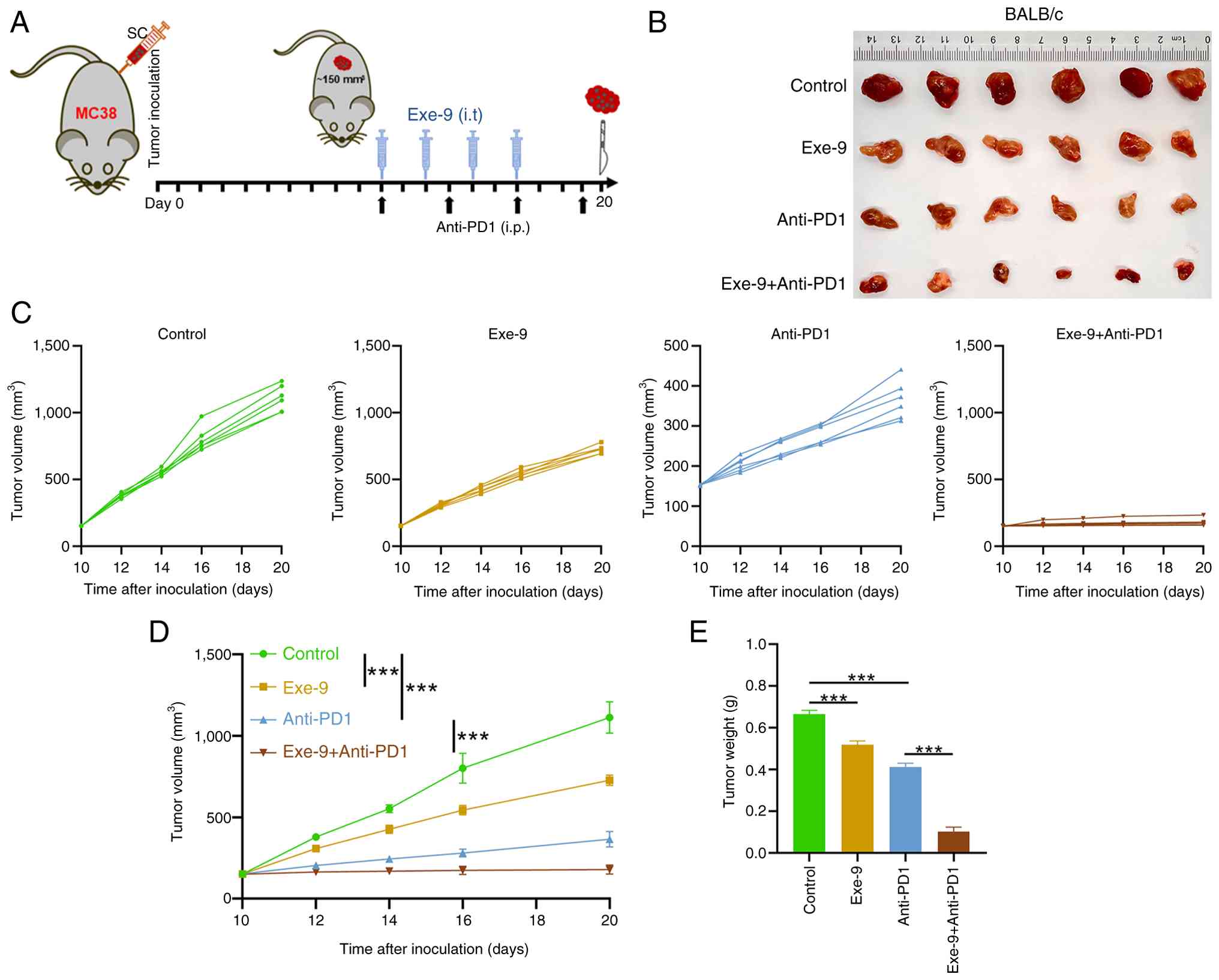
A combination treatment experiment with a GLP-1R
antagonist and an ICI was performed in mice bearing MC38 tumors.
