Introduction
Cervical cancer remains one of the leading causes of
cancer-related mortalities among women, ranking fourth in both
incidence and mortality rates for malignancies in women globally
(1,2). Cervical cancer accounts for ~9% of
novel cancer cases worldwide and 8% of all cancer mortalities
(3). Current prevention strategies
primarily rely on vaccination and molecular screening programs
(4). However, large-scale vaccine
implementation remains constrained by socioeconomic disparities and
uneven human development levels, hindering universal coverage
(5). Standard treatments for
cervical cancer include surgery, radiotherapy and platinum-based
chemotherapy. In recent years, stimulus-responsive hydrogel therapy
and 5-aminolevulinic acid photodynamic therapy (5-ALA-PDT) have
emerged as promising systemic therapeutic options (6–8).
Notably, the application of immune checkpoint inhibitors has
markedly improved survival outcomes for advanced-stage patients
(9). Despite these advances, there
remains an urgent need to identify more reliable biomarkers to
potentially guide the development of novel targeted therapeutic
strategies in the future.
Cancer is characterized by somatic genomic
instability and metabolic reprogramming of cancer cells serves a
key role in driving tumorigenesis (10). Metabolic enzymes and their
accumulated metabolites often serve as substrates for
post-translational modifications-such as lactylation, acetylation
and succinylation-which in turn induce epigenetic remodeling and
influence cancer progression (11,12).
The extracellular matrix (ECM), a key component of the tumor
microenvironment (TME), can be remodeled by metabolic byproducts
such as lactate. These substances promote tumor cell invasion and
metastasis by altering cellular metabolic states (13). This metabolic adaptation mechanism
enables tumor cells to meet the energy and biosynthetic demands
required for sustained growth and proliferation (14). Recently, lactylation has emerged as
a novel post-translational modification with notable implications
in cancer biology. Previous studies revealed that abnormal protein
lactylation in multiple malignancies, including breast (15), gastric (16) and non-small cell lung cancer
(17), suggesting that
dysregulation of lactylation pathways may promote tumor progression
and constitute potential therapeutic targets in the future
(11).
The present study comprehensively investigated the
role of lactylation-associated genes (LAGs) in cervical cancer by
integrating multi-omics data from The Cancer Genome Atlas (TCGA)
and the Gene Expression Omnibus (GEO) alongside single-cell
transcriptomics analysis. Candidate LAGs were screened using
single-cell RNA-sequencing (scRNA-seq) data and a prognostic model
was constructed using multiple machine learning algorithms in
combination with GEO datasets. The predictive efficacy of the
LAGs-based model for cervical squamous cell carcinoma (CESC) was
systematically evaluated. Functional enrichment analysis explored
biological relevance, immune microenvironment associations and
intercellular communication patterns. Furthermore, Mendelian
randomization (MR) analysis was employed to assess the causal
relationship between key prognostic LAGs and cervical cancer risk.
Lastly, in vitro experiments using SiHa and HeLa cell lines
validated the role of high-mobility group nucleosome-binding
protein 1 (HMGN1) in cervical cancer cell proliferation and
progression. Collectively, these findings provide novel insights
for cervical cancer prognosis assessment and pave potential
pathways for personalized therapeutic interventions in the future.
The overall study design is illustrated in Fig. 1.
Materials and methods
Data collection and processing
The TCGA-CESC dataset (accessed December 20, 2024)
contains 306 CC tumor tissues and survival data from 293 samples.
(https://portal.gdc.cancer.gov/)
(18). Samples with incomplete or
missing clinicopathological information were excluded. Differential
gene expression analysis was performed using the R package ‘limma’
(version 3.64.3) (19). All
datasets were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/), including
three microarray datasets [GSE122697 (20), GSE146114 (21) and GSE63514 (22)] for WGCNA analysis, the scRNA-seq
dataset GSE44001 (23) and the
validation set GSE30760 (24). The
GSE7803 (25) dataset was used for
independent external validation of the prognostic model. At
present, no other publicly available scRNA-sequencing datasets with
sufficient sample size, annotation quality and relevance to
cervical cancer are available; therefore, GSE44001 was selected as
the only suitable dataset for single-cell analysis in the present
study. Detailed information about the datasets is provided in
Table SI. Batch effects were
mitigated using the ‘sva’ package (version 3.56.0) (26). Furthermore, 325 LAGs were collected
from previously published studies, as detailed in Table SII (27–29).
To further assess the association between the
prediction model and the immune response of the patients, the
present study integrated data from the IMvigor210 cohort using the
‘IMvigor210CoreBiologies’ R package (version 1.15.0.) (30). The present study also obtained
cervical cancer genome-wide association studies (GWAS) data and LAG
expression quantitative trait loci (eQTL) GWAS data from the
FinnGen database (https://risteys.finngen.fi/) for MR analysis (31). The cervical cancer GWAS dataset,
‘FinnGen R12 CD2 INSITU CERVIX UTERI EXALLC’ contains data on
20,104,836 single nucleotide polymorphisms (SNPs).
Single-cell transcriptome data
analysis and processing
scRNA-seq data from both normal and cervical cancer
tissues were obtained from the GSE168652 dataset. Data
preprocessing and quality control were performed using the ‘Seurat’
R package (version 5.3.1; http://satijalab.org/seurat/) (32). Cells with low gene counts or high
mitochondrial gene percentages were filtered out to ensure data
integrity. Batch effects across samples were corrected using the
‘Harmony’ package (version 1.2.4; http://github.com/immunogenomics/harmony) (33). Following normalization and scaling,
unsupervised cell clustering was performed at a resolution of 0.8
and dimensionality reduction was visualized using t-distributed
stochastic neighbor embedding. To assess the activity of
lactylation-related gene sets in individual cells, the
‘AddModuleScore’ function in Seurat was applied. In addition,
intercellular communication networks were analyzed using the
‘CellChat’ R package to explore signaling interactions among
different cell populations (version 2.2.0; http://github.com/jinworks/CellChat) (34).
Differential gene expression analysis
and WGCNA construction
Differentially expressed genes (DEGs) were
identified using three GEO datasets: GSE122697, GSE146114 and
GSE63514. These datasets were chosen for their high-quality,
well-annotated expression profiles and adequate sample sizes,
ensuring data reliability and reproducibility. The grouping details
for each dataset are provided in Table
SI. To minimize batch effects, the ComBat function from the
‘sva’ R package was applied (version 3.22) (26). Subsequently, a disease-specific
co-expression network was constructed using the ‘WGCNA’ R package
(version 1.73) (35). The optimal
soft-thresholding power (β) was selected to ensure scale-free
topology and modules most correlated with cervical cancer were
identified for downstream analyses.
Construction of LAGs-related
prognostic model
The highly disease-associated genes identified by
WGCNA were cross-analyzed with differentially expressed genes and
lactation-related genes to identify potential LAG-associated
prognostic candidate genes. Subsequently, these genes were
subjected to feature selection and prognostic modeling using
multiple machine learning algorithms, including least absolute
shrinkage and selection operator (LASSO) (36), Stepwise Cox (Backward), CoxBoost
(37), random survival forest (RSF)
(38), generalized boosted
regression model and survival support vector machine (18–21).
Each algorithm was independently evaluated for the prognostic
performance of the candidate genes in TCGA training cohort and
GSE30760 validation cohort. The concordance index (C-index) was
used to quantify model performance and the models were ranked
according to their average C-index values. The StepCox (Backward)
model achieved the highest predictive performance [area under the
curve (AUC)=0.82; C-index=0.79], outperforming CoxBoost and
survival SVM while maintaining simplicity and interpretability. To
minimize overfitting and ensure robustness, the StepCox (Backward)
model was lastly selected for downstream analysis, which is
consistent with previous studies demonstrating the stability and
strong generalization ability of this model in cancer prognosis
modeling (39).
Evaluation of predictive model
performance and independent prognostic analysis
After obtaining the LAGs risk score of each patient,
patients were divided into high-risk and low-risk groups based on
the median of the LAGs risk score distribution within the cohort.
Survival outcomes, including progression-free survival (PFS),
disease-specific survival (DSS) and disease-free interval (DFI),
were compared between the two groups using Kaplan-Meier analysis
implemented in the ‘survminer’ R package (version 0.5.1; http://rpkgs.datanovia.com/survminer/index.html).
The predictive accuracy of the model was further assessed by
calculating the AUC for 1-, 3- and 5-year overall survival (OS)
using the ‘timeROC’ R package (version 0.4) (40). Differences in risk scores across
clinical subgroups, such as tumor (T), lymph node (N) and
metastasis (M) stage and histological grade (41), were examined using TCGA-CESC
clinical data. To determine whether the LAGs-based risk model
serves as an independent prognostic factor, univariate and
multivariate Cox regression analyses were performed. Lastly, a
nomogram integrating the LAGs risk score and key clinical variables
was constructed to predict individual patient survival
probabilities and enhance the model's clinical applicability.
Simultaneously, the GEPIA2.0 database (http://gepia2.cancer-pku.cn/) (42) was used to validate the survival
analysis.
Functional enrichment analysis of the
LAGs-based risk model
To explore the biological pathways underlying
differences between the high- and low-risk groups, Gene Set
Enrichment Analysis (GSEA) was performed (43). Additionally, enrichment scores were
quantified at the individual sample level using single-sample gene
set enrichment analysis (ssGSEA) implemented in the R package GSVA
(version 2.2.1) on the TCGA-CESC (Cervical squamous cell carcinoma
and endocervical adenocarcinoma) dataset downloaded from The Cancer
Genome Atlas (TCGA) via the GDC Data Portal, to assess pathway
activity (44). Functional
annotations were further investigated using Gene Ontology (GO) and
Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses
to identify biological processes and signaling pathways associated
with the LAGs risk model. Pathways with P<0.05 were considered
statistically significant.
Immune landscape characterization
To examine immune infiltration patterns across LAGs
risk subgroups, the ESTIMATE analysis was performed to determine
the malignant tumor stroma and immune cell (ESTIMATE) scores of
patients (version 4.7.0) (45). The
CIBERSORT algorithm (https://cibersortx.stanford.edu/) was then used to
estimate the relative abundance of immune cell populations, while
ssGSEA was applied to assess immune cell signature enrichment and
immune-related functions. Concurrently, ssGSEA was applied to
assess the enrichment of immune cell signatures and immune-related
functions. Statistical differences between subgroups were evaluated
using the Wilcoxon rank-sum test. In addition, to assess
intratumoral heterogeneity, the Mutant-Allele Tumor Heterogeneity
(MATH) algorithm was utilized to calculate mutation-derived
heterogeneity scores (version 2.4.6) (46). These MATH scores were subsequently
incorporated into survival analyses to examine their association
with patient prognosis and genomic instability.
Drug sensitivity analysis of LAGs risk
subgroups
To evaluate potential therapeutic implications of
the LAGs-based risk model, drug response prediction was performed
using the ‘pRRophetic’ R package (version 0.5.1) (47). The half-maximal inhibitory
concentration (IC50) values of commonly used
chemotherapeutic and targeted agents were estimated for each
patient. Differences in predicted drug sensitivity between high-
and low-risk groups were compared using the Wilcoxon rank-sum test,
with P<0.05 considered statistically significant. These analyses
aimed to identify potential compounds with differential efficacy
across risk subgroups, providing a foundation for personalized
treatment strategies in CESC.
Two-sample MR analysis of cervical
cancer and LAGs prediction models
To further elucidate the causal relationship between
LAGs prediction models and cervical cancer, two-sample MR analysis
was employed to assess whether the expression levels of prognostic
LAGs exert a causal effect on cervical cancer susceptibility. SNPs
were selected as instrumental variables (IVs) if they demonstrated
significant association with the exposure variable
(P<5×10−8). To ensure variant independence and
minimize linkage disequilibrium (LD) effects, SNPs were clustered
using an LD threshold of R2<0.001 and a 10,000 kb
clustering window. Variants with minor allele frequency <0.01 or
F-statistic <10 were excluded to avoid weak instrument bias. The
inverse variance weighting (IVW) method was employed as the primary
analysis to estimate causal effects between LAGs and cervical
cancer. Heterogeneity among IVs was assessed using the Cochran Q
test, while potential horizontal pleiotropy and directional bias
were assessed via MR-Egger regression. Sensitivity analyses,
including stepwise exclusion tests, validated the robustness of
causal estimates. All MR analyses were performed using the
‘TwoSampleMR’ and ‘MRPRESSO’ R packages (version 1.0) (48,49).
Sensitivity analyses were conducted for the two-sample MR studies,
including leave-one-out tests, pleiotropy analysis via the MR-Egger
intercept test and heterogeneity analysis using Cochran's Q test
(50). These analyses ensure the
robustness and validity of the MR findings by identifying potential
biases or heterogeneity in the genetic instruments used. Final
causal estimates were visualized with forest plots and scatterplots
to display the direction and magnitude of the association.
Cell culture and transfection
Human cervical squamous cell carcinoma cell lines
SiHa (cat. no. HTB-35) and HeLa (cat. no. CRM-CCL-2) were obtained
from the American Type Culture Collection and the human
keratinocyte cell line HaCaT was obtained from The Cell Bank of
Type Culture Collection of the Chinese Academy of Sciences
(SCSP-5091; Beijing, China). Although short tandem repeat analysis
was not performed, the HaCaT cells were purchased from a certified
national cell bank with traceable origins and their authenticity
was confirmed by morphological observation and mycoplasma testing.
