Histone deacetylases (HDACs), acting as key
epigenetic regulators, control chromatin structure and gene
expression by removing acetyl groups from histones. HDACs
participate in fundamental cellular processes such as proliferation
and apoptosis. Dysregulation of their activity is associated with
tumorigenesis and progression, endowing them with dual importance
both as therapeutic targets and biomarkers (1,2).
Within the HDAC family, functional roles and expression patterns
exhibit tumor-specific variability. For instance, upregulation of
HDAC1 and HDAC2 enhances tumor invasiveness and chemoresistance
(3,4). HDACs interact with other enzymes, such
as lysine acetyltransferases and DNA methyltransferases (DNMTs),
which renders the analysis of the relevant mechanisms more complex
(5,6). HDAC inhibitors (HDACis) represent
promising therapeutic agents. Notably, several HDACis have gained
approval from the USA Food and Drug Administration (FDA) for
hematological malignancies. However, their efficacy in solid tumors
remains limited due to off-target effects, poor sensitivity and
acquired resistance (7,8). Current research priorities include
investigating resistance mechanisms and exploring combination
therapies, approaches whose potential have already been
demonstrated across multiple tumor types. The development of novel
HDACis with enhanced specificity and reduced toxicity is actively
being investigated (9–13). The present review comprehensively
examines the functions, mechanisms and therapeutic potential of
HDACs to inform ongoing research and strategic development.
The HDAC family is classified into class I, IIa, IIb
and IV, with each subclass exhibiting distinct cellular
localization, substrate specificity and functional roles. Members
of class I, including HDAC1, HDAC2 and HDAC3, are predominantly
localized in the nucleus and serve key roles in regulating gene
expression, cell cycle progression and apoptosis. Notably, HDAC3
markedly influences both cell cycle regulation and apoptotic
pathways, emerging as a key contributor to tumorigenesis and
progression (14). The class IIa
subgroup comprises HDAC4, HDAC5, HDAC7 and HDAC9. These enzymes
shuttle dynamically between the nucleus and cytoplasm to
participate in the modulation of diverse signaling cascades. For
instance, HDAC5 and HDAC7 engage with pathways implicated in cancer
metastasis, underscoring their oncological relevance (15–17).
Class IIb HDACs, represented by HDAC6 and HDAC10, reside primarily
in the cytoplasm, where they mediate non-histone deacetylation
events, affecting processes such as cell motility and stress
responses. Among these, HDAC6 has garnered attention as a promising
therapeutic target due to its association with cancer and
neurodegenerative disorders (18–20).
Class IV histone deacetylase comprises only one isoform, HDAC11.
Although the development of selective inhibitors faces numerous
challenges due to the lack of crystal structure data, accumulating
evidence indicates that this protein is involved in the initiation
and progression of tumors and inflammatory diseases. The chemical
space, scaffold diversity and key structural features of HDAC11
have been systematically characterized, providing core structural
basis and a foundation for the design of selective inhibitors
targeting this protein (21). The
enzymatic activity of HDAC11 mediates resistance to MEK inhibitors
in uveal melanoma, serving as an important molecule driving
malignant progression of this tumor (22). In Hodgkin lymphoma cells, HDAC11
plays a central regulatory role in the expression of OX40 ligand,
acting as a key factor involved in the formation of the tumor
inflammatory microenvironment (23). Structural divergence among HDAC
isoforms markedly impacts their interaction profiles with
substrates and enzymatic activity. Conserved motifs and specialized
domains dictate substrate-binding affinity and specificity,
features that are fundamental to their cellular functions.
Comprehensive understanding of these classification schemes and
structural characteristics is essential in designing selective
HDACis, enabling tumor-specific targeting for cancer therapy and
other pathologies. Elucidating their precise mechanisms will
establish a robust foundation for novel therapeutic interventions
in the future (24,25).
HDACs exert a central role in regulating chromatin
structure and gene expression by catalyzing the deacetylation of
lysine residues on histones. This process induces compaction of
chromatin conformation, thereby reducing the accessibility of
transcriptional machinery to DNA templates and subsequently
repressing the transcriptional activity of tumor suppressor genes.
HDAC-mediated histone deacetylation enhances the electrostatic
interaction between histones and DNA, rendering the chromatin
structure more compact and thereby restricting the binding of
transcription factors to gene promoters. This process is one of the
key epigenetic mechanisms that regulate gene expression in cancer
cells (26).
Aberrant expression of HDACs is implicated in
various malignant neoplasms. For example, in hepatocellular
carcinoma (HCC), dysregulated histone acetylation drives
tumorigenesis and progression by silencing tumor suppressor genes
(27,28). Furthermore, HDACs extend their
regulatory influence beyond histones through deacetylation of
non-histone proteins, including transcription factors, thereby
modulating cell signaling pathways and altering transcriptional
activity. Such post-translational modifications can either enhance
or repress target gene expression involved in cell survival,
proliferation and metastasis (29,30).
This dual regulatory capacity underscores the pivotal role of HDACs
in maintaining chromatin dynamics and cellular signaling networks,
establishing them as compelling therapeutic targets for cancer
treatment. Additionally, HDACs interact with chromatin-remodeling
complexes such as switch defective/sucrose non-fermentable; these
interactions are key to preserving chromatin architecture and
governing gene expression programs. Notably, recruitment of HDACs
to specific genomic loci can displace chromatin remodelers, thereby
reinforcing transcriptionally repressive environments (31).
Recent studies have revealed the synergistic role of
combinatorial histone modifications in chromatin regulation,
offering a novel dimension in understanding HDAC function (32,33).
Notably, histone modifications can exert synergistic effects
through combinatorial modification coding, while HDAC-mediated
deacetylation may disrupt the balance of the acetyl-methyl lysine
dual modification, thereby altering chromatin accessibility at
transcription start sites. The existence of this acetyl-methyl
lysine modification has been confirmed in relevant studies
(34). For example, in endometrial
carcinoma, HDAC1 forms a complex with enhancer of zeste homolog 2.