Tumors were established to an average volume of ~150 mm3
by day 10 post-injection. Mice were treated intratumorally with
Exe-9 on days 10, 12, 14 and 16 or injected intraperitoneally with
anti-PD-1 on days 10, 13, 16 and 19. A combination of both
treatments or a control with saline and IgG, was administered at
the same time points. The tumors were collected on day 20 for
further analysis (Fig. 4A). The
control group exhibited the largest tumors, followed by the Exe-9,
then the Anti-PD-1 group and the Exe-9 + Anti-PD-1 group with the
smallest tumors (Fig. 4B). While
the tumor volumes in the control, Exe-9 and Anti-PD-1 groups
continuously increased throughout the treatment period, the
combination of Exe-9 and Anti-PD-1 resulted in effective tumor
control, with no significant changes in tumor volume observed
(Fig. 4C). By day 20, the tumor
volumes were quantified as shown in Fig. 4D and revealed the same trend as
shown in the representative images in Fig. 2B. Consistent with these
observations, the tumor weights at the endpoint also demonstrated
significant differences among groups. Specifically, the tumor
weight in the combination treatment group (Exe-9 + Anti-PD-1) was
significantly reduced compared with that in the Anti-PD-1
monotherapy group (P<0.001; Fig.
4E). These results illustrate that Exe-9 enhanced the antitumor
efficacy of anti-PD-1 in vivo, indicating a potential
synergistic interaction between GLP-1R antagonism and immune
checkpoint inhibition in suppressing colon cancer growth.
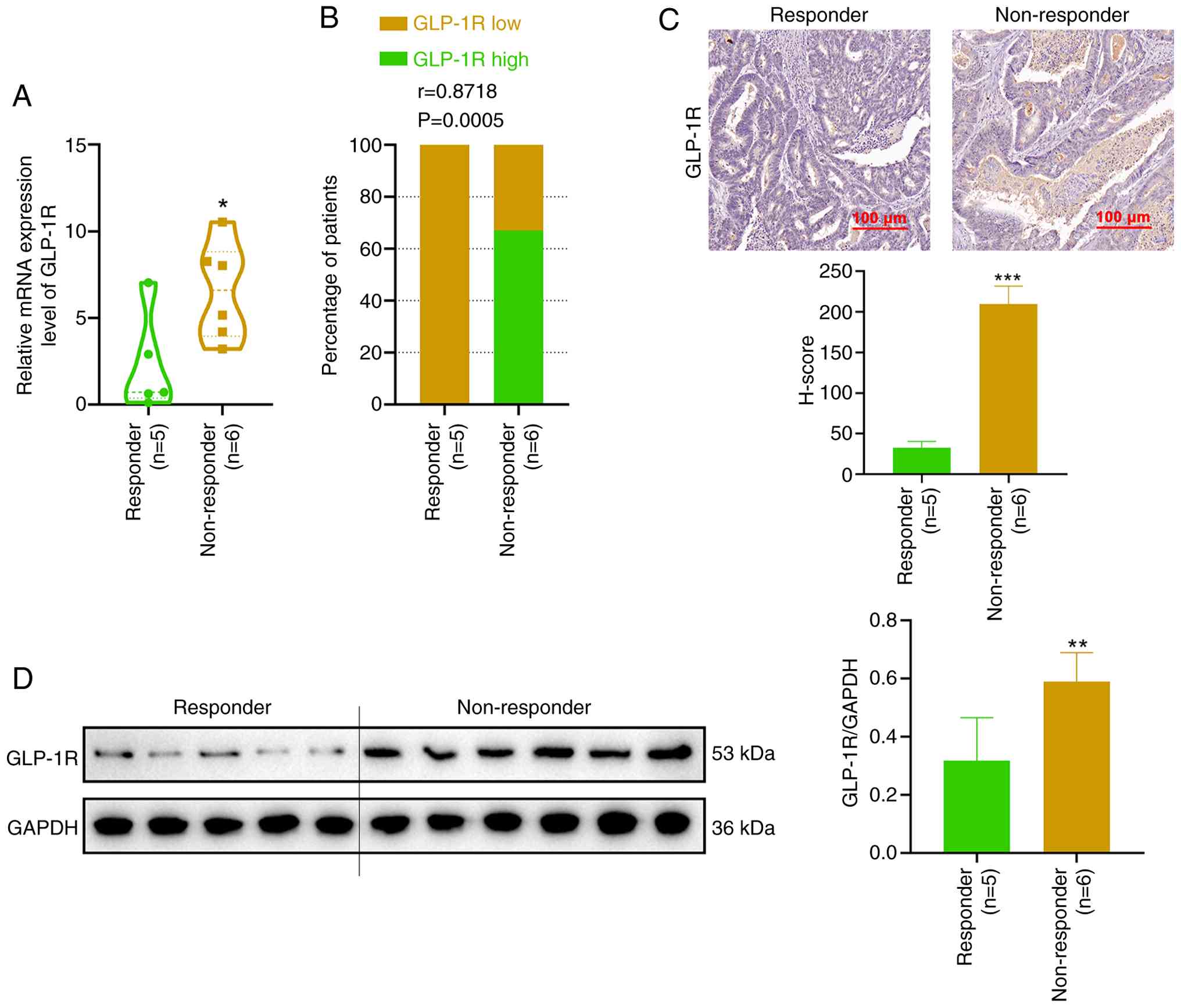
High expression of GLP-1R correlates
with reduced efficacy of anti-PD-1 immunotherapy
To investigate the clinical relevance of GLP-1R
expression in tumor immunotherapy, tumor samples from 11 patients
with CRC treated with ICIs were collected and categorized into
responder and non-responder groups based on therapeutic outcomes.