All cell lines were cultured in DMEM (cat. no. KGL1201-500; Jiangsu
KeyGen Biotech Co., Ltd.) containing 10% FBS (cat. no. C04001-500;
Shanghai Xiaopeng Biotechnology Co., Ltd.) and 1%
penicillin-streptomycin-amphotericin B (cat. no. P7630; Beijing
Solare Technology Co., Ltd.) at 37°C in a humidified incubator with
5% CO2.
SiHa and HeLa cells were seeded into 6-well plates
and transfected with HMGN1-specific small interfering RNA (siRNA)
or non-targeting control (NC) siRNA using siTran 2.0™ (cat. no.
TT320002; Origene Technologies, Inc.) according to the
manufacturer's instructions. The sequences and catalog numbers of
the HMGN1-specific siRNAs and NC siRNA are provided in Table SIII (Tsigke). Cells were cultured
to approximately 60–70% confluence prior to transfection. A final
concentration of 50 nM siRNA was used for each transfection. For
each well of a 6-well plate, the appropriate amount of siRNA was
gently mixed with 2.4 µl siTran 2.0™ reagent and 100 µl
transfection buffer, followed by incubation at room temperature for
15 min to allow complex formation. The complexes were then added
dropwise to the cell culture medium. After incubating the cells at
37°C for 12 h, the medium was replaced with fresh growth medium.
Cells were collected after an additional 12 or 24 h incubation. RNA
and proteins were extracted separately for subsequent analysis.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from SiHa and HeLa cells
using the RNeasy™ Kit (cat no. R0027; Beyotime Biotechnology)
according to the manufacturer's instructions. The extracted total
RNA was then reverse transcribed into complementary DNA (cDNA)
using SuperMix (R323-01; Vazyme Biotech Co., Ltd.). After cDNA
synthesis, qPCR was performed on QuantStudio 5 instrumentation
using SYBR® Green PCR Master Mix (cat. no. Q311-02;
Vazyme Biotech Co., Ltd.). The thermocycling conditions were as
follows: An initial pre-denaturation at 95°C for 10 min, followed
by 40 cycles of denaturation at 95°C for 10 sec, annealing at 58°C
for 30 sec and extension at 72°C for 30 sec. GAPDH was used as the
internal reference gene for normalization. Relative gene expression
levels were calculated using the 2−∆∆Cq method (51). The human-derived mRNA sequences of
the 14 genes for the lactylation prediction model are shown in
Table SIV. All mRNA sequences were
provided by Beijing Qingke Biotechnology Co., Ltd.
Western blotting analysis
Cells were collected and the medium was discarded.
Total protein was extracted using RIPA lysis buffer (cat no.
P0013B; Beyotime Biotechnology) supplemented with protease and
phosphatase inhibitors (cat no. P1045; Beyotime Biotechnology).
Samples were centrifuged at 12,000 × g for 15 min at 4°C and the
supernatant was collected for subsequent analysis. Protein
concentration was quantified using the BCA Protein Assay Kit (cat
no. 20201ES86, Shanghai Yeasen Biotechnology Co., Ltd). Equal
amounts (10 µg) of protein were separated by 10% SDS-PAGE and
transferred onto PVDF membranes (cat no. IPVH00010; MerckKGaA).
Membranes were incubated in Rapid Blocking Buffer (cat. no. P30500;
Suzhou Xinsaimai Biotechnology Co., Ltd.) for 20 min at room
temperature to minimize non-specific binding. After blocking,
membranes were incubated overnight at 4°C with anti-HMGN1 (1:1,000;
cat no. PK87656; Abmart, Inc.) and anti-β-actin (1:1,000; cat no.
A17910; ABclonal, Inc.; internal control). After washing with 0.1%
TBS-Tween, membranes were incubated with HRP-conjugated anti-rabbit
secondary antibody (cat. no. ZB-2306; ZSGB-Bio, Inc.) for 1 h at
room temperature. The membranes were detected using an
ultra-sensitive ECL kit (cat no. P10100; Suzhou CellPro
Biotechnology Co., Ltd.) and quantified using ImageJ software
(version 1.0; National Institutes of Health).
Cell Counting Kit-8 (CCK-8) assay
Cell proliferation was assessed using the CCK-8
Assay Kit (cat. no. C0005; TargetMol Chemicals Inc.). Cells were
seeded in 96-well plates and absorbance was measured at 24, 48 and
72 h after adhesion. To assess cell proliferation, 10 µl of CCK-8
reagent was added to each well and the cells were then incubated at
37°C and 5% CO2 for 1 h. The absorbance at 450 nm was
then measured using a microplate reader to determine cell
viability.
Flow cytometry analysis
Cell cycle distribution was analyzed using propidium
iodide (PI) staining. Transfected SiHa and HeLa cells were seeded
into 6-well plates and cultured under standard conditions. Cells
were collected, washed twice with PBS (cat. no. C0221A; Beyotime
Biotechnology) and fixed in pre-cooled 70% ethanol (cat. no.
Y263010; Beyotime Biotechnology) at 4°C for 8 h. Fixed cells were
centrifuged, washed and resuspended in staining buffer containing
RNase A (cat. no. ST576; Beyotime Biotechnology) and 5 µg/ml PI
(cat. no. G1021; Wuhan Servicebio Technology Co. Ltd.). After
incubation for 20 min in the dark at room temperature, samples were
analyzed using a FACSCalibur flow cytometer (BD Biosciences) and
data were processed with CytExpert 2.0 software (Becton; Dickinson
and Company).
Statistical analysis
All statistical analyses were conducted using R
software (version 4.4.1; Posit Software, PBC) and GraphPad Prism
(version 10.6.1; Dotmatics). All results represent the mean ±
standard deviation of at least three independent experiments.
Intergroup comparisons were performed using the two-tailed unpaired
Student's t-test, while comparisons among multiple groups employed
one-way ANOVA followed by Tukey's post hoc test. The relationship
between two continuous variables was assessed using Wilcoxon
rank-sum. To compare differences in Kaplan-Meier survival curves,
the two-tailed log-rank test was applied. P<0.05 was considered
to indicate a statistically significant difference, unless
otherwise specified.
Results
Integration of single-cell
transcriptomics and WGCNA identifies LAGs
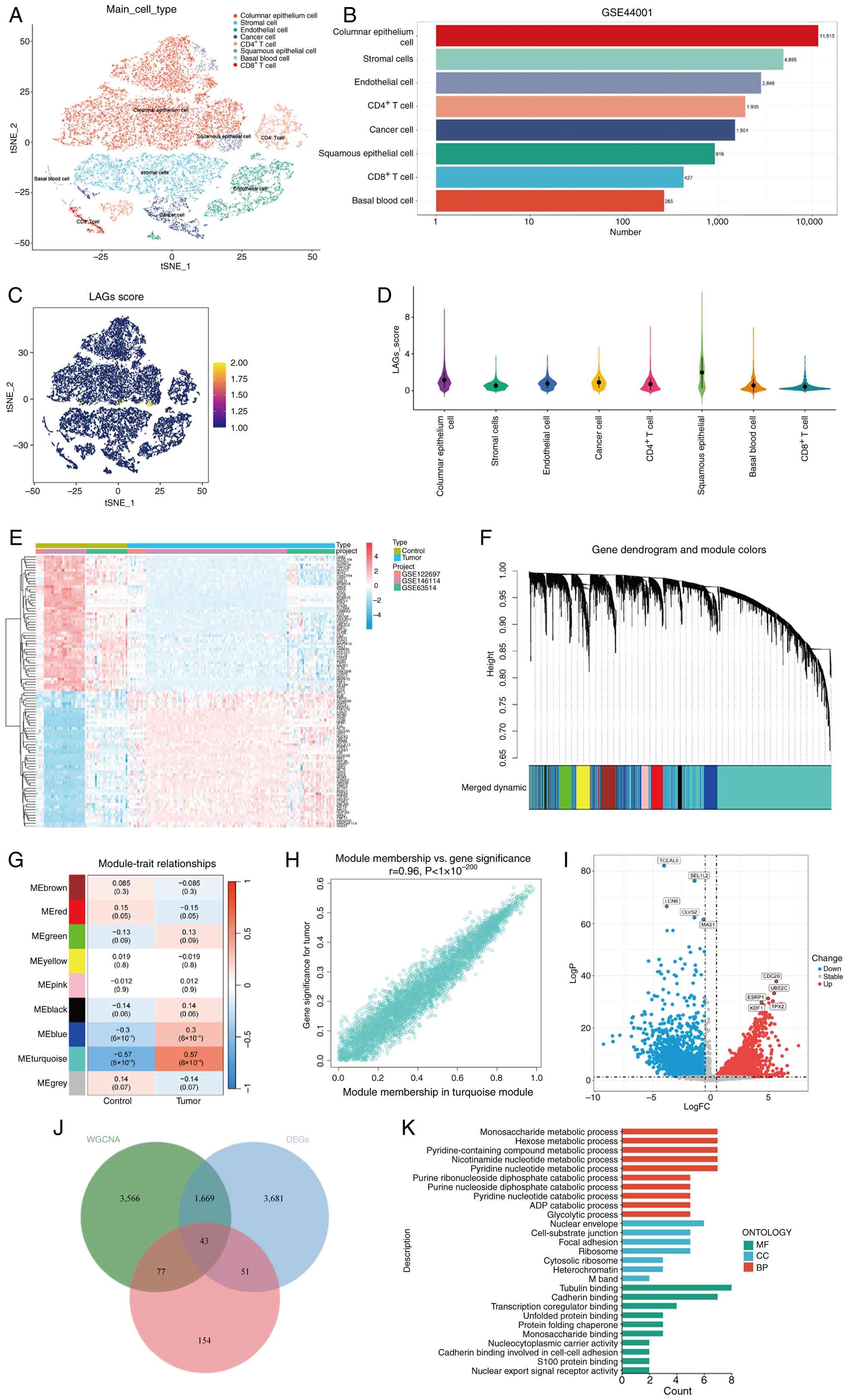
The GSE44001 dataset, comprising transcriptomic data
from 300 patients with cervical cancer, included scRNA-seq data for
a total of 22,702 cells. After rigorous quality control and
filtering, cells were categorized into eight major clusters:
Columnar and squamous epithelial cells, CD8+ T cells,
CD4+ T cells, stromal cells, cancer cells, basal blood
cells, and endothelial cells (Fig.
2A). The proportions of these cell populations are shown in a
bar chart (Fig. 2B). To evaluate
the activity of LAGs across different cell types, the
‘AddModuleScore’ function of the ‘Seurat’ package was applied. The
resulting scores demonstrated that squamous and columnar epithelial
cells exhibited higher LAG activity compared with other clusters
(Fig. 2C and D), suggesting that
LAGs play a key role in cervical cancer pathogenesis, as these
epithelial cells are involved in tumor initiation and progression.
Elevated LAG levels in these cells may be associated with their
influence on cervical cancer progression. Based on the distribution
of LAG activity, cells were stratified into high- and low-LAG
groups, yielding 5,443 DEGs for subsequent analysis. To validate
these findings, three GEO datasets (GSE122697, GSE146114 and
GSE63514) were integrated after batch correction using the ComBat
algorithm (Fig. S1A-D). From these
datasets, disease-related DEGs were identified and the top 50 genes
were visualized in a heatmap (Fig.
2E).
Next, WGCNA was performed to identify gene modules
associated with cervical cancer. After removing outlier samples,
hierarchical clustering and correlation analyses between normal and
tumor groups were performed using the ssGSEA algorithm. With a
merging threshold of 0.25, a total of nine co-expression modules
were identified (Fig. 2F and G).
Among them, the turquoise module (MEturquoise) revealed the
strongest positive correlation with cervical cancer progression
(r=0.57; P<0.001). The strong association between gene
significance and module membership within this module (r=0.96;
P<0.001; Fig. 2H) further
confirmed its biological relevance. Genes within the turquoise
module were thus selected for further analysis. Differential
expression analysis between tumor and normal tissues revealed
significant gene expression changes (adjusted P<0.05;
log2FC >0.5), which were visualized in a volcano plot
(Fig. 2I). By intersecting
turquoise module genes, DEGs and LAGs, a total of 43 overlapping
genes were identified as LAGs (Fig.
2J). GO enrichment analysis revealed that these LAGs were
predominantly involved in ‘monosaccharide metabolic process’,
‘hexose metabolic process’ and ‘pyridine-containing compound
metabolic process’. They were also enriched in cellular components
such as the ‘nuclear envelope’ and ‘cell-substrate junction’ and
exhibited molecular functions associated with ‘tubulin binding’
(Fig. 2K; Table SV).
Construction and validation of a
prognostic model based on CESC-related LAGs
To elucidate the prognostic relevance of LAGs in
CESC, multiple machine learning algorithms were employed to
construct and evaluate prognostic models based on 43 differentially
expressed LAGs identified in prior analyses. TCGA-CESC cohort was
used as the training dataset, whereas GSE30760 served as the
independent validation cohort. A total of 101 predictive models
were generated via cross-validation and their predictive efficacy
was systematically compared using the C-index across both training
and validation datasets (Fig. 3A).
After excluding models exhibiting overfitting tendencies, the
StepCox (Backward) model demonstrated the most stable and robust
performance and was therefore selected as the optimal prognostic
model. This refined model incorporated 14 feature genes with
significant prognostic relevance. For each patient, a risk score
was calculated by summing the expression levels of these 14 genes
weighted by their respective Cox regression coefficients (Fig. 3B). Based on the median risk score,
patients were stratified into high-risk and low-risk groups.