Through deacetylation-methylation, this complex silences tumor
suppressor genes such as p21. HDAC1-mediated histone deacetylation
compacts chromatin to create conditions for methylation, therefore
blocking cell cycle regulatory pathways (35). Furthermore, HDACs do not act
independently in epigenetic regulation, but exhibit a close
crosstalk with histone methyltransferases and DNMTs. In acute
myeloid leukemia (AML), combining HDACis with DNMT inhibitors
(DNMTis) disrupts the epigenetic repressive network, reactivates
tumor suppressor genes and inhibits cancer cell proliferation
(36). In lung cancer models, the
interaction between HDAC3 and lysine-specific demethylase 1 affects
enhancer activity by regulating histone H3 lysine 4 monomethylation
levels, thus highlighting the key role of crosstalk between HDACs
and other epigenetic regulators in tumorigenesis (37).
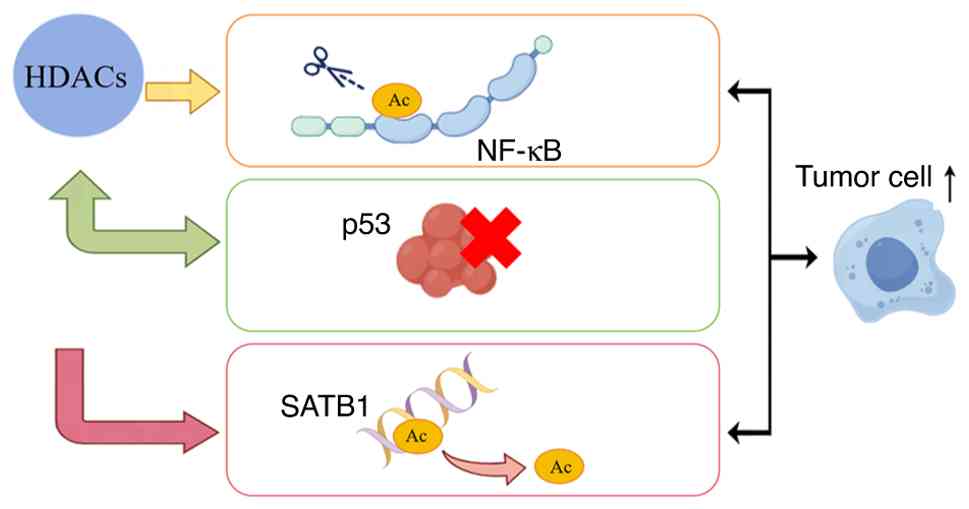
HDACs interact with key signaling pathways involving
factors such as nuclear factor-κB (NF-κB), p53 and heat-shock
protein 90 (HSP90), thereby modulating cellular responses to stress
and proliferation signals. For instance, within the constitutively
activated NF-κB pathway frequently observed in cancer, HDACs exert
post-translational modifications on pathway components via
deacetylation, which is an action that fosters tumor cell survival
and proliferation (38,39). Similarly, the tumor-suppressive
functionality of the classical guardian protein p53 undergoes
negative regulation by HDACs. Inhibition of p53 transcriptional
activity through deacetylation enables cancer cells to evade
apoptosis (5,40). A specific example during cancer
progression involves crosstalk between HDAC5 and special AT-rich
sequence-binding protein 1 (SATB1), a chromatin architect
regulating tumor suppressor gene expression. Previous studies have
demonstrated that, in lung adenocarcinoma, HDAC5-mediated
deacetylation of SATB1 represses tumor suppressor genes thus not
only promoting neoplastic growth and metastasis but also conferring
chemoresistance (41,42). The aberrant expression patterns of
HDACs across multiple malignancies, including lung cancer,
underscore their dual role in oncogenic signaling, acting both as
drivers of carcinogenesis and promising therapeutic targets
(Fig. 1). Deciphering the intricate
interactions between HDACs and these key regulators along with
associated signaling cascades offers promising avenues for the
development of enhanced cancer treatment modalities. Notably,
combining HDACis with conventional therapies represents a strategic
approach to overcome drug resistance and improve patient outcomes
(19,43,44).
In summary, various subtypes of the HDAC family form
a mechanism-phenotype regulatory network through chromatin
remodeling, non-histone modification and regulation of signaling
pathways (such as the NF-κB and p53 pathways). Aberrant activation
of class I HDACs (such as HDAC1/3) can promote cell proliferation
by silencing tumor suppressor genes. Class II HDACs (such as
HDAC5/7) are involved in tumor metastasis through regulating
epithelial-mesenchymal transition (EMT) and angiogenesis. The
bidirectional role of class IV HDAC11 is dependent on the
specificity of the tumor microenvironment. These mechanistic
foundations provide a core theoretical framework for subsequent
analysis of the functional differences of HDACs in diverse tumor
types.
This section focuses on ovarian cancer, endometrial
cancer, glioma, osteosarcoma and multiple myeloma (MM). These
specific cancers were selected to provide a broad yet distinct
perspective on HDAC biology across diverse tumor contexts. They
represent major malignancies from different organ systems
(gynecologic, central nervous system, bone/bone marrow) with unique
etiologies, clinical challenges and patterns of HDAC
expression/function. This comparative approach allows us to analyze
conserved vs. tumor-specific roles of HDACs and their inhibitors,
which is crucial for informing future cross-tumor targeting
strategies. This section aims to analyze in detail the
tumor-specific expression patterns of HDAC subtypes in each cancer
type (including the subtype-specific functions of HDAC9 in ovarian
cancer and the mechanism by which HDACs regulate ferroptosis in
glioma). Additionally, this section aims to compare the core
oncogenic pathways mediated by HDACs and the differences in
therapeutic responses to HDACis across these different tumor types,
thereby providing a reference for cross-tumor targeting strategies
in the future.
HDACs have emerged as pivotal regulatory factors in
the pathogenesis of ovarian cancer, particularly implicated in
late-stage diagnosis and chemoresistance (45,46).