Quantitative PCR analysis revealed that GLP-1R expression levels
were significantly higher in tumor tissues from non-responders
compared with those from responders (P<0.05; Fig. 5A). Stratifying patients into high
and low GLP-1R expression groups based on the median expression
level revealed a positive correlation between high GLP-1R
expression and non-responsiveness to therapy (r=0.8718; P=0.0005;
Fig. 5B). These findings were
corroborated by IHC and western blotting analyses, revealing that
GLP-1R protein expression was significantly higher in tumor tissues
from non-responders compared with those from responders (Fig. 5C and D). These results indicate that
elevated GLP-1R expression is associated with reduced efficacy of
anti-PD-1 immunotherapy in patients with CRC.
Discussion
The incidence of CRC is rapidly increasing and is
associated with high mortality. However, the clinical efficacy of
ICIs remains limited, highlighting the urgent need to explore novel
complementary therapeutic strategies (37). In the present study, a multitiered
experimental approach was employed to comprehensively assess the
application of the GLP-1R antagonist Exe-9 and its potential as an
adjunct in CRC immunotherapy.
GLP-1R antagonists competitively occupy the binding
site of GLP-1R, inhibiting downstream GLP-1 signal transduction.
In vitro experiments revealed that under co-culture
conditions with activated T cells, Exe-9 significantly decreased
cancer cell viability in a dose-dependent manner, whereas no
cytotoxic effect was observed in the absence of T cells. This
finding was corroborated by in vivo studies using a CRC
mouse model in which BALB/c mice treated with Exe-9 exhibited
significantly attenuated tumor growth and increased intratumoral
infiltration of CD8+ T cells, which were absent in nude
mice lacking functional T cells. These results indicate that the
cytotoxic effects induced by GLP-1R antagonism were mediated by
T-cell activity. This observation is consistent with previous
reports revealing that GLP-1R signaling exerts a negative
modulatory effect on T-cell function and antagonism of this
receptor can potentiate antitumor immune responses (38,39).
However, other studies have reported conflicting
findings regarding the role of GLP-1R in tumor biology, reflecting
its diverse functions across various cancer types, experimental
models and immune microenvironments. For instance, the GLP-1R
agonist semaglutide promotes tumor cell proliferation in
neuroendocrine cancer (40),
whereas a meta-analysis encompassing eight original studies
revealed that GLP-1R agonists may confer protective effects against
liver cancer (24,41). Furthermore, in CRC cell lines,
GLP-1R activation inhibits tumor cell proliferation (42). These discrepancies underscore the
complex and context-dependent nature of GLP-1R signaling. The
present study provided direct laboratory evidence supporting the
antitumor effects of GLP-1R antagonism in CRC, as opposed to
agonism; however, further investigations are warranted. Future
researchers should aim to delineate the regulatory mechanisms and
dose-response relationships of the GLP-1R pathway under more
refined experimental conditions, facilitating the development of
precise immunotherapeutic strategies.
Furthermore, Exe-9 treatment was associated with a
dose-dependent increase in tumor cell death, concomitant with an
increase in IFN-γ and TNF-α levels in the culture supernatant.