Kaplan-Meier survival analysis revealed that patients in the
high-risk group exhibited significantly worse OS compared with
those in the low-risk group (P<0.001; Fig. 3C). Consistent findings were observed
for DSS and PFI, with low-risk patients demonstrating notably
increased clinical outcomes (P<0.0001; Fig. 3D and E). To further assess the
discriminatory capability of the model, time-dependent receiver
operating characteristic (ROC) curve analyses were conducted. In
TCGA training cohort, the model achieved AUC values of 0.80, 0.78
and 0.77 for 1-, 3- and 5-year survival, respectively (Fig. 3F). Similar results were obtained in
the GSE30760 validation cohort, yielding AUC values of 0.76, 0.67
and 0.67, indicating moderate generalization ability and
demonstrating the acceptable performance of the model in short-term
survival prediction (Fig. 3G).
Furthermore, external validation using the GSE7803 dataset further
confirmed the predictive reliability of the 14-gene model, with AUC
values >0.5 for all included genes except PFKM. However, due to
sample limitations, the predictive capability of genes other than
PAK2 and TCOF1 may be limited (Fig.
3H). These findings indicate that LAG-based predictive models
may be able to evaluate the prognosis of patients with cervical
cancer and provide important reference for clinical stratification
management.
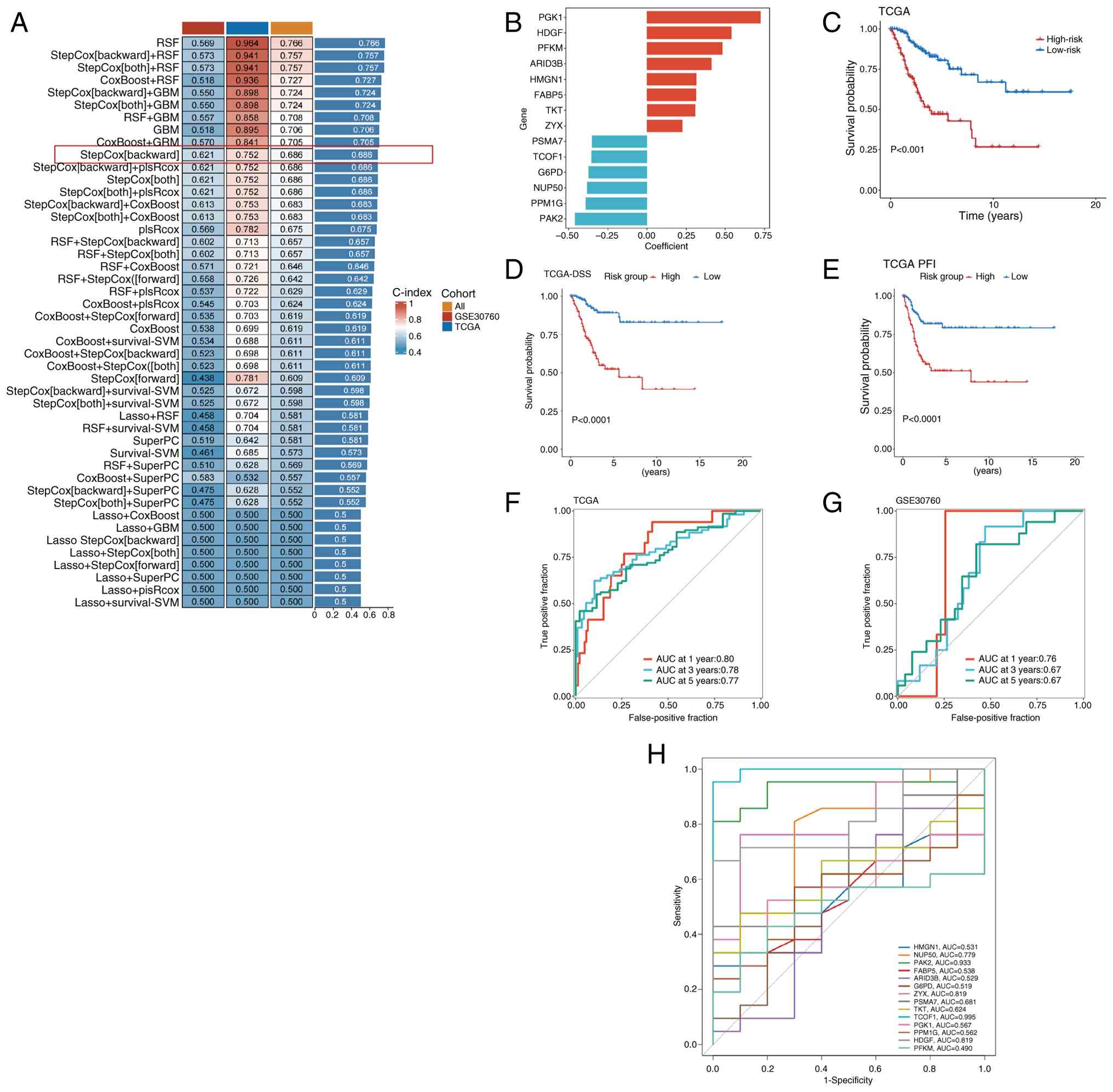 | Figure 3.Characterized LAG genes were screened
using machine algorithms. (A) Develop predictive models using the
10-fold cross-validation method and calculate the C-index for each
model for the training and validation data sets. (B) Bar graph
depicting regression coefficients for 14 genes determined by Cox
regression analysis. (C) Kaplan-Meier curves demonstrating the risk
score and overall survival of patients in TCGA-CESC cohort.
Kaplan-Meier curves for (D) DSS and (E) PFI. ROC curves of the LAGs
prognostic model at 1, 3 and 5 years in (F) TCGA training set and
(G) GSE30760 validation set. (H) ROC curves indicating the AUC
values of 14 prognostic genes in the GSE7803 dataset. LAGs,
lactylation-associated genes; C-index, concordance-index; TCGA, The
Cancer Genome Atlas; DSS, disease-specific survival; PFI,
progression-free interval; CESC, cervical squamous cell carcinoma;
AUC, area under the curve; ROC, receiver operating characteristic;
HMGN1, high-mobility group nucleosome-binding protein 1; TKT,
transketolase; ZYX, zyxin; PAK2, p21 (RAC1) activated kinase 2;
ARID3B, AT-rich interaction domain 3B; FABP5, fatty acid binding
protein 5; HDGF, hepatoma-derived growth factor. |
Clinical prognostic stratification and
independent prognostic analysis of the LAGs-based model
To further evaluate the clinical applicability and
prognostic independence of the LAGs risk model in cervical cancer,
the present study examined the relationship between the
LAGs-derived risk score and diverse clinicopathological
characteristics using stratified and Cox regression analyses. This
comprehensive evaluation aimed to assess the ability of the model
to predict patient outcomes across different clinical subgroups.
Patients were stratified according to clinical parameters,
including age (>60 vs. ≤60 years), T stage (T1; T2; T3 and T4),
N stage (N0, N1 and NX), M stage (M0, M1 and MX) and tumor stage
(I; II; III and IV), as illustrated in the clinical distribution
plot (Fig. 4A and B). Notably,
patients in the stage M1, III–IV and T3-4 subgroups exhibited
significantly higher risk scores compared with those patients in
the M0, I–II and T1-2 subgroups, respectively (Fig. 4C and D). These findings indicate a
strong association between elevated LAGs risk scores and advanced
tumor stage as well as unfavorable prognosis in CESC.
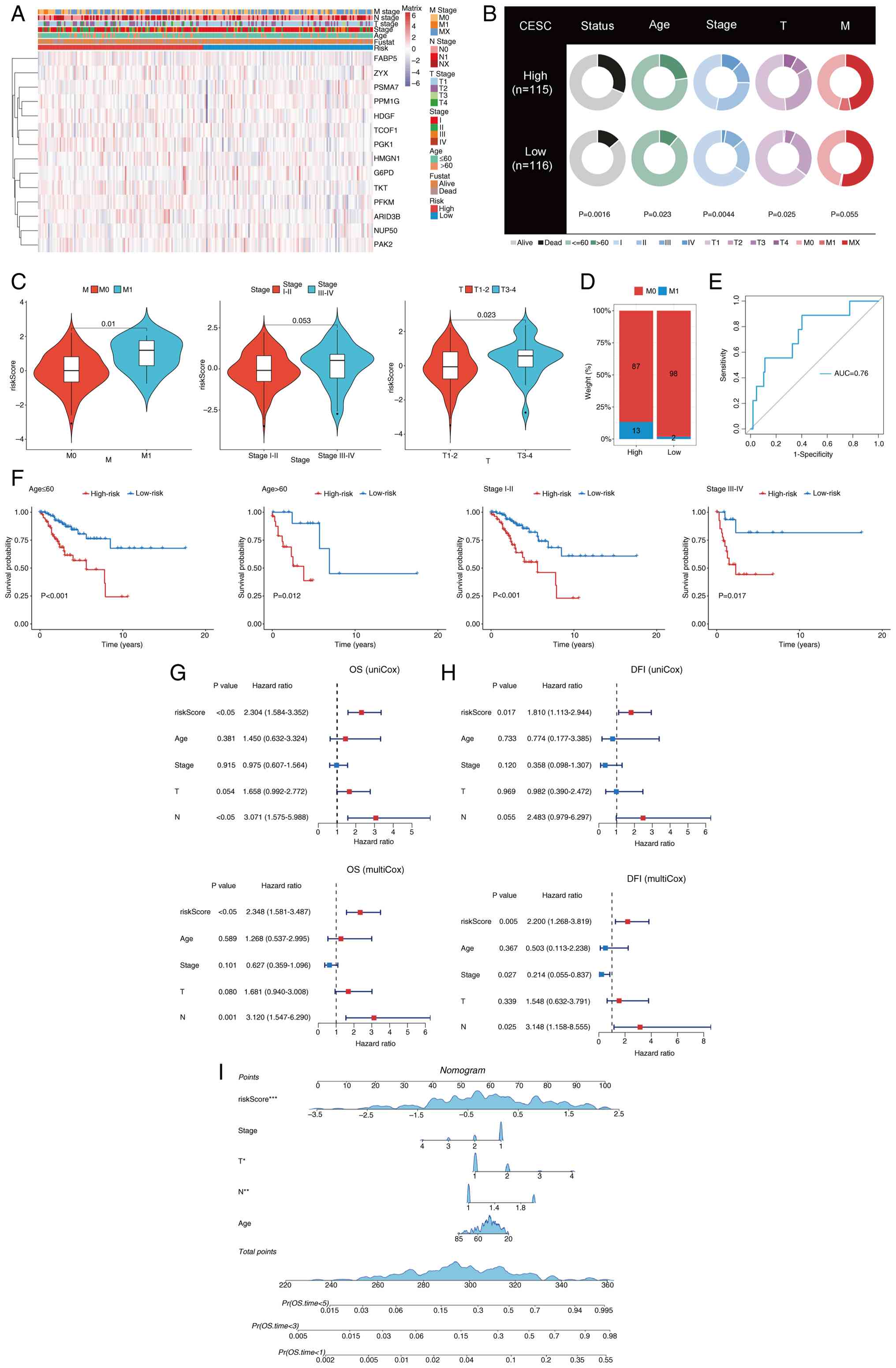 | Figure 4.Predictive level of LAGs was analyzed
based on clinical characteristics. (A) Distribution of clinical
features based on LAGs risk scores and expression levels of model
genes Purple indicates low expression, red indicates high
expression. (B) Relationship between high- and low-LAGs risk groups
and various clinical features. (C) Comparative analysis of risk
scores across different M stage and T staging categories. (D)
Distribution of M stage within LAGs risk groupings. (E) Receiver
operating characteristic curves for the LAGs risk model in
predicting M stage. (F) Kaplan-Meier survival curves demonstrating
the consistent performance of LAGs across subgroups of patients
with CESC, including age and stage. Univariate and multivariate Cox
regression analyses of clinical characteristics of (G) OS and (H)
DFI with LAGs in patients in The Cancer Genome Atlas-CESC cohort.
(I) Column-line plots of the survival probabilities of patients at
1, 3 and 5 years were constructed in conjunction with the risk
score of LAGs. *P<0.05, **P<0.01 and ***P<0.001. LAGs,
lactylation-associated genes; CESC, cervical squamous cell
carcinoma; OS, overall survival; DFI, disease-free interval; AUC,
area under the curve; HMGN1, high-mobility group nucleosome-binding
protein 1; TKT, transketolase; ZYX, zyxin; PAK2, p21 (RAC1)
activated kinase 2; ARID3B, AT-rich interaction domain 3B; FABP5,
fatty acid binding protein 5; HDGF, hepatoma-derived growth factor;
T, tumor stage; N, lymph node stage. |
ROC curve analysis further confirmed the predictive
utility of the LAGs-based model, with an AUC value of 0.76 in
distinguishing the prognosis of patients with different M stages,
indicating moderate predictive capability (Fig. 4E). In addition, stratified analyses
by age and tumor stage demonstrated that high-risk patients
consistently exhibited worse survival outcomes (Fig. 4F). Kaplan-Meier analyses of the 14
model genes using the GEPIA2 database that low expression levels of
transketolase (TKT) and phosphoglycerate kinase 1 (PGK1) were
significantly associated with improved prognosis (P<0.05), while
no significant associations were observed for other genes (Fig. S2A). To further validate the
prognostic independence of the model, both univariate and
multivariate Cox regression analyses were performed for OS, DFI,
PFI and DSS in the training cohort. The results demonstrated that
LAGs were independent prognostic factors (all P<0.05; Figs. 4G and H; and S2B and C). Lastly, a nomogram integrating
the risk score with key clinical parameters was constructed to
quantitatively predict survival outcomes in patients with CESC
(Fig. 4I). The nomogram analysis
confirmed that the risk score significantly contributed to the
highest prognostic weight (P<0.001), underscoring its clinical
utility as a robust and independent predictor of cervical cancer
prognosis.