Dysregulated expression of HDACs is associated with tumor
aggressiveness. Their hyperactivation induces chromatin
condensation and transcriptional repression of tumor suppressor
genes (47,48). This dysregulation markedly
contributes to poor clinical outcomes in patients with advanced
ovarian cancer by enhancing cancer cell survival under conventional
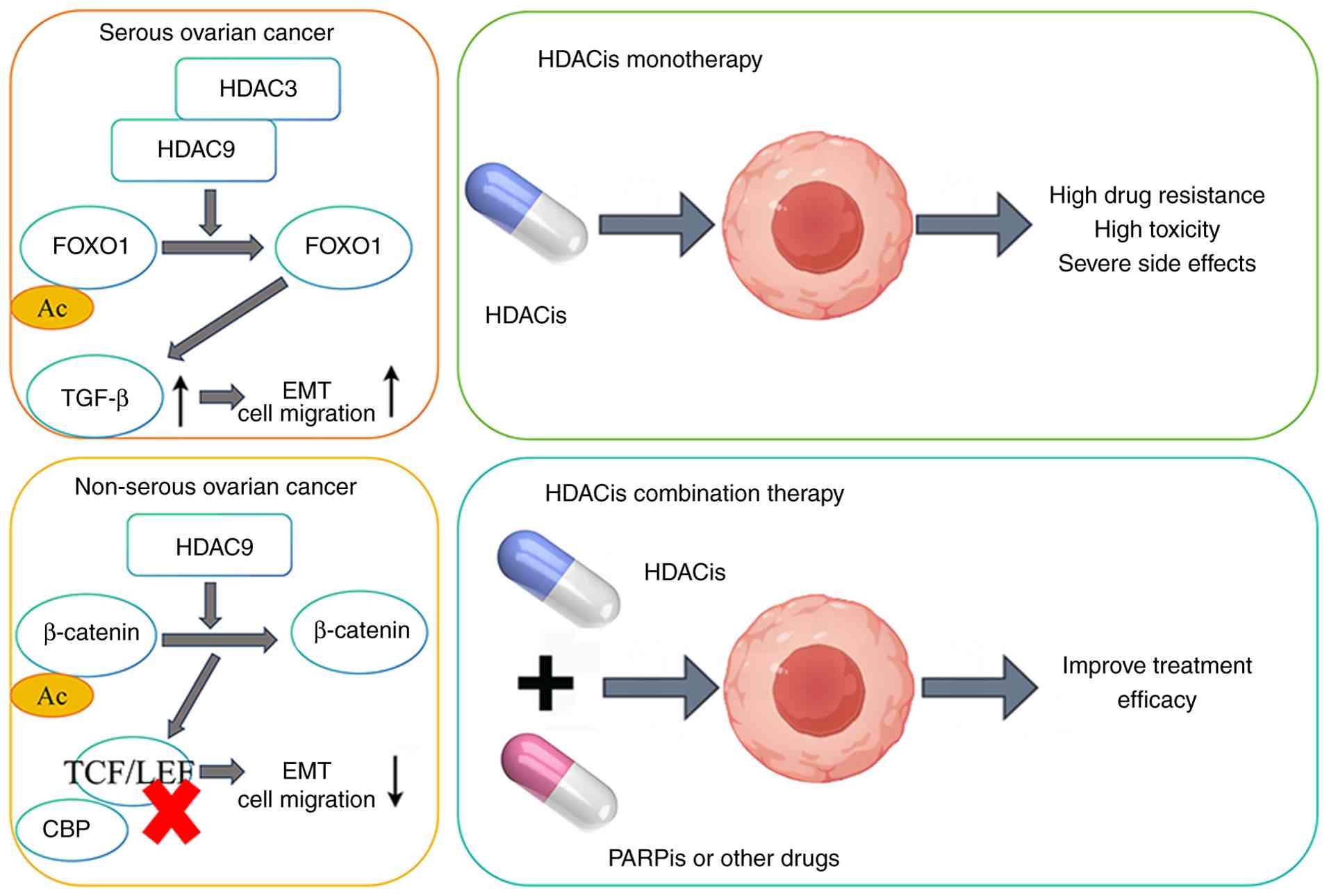
chemotherapy regimens. Notably, elevated expression levels of HDAC9
are associated with adverse prognosis in patients with serous
ovarian carcinoma (49). By
contrast, this enzyme exhibits tumor-suppressive effects in
non-serous subtypes, highlighting histological subtype-specific
functionalities among HDAC family members (49). Furthermore, interactions between
HDACs and signaling pathways involving forkhead box protein O1 or
transforming growth factor-β underscore their role in promoting
cell migration and invasion, which are key hallmarks of metastatic
progression in ovarian malignancy (50).
At the therapeutic strategy level, HDACis have
demonstrated promising potential in pre-clinical studies,
exhibiting notable anticancer activity against ovarian cancer cells
(51–53). However, clinical trials have
reported limitations in the efficacy of HDACis when administered as
monotherapy, which is a finding that has driven research into
combination regimens to enhance therapeutic outcomes. For example,
combining HDACis with other anticancer agents has emerged as a
highly prospective approach capable of overcoming the constraints
associated with single-agent treatments. This strategy leverages
synergistic effects between distinct drugs to amplify therapeutic
efficacy while potentially mitigating toxicity associated with
high-dose monotherapy (54,55). Recent research has focused on
elucidating the combinatorial potential of HDACis with
poly(ADP-ribose) polymerase inhibitors (PARPis) and other agents;
such regimens have demonstrated enhanced cytotoxic effects in
ovarian cancer cell lines (56,57).
Furthermore, novel dual-target inhibitors targeting both HDAC and
complementary signaling pathways, such as the phosphoinositide
3-kinase (PI3K) pathway, are under development, reflecting an
evolutionary trend towards increasingly personalized and efficient
treatment modalities for ovarian cancer (Fig. 2) (58).
HDACs serve a key role in the pathogenesis of
endometrial carcinoma, with their mediated histone deacetylation
processes closely associated with tumor invasion and poor clinical
outcomes (35). As the most common
malignant tumor in the female genital tract, endometrial cancer has
a global incidence of ~20.2 per 100,000 women, accounting for
30–40% of all female genital tract malignancies. This tumor
typically exhibits an aberrant histone acetylation pattern. Among
histone deacetylases, HDAC6 acts as a key member and is highly
expressed in 76.8% of endometrial cancer tissues. Such aberrant
acetylation serves as a critical factor driving aggressive
phenotypes including deep myometrial invasion and lymph node
metastasis, as well as advanced-stage disease (46.0% at stages
III–IV). It is also closely associated with reduced 5-year
disease-free survival (41.3%), representing a key molecular feature
underlying poor prognosis (59).
Disrupted histone acetylation is strongly associated with
high-grade tumors, which are characterized by enhanced invasiveness
and metastatic propensity (60).