Consistently, a significant increase in IFN-γ-secreting splenocytes
was observed in CRC mouse models and this parameter was inversely
correlated with tumor volume. This finding aligned with previous
reports indicating that intratumoral IFN-γ administration enhances
immune cell infiltration and suppresses tumor growth (26,43–46).
Furthermore, the capacity for IFN-γ secretion, a key T-cell
activation marker (47), supports
the notion that Exe-9 promotes tumor-specific CD8+
T-cell activation and expansion. Collectively, these findings
indirectly corroborated that Exe-9 enhances T-cell-mediated
cytotoxicity in tumor cells. The present study findings support a
T-cell-dependent mechanism based on CD8 infiltration, IFN-γ ELISpot
responses and the lack of effect in nude mice; however, the present
study acknowledges that detailed flow cytometry was not performed
to assess T-cell activation, exhaustion markers or comprehensive
tumor immune microenvironment profiling. Future studies should
incorporate multiparameter flow cytometry and immune cell profiling
to more precisely delineate how GLP-1R antagonism modulates T-cell
functional states and tumor-infiltrating immune subsets.
Notwithstanding this limitation, the present study findings provide
indirect evidence that Exe-9 enhances T-cell-mediated
cytotoxicity.
In addition to demonstrating the antitumor efficacy
of GLP-1R antagonists as monotherapy for CRC, to the best of our
knowledge, the present study provided the first direct evidence
that combining a GLP-1R antagonist with anti-PD-1 therapy produces
a synergistic antitumor effect. Specifically, mice receiving the
combination treatment exhibited significantly reduced tumor volumes
and weights compared with those treated with either agent alone.
The therapeutic rationale behind the use of ICIs, such as
anti-PD-1/PD-L1 or anti-cytotoxic T-lymphocyte-associated antigen-4
antibodies, is based on their ability to block inhibitory signals
from tumor cells that dampen T-cell activity, restoring
T-cell-mediated antitumor responses. However, immunotherapy has
exhibited limited efficacy in CRC primarily due to the reduced
neoantigen burden of tumor cells, which hampers CD8+
T-cell activation and expansion as well as immunosuppressive
factors present within the tumor microenvironment (48,49).
Therefore, enhancing T-cell activity is considered a promising
strategy in augmenting the effects of ICIs. For instance, CXCL10
overexpression increases CD8+ T-cell infiltration,
sensitizing colorectal tumors to combined cetuximab and anti-PD-1
therapy (50). N-acetylcysteine
induces differentiation of CD8+ T cell subpopulations,
synergizing with anti-PD-1 antibodies to inhibit CRC progression in
murine models (51). Similarly, the
present study findings revealed that Exe-9, by antagonizing GLP-1R,
enhances CD8+ T cell infiltration, activation and
cytotoxic efficacy. This mechanism may help in overcoming the
limitations of ICIs in treating CRC, particularly in ‘cold tumor’
settings and suggests a novel therapeutic target. Furthermore, the
present in vitro and in vivo data were corroborated
by clinical sample analyses, which revealed that high GLP-1R
expression in patients with CRC undergoing ICI therapy was closely
associated with a poor treatment response, underscoring the
potential of GLP-1R as a negative predictive biomarker in
immunotherapy. These findings are consistent with emerging reports
identifying GLP-1R as a negative costimulatory signal in T cells
(26,52), providing biological plausibility for
its role as a therapeutic target and predictive biomarker.
Beyond T-cell co-stimulation, GLP-1R signaling has
been associated with inflammatory regulation. Multiple studies have
reported that GLP-1R activation dampens pro-inflammatory signaling
pathways (53,54) and studies have identified GLP-1R as
a negative costimulatory molecule in T cells, suggesting that
antagonism may relieve this suppression and enhance effector T cell
[T helper 1 cell (Th1)-type] responses (26,38,39).
Consistent with this, the present study findings of increased IFN-γ
secretion suggest that GLP-1R blockade may promote a Th1-skewed
immune milieu, which is key to effective antitumor immunity.
Despite these promising results, the present study
had certain limitations. First, the relatively small sample size in
animal and clinical analyses may limit the generalizability of the
present study findings. Second, exploratory analyses of The Cancer
Genome Atlas CRC bulk RNA-sequencing did not reveal a strong
correlation between GLP-1R expression and immune infiltration due
to the low GLP-1R signal in these datasets (55,56).