Molecular mechanisms underlying
LAGs-based subtype classification
To further elucidate the molecular mechanisms that
associate LAGs with the prognosis of cervical cancer, functional
annotation analyses were performed across the identified
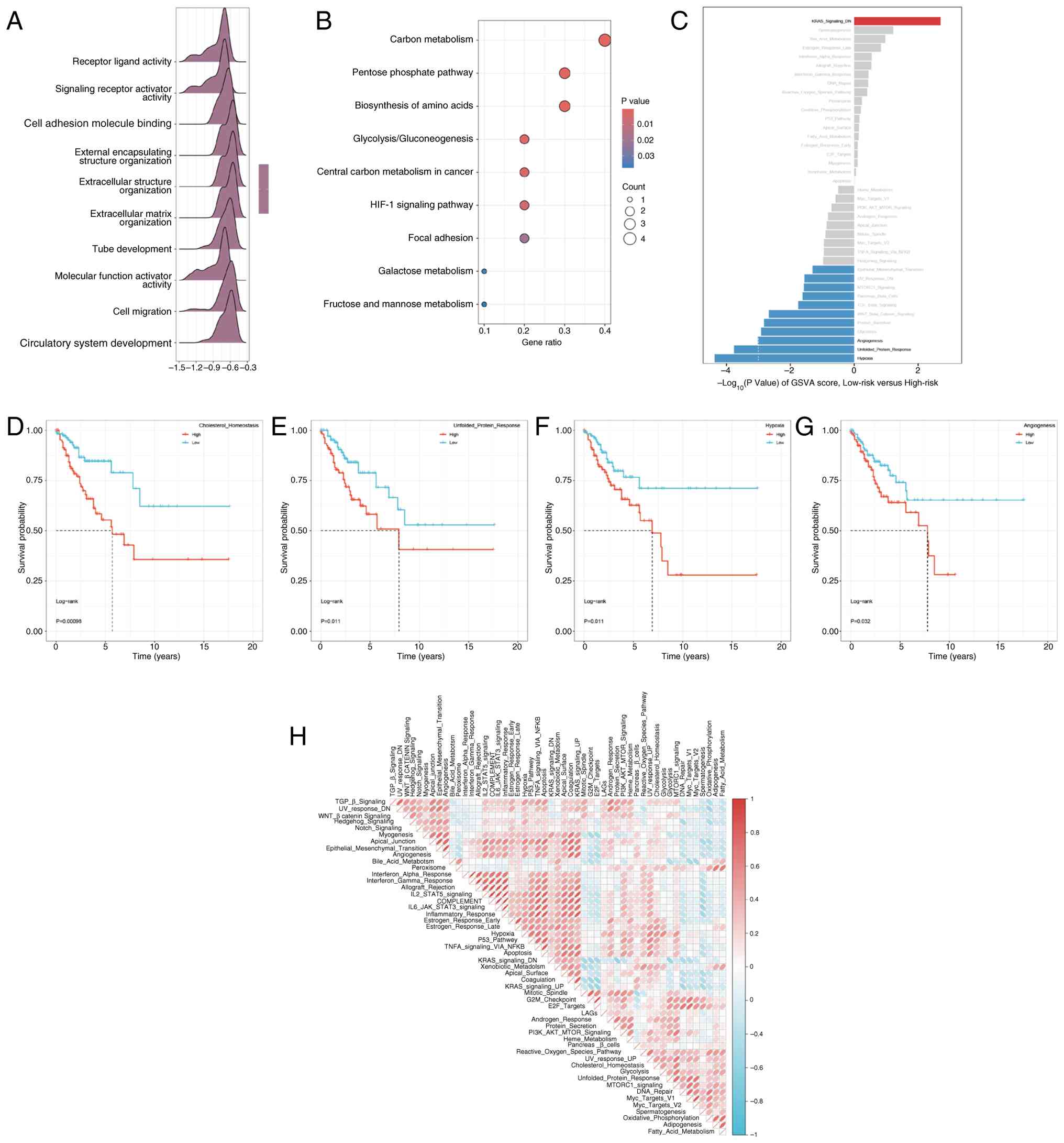
LAGs-derived molecular subtypes. In the low-risk group, Gene Set
Variation Analysis (GSVA) revealed significant enrichment of
biological pathways associated with ‘receptor ligand activity,
‘signaling receptor activator activity’ and ‘cell adhesion molecule
binding’ (Fig. 5A). KEGG pathway
enrichment further demonstrated that the most enriched pathways
were predominantly involved in energy metabolism, including
‘central carbon metabolism in cancer’, ‘biosynthesis of amino
acids’ and ‘glycolysis/gluconeogenesis’ (Fig. 5B). Comparative GSVA between risk
subgroups (Table SVI) indicated
distinct molecular signatures: The high-risk group exhibited
stronger activation of oncogenic and proliferative pathways, such
as KRAS signaling downregulation, whereas the low-risk group
displayed higher activity in immune- and stress-related pathways,
including ‘hypoxia’, ‘unfolded protein response’ and ‘angiogenesis’
(Fig. 5C). Correlation analysis
further confirmed that LAGs risk scores were significantly
associated with these key signaling pathways (Fig. 5H). To assess the prognostic
relevance of these pathways, Kaplan-Meier survival analyses were
performed. Pathways negatively correlated with LAGs expression,
such as cholesterol homeostasis, unfolded protein response, hypoxia
and angiogenesis, were associated with favorable survival outcomes
(Fig. 5D-G). By contrast, KRAS
signaling downregulation, which was positively correlated with
LAGs, was associated with poor prognosis (Fig. S2D). Collectively, these findings
suggest that differential pathway activation between LAGs-defined
subgroups reflects distinct metabolic and signaling landscapes,
thereby elucidating potential molecular mechanisms by which LAGs
contribute to cervical cancer progression.
Correlation between LAGs and
single-cell characteristics
To investigate the role of LAG in the tumor immune
microenvironment at the single-cell transcriptome level, the
expression profiles of LAG prognostic genes were analyzed across
distinct cell populations (Fig.
6A). Based on the LAGs prognostic model, tumor cells were
categorized into high- and low-risk groups, followed by
differential interaction analysis to examine intercellular
communication patterns within the TME. Distinct cell-cell
communication networks were observed between the two risk groups
(Fig. 6B and C). Notably,
intercellular signaling occurred among nearly all cell types
(Fig. S2E), with particularly
strong interactions detected between columnar and squamous
epithelial cells. Functional annotation of ligand-receptor
signaling revealed that the collagen, laminin and Notch pathways
were predominantly enriched in highly cancerous cells, squamous and
columnar epithelial cells, CD4+ T cells and endothelial
cells (Fig. 6D-F). These
intercellular interactions are likely to regulate cell adhesion,
migration and differentiation, thereby contributing to tumor
progression and microenvironmental remodeling.
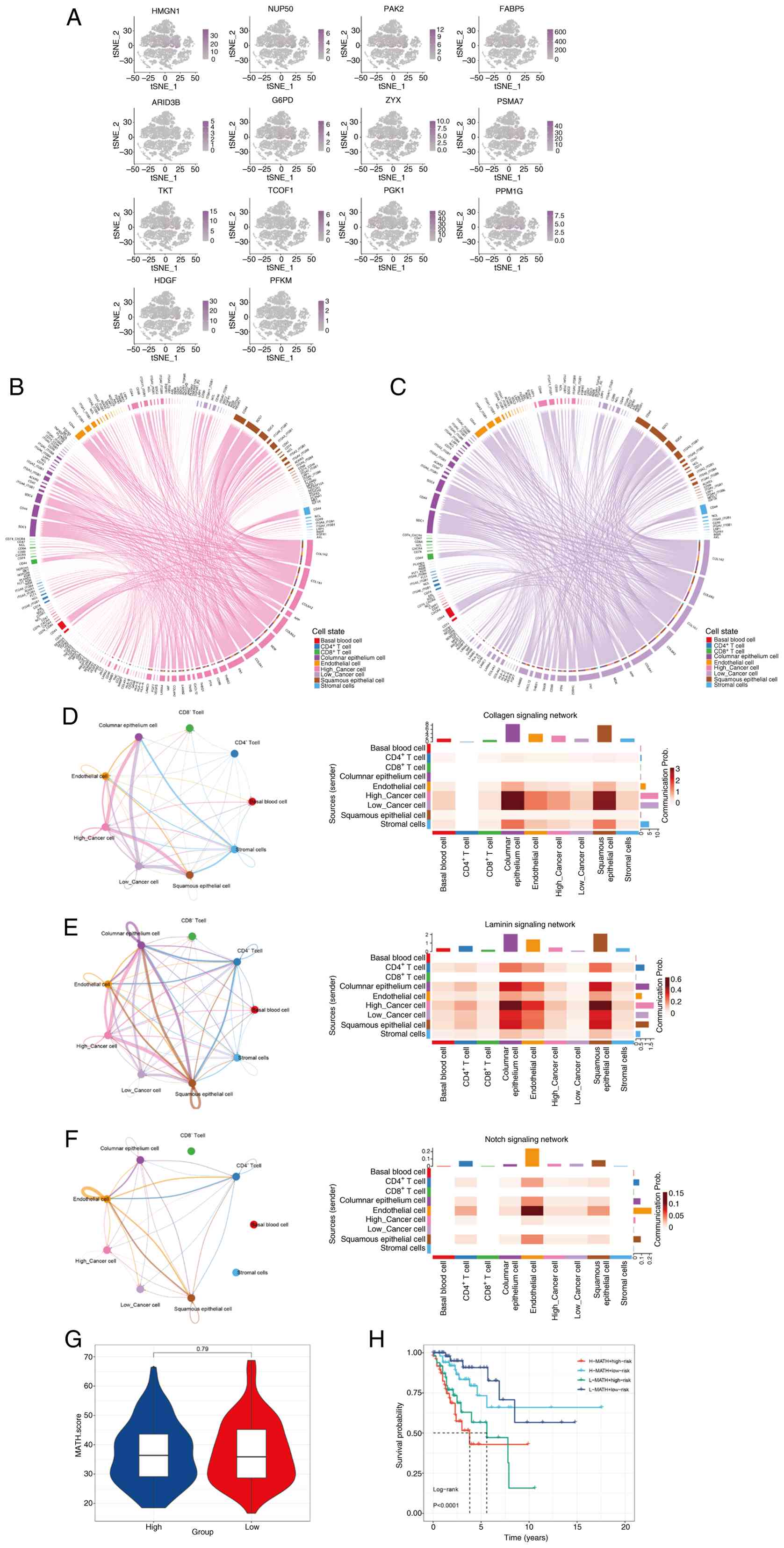 | Figure 6.Correlation of the LAGs with
single-cell characteristics. (A) Distribution of prognostic gene
expression of LAGs in the single-cell transcriptome. The shade of
color indicates the distribution level of LAGs within cells,
ranging from light purple (low expression) to dark purple (high
expression). (B) Cellular communication network diagram for
high-risk tumor cells. (C) Cellular communication network diagram
for low-risk tumor cells. Network diagrams of the (D) collagen, (E)
laminin and (F) notch signaling pathways, where the circle plots
illustrate the communication network and the heatmaps indicate the
proportion of different cell types within the network. (G)
Comparison of MATH tumor heterogeneity scores between high-risk and
low-risk groups. (H) Overall survival analysis using MATH scores
combined with LAGs. LAGs, lactylation-associated genes; HMGN1,
high-mobility group nucleosome-binding protein 1; TKT,
transketolase; ZYX, zyxin; MATH, mutant-allele tumor heterogeneity;
PAK2, p21 (RAC1) activated kinase 2; ARID3B, AT-rich interaction
domain 3B; FABP5, fatty acid binding protein 5; HDGF,
hepatoma-derived growth factor. |
Since intratumoral heterogeneity is a major driver
of tumorigenesis, progression, metastasis, therapeutic resistance
and immune evasion, the present study next evaluated heterogeneity
using the MATH score. Tumors with high LAGs risk exhibited markedly
elevated MATH scores, indicating greater genomic instability
(Fig. 6G). Furthermore, integrative
analysis combining LAGs and MATH metrics revealed that patients in
the high-risk and high-MATH score subgroup experienced
significantly worse survival outcomes compared with those in the
low-risk and low-MATH score subgroup (Fig. 6H; P<0.001). Collectively, these
findings suggest that LAG activity is closely associated with
alterations in cellular communication, tumor heterogeneity and the
immune microenvironment, potentially influencing cervical cancer
progression and patient prognosis.