Elevation in HDAC levels is markedly associated with advanced
disease stages and diminished survival rates, positioning them as
potential prognostic biomarkers (61). Furthermore, upregulation of specific
HDAC isoforms (such as HDAC1 and HDAC6) promotes EMT, which
enhances cancer cell invasive capabilities. Based on these
mechanistic insights, therapeutic strategies targeting HDAC
activity emerge as key interventions in restraining tumor
progression in endometrial carcinoma (35,62).
The therapeutic potential of HDACis in endometrial
carcinoma has garnered notable attention, primarily due to their
capacity to induce cell-cycle arrest and apoptosis in cancerous
cells. Compounds such as suberoylanilide hydroxamic acid and
romidepsin, which are prominent members of the HDACis family, have
demonstrated efficacy in pre-clinical models by restoring
acetylation levels and reactivating epigenetically silenced tumor
suppressor genes (35,60,63).
These agents not only suppress tumor growth but also sensitize
endometrial cancer cells to conventional chemotherapeutic drugs,
thus generating synergistic cytotoxic effects. For instance,
combinatorial regimens incorporating HDACis with standard
chemotherapy have been reported to enhance DNA damage responses and
promote apoptotic pathways in endometrial cancer cell lines
(35). Ongoing clinical trials
evaluating such combination therapies in patients have yielded
preliminary results indicating improved tumor regression rates and
overall survival outcomes (35,60,64,65).
The strategic deployment of HDACis, particularly when integrated
with other treatment modalities, represents a promising approach to
enhance therapeutic efficacy and overcome drug resistance in
endometrial carcinoma (38,66).
HDACs occupy a central position in the regulatory
networks governing glioma cells, particularly regarding ferroptosis
(a form of regulated cell death driven by iron-dependent lipid
peroxidation) (67). As integral
components of this pathway, HDACs directly influence tumor
progression and therapeutic responses. Inhibition of HDAC activity
elevates both histone and non-histone protein acetylation levels,
disrupting cellular homeostasis and promoting ferroptotic cell
death in glioma cells (68). This
mechanism holds particular importance for glioblastoma, which is
characterized by high invasiveness and resistance to conventional
therapies. The prevalent dysregulation of iron metabolism observed
in gliomas is exacerbated by aberrant upregulation of HDACs,
thereby sustaining tumor cell survival and proliferation (69,70).
Targeting HDACs to induce ferroptosis represents a
promising therapeutic strategy, as previous studies have
demonstrated that HDACis enhance the sensitivity of glioma cells to
this form of cell death, simultaneously suppressing tumor growth
and potentiating the efficacy of existing treatments (67,71).
HDACis have emerged as potential therapeutic agents for glioma due
to their ability to alter acetylation status in both histone and
non-histone proteins, thereby modulating gene expression profiles
and cellular behavior. For example, valproic acid (VPA) exhibits
notable antineoplastic effects in in vitro and in
vivo models of glioma, demonstrating capabilities to inhibit
cell proliferation while increasing responsiveness to radiotherapy
and chemotherapy, effects that are likely mediated through the
regulation of survival/apoptosis signaling cascades (8,72,73).
Mechanistically, HDACis operate via multiple pathways, including
reactivating epigenetically silenced tumor suppressor genes,
downregulating oncogenic drivers to alter cellular dynamics and
driving tumor cell death. Additionally, HDACis contribute to
enhancing antitumor immune responses by remodeling the tumor
microenvironment and facilitating infiltration of immune effector
cells (74,75). Clinically, HDACis have demonstrated
promise in improving patient outcomes, particularly for isocitrate
dehydrogenase (IDH)-mutant glioma subtypes, where malignant cells
display increased sensitivity to HDAC inhibition (76,77).
Their therapeutic synergy with immune checkpoint inhibitors further
underscores their value in glioma management, while highlighting
the increasing demand for personalized medicine based on
individualized molecular profiling of tumors.
Osteosarcoma, a highly aggressive bone malignancy
predominantly affecting children and adolescents, exhibits
HDAC-driven progression through enhanced tumor cell migration and
invasion, primarily via regulation of EMT. For example, HDAC6 is
markedly upregulated in doxorubicin- and cisplatin-resistant
osteosarcoma cells, directly associating with their increased
metastatic potential (78).
Mechanistically, HDAC6 interacts with estrogen receptor-related
proteins to modulate their acetylation status and stability,
thereby elevating cancer-cell survival rates and conferring
apoptosis resistance (79,80). Furthermore, HDACs contribute to the
development of chemoresistance and, therefore, inhibiting their
activity restores sensitivity to conventional chemotherapeutics,
which is a key finding due to the poor prognosis associated with
this aggressive disease despite intensive treatment protocols
(81). Elevation of HDAC expression
in drug-resistant cell lines underscores their promise as
therapeutic targets (79,82). Notably, HDACis such as vorinostat
and entinostat synergize with doxorubicin in pre-clinical models,
enhancing treatment efficacy (83).
These agents exert antitumor effects by altering
histone/non-histone acetylation landscapes, including reactivating
silenced tumor suppressor genes and activating pro-apoptotic
pathways; furthermore, when combined with chemotherapy, HDACis
exhibit potent combination therapeutic effects, thereby markedly
reducing the viability of osteosarcoma cell lines and inducing
their apoptosis (84).
Additionally, HDACis impede tumor cell motility and invasion
(85), while promoting
autophagy-induced autonomous cell death (86). Ongoing clinical trials evaluating
the safety and efficacy of HDACis combined with standard
chemotherapy aim to overcome chemoresistance, improve response
rates and, ultimately, enhance patient outcomes in the future
(87,88).