Hence, validation relies on larger IHC/qPCR-based cohorts and
high-resolution single-cell or spatial datasets. Furthermore, the
precise molecular mechanisms underlying the observed synergistic
interaction between GLP-1R antagonism and immune checkpoint
inhibition remain to be elucidated. Lastly, the long-term efficacy
and safety of this combinatorial approach require further
investigation in larger preclinical studies and clinical
trials.
Translational challenges need to be considered. In
the present preclinical study, Exe-9 was administered
intratumorally, which is not a practical clinical route for most
patients with CRC. Therefore, future studies should explore
systemic delivery strategies, such as intravenous or subcutaneous
administration and evaluate their pharmacokinetics and
biodistribution. In addition, because GLP-1 signaling is closely
associated with glucose metabolism and gastrointestinal function,
GLP-1R antagonism may have metabolic side effects, including
hyperglycemia or altered insulin responses. Careful preclinical
safety assessments and early-phase clinical trials are required to
determine tolerability. Notably, whether these effects extend to
advanced or metastatic CRC remains to be elucidated, underscoring
the need for validation in larger late-stage patient cohorts to
determine the true clinical applicability. Lastly, future
directions include the development of clinically feasible GLP-1R
antagonists, the identification of predictive biomarkers to guide
patient selection and the integration of this approach into
rational combinatorial immunotherapy regimens.
In summary, the present study revealed that the
GLP-1R antagonist Exe-9 significantly enhances T-cell-mediated
cytotoxicity against CRC and when combined with ICIs, producing a
synergistic antitumor effect. Clinical correlations indicate a
negative association between GLP-1R expression and the efficacy of
anti-PD-1 therapy. These findings highlight the potential of GLP-1R
antagonism as a complementary strategy in improving immunotherapy
outcomes in patients with CRC in the future.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Guangzhou Health Science
and Technology Project (grant no. 20251A010102).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
ZZ and FC designed the experiments. CZ and LL
conducted the experiments. DL and GM analyzed the experimental
results. ZZ wrote the manuscript. FC revised the manuscript
accordingly. ZZ and CZ confirm the authenticity of all the raw
data. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
All experimental procedures involving animals and
human tissues were approved by the Ethics Committee of Guangzhou
Development District Hospital (approval no. F2024-030; Guangzhou,
China). All animal experiments were approved by the Institutional
Animal Care and Use Committee of Guangzhou Development District
Hospital, Guangzhou, China. For human studies (approval no.
F2024-030), peripheral blood or tumor tissues were collected from
healthy donors and patients, respectively, after obtaining written
informed consent, following the Declaration of Helsinki. The
Institutional Review Board of the Guangzhou Development District
Hospital approved the present study protocol.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bin JF, Chen LF, Wang Y, Ge H and Chen W:
Bibliometric and visual analysis of global crc circular RNA
research 2015–2023. Front Immunol. 16:15804052025. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tsukanov VV, Vasyutin AV, Kasparov EV and
Tonkikh JL: Is the use of artificial intelligence the main stage
for detecting polyps during colonoscopy? World J Gastroenterol.
31:1065002025. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Saha P, Ravanan P and Talwar P: A
multi-omics exploration of PPARG activation in colon cancer:
Kinases featuring a PPRE sequence within regulatory regions. Biol
Direct. 20:682025. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kamara AA, He S, Joseph Fofanah A, Xu R
and Chen Y: MDPNet: Multiscale dynamic Polyp-focus network for
enhancing medical image polyp segmentation. IEEE Trans Med Imaging.
44:5208–5220. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Araghi M, Soerjomataram I, Jenkins M,
Brierley J, Morris E, Bray F and Arnold M: Global trends in
colorectal cancer mortality: Projections to the year 2035. Int J
Cancer. 144:2992–3000. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ghorbaninezhad F, Nour MA, Farzam OR,
Saeedi H, Vanan AG, Bakhshivand M, Jafarlou M, Hatami-Sadr A and
Baradaran B: The tumor microenvironment and dendritic cells:
Developers of pioneering strategies in colorectal cancer
immunotherapy? Biochim Biophys Acta Rev Cancer. 1880:1892812025.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Steup C, Kennel KB, Neurath MF,
Fichtner-Feigl S and Greten FR: Current and emerging concepts for
systemic treatment of metastatic colorectal cancer. Gut.