Analysis of the tumor immune
microenvironment in high- and low-risk LAGs groups
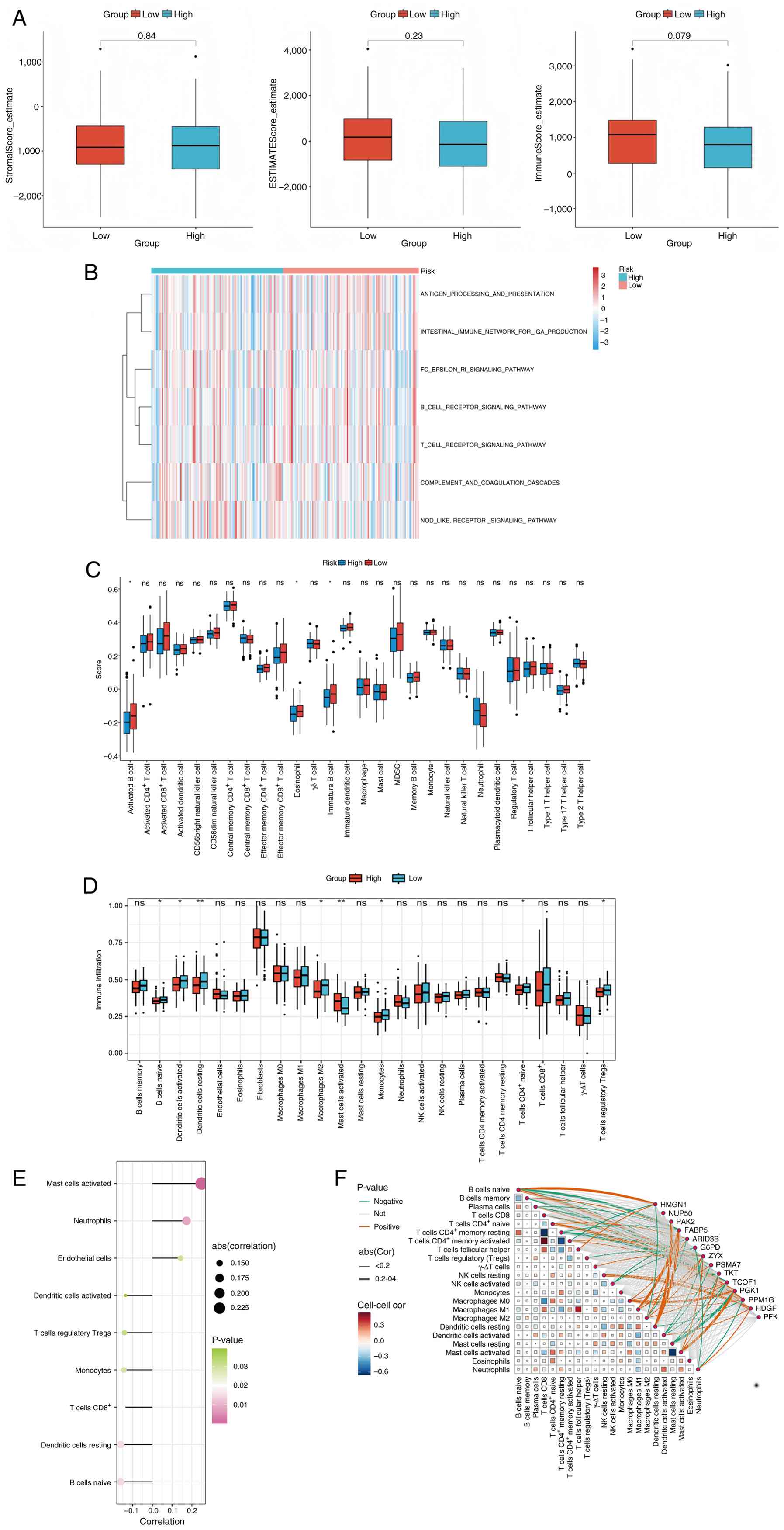
Immune infiltration serves a key role in tumor
initiation, progression and therapeutic response, profoundly
influencing the clinical outcomes of patients with cancer. To
characterize the immune landscape of cervical cancer, the present
study calculated the immune, stromal and ESTIMATE scores for each
sample (Fig. 7A). Although the
high-risk group exhibited slightly elevated immune and stromal
scores compared with the low-risk group, these differences did not
reach statistical significance. Subsequently, ssGSEA was performed
to evaluate immune pathway activities. The results demonstrated
significant enrichment of the ‘antigen processing and presentation’
pathway and the ‘NOD-like receptor signaling’ pathway in the
high-risk group (Fig. 7B). To
further elucidate the immune cell composition, the abundance of
tumor-infiltrating immune cells was quantified across samples.
Activated B cells, eosinophils and immature B cells were
predominantly enriched in the low-risk group, displaying
significant intergroup differences (Fig. 7C). These findings were independently
validated using the CIBERSORT algorithm, which yielded consistent
results (Fig. 7D). Spearman
correlation analysis identified nine immune cell subtypes
significantly associated with LAGs risk scores (Fig. 7E). Furthermore, correlation analyses
between LAGs prognostic genes and specific immune cell populations
revealed that AT-rich interaction domain 3B, phosphoglycerate
kinase 1 (PGK1) and protein phosphatase,
Mg2+/Mn2+ dependent 1G (PPM1G) were
positively correlated with M0 macrophages and activated mast cells
and indicated widespread associations with monocyte-related gene
expression (Figs. 7F and S2F). Lastly, the relationship between TME
infiltration and OS was examined and six immune cell subtypes were
significantly associated with patient prognosis (log-rank test;
P<0.05; Fig. S2G-L), including
M0 and M1 macrophages, resting mast cells, CD8, γΔ and regulatory T
cells. Collectively, these findings indicate that the LAGs risk
signature is closely associated with alterations in the immune
landscape of cervical cancer, potentially influencing both tumor
immunity and clinical outcomes.
Association between LAGs risk
subgroups, drug sensitivity and immunotherapy response
To further assess the clinical utility of the
LAGs-based risk model in guiding treatment strategies, the present
study explored its association with immunotherapy response and drug
sensitivity. Using the IMvigor210 cohort, the present study
evaluated the predictive value of the LAGs risk score for
immunotherapy efficacy. The distribution of clinical
responses-including complete remission (CR), partial remission
(PR), stable disease (SD) and progressive disease (PD)-was compared
between risk subgroups using χ2 tests. The high-risk
group exhibited a markedly higher proportion of PD/SD cases,
whereas CR/PR responses were significantly more frequent in the
low-risk group (Fig. 8A).
Consistently, patients achieving CR/PR demonstrated significantly
lower risk scores compared with those with SD/PD, suggesting that
lower LAGs scores are associated with more favorable
immunotherapeutic outcomes (Fig. 8B and
C). To evaluate chemotherapeutic drug sensitivity, the
IC50 values of various agents were compared between
high- and low-risk subgroups. The risk score exhibited a
significant positive correlation with the IC50 values of
paclitaxel (R=0.16; P<0.05) and rapamycin (R=0.26; P<0.05)
(Fig. 8D and E) and a negative
correlation with midostaurin (Fig.
8F; R=−0.32; P<0.05). Specifically, paclitaxel and rapamycin
displayed lower IC50 values in the low-risk group,
indicating higher predicted sensitivity, while midostaurin
demonstrated higher IC50 values in the high-risk group,
suggesting potential efficacy in these patients (Fig. 8G-I).
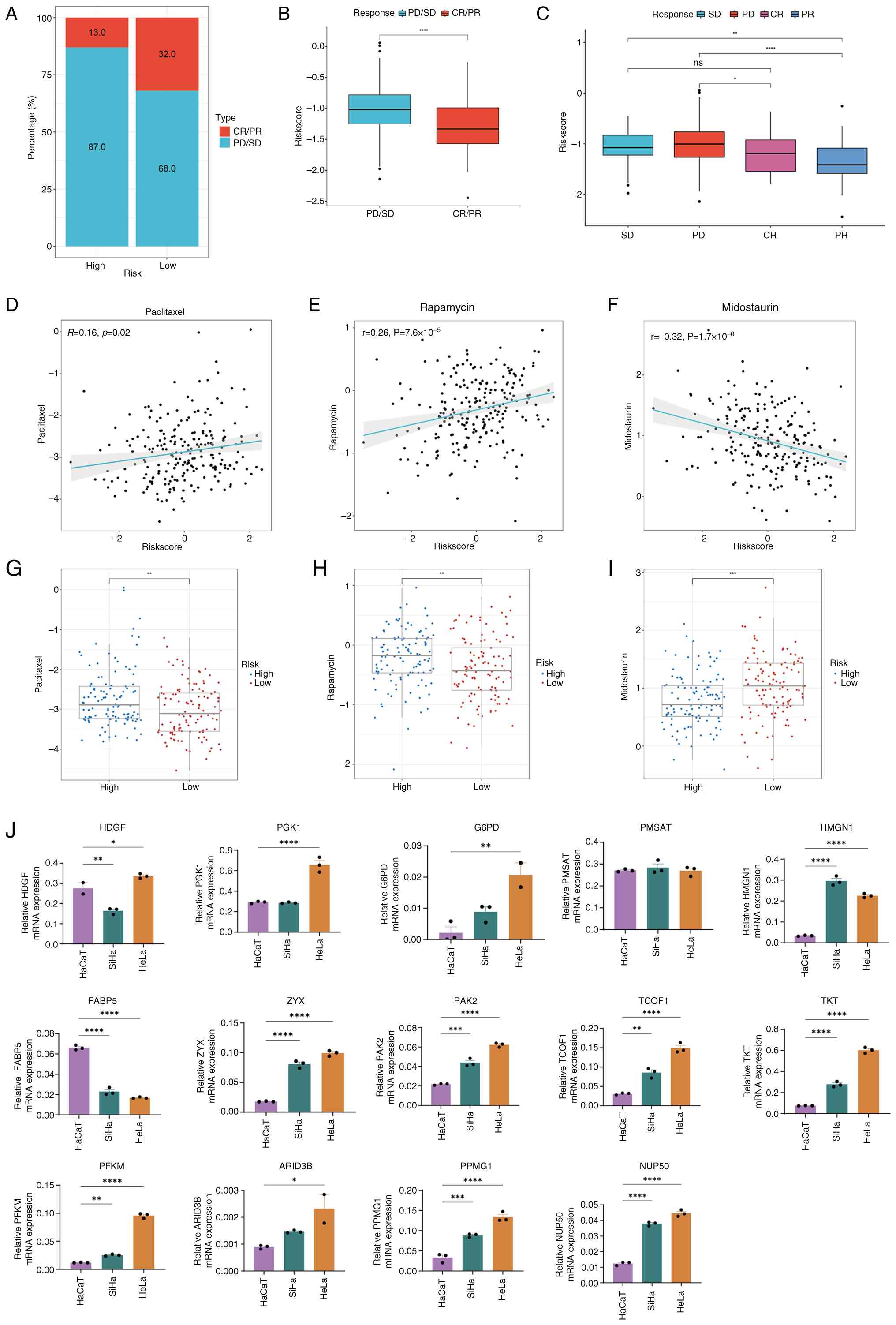 | Figure 8.Analysis of LAGs risk subgroups in
relation to drug sensitivity and immunotherapy response. (A)
Proportion of patients with CR/PR or SD/PD receiving immunotherapy
in the high- and low-risk groups of the IMvigor210 cohort. (B) Box
plot illustrating the difference in risk scores between patients
with CR/PR and those with SD/PD in the IMvigor210 cohort. (C) Box
plot depicting the distribution of risk scores among CR, PR, SD and
PD patients in the IMvigor210 cohort. Correlation between
LAGs-related risk scores and half-maximal inhibitory concentrations
of (D) paclitaxel, (E) rapamycin and (F) midostaurin. Comparison of
drug sensitivity between high- and low-risk groups for (G)
paclitaxel, (H) rapamycin and (I) midostaurin. (J) mRNA expression
levels of genes associated with the LAGs prediction model.
*P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001. ns, not
significant; LAGs, lactylation-associated genes; CR, complete
remission; PR, partial remission; SD, stable disease; PD,
progressive disease; HMGN1, high-mobility group nucleosome-binding
protein 1; TKT, transketolase; ZYX, zyxin; PAK2, p21 (RAC1)
activated kinase 2; ARID3B, AT-rich interaction domain 3B; FABP5,
fatty acid binding protein 5; HDGF, hepatoma-derived growth
factor. |
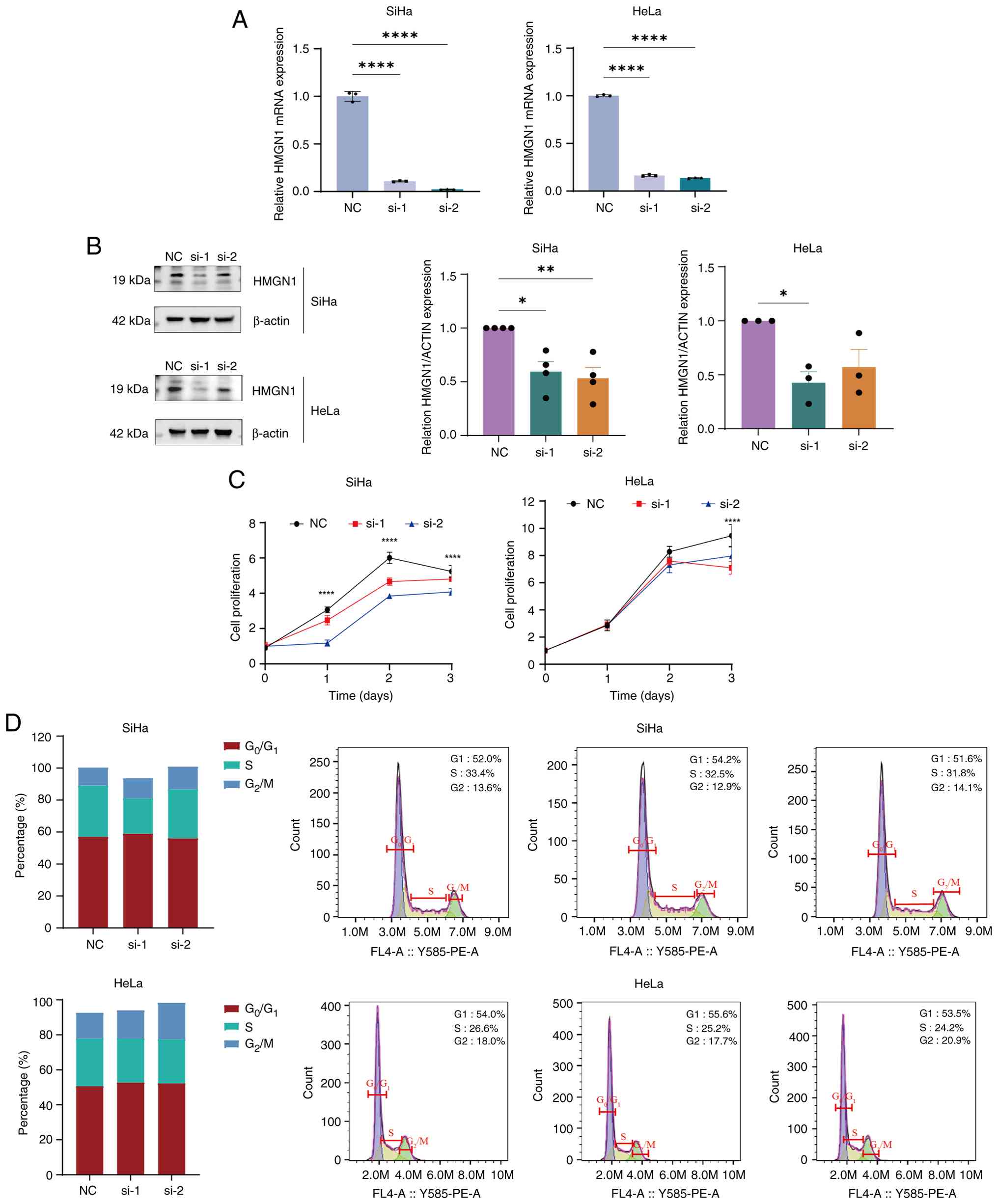
Furthermore, to validate the expression of LAGs
in vitro, the present study measured mRNA expression levels
of the 14 LAGs included in the prognostic model using RT-qPCR in
HaCaT epithelial cells and cervical squamous carcinoma cell lines
(SiHa and HeLa). The results revealed that the majority of genes,
including G6PD, HMGN1, ZYX, PAK2, TCOF1 and TKT, were significantly
upregulated in cervical cancer cells compared with normal
epithelial cells (Fig. 8J).