HDACis represent a cornerstone therapeutic class for
MM, with regulatory approval from the USA FDA for the clinical use
of vorinostat, belinostat and romidepsin. Their mechanism of action
involves HDAC suppression to disrupt key pathways governing tumor
growth and survival, including inhibition of NF-κB signaling
cascades, upregulation of cell cycle regulators (including p21 and
p53), downregulation of the anti-apoptotic protein B-cell
lymphoma-2 (Bcl-2) and induction of apoptotic programs in myeloma
cells (89). Concurrently, HDACis
enhance antitumor immunity and promote autophagy-mediated cancer
cell clearance (90,91). Therapeutic synergy emerges when
HDACis are combined with immunomodulatory agents, conventional
chemotherapy or targeted therapies, which markedly improves
treatment outcomes in patients with MM. For instance, combinatorial
regimens incorporating HDACis with proteasome inhibitors (such as
bortezomib) demonstrated increased antimyeloma activity and
improved survival rates (92),
effectively mitigating drug resistance while simultaneously
targeting multiple pathogenic pathways implicated in MM
pathogenesis. As a classic HDAC inhibitor, vorinostat in
combination with the immunomodulatory drug lenalidomide can enhance
lenalidomide-mediated CRBN pathway activity through epigenetic
regulation, upregulate the expression of NKG2D ligands and promote
the cytotoxic function of NK cells, thereby synergistically
inhibiting the proliferation of MM cells (93). When combined with the conventional
chemotherapeutic agent bortezomib, vorinostat blocks HDAC-mediated
protein deacetylation and jointly inhibits the proteasome/aggregate
degradation system with bortezomib, thus overcoming bortezomib
resistance and inducing synergistic apoptosis in MM cells (94). The HDAC6 inhibitor ACY-241 in
combination with the anti-CD38 targeted therapeutic daratumumab
shows unique advantages: It can upregulate CD38 expression on the
surface of MM cells, significantly enhance the antibody-dependent
cellular cytotoxicity effect of daratumumab and effectively improve
the efficiency of targeted elimination of MM cells (95).
HDAC3 is a key regulatory factor in cancer biology.
By catalyzing histone deacetylation, it promotes chromatin
condensation and represses the transcription of genes associated
with the cell cycle and apoptosis, thus disrupting the balance
between cell proliferation and death. In colorectal cancer (CRC),
HDAC3 regulates cancer stem cell-associated genes, thereby
influencing tumor cell plasticity and chemoresistance.
Ubiquitin-specific peptidase 38 (USP38) modulates HDAC3 stability,
while pharmacological or genetic inhibition of USP38 induces HDAC3
degradation and promotes the development of aggressive tumor
phenotypes. These findings suggest that targeting the HDAC3-USP38
axis may be an effective strategy to overcome chemoresistance
(96–98). HDAC3 also impacts the tumor
microenvironment via an NF-κB/p65-dependent mechanism, which
silences the chemokine gene C-X-C motif chemokine ligand 10
(CXCL10), thus impeding CD8+ T-cell infiltration and
fostering an immunosuppressive microenvironment. This mechanism is
particularly prominent in KRAS-mutant lung cancer and represents a
major cause of its low response rate to immune checkpoint
inhibitors (99). Inhibiting HDAC3
increases CXCL10 expression and its combination with
anti-programmed cell death protein-1 (PD-1) antibodies markedly
enhances tumor regression and T-cell infiltration (37,100).
Additionally, HDAC3 inhibition directly induces apoptosis in cancer
cells (including breast and lung cancer). Selective HDAC3
inhibitors have demonstrated notable preclinical efficacy; compared
with pan-HDAC inhibitors, HDAC3 inhibitors exhibit fewer off-target
effects and optimize the therapeutic index (101–103).
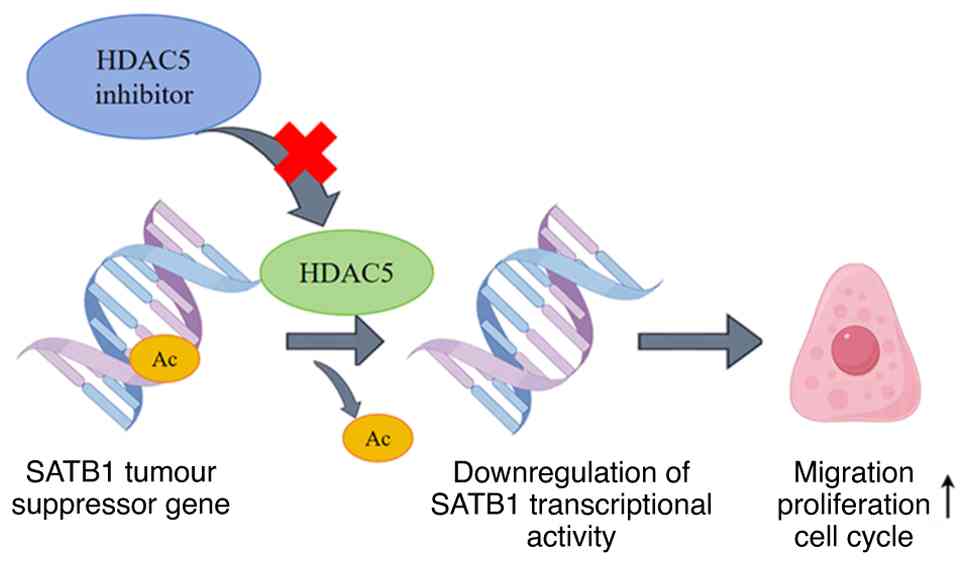
HDAC5 serves a key role in transcriptional
regulation and is closely associated with the pathogenesis of
various cancer types, including lung adenocarcinoma. The core
mechanism is that HDAC5 reduces the transcriptional activity of
SATB1 via deacetylation at the K411 residue in lung adenocarcinoma,
thereby suppressing the expression of tumor suppressor genes and
promoting cancer cell migration (42). Downregulation of SATB1 activity
enhances metastatic potential by promoting lung adenocarcinoma cell
migration. Pharmacological or genetic inhibition of HDAC5 reverses
these effects; this not only impedes tumor cell migration, but also
reactivates silenced tumor suppressor genes (42). This positions HDAC5 as a promising
therapeutic target to prevent invasiveness and restore key
antitumor pathways in lung adenocarcinoma. Furthermore, HDAC5
suppression induces cell-cycle arrest, effectively blocking the
uncontrolled proliferation of malignant cells (104–106). This dual functionality
(simultaneously blocking metastasis and restricting tumor growth)
highlights the therapeutic potential of HDAC5 inhibitors. Since
aberrant cell cycle progression constitutes a hallmark of
malignancy, targeting HDAC5 offers notable value in cancer therapy
(Fig. 3). Such intervention could
enhance existing treatment modalities and improve patient outcomes,
underscoring the need for further investigation into HDAC5
inhibitors as adjunctive therapeutic agents in oncology (107).