74:2070–2095. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hossain MS, Karuniawati H, Jairoun AA,
Urbi Z, Ooi DJ, John A, Lim YC, Kibria KK, Mohiuddin A and Ming LC:
Colorectal cancer: A review of carcinogenesis, global epidemiology,
current challenges, risk factors, preventive and treatment
strategies. Cancers. 14:17322022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nors J, Iversen LH, Erichsen R, Gotschalck
KA and Andersen CL: Incidence of recurrence and time to recurrence
in stage I to III colorectal cancer: A nationwide Danish cohort
study. JAMA Oncol. 10:54–62. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
American Cancer Society, . Cancer Facts
& Figures 2024. Atlanta, GA: American Cancer Society; 2024
|
|
11
|
Sabbatino F, Liguori L, Pepe S and Ferrone
S: Immune checkpoint inhibitors for the treatment of melanoma.
Expert Opin Biol Ther. 22:563–576. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou F, Qiao M and Zhou C: The
cutting-edge progress of immune-checkpoint blockade in lung cancer.
Cell Mol Immunol. 18:279–293. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Makaremi S, Asadzadeh Z, Hemmat N,
Baghbanzadeh A, Sgambato A, Ghorbaninezhad F, Safarpour H,
Argentiero A, Brunetti O, Bernardini R, et al: Immune checkpoint
inhibitors in colorectal cancer: Challenges and future prospects.
Biomedicines. 9:10752021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Emambux S, Tachon G, Junca A and Tougeron
D: Results and challenges of immune checkpoint inhibitors in
colorectal cancer. Expert Opin Biol Ther. 18:561–573. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ros J, Balconi F, Baraibar I, Saoudi
Gonzalez N, Salva F, Tabernero J and Elez E: Advances in immune
checkpoint inhibitor combination strategies for microsatellite
stable colorectal cancer. Front Oncol. 13:11122762023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Trujillo JM, Nuffer W and Smith BA: GLP-1
receptor agonists: An updated review of head-to-head clinical
studies. Ther Adv Endocrinol Metab. 12:20420188219973202021.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zheng Z, Zong Y, Ma Y, Tian Y, Pang Y,
Zhang C and Gao J: Glucagon-like peptide-1 receptor: Mechanisms and
advances in therapy. Signal Transduct Target Ther. 9:2342024.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
del Olmo-Garcia MI and Merino-Torres JF:
GLP-1 receptor agonists and cardiovascular disease in patients with
type 2 diabetes. J Diabetes Res. 2018:40204922018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yabut JM and Drucker DJ: Glucagon-like
peptide-1 receptor-based therapeutics for metabolic liver disease.
Endocr Rev. 44:14–32. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Halloum W, Dughem YA, Beier D and Pellesi
L: Glucagon-like peptide-1 (GLP-1) receptor agonists for headache
and pain disorders: A systematic review. J Headache Pain.
25:1122024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gala K, Camilleri M, Goyal M, Ohri A,
Marek G and Ravi K: Glucagon-like Peptide-1 receptor agonist use is
associated with and may lead to esophageal motility abnormalities.
Clin Gastroenterol Hepatol. Nov 8–2025.(Epub ahead of print). doi:
10.1016/j.cgh.2025.11.003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Saha B, Kamalumpundi V and Codipilly DC:
GLP1 and GIP receptor agonists: Effects on the gastrointestinal
tract and management strategies for primary care physicians. Mayo
Clin Proc. Dec 1–2025.(Epub ahead of print). doi:
10.1016/j.mayocp.2025.09.017. View Article : Google Scholar
|
|
23
|
Chen J, Mei A, Wei Y, Li C, Qian H, Min X,
Yang H, Dong L, Rao X and Zhong J: GLP-1 receptor agonist as a
modulator of innate immunity. Front Immunol. 13:9975782022.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
He Y, Xu B, Zhang M, Chen D, Wu S, Gao J,
Liu Y, Zhang Z, Kuang J and Fang Q: Advances in GLP-1 receptor
agonists for pain treatment and their future potential. J Headache
Pain. 26:462025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Valencia-Rincón E, Rai R, Chandra V and
Wellberg EA: GLP-1 receptor agonists and cancer: Current clinical
evidence and translational opportunities for preclinical research.