Therefore, these findings demonstrate that the LAGs-based
prognostic model not only stratifies patients by survival risk but
also provides notable insights into immunotherapy responsiveness
and chemotherapeutic sensitivity, thereby offering a potential
framework for personalized treatment optimization in cervical
cancer in the future.
MR analysis demonstrates a causal
relationship between the LAGs prognostic model and cervical
cancer
To further investigate the relationship between the
LAG prognostic model and cervical cancer and to determine whether
this relationship is causal or merely correlative, the present
study performed a two-sample MR analysis. In this analysis,
prognostic genes were used as exposure factors and cervical cancer
was the outcome. The present study was unable to obtain identifiers
for several genes, including glucose-6-phosphate dehydrogenase,
proteasome subunit α type 7, treacle ribosome biogenesis factor 1,
PGK1, PPM1G and PFKM, as certain genes lacked sufficient SNPs,
leading to their exclusion from the analysis. Using the IVW method
(Table I), the present study
identified HMGN1 as a protective factor, with an odds ratio of 0.81
(95% CI, 0.70–0.94; P=0.0051; Fig. 9A
and D). However, no significant causal relationship was
observed between zyxin (ZYX) and TKT (P>0.05; Fig. 9B and C, E and F).
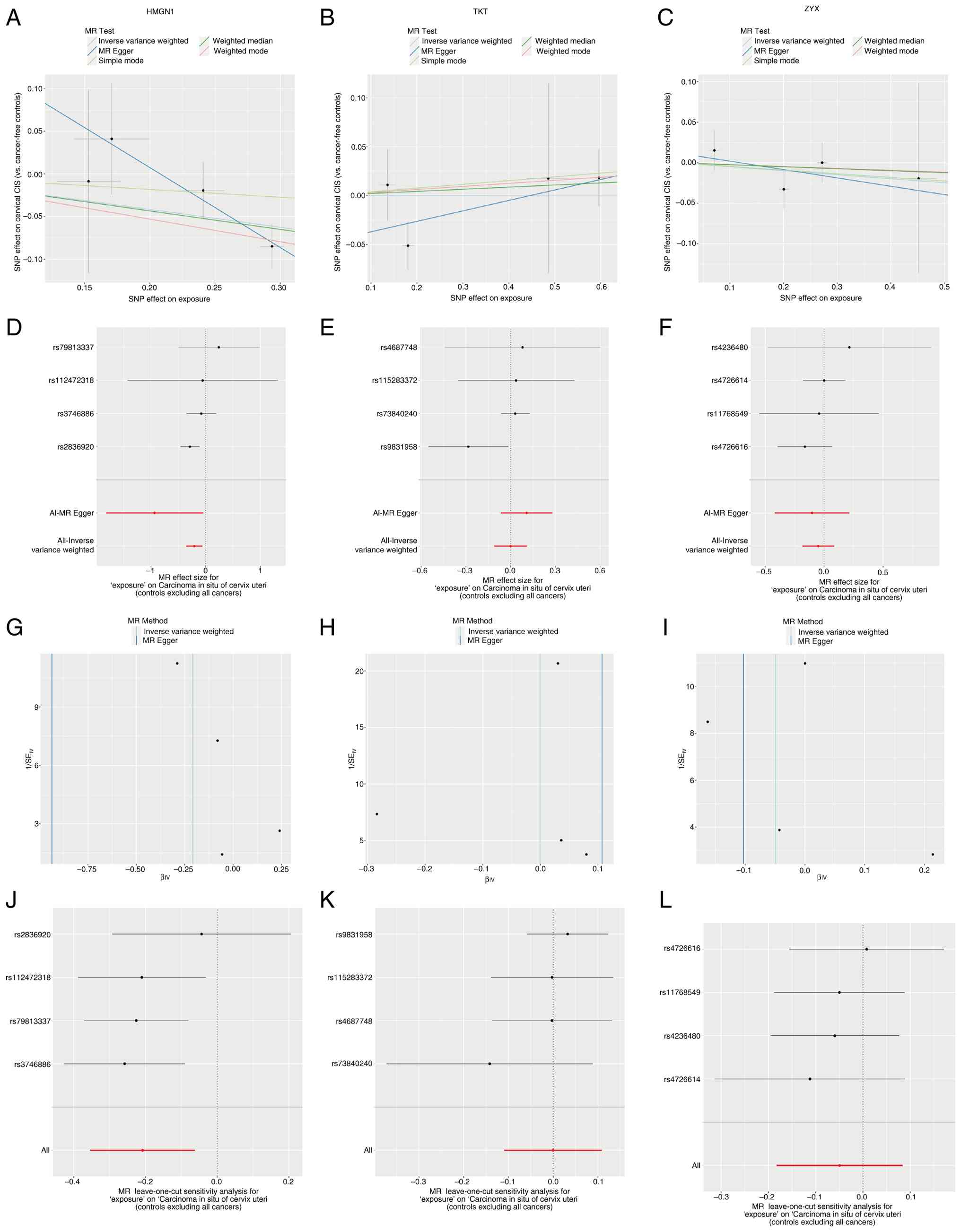 | Figure 9.MR analysis demonstrates a causal
relationship between the LAGs prognostic model and cervical cancer.
MR scatter plots display the association between (A) HMGN1, (B)
TKT, (C) ZYX and patients with cervical cancer. Forest plots
depicting the overall effect of (D) HMGN1, (E) TKT and (F) ZYX on
cervical cancer. Funnel plots for (G) HMGN1, (H) TKT and (I) ZYX in
cervical cancer. Leave-one-out analysis of the remaining stability
after excluding (J) HMGN1, (K) TKT and (L) ZYX SNPs. SNPs, single
nucleotide polymorphisms; HMGN1, high-mobility group
nucleosome-binding protein 1; TKT, transketolase; ZYX, zyxin; MR,
Mendelian randomization. |
 | Table I.MR analysis of the LAGs prognostic
model from the finngen_R12_CD2_INSITU_CERVIX_UTERI_EXALLC data
source. |
Table I.
MR analysis of the LAGs prognostic
model from the finngen_R12_CD2_INSITU_CERVIX_UTERI_EXALLC data
source.
| Exposure | Gene | Method | Number of SNPs | β | SE | P-value | OR | or_lci95 | or_uci95 |
|---|
| Carcinoma in
situ of cervix | HMGN1 | Inverse variance
weighted | 20 | −0.208311264 | 0.074517941 | 0.005182733 | 0.811954265 | 0.701617827 | 0.939642214 |
| uteri |
| MR Egger |
| −0.935886258 | 0.451742896 | 0.174083585 | 0.392238087 | 0.161814877 | 0.950782279 |
|
|
| Simple mode |
| −0.090818216 | 0.157267515 | 0.604106442 | 0.913183698 | 0.670948735 | 1.242873595 |
|
|
| Weighted mode |
| −0.264655633 | 0.086377683 | 0.054823054 | 0.767470196 | 0.647940834 | 0.909049825 |
|
|
| Weighted
median |
| −0.216692908 | 0.077847357 | 0.005376567 | 0.805177195 | 0.691236174 | 0.937899867 |
|
| NUP50 | Inverse variance
weighted | 2 | −0.004228927 | 0.094542495 | 0.964322174 | 0.995780002 | 0.827346061 | 1.198504301 |
|
| TKT | Inverse variance
weighted | 20 | −0.000368019 | 0.0557443 | 0.994732468 | 0.999632048 | 0.89616848 | 1.115040592 |
|
|
| MR Egger |
| 0.107180793 | 0.08810701 | 0.347880425 | 1.113135483 | 0.936590667 | 1.322958522 |
|
|
| Simple mode |
| 0.038402893 | 0.11447086 | 0.759352482 | 1.039149815 | 0.830306813 | 1.300522073 |
|
|
| Weighted mode |
| 0.031367274 | 0.05042089 | 0.577947782 | 1.031864411 | 0.934767287 | 1.139047309 |
|
|
| Weighted
median |
| 0.021688186 | 0.047296271 | 0.646550039 | 1.021925085 | 0.931450245 | 1.121188044 |
|
| ZYX | Inverse variance
weighted | 20 | −0.049281896 | 0.06806318 | 0.469028271 | 0.951912752 | 0.833029912 | 1.087761524 |
|
|
| MR Egger |
| −0.103085931 | 0.160727915 | 0.586973835 | 0.902049456 | 0.658288072 | 1.236074684 |
|
|
| Simple mode |
| −0.045157699 | 0.118377715 | 0.728268287 | 0.955846734 | 0.75791956 | 1.205461669 |
|
|
| Weighted mode |
| −0.022977045 | 0.091040541 | 0.817049205 | 0.977284917 | 0.817571837 | 1.16819803 |
|
|
| Weighted
median |
| −0.024763227 | 0.07300978 | 0.73447651 | 0.975540867 | 0.845470167 | 1.125622191 |
|
| PAK2 | Inverse variance
weighted | 2 | 0.10067654 | 0.252150728 | 0.689693339 | 1.105918863 | 0.674665081 | 1.812835086 |
|
| ARID3B | Wald ratio | 1 | 0.047089984 | 0.132516165 | 0.722325339 | 1.048216328 | 0.808445752 | 1.359098576 |
|
| FABP5 | Wald ratio | 1 | 0.072237589 | 0.073148103 | 0.32337188 | 1.0749107 | 0.931338302 | 1.240615801 |
|
| HDGF | Wald ratio | 1 | 0.425897326 | 0.181286783 | 0.018808666 | 1.530963576 | 1.073125298 | 2.18413402 |
To assess the robustness of the present analysis,
funnel plots were constructed using the β coefficients and SEs for
each IV associated with the three genes studied (Fig. 9G-I). The distributions of these
plots revealed certain asymmetry, possibly due to the limited
number of IVs, which may introduce heterogeneity and potential
bias. To account for this, the present study performed
heterogeneity tests, all of which yielded P>0.05, indicating
that heterogeneity did not significantly affect the present study
results (Table SVII). Furthermore,
the present study conducted leave-one-out analyses to assess the
stability of the present study findings. The results remained
consistent even after excluding individual SNPs, further supporting
the robustness of the associations (Fig. 9J-L). The present study also
performed a test for horizontal pleiotropy on HMGN1, with
P>0.05, indicating no significant evidence of horizontal
pleiotropy influencing the present study results (Table SVIII). The present study results
provide strong evidence for a causal relationship between HMGN1 and
cervical cancer, while no significant MR associations were observed
for the other analyzed genes. These findings highlight the
potential role of HMGN1 in the pathogenesis of cervical cancer.
HMGN1 can serve as a therapeutic
target for patients with CESC
To further investigate the role of HMGN1 in cervical
cancer, the present study designed HMGN1-specific siRNA sequences
using siRNA technology and transfected them into SiHa and HeLa
cells. Subsequently, 24 h after transfection, the present study
assessed the knockdown efficiency of HMGN1 using RT-qPCR, which
confirmed that HMGN1 was successfully silenced in both cell lines
(Fig. 10A). Subsequent western
blotting was performed to assess the protein expression of HMGN1,
further validating the effectiveness of the knockdown (Fig. 10B). Functional assays revealed that
inhibition of HMGN1 significantly suppressed the proliferation of
SiHa and HeLa cells (Fig. 10C),
reduced the proportion of S-phase cells and prolonged the
G2 phase of the cell cycle (Fig. 10D). Therefore, these findings
suggest that HMGN1 may be a risk factor for CESC.
Discussion
Cervical cancer is a prevalent and aggressive
malignancy in women, marked by high recurrence and metastasis rates
that lead to poor outcomes (52).
Although TNM staging and histopathological grading are extensively
used, their prognostic accuracy and value for individualized
therapy remain limited (53,54).