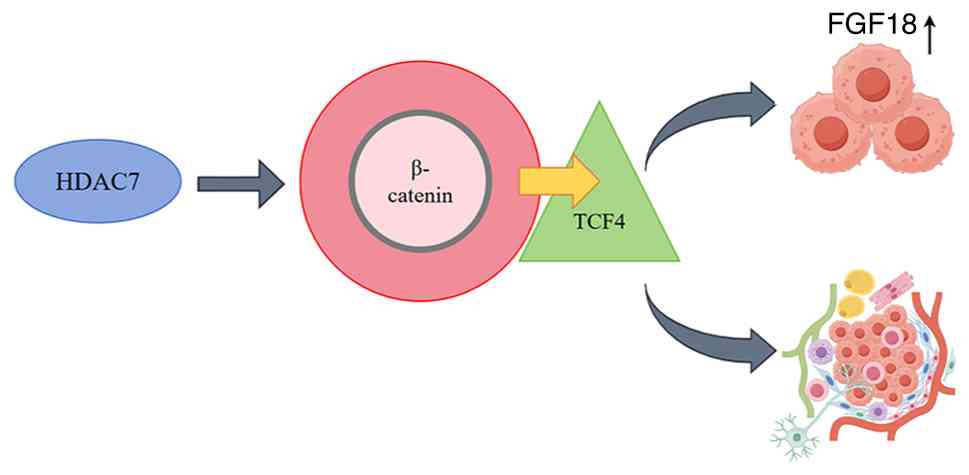
HDAC7 is a key regulator driving tumor growth,
metastasis and drug resistance through mechanisms intrinsically
associated with angiogenic microenvironment modulation. Evidence
has demonstrated that HDAC7 is often upregulated in malignancies
such as non-small cell lung cancer (NSCLC) and CRC, where these
elevated levels are associated with advanced disease stages and
poor clinical outcomes (15,108).
In NSCLC specifically, HDAC7 potentiates oncogenic signaling by
intersecting with fibroblast growth factor 18 (FGF18) pathways,
thus enhancing both proliferative capacity and metastatic spread
(15). Mechanistically, HDAC7
stabilizes β-catenin (a central mediator of Wnt signaling),
facilitating its nuclear translocation and transcriptional complex
formation with transcription factor 4 to activate FGF18 expression
(Fig. 4) (15). Targeting this axis could disrupt
pro-tumorigenic cascades, highlighting the therapeutic
vulnerability of HDAC7.
Beyond cellular autonomy, HDAC7 orchestrates
extracellular niche formation via dual regulation of angiogenesis
and antitumor immunity within the microenvironment. For instance,
its role in macrophage polarization and inflammatory reprogramming
notably supports metastatic progression (109,110). By engineering pro-vasculature
ecosystems conducive to tumor expansion, HDAC7 creates therapeutic
challenges through desmoplastic stroma development. Clinically,
HDAC7 can serve as both a prognostic biomarker and a therapeutic
target. Its high expression predicts poor survival outcomes in
patients with various malignancies, including diffuse large B-cell
lymphoma (DLBCL), NSCLC, CRC and breast cancer, and can be used for
patient risk stratification (108,111). The core mechanism is that high
expression of HDAC7 promotes tumor cell proliferation,
anti-apoptosis, invasion and metastasis by activating oncogenic
pathways such as NF-κB, PI3K/AKT and β-catenin-FGF18, regulating
the EMT process and cell cycle-related proteins. Meanwhile, it
remodels the immunosuppressive tumor microenvironment and enhances
resistance to cancer therapy. Consequently, high HDAC7 expression
is closely associated with adverse pathological features including
advanced clinical stage, lymph node metastasis and distant
metastasis, ultimately leading to shortened overall survival and
progression-free survival in patients with DLBCL, NSCLC, CRC and
other malignancies, making it a key molecular marker for predicting
poor survival outcomes (108,111). Preclinical models have
demonstrated that selective inhibition of HDAC7 can sensitize
cancer cells to chemotherapy and immunotherapy by disrupting cell
survival-related signaling pathways including PI3K/AKT, NF-κB and
Wnt/β-catenin, providing a highly promising combination therapeutic
strategy to overcome treatment resistance (2,112).
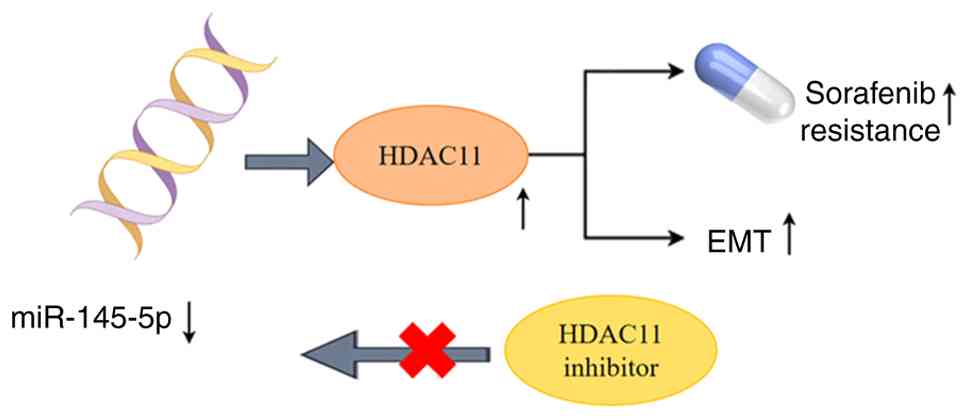
Previous research has revealed the dual role of
HDAC11 in oncology, functioning as either an oncogene or tumor
suppressor gene depending on the cancer type and microenvironmental
context. In HCC, HDAC11 maintains cancer stemness and confers
resistance to sorafenib therapy via the microRNA
(miR)-145-5p/HDAC11 axis. Downregulation of miR-145-5p elevates
HDAC11 expression, thereby enhancing HCC cell survival and
proliferative capacity under therapeutic stress (113,114). Within sorafenib-resistant HCC cell
lines, dysregulated HDAC11 is associated with augmented drug
metabolism and altered signaling pathways driving metastasis.