J Clin Invest. 135:e1947432025. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rode AK, Buus TB, Mraz V, Al-Jaberi FAH,
Lopez DV, Ford SL, Hennen S, Eliasen IP, Klewe IV, Gharehdaghi L,
et al: Induced human regulatory T cells express the glucagon-like
peptide-1 receptor. Cells. 11:25872022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Paul MS and Ohashi PS: The roles of CD8+ T
cell subsets in antitumor immunity. Trends Cell Biol. 30:695–704.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu YT and Sun ZJ: Turning cold tumors
into hot tumors by improving T-cell infiltration. Theranostics.
11:53652021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vafaei S, Zekiy AO, Khanamir RA, Zaman BA,
Ghayourvahdat A, Azimizonuzi H and Zamani M: Combination therapy
with immune checkpoint inhibitors (ICIs); a new frontier. Cancer
Cell Int. 22:22022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chiang CH, Song J, Chi KY, Chang YC,
Xanthavanij N, Chang Y, Hsia YP, Chiang CH, Ghamari A, Reynolds KL,
et al: Glucagon-like Peptide-1 agonists reduce cardiovascular
events in cancer patients on immune checkpoint inhibitors. Eur J
Cancer. 216:1151702025. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Quagliariello V, Canale ML, Bisceglia I,
Iovine M, Giordano V, Giacobbe I, Scherillo M, Gabrielli D, Maurea
C, Barbato M, et al: Glucagon-like peptide 1 receptor agonists in
Cardio-oncology: Pathophysiology of cardiometabolic outcomes in
cancer patients. Int J Mol Sci. 25:112992024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vignarajah A, Kim S, Albliwi M, Ahn HM,
Izda A, Naffa F, Vigneswaramoorthy N, Barot S and Shah G: Role of
GLP-1 receptor agonists in managing cancer therapy-related cardiac
dysfunction. J Am Heart Assoc. 14:e0409192025. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Borner T, Pataro AM, Doebley SA, Furst CD,
White AD, Gao SX, Chow A, Sanchez-Navarro MJ, Ghidewon MY, Halas
JG, et al: Hypophagia and body weight loss by tirzepatide are
accompanied by fewer GI adverse events compared to semaglutide in
preclinical models. Sci Adv. 11:eadu15892025. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Eisenhauer EA, Therasse P, Bogaerts J,
Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S,
Mooney M, et al: New response evaluation criteria in solid tumours:
Revised RECIST guideline (version 1.1). Eur J Cancer. 45:228–247.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wen Z, Luo D, Wang S, Rong R, Evers BM,
Jia L, Fang Y, Daoud EV, Yang S, Gu Z, et al: Deep Learning-Based
H-Score quantification of Immunohistochemistry-stained images. Mod
Pathol. 37:1003982024. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using Real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Islam MR, Akash S, Rahman MM, Nowrin FT,
Akter T, Shohag S, Rauf A, Aljohani AS and Simal-Gandara J: Colon
cancer and colorectal cancer: Prevention and treatment by potential
natural products. Chem Biol Interact. 368:1101702022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ben Nasr M, Usuelli V, Dellepiane S,
Seelam AJ, Fiorentino TV, D'Addio F, Fiorina E, Xu C, Xie Y,
Balasubramanian HB, et al: Glucagon-like peptide 1 receptor is a T
cell-negative costimulatory molecule. Cell Metab. 36:1302–1319.e12.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wong CK, Yusta B, Koehler JA, Baggio LL,
McLean BA, Matthews D, Seeley RJ and Drucker DJ: Divergent roles
for the gut intraepithelial lymphocyte GLP-1R in control of
metabolism, microbiota, and T cell-induced inflammation. Cell
Metab. 34:1514–1531.e7. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shilyansky JS, Chan CJ, Xiao S,
Gribovskaja-Rupp I, Quelle DE, Howe JR, Dillon JS and Ear PH:
GLP-1R agonist promotes proliferation of neuroendocrine neoplasm
cells expressing GLP-1 receptors. Surgery. 179:1089432025.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shabil M, Khatib MN, Ballal S, Bansal P,
Tomar BS, Ashraf A, Kumar MR, Sinha A, Rawat P, Gaidhane AM, et al:
Risk of hepatocellular carcinoma with Glucagon-like Peptide-1
receptor agonist treatment in patients: A systematic review and
meta-analysis. BMC Endocr Disord. 24:2462024. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Koehler JA, Kain T and Drucker DJ:
Glucagon-like peptide-1 receptor activation inhibits growth and
augments apoptosis in murine CT26 colon cancer cells.