Therefore, identifying novel prognostic biomarkers is key to
improve risk stratification and advance precision medicine in
cervical cancer management in the future.
Lactylation, an epigenetic post-translational
modification on lysine residues, regulates protein function and
participates in key cellular processes such as metabolism, immune
regulation and signal transduction (24–26).
Increasing evidence associate abnormal lactylation with
tumorigenesis and cancer progression (55–57).
In the present study, TCGA-CESC and GEO datasets were integrated to
identify 43 LAGs associated with cervical cancer prognosis and a
14-gene prognostic model was developed using multiple machine
learning algorithms (CoxBoost, StepCox, RSF and LASSO). The model
demonstrated independent predictive performance for OS, DFI, PFI
and DSS, surpassing conventional clinical factors such as age,
stage and T classification. To ensure generalizability and reduce
overfitting, it was externally validated in the GSE30760 cohort and
key genes were further confirmed in GSE7803 using AUC analysis.
Consistent performance across datasets demonstrated its robustness
and clinical utility. This risk scoring system provides a potential
practical framework for patient stratification-high-risk patients
may require intensive therapy or closer follow-up, whereas low-risk
patients could benefit from less aggressive management.
Functional enrichment analysis elucidated the
biological mechanisms underlying the prognostic relevance of LAGs
in cervical cancer. GSVA analysis revealed that the low-risk group
was enriched in pathways associated with ‘TNFA signaling via
NF-KB’, ‘hypoxia’, ‘unfolded protein response’ and ‘angiogenesis’,
while the high-risk group exhibited activation of the KRAS
signaling pathway. These pathways are key mediators of tumor cell
proliferation, invasion and immune regulation (27–29).
Previous studies have indicated that human papilloma virus (HPV)
type 16 E2 protein enhances TNF-α-induced NF-κB activation,
promoting cervical tumor progression; whereas hypoxia-inducible
factor-1α and VEGF drive hypoxia-induced angiogenesis and
metastasis (58,59). Similarly, mutations in the MAPK/ERK
cascade (common in gynecological cancer) also support the
involvement of these oncogenic pathways (60). KEGG and GO analyses further revealed
enrichment in ‘carbon metabolism’, ‘glycolysis/gluconeogenesis’ and
‘amino acid biosynthesis’, highlighting the role of metabolic
reprogramming in LAG-mediated tumor progression. Consistent with a
previous study by Tong et al (8), the present study findings indicated
that lactylated histones both promote glycolytic metabolism and
regulate immune homeostasis, suggesting LAGs may cascade metabolic
activation with immune evasion in cervical cancer. Notably, aerobic
glycolysis enhances tumor metastasis and chemotherapy resistance
(61). The myosin regulatory light
polypeptide 9 protein promotes glycolysis (62), while celecoxib combined with
cisplatin or paclitaxel inhibits tumor growth by disrupting energy
metabolism (63). Collectively,
these findings suggest that LAGs may drive cervical cancer
progression by coordinating metabolic and immune regulatory
pathways, providing a theoretical basis for metabolic and
immune-targeting therapeutic strategies.
Single-cell transcriptomic analysis further revealed
the expression of LAGs in various cell types, including columnar
and squamous epithelial cells, CD4+ T cells and tumor
cells, highlighting their involvement in cervical cancer
progression and the immune response. Cell-cell communication
analysis further identified significant associations between Notch,
laminin and collagen signaling pathways and LAGs-based risk
subgroups. These pathways modulate ECM interactions, influence
tumor cell adhesion and migration, and impede immune cell
communication, thereby promoting tumor progression (64). Among them, Notch signaling is a key
driver of tumorigenesis and is associated with poor prognosis in
cervical cancer (65). Laminin
enhances migration and differentiation through PI3K/AKT and MAPK
pathways, which are dysregulated by HPV oncoproteins (E5, E6 and
E7) (66). Similarly, aberrant MAPK
activation promotes proliferation and survival through the
AKT/mTOR/pyruvate dehydrogenase kinase 1 axis (67). Collectively, these findings
underscore that integrating single-cell transcriptomic data with
molecular profiling provides key insights into the
microenvironmental mechanisms underlying cervical cancer and
informs potential therapeutic strategies.
Immunotherapy has emerged as a promising therapeutic
strategy for cervical cancer by reshaping the tumor immune
microenvironment and addressing tumor heterogeneity, thereby
enabling early molecular stratification and personalized
intervention (52). In the present
study, immune infiltration analysis revealed that the low-risk
group exhibited higher levels of activated immune cells, including
B and mast cells, M2 macrophages and monocytes, compared with the
high-risk group, indicating a more active antitumor immune
response. By contrast, the high-risk group displayed an
immunosuppressive microenvironment, which may contribute to worse
clinical outcomes. These findings are consistent with previous
studies that reported M2 macrophage infiltration is associated with
immune escape and unfavorable prognosis in cervical cancer
(68,69). Furthermore, analysis of the
IMvigor210 real-world immunotherapy cohort supported these
observations: Low-risk patients achieved higher rates of CR/PR,
while high-risk patients were more likely to experience disease
SD/PDs. Drug sensitivity analysis further revealed that paclitaxel
and rapamycin exhibited greater predicted efficacy in the low-risk
group, whereas midostaurin was more effective in the high-risk
group. Mechanistically, the paclitaxel-platinum combination remains
the standard first-line regimen for cervical cancer and
rapamycin-an mTOR pathway inhibitor-may suppress tumor growth
through metabolic reprogramming (70).
Notably, the LAGs-based model also provides
mechanistic insight into how immune cell composition influences
therapeutic response. In the high-risk group, the TME is dominated
by immunosuppressive M2 macrophages that inhibit CD8+ T
cell activation, thereby reducing cytotoxicity and promoting
resistance to immune checkpoint inhibitors. These findings suggest
that high-risk patients might benefit more from combination
regimens that both block immune checkpoints (for example,
programmed cell death protein-1/programmed cell death-ligand 1
inhibitors) and modulate macrophage polarization to restore an
inflammatory (M1-like) phenotype. By contrast, the low-risk group
demonstrated higher infiltration of activated CD8+ T
cells, which are key to antitumor cytotoxicity. Such patients may
be more responsive to ICI monotherapy or therapeutic strategies
aimed at further enhancing CD8+ T cell activity.
Collectively, these findings demonstrate that the LAGs-based
prognostic model not only stratifies patients by survival risk but
also provides key guidance for immunotherapy selection and
chemotherapy optimization. By integrating immune and metabolic
profiling, the present study model supports the development of
personalized treatment strategies and precision medicine frameworks
for CESC.
Using MR analysis, the present study investigated
the causal relationship between lactose-associated genes (LAGs) and
cervical cancer. Using IVW, weighted median and MR-Egger regression
analyses, the present study identified a significant causal
association between HMGN1 expression and cervical cancer risk.
Other prognostic genes demonstrated no causal effects, potentially
due to genetic or environmental influences. Previous studies
confirm the key role of HMGN1 in DNA repair, genomic stability and
tumor immune microenvironment regulation (39–42).
In vitro experiments further validated that silencing HMGN1
in SiHa and HeLa cells inhibits cell proliferation and S-phase
progression, highlighting its potential as a therapeutic and
prognostic target for cervical cancer. Several limitations warrant
attention: In vitro validation was limited to HPV-positive
cell lines and primarily focuses on proliferation studies, without
addressing other carcinogenic processes such as cell migration and
invasion. Future research should extend to HPV-negative models and
patient-derived models to conduct more detailed investigations.
Larger multi-ethnic GWAS and comprehensive eQTL datasets will
enhance causal inference. The proposed prognostic model stratifies
patients into high-risk and low-risk groups to guide treatment
intensity-high-risk patients may require more aggressive therapy or
closer monitoring, while low-risk patients can receive
lower-intensity management. External validation in independent
cohorts and optimization of risk thresholds are essential prior to
clinical implementation. Integration into hospital information
systems could enable automated risk prediction, while prospective
clinical trials are needed to confirm its impact on survival rates
and quality of life. Collaboration with diagnostic reagent
developers to establish standardized LAG testing protocols will
further advance clinical translation and promote precision
management of cervical cancer.
The present study established a 14-gene prognostic
model based on LAGs using an integrative multi-omics analysis of
cervical cancer. The model accurately predicts patient outcomes and
exhibits strong associations with immune cell infiltration, tumor
heterogeneity, cell-cell communication and drug sensitivity.
Furthermore, HMGN1 was identified as a key gene with a causal role
in cervical cancer and potential as a therapeutic target,
emphasizing its importance in tumor progression. Collectively,
these findings provide novel insights into the molecular mechanisms
underlying cervical cancer and highlight the clinical utility of
LAGs for both prognostic assessment and personalized treatment
strategies in the future.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by The National Natural Science
Foundation of China (grant no. 82074478), Key program of
Administration of Traditional Chinese Medicine of Jiangsu Province,
China (grant no. ZX202102), Jiangsu Province Leading Talents
Cultivation Project for Traditional Chinese Medicine (grant no.
SLJ0307).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
RW contributed to writing, conceptual design and
methodology development. LN was involved in writing, draft
preparation, bioinformatics analysis, and experimental research. XL
contributed to writing, software application and data collection.
YX and YS participated in methodology development and
visualization. QR and HZ contributed to conceptual design, writing,
review, and editing. RW also contributed to software application
and data acquisition. RW and LN participated in data validation and
analysis. QR contributed to study design and supervision. LN and RW
confirm the authenticity of the raw data. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Laversanne M, Sung H, Ferlay J,
Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 74:229–263.
2024.PubMed/NCBI
|
|
2
|
Wisniak A, Yakam V, Bolo SE, Yakam V,
Schmidt NC, Kenfack B and Petignat P: Fertility and miscarriage
incidence after cervical intraepithelial neoplasia treatment by
thermal ablation: A cohort study. BMC Womens Health. 132:167–177.
2025.
|
|
3
|
Duenas-Gonzalez A, Serrano-Olvera A,
Cetina L and Coronel J: New molecular targets against cervical
cancer. Int J Women Health. 6:1023–1031. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wei F, Georges D, Man I, Baussano I and
Clifford GM: Causal attribution of human papillomavirus genotypes
to invasive cervical cancer worldwide: A systematic analysis of the
global literature. Lancet. 404:435–444. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Malagón T, Franco EL, Tejada R and
Vaccarella S: Epidemiology of HPV-associated cancers past, present
and future: Towards prevention and elimination. Nat Rev, Clin
Oncol. 21:522–538. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Caruso G, Wagar MK, Hsu HC, Hoegl J, Rey
Valzacchi GM, Fernandes A, Cucinella G, Sahin Aker S, Jayraj AS,
Mauro J, et al: Cervical cancer: A new era. Int J Gynecol Cancer.
34:1946–1970. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang Y, Lu Y, Li S, Zheng F, Dong Y, Tang
H, Wang X and Wang J: Precision theranostics in cervical Cancer:
Harnessing stimuli-responsive hydrogels for tumor
microenvironment-targeted therapy and diagnosis. Materials Today
Bio. 35:1023922025. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tong H, Jiang Z, Song L, Tan K, Yin X, He
C, Huang J, Li X, Ling X, et al: Dual impacts of
serine/glycine-free diet in enhancing antitumor immunity and
promoting evasion via PD-L1 lactylation. Cell Metab. 36:2493–2510.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sia TY, Wan V, Finlan M, Zhou QC, Iasonos
A, Zivanovic O, Sonoda Y, Chi DS, Long Roche K, Jewell E, et al:
Procedural interventions for oligoprogression during treatment with
immune checkpoint blockade in gynecologic malignancies: A case
series. Int J Gynecol Cancer. 34:594–601. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pinheiro C, Garcia EA, Morais-Santos F,
Moreira MA, Almeida FM, Jubé LF, Queiroz GS, Paula ÉC, Andreoli MA,
Villa LL, et al: Reprogramming energy metabolism and inducing
angiogenesis: Co-expression of monocarboxylate transporters with
VEGF family members in cervical adenocarcinomas. BMC Cancer.
15:8352015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gao Y, Siyu Zhang, Zhang X, Du Y, Ni T and
Hao S: Crosstalk between metabolic and epigenetic modifications
during cell carcinogenesis. iScience. 27:1113592024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lin Y, Li L, Yuan B, Luo F, Zhang X, Yang
Y, Luo S, Lin J, Ye T, Zhang Y, et al: Phosphorylation determines
the glucose metabolism reprogramming and tumor-promoting activity
of sine oculis homeobox 1. Signal Transduct Target Ther. 9:3372024.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ippolito L, Duatti A, Iozzo M, Comito G,
Pardella E, Lorito N, Bacci M, Pranzini E, Santi A, Sandrini G, et
al: Lactate supports cell-autonomous ECM production to sustain
metastatic behavior in prostate cancer. EMBO Rep. 25:3506–3531.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhao Y, Liu MJ, Zhang L, Yang Q, Sun QH,
Guo JR, Lei XY, He KY, Li JQ, Yang JY, et al: High mobility group
A1 (HMGA1) promotes the tumorigenesis of colorectal cancer by
increasing lipid synthesis. Nat Commun. 15:99092024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Patil K, Johnston E, Novack J, Wallace G,
Lin M and Pai SB: Multifaceted impact of HIV inhibitor dapivirine
on triple negative breast cancer cells reveals potential entities
as targets for novel therapy. Sci Rep. 14:301032024. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lin C, Ye J, Xu C, Zheng Y, Xu Y, Chen Y,
Chi L, Lin J, Li F, Lin Y and Wang Q: Evaluating lactate metabolism
for prognostic assessment and therapy response prediction in
gastric cancer with emphasis on the oncogenic role of SLC5A12.