Furthermore, the regulatory influence of HDAC11 over EMT
underscores its importance in metastatic progression across
multiple malignancies. Its crosstalk with non-coding RNAs,
particularly miR-145-5p, forms a key determinant of treatment
response in HCC cells, suggesting that the therapeutic targeting of
this axis could improve efficacy and overcome chemoresistance
(114,115). Clinically, HDAC11 upregulation is
associated with poor prognosis in HCC by sustaining stem-like
properties and enabling metabolic adaptation for survival under
unfavorable conditions. Mechanistically, miR-145-5p suppression
elevates HDAC11 levels, which reinforce sorafenib resistance and
metastatic potential; by contrast, pharmacological inhibition of
HDAC11 restores miR-145-5p abundance, resensitizing HCC cells to
sorafenib while attenuating their metastatic abilities (Fig. 5) (113,116). Adding complexity to its functional
repertoire, HDAC11 modulates immune evasion pathways and
tumor-stroma interactions within the microenvironment, implying
that multidimensional therapeutic strategies against HDAC11 may
yield notable clinical benefits (116–118).
HDACis exert antitumor effects by targeting the
active site of HDACs or regulating their interaction with
substrates to reverse aberrant epigenetic modifications. Marked
differences exist in the mechanisms of action and subtype
selectivity among different types of HDACis. Classified by chemical
structure and mechanism of action, HDACis mainly fall into four
categories: i) Hydroxamic acid derivatives (such as vorinostat),
which achieve pan-subtype inhibition of class I and II HDACs by
chelating zinc ions in the active site of HDACs. In cutaneous T
cell lymphoma, hydroxamic acid derivatives can induce excessive
histone acetylation to activate the transcription of tumor
suppressor genes such as p21 (119,120); ii) benzamide derivatives (such as
entinostat), which exhibit higher selectivity for class I HDACs
(HDAC1/2/3). In osteosarcoma models, benzamide derivatives enhance
the cytotoxicity of the chemotherapeutic drug doxorubicin by
specifically inhibiting HDAC3 (83); iii) cyclic peptide derivatives (such
as romidepsin), which bind to the active pocket of HDACs via their
unique cyclic peptide structure. In MM, cyclic peptide derivatives
can downregulate the expression level of the anti-apoptotic protein
Bcl-2 by inhibiting the NF-κB signaling pathway (89); and iv) fatty acid derivatives (such
as VPA), which inhibit HDAC activity in a non-competitive manner.
In glioma, fatty acid derivatives can promote the expression level
of ferroptosis-related genes and enhance the radiosensitivity of
tumor cells (67,71). Beyond regulating histone
acetylation, HDACis also exert pleiotropic effects through
non-histone modifications: i) In lung adenocarcinoma, HDACis can
acetylate the transcription factor SATB1, enhancing its regulatory
activity on tumor suppressor genes and reversing the pro-metastatic
effect mediated by HDAC5 (42); and
ii) in CRC, HDACis disrupt the interaction between HSP90 and its
target proteins [namely, protein kinase B (AKT)] by acetylating
HSP90, thereby inhibiting cell proliferation signaling pathways
(96). These mechanisms
collectively form the molecular basis for the antitumor activity of
HDACis and provide a target basis for the development of
subtype-selective inhibitors.
The clinical efficacy of HDACis differs notably
between hematological malignancies and solid tumors, with marked
limitations in monotherapy and combination therapy has become the
core strategy to improve therapeutic outcomes. In terms of
monotherapy, the USA FDA has approved five HDACis for clinical use,
including: i) Vorinostat, which is approved for cutaneous T cell
lymphoma, with an objective response rate (ORR) of 30–40%, but it
is associated with dose-limiting toxicities such as fatigue and
gastrointestinal disturbances (119,120); ii) belinostat and romidepsin,
which are used for peripheral T cell lymphoma, with ORRs of 25 and
35%, respectively, but their survival benefits for advanced
patients are limited (121,122);
iii) panobinostat, which is combined with bortezomib for MM and as
monotherapy, its ORR is <20%; therefore, it is only used as a
salvage treatment option for drug-resistant patients (89,92);
and iv) VPA. Phase II clinical trials of VPA in glioma have
reported that monotherapy achieves a disease control rate (DCR) of
~40%, but it fails to markedly prolong the progression-free
survival (PFS) of patients (74,75).
Overall, HDACi monotherapy has poor efficacy in solid tumors. For
example, the ORR of vorinostat monotherapy in ovarian cancer is
only 12% and the DCR of romidepsin monotherapy in endometrial
cancer is <30% (35,54,55,62).
Combination therapy strategies have demonstrated synergistic
effects in various tumors. In ovarian cancer, the combination of
HDACis and PARPis can inhibit the DNA damage repair pathway,
increasing the ORR of patients with wild-type BRCA from 15 to 45%
without markedly increasing the risk of myelosuppression (56,57).
In osteosarcoma, the combination of vorinostat and doxorubicin can
downregulate HDAC6 expression, reverse chemoresistance in tumor
cells and increase the DCR from 38 to 65% (83,85).
In MM, the triple regimen of panobinostat, bortezomib and
dexamethasone can markedly prolong the PFS of patients from 9.5 to
12.5 months (92,123). In glioma, the combination of VPA
and immune checkpoint inhibitors (anti-PD-1 antibodies) can enhance
the infiltration of CD8+ T cells in the tumor
microenvironment, increasing the ORR from 18 to 35% (76,77).
Furthermore, dual-target inhibitors (for example CUDC-907) that
concurrently target HDACs and pathways such as PI3K have exhibited
enhanced antitumor activity in preclinical models of ovarian and
breast cancer, offering a novel avenue for combination therapy
(58,124).