Endocrinology. 152:3362–3372. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tang Y, Wei J, Ge X, Yu C, Lu W, Qian Y,
Yang H, Fu D, Fang Y, Zhou X, et al: Intratumoral injection of
interferon gamma promotes the efficacy of anti-PD1 treatment in
colorectal cancer. Cancer Lett. 588:2167982024. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhong FM, Yao FY, Yang YL, Liu J, Li MY,
Jiang JY, Zhang N, Xu YM, Li SQ, Cheng Y, et al: Molecular subtypes
predict therapeutic responses and identifying and validating
diagnostic signatures based on machine learning in chronic myeloid
leukemia. Cancer Cell Int. 23:612023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xie Y, Lv Z, Wang Y, Ma J, Wei X, Zheng G
and Wu J: Study on the efficacy of IFN-γ- and sPD-1-overexpressing
BMSCs in enhancing immune effects for the treatment of lung
adenocarcinoma. Front Immunol. 16:15544672025. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hogg GD, Weinstein AG, Kingston NL, Liu X,
Dres OM, Kang LI, Lander VE, Kao YL, Ahmad F, Knolhoff BL, et al:
Combined Flt3L and CD40 agonism restores dendritic cell-driven T
cell immunity in pancreatic cancer. Sci Immunol. 10:eadp39782025.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Andrews LP, Butler SC, Cui J, Cillo AR,
Cardello C, Liu C, Brunazzi EA, Baessler A, Xie B, Kunning SR, et
al: LAG-3 and PD-1 synergize on CD8+ T cells to drive T cell
exhaustion and hinder autocrine IFN-γ-dependent anti-tumor
immunity. Cell. 187:4355–4372.e22. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhou C, Cheng X and Tu S: Current status
and future perspective of immune checkpoint inhibitors in
colorectal cancer. Cancer Lett. 521:119–129. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sahin IH, Ciombor KK, Diaz LA, Yu J and
Kim R: Immunotherapy for microsatellite stable colorectal cancers:
Challenges and novel therapeutic avenues. Am Soc Clin Oncol Educ
Book. 42:1–12. 2022.PubMed/NCBI
|
|
50
|
Yan W, Qiu L, Yang M, Xu A, Ma M, Yuan Q,
Ma X, Liang W, Li X and Lu Y: CXCL10 mediates CD8+ T cells to
facilitate vessel normalization and improve the efficacy of
cetuximab combined with PD-1 checkpoint inhibitors in colorectal
cancer. Cancer Lett. 567:2162632023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhou W, Qu M, Yue Y, Zhong Z, Nan K, Sun
X, Wu Q, Zhang J, Chen W and Miao C: Acetylcysteine synergizes PD-1
blockers against colorectal cancer progression by promoting TCF1+
PD1+ CD8+ T cell differentiation. Cell Commun Signal. 22:5032024.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhu C, Lai Y, Liu C, Teng L, Zhu Y, Lin X,
Fu X, Lai Q, Liu S, Zhou X and Fang Y: Comprehensively prognostic
and immunological analyses of GLP-1 signaling-related genes in
pan-cancer and validation in colorectal cancer. Front Pharmacol.
15:13872432024. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Alharbi SH: Anti-inflammatory role of
glucagon-like peptide 1 receptor agonists and its clinical
implications. Ther Adv Endocrinol Metab. 15:204201882312223672024.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wong S, Le GH, Dri CE, Teopiz KM and
McIntyre RS: Evaluating biased agonism of glucagon-like peptide-1
(GLP-1) receptors to improve cellular bioenergetics: A systematic
review. Diabetes Obes Metab. 27:6105–6115. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wong CK, Yusta B, Tong JCL, Broichhagen J,
Hodson DJ and Drucker DJ: Reassessment of antibody-based detection
of the murine T cell GLP-1 receptor. Cell Metab. 37:1783–1788.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Pang J, Feng JN, Ling W and Jin T: The
anti-inflammatory feature of glucagon-like peptide-1 and its based
diabetes drugs-Therapeutic potential exploration in lung injury.
Acta Pharm Sin B. 12:4040–4055. 2022. View Article : Google Scholar : PubMed/NCBI
|