Biochim Biophys Acta Gen Subj. 1869:1307392024. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun D, Lu J, Zhao W, Chen X, Xiao C, Hua
F, Hydbring P, Gabazza EC, Tartarone A, Zhao X and Yang W:
Construction and validation of a prognostic model based on
oxidative stress-related genes in non-small cell lung cancer
(NSCLC): Predicting patient outcomes and therapy responses. Transl
Lung Cancer Res. 13:3152–3174. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang H, Yang Y, Xing W, Li Y and Zhang S:
Expression and gene regulatory network of S100A16 protein in
cervical cancer cells based on data mining. BMC Cancer.
23:11242023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: Limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Roychowdhury A, Samadder S, Das P,
Mazumder DI, Chatterjee A, Addya S, Mondal R, Roy A, Roychoudhury S
and Panda CK: Deregulation of H19 is associated with cervical
carcinoma. Genomics. 112:961–970. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fjeldbo CS, Hompland T, Hillestad T,
Aarnes EK, Günther CC, Kristensen GB, Malinen E and Lyng H:
Combining imaging- and gene-based hypoxia biomarkers in cervical
cancer improves prediction of chemoradiotherapy failure independent
of intratumour heterogeneity. EBioMedicine. 57:1028412020.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
den Boon JA, Pyeon D, Wang SS, Horswill M,
Schiffman M, Sherman M, Zuna RE, Wang Z, Hewitt SM, Pearson R, et
al: Molecular transitions from papillomavirus infection to cervical
precancer and cancer: Role of stromal estrogen receptor signaling.
Proc Natl Acad Sci USA. 112:E3255–E3264. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee YY, Kim TJ, Kim JY, Choi CH, Do IG,
Song SY, Sohn I, Jung SH, Bae DS, Lee JW and Kim BG: Genetic
profiling to predict recurrence of early cervical cancer. Gynecol
Oncol. 131:650–654. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Teschendorff AE, Jones A and Widschwendter
M: Stochastic epigenetic outliers can define field defects in
cancer. BMC Bioinformatics. 17:1782016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhai Y, Kuick R, Nan B, Ota I, Weiss SJ,
Trimble CL, Fearon ER and Cho KR: Gene expression analysis of
preinvasive and invasive cervical squamous cell carcinomas
identifies HOXC10 as a key mediator of invasion. Cancer Res.
67:10163–10172. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Leek JT, Johnson WE, Parker HS, Jaffe AE
and Storey JD: The sva package for removing batch effects and other
unwanted variation in high-throughput experiments. Bioinformatics.
28:882–883. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cheng Z, Huang H, Li M, Liang X, Tan Y and
Chen Y: Lactylation-related gene signature effectively predicts
prognosis and treatment responsiveness in hepatocellular carcinoma.
Pharmaceuticals. 16:6442023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang D, Tang Z, Huang H, Zhou G, Cui C,
Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, et al: Metabolic
regulation of gene expression by histone lactylation. Nature.
574:575–580. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Moreno-Yruela C, Zhang D, Wei W, Bæk M,
Liu W, Gao J, Danková D, Nielsen AL, Bolding JE, Yang L, et al:
Class I histone deacetylases (HDAC1-3) are histone lysine
delactylases. Sci Adv. 8:eabi66962022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mariathasan S, Turley SJ, Nickles D,
Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita
JL, Cubas R, et al: TGFβ attenuates tumour response to PD-L1
blockade by contributing to exclusion of T cells. Nature.
554:544–548. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kurki MI, Karjalainen J, Palta P, Sipilä
TP, Kristiansson K, Donner KM, Reeve MP, Laivuori H, Aavikko M,
Kaunisto MA, et al: FinnGen provides genetic insights from a
well-phenotyped isolated population. Nature. 613:508–518. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Stuart T, Butler A, Hoffman P, Hafemeister
C, Papalexi E, Mauck WM III, Hao Y, Stoeckius M, Smibert P and
Satija R: Comprehensive Integration of Single-Cell Data. Cell.
177:1888–1902.e21. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Korsunsky I, Millard N, Fan J, Slowikowski
K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR and Raychaudhuri
S: Fast, sensitive and accurate integration of single-cell data
with Harmony. Nat Methods. 16:1289–1296. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jin S, Guerrero-Juarez CF, Zhang L, Chang
I, Ramos R, Kuan CH, Myung P, Plikus MV and Nie Q: Inference and
analysis of cell-cell communication using CellChat. Nat Commun.
12:10882021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wyss R, Van Der Laan M, Gruber S, Shi X,
Lee H, Dutcher SK, Nelson JC, Toh S, Russo M, Wang SV, et al:
Targeted learning with an undersmoothed LASSO propensity score
model for large-scale covariate adjustment in health-care database
studies. Am J Epidemiol. 193:1632–1640. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Binder H and Schumacher M: Allowing for
mandatory covariates in boosting estimation of sparse
high-dimensional survival models. BMC Bioinformatics. 9:142008.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Friedman J, Hastie T and Tibshirani R:
Regularization paths for generalized linear models via coordinate
descent. J Stat Soft. 33:1–22. 2010.umerical. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen D, Lin D, Li H, Yang J, Liu L, Zhang
H, Tang D and Wang K: The glycolytic characteristics of
hepatocellular carcinoma and its interaction with the
microenvironment: A comprehensive omics study. J Transl Med.
23:4242025. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Blanche P, Dartigues J and Jacqmin-Gadda
H: Estimating and comparing time-dependent areas under receiver
operating characteristic curves for censored event times with
competing risks. Statistics in Medicine. 32:5381–5397. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Restaino S, Pellecchia G, Arcieri M,
Bogani G, Taliento C, Greco P, Driul L, Chiantera V, Ercoli A,
Fanfani F, et al: Management for cervical cancer patients: A
comparison of the guidelines from the international scientific
societies (ESGO-NCCN-ASCO-AIOM-FIGO-BGCS-SEOM-ESMO-JSGO). Cancers.
16:25412024. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tang Z, Kang B, Li C, Chen T and Zhang Z:
GEPIA2: An enhanced web server for large-scale expression profiling
and interactive analysis. Nucleic Acids Res. 47:W556–W560. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hänzelmann S, Castelo R and Guinney J:
GSVA: Gene set variation analysis for microarray and RNA-seq data.
BMC Bioinf. 14:72013. View Article : Google Scholar
|
|
45
|
Yoshihara K, Shahmoradgoli M, Martínez E,
Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW,
Levine DA, et al: Inferring tumour purity and stromal and immune
cell admixture from expression data. Nat Commun. 4:26122013.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mroz EA and Rocco JW: MATH, a novel
measure of intratumor genetic heterogeneity, is high in
poor-outcome classes of head and neck squamous cell carcinoma. Oral
Oncology. 49:211–215. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Geeleher P, Cox N and Huang RS:
pRRophetic: An R package for prediction of clinical
chemotherapeutic response from tumor gene expression levels. PLoS
One. 9:e1074682014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Walker VM, Davies NM, Hemani G, Zheng J,
Haycock PC, Gaunt TR, Davey Smith G and Martin RM: Using the
MR-Base platform to investigate risk factors and drug targets for
thousands of phenotypes. Wellcome Open Res. 4:1132019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Verbanck M, Chen CY, Neale B and Do R:
Detection of widespread horizontal pleiotropy in causal
relationships inferred from Mendelian randomization between complex
traits and diseases. Nat Genet. 50:693–698. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cho Y, Haycock PC, Sanderson E, Gaunt TR,
Zheng J, Morris AP, Davey Smith G and Hemani G: Exploiting
horizontal pleiotropy to search for causal pathways within a
mendelian randomization framework. Nat Commun. 11:10102020.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yordanov A, Damyanova P, Vasileva-Slaveva
M, Hasan I, Kostov S and Shivarov V: Integrated analysis of
phagocytic and immunomodulatory markers in cervical cancer reveals
constellations of potential prognostic relevance. Int J Mol Sci.
25:91172024. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Allemani C, Minicozzi P, Morawski B, Lima
CA, Bennett D, Pongnikorn D, Petrova D, Innos K, Girardi F, Galán
Alvarez Y, et al: Global variation in patterns of care and time to
initial treatment for breast, cervical, and ovarian cancer from
2015 to 2018 (VENUSCANCER): A secondary analysis of individual
records for 275 792 women from 103 population-based cancer
registries in 39 countries and territories. Lancet. 406:2325–2348.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Andersen K, Bonde J, Waldstrøm M, Jakobsen
MV, Lamy P, Pedersen H, Bønløkke S, Stougaard M and Steiniche T:
Evaluation of targeted next-generation sequencing for detection of
HPV genotypes and sublineages in cervical liquid-based cytology
SurePath samples from the Danish screening program. Int J Cancer.
158:193–201. 2026. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Jian X, Cheng C, Lu W, Peng H and Yang D:
Histone lactylation: Unveiling a novel pathway for the impact of
lactate on physiological and pathological processes (review). Int J
Mol Med. 57:1–12. 2025. View Article : Google Scholar
|
|
56
|
Dang T, You Y, Wei L, Li Q, Sun H, Sun M,
Li X, Yang S, Zeng T, Zhang L, et al: ICAT drives lactylation of
tumor-associated macrophages via the c-myc-ENO1 axis to promote
cervical cancer progression. Free Radic Biol Med. 241:316–329.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Xue ZR, Xin YY and Jin WL: Exploiting
metabolic vulnerabilities in cancer: From mechanisms to therapeutic
opportunities. Cancer Lett. 634:2180672025. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Sarwar F, Ashhad S, Vimal A and
Vishvakarma R: Small molecule inhibitors of the VEGF and tyrosine
kinase for the treatment of cervical cancer. Med Oncol. 41:1992024.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mokoala KMG, Lawal IO, Maserumule LC, Bida
M, Maes A, Ndlovu H, Reed J, Mahapane J, Davis C, Van de Wiele C,
et al: Correlation between [68Ga]Ga-FAPI-46 PET imaging and HIF-1α
immunohistochemical analysis in cervical cancer: Proof-of-concept.
Cancers. 15:39532023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Nasioudis D, Fernandez ML, Wong N, Powell
DJ Jr, Mills GB, Westin S, Fader AN, Carey MS and Simpkins F: The
spectrum of MAPK-ERK pathway genomic alterations in gynecologic
malignancies: Opportunities for novel therapeutic approaches.
Gynecol Oncol. 177:86–94. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Del Dotto V, Grillini S, Righetti R,
Grandi M, Giorgio V, Solaini G and Baracca A: Bioenergetics of
cancer cells: Insights into the warburg effect and regulation of
ATP synthase. Mol Med. 31:3112025. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wen B, Luo L, Zeng Z and Luo X: MYL9
promotes squamous cervical cancer migration and invasion by
enhancing aerobic glycolysis. J Int Med Res.
51:30006052312085822023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Robledo-Cadena DX, Pacheco-Velázquez SC,
Vargas-Navarro JL, Padilla-Flores JA, López-Marure R, Pérez-Torres
I, Kaambre T, Moreno-Sánchez R and Rodríguez-Enríquez S:
Synergistic celecoxib and dimethyl-celecoxib combinations block
cervix cancer growth through multiple mechanisms. PLoS One.
19:e03082332024. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sheng B, Pan S, Ye M, Liu H, Zhang J, Zhao
B, Ji H and Zhu X: Single-cell RNA sequencing of cervical
exfoliated cells reveals potential biomarkers and cellular
pathogenesis in cervical carcinogenesis. Cell Death Dis.
15:1302024. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Limones-Gonzalez JE, Aguilar Esquivel P,
Vazquez-Santillan K, Castro-Oropeza R, Lizarraga F, Maldonado V,
Melendez-Zajgla J, Piña-Sanchez P and Mendoza-Almanza G: Changes in
the molecular nodes of the notch and NRF2 pathways in cervical
cancer tissues from the precursor stages to invasive carcinoma.
Oncol Lett. 28:5222024. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Hazazi A, Khan FR, Albloui F, Arif S,
Abdulaziz O, Alhomrani M, Sindi AAA, Abu-Alghayth MH, Abalkhail A,
Nassar SA and Binshaya AS: Signaling pathways in HPV-induced
cervical cancer: Exploring the therapeutic promise of RNA
modulation. Pathol Res Pract. 263:1556122024. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Yan Y, Dai T, Guo M, Zhao X, Chen C, Zhou
Y, Qin M, Xu L and Zhao J: A review of non-classical MAPK family
member, MAPK4: A pivotal player in cancer development and
therapeutic intervention. Int J Biol Macromol. 271:1326862024.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zheng X, Tong T, Duan L, Ma Y, Lan Y, Shao
Y, Liu H, Chen W, Yang T and Yang L: VSIG4 induces the
immunosuppressive microenvironment by promoting the infiltration of
M2 macrophage and Tregs in clear cell renal cell carcinoma. Int
Immunopharmacol. 142:1131052024. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Miao C, You X, Zhang Z, Jiang Z, Liu L,
Jia Y, Bai J, Gao Y, Ye L, Cao Y, et al: SCG2 mediates HNSCC
progression with CCL2/TGFβ1 M2 macrophage infiltration. Oral Dis.
31:782–795. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Feng L, Shi Q, Wang S, Zhao Y, Wu H, Wei
L, Hao Q, Cui Z, Wang L, Zhang J, et al: The outcome of advanced
and recurrent cervical cancer patients treated with First-line
platinum and paclitaxel with or without indication for immune
checkpoint inhibitors: The comparative study. BMC Cancer.
24:12672024. View Article : Google Scholar : PubMed/NCBI
|