Although HDACis exhibit potential in combination
therapy, their clinical translation is limited by three core
challenges, namely: i) Drug resistance; ii) off-target toxicity;
and iii) a lack of biomarkers. Drug resistance is the primary cause
of HDACi treatment failure, with three main mechanisms, including:
i) Compensatory upregulation of HDAC subtypes. For example, in AML,
prolonged use of pan-HDACis leads to increased HDAC3 expression,
which maintains tumor cell survival via enhancement of NF-κB
pathway activity (98,125). In osteosarcoma, HDAC6 expression
is markedly higher in doxorubicin-resistant cell lines compared
with that in doxorubicin-sensitive cells, thus contributing to
doxorubicin resistance. This phenotype can be reversed by
combination with HDAC6-selective inhibitors (79,85);
ii) adaptive activation of signaling pathways. For instance, HDACi
treatment induces PI3K/AKT pathway activation in lung cancer
(102) and triggers compensatory
NF-κB pathway activation in MM (89,92);
and iii) tumor microenvironment remodeling, such as increased
infiltration of myeloid-derived suppressor cells (MDSCs) in
pancreatic cancer following HDACi treatment (126). Furthermore, tumor genomic
heterogeneity driven by abnormal mutagenic processes exacerbates
HDACi resistance; HDAC dysregulation may impair DNA repair
capacity, forming an epigenetic dysregulation, which leads to
genomic instability, and in turn induces the drug resistance
cascade (127). Off-target
toxicity limits dose escalation of HDACis. Due to a lack of subtype
selectivity, pan-HDACis cause multi-organ adverse effects,
including thrombocytopenia (30%) and neutropenia (25%) in the
hematological system (128,129);
gastrointestinal disturbances (45%) in the digestive system
(130); and grade 3+ fatigue (30%)
in systemic reactions (131). The
grading and classification of adverse events in this article were
based on the Common Terminology Criteria for Adverse Events, the
universal standard in clinical oncology. Clinically, adverse
effects are mitigated by adjusted dosing regimens (such as
twice-weekly vorinostat, low initial dose with gradual escalation
of VPA) (74,128) or combination with symptomatic
drugs. Certain HDACis (such as vorinostat) also carry
cardiotoxicity risk (QT interval prolongation) (120). The absence of biomarkers hinders
precise patient stratification. No clear molecular biomarkers for
the prediction of HDACi efficacy exist. The correlation between
HDAC9 expression and HDACi sensitivity in ovarian cancer (49,50),
IDH mutation status and HDACi efficacy in glioma (76,77)
and p53 mutation and HDACi-bortezomib synergy in MM (89) remain unconfirmed, leading to
treatment blindness and increased ineffective treatment rates.
To address the challenges in the clinical
translation of HDACis, current research focuses on three key
directions: i) Structural modification; ii) targeted delivery; and
iii) optimization of combination strategies, aiming to enhance the
selectivity and efficacy of HDACis while reducing toxicity.
Structural modification is key to developing
subtype-selective HDACis. HDAC3 inhibitors, with an introduced
isoquinoline structure, inhibit tumor growth in CRC models without
affecting platelet production (103,132). HDAC6 inhibitors, with optimized
hydroxamic acid side chain lengths, reduce metastasis rates in
triple-negative breast cancer (TNBC) without causing notable
gastrointestinal toxicity (18,133).
Thiophene derivatives (as HDAC11 inhibitors) can reverse sorafenib
resistance in HCC (21,113,114). By contrast, the novel mechanism by
which kelch repeat and BTB domain-containing 4 mutations disrupt
the ubiquitin-dependent regulation of HDACs suggests that HDAC drug
development should consider both HDAC catalytic activity and
regulatory networks (134).
Furthermore, proteolysis-targeting chimeras (PROTACs) technology
enables specific degradation of HDAC subtypes. For example, HDAC7
PROTACs inhibit tumors with low toxicity in lymphoma and overcome
compensatory HDAC upregulation in solid tumors (112,134,135).
Targeted delivery focuses on nanocarriers. Liposomes
increase drug concentration in osteosarcoma by 8-fold via the
enhanced permeability and retention effect (136,137). Epidermal growth factor receptor
(EGFR)-targeted polymeric nanocarriers cross the blood-brain
barrier, boosting drug concentration in glioma brains by 12-fold
(74,138). Inorganic nanocarriers combined
with photothermal effects achieve a 55% ORR in breast cancer
(135,139). Liver-specific carriers enhance
intrahepatic drug concentration in HCC by 6-fold (140,141).
Combination strategies rely on mechanistic synergy.
In epigenetic combinations, HDACis plus DNMTis increase ORR from 30
to 60% in AML (36,142). In immune combinations, HDACis plus
anti-PD-1 antibodies raise ORR from 25 to 50% in melanoma (143,144). In targeted combinations, HDACis
plus PI3K inhibitors improve ORR from 20 to 45% in patients with
ovarian cancer who exhibit PI3K pathway activation (58,145).
Based on the aforementioned analysis, three
unresolved gaps remain in current HDAC research, including: i)
Mechanisms of subtype-specific functions in more solid tumors,
which have not been clarified; ii) lack of biomarkers in predicting
the efficacy of combination therapy; and iii) relatively low
clinical translation efficiency of novel inhibitors. Considering
this, the following section proposes future research directions
from four dimensions: i) Precise targeting; ii) multi-target
combination; iii) immune microenvironment regulation; and iv) drug
development.
The development of HDAC isoform-selective inhibitors
represents a novel research direction in cancer therapy. As core
molecules in epigenetic regulation, HDACs modulate key cellular
processes through deacetylation. Notably, different HDAC isoforms
exhibit distinct expression patterns and functions across tumors.
For instance, the upregulation of class I HDACs in TNBC is
associated with poor prognosis and therapeutic resistance, making
them a precise target for intervention. This therapeutic approach
not only enhances efficacy but also avoids the off-target effects
commonly observed with pan-HDACis (146).
The mechanisms underlying acquired resistance to
HDACis are complex, such as the compensatory upregulation of
alternative HDAC isoforms in AML (125). Researchers are accelerating the
development of novel drugs through various strategies, including
combining HDACis with chemotherapy/targeted agents, dynamically
adjusting treatment regimens via biomarker monitoring and
structure-based drug design or dual-target inhibitors.
Additionally, multi-target compounds provide a novel pathway in
preventing drug resistance (145,147).
To advance clinical translation, three key research
areas should be prioritized over the next 3–5 years: i) Developing
HDAC11-selective inhibitors by using structural biology to
characterize its active site, addressing sorafenib resistance in
HCC and reducing off-target toxicity; ii) establishing patient
stratification models based on HDAC isoform expression profiles;
for example, designing screening protocols for populations with
high class I HDAC expression in TNBC, constructing a
multi-dimensional classification system and developing convenient
detection kits; and iii) exploring interactions between HDAC
isoforms and non-coding RNAs, analyzing key mechanisms such as the
miR-145-5p/HDAC11 axis and identifying novel therapeutic targets
and candidate biomarkers. These efforts will improve the
therapeutic system from three different perspectives.
The oncology field has increasingly prioritized
combinatorial regimens involving HDACis, DNMTis and histone
methyltransferase inhibitors (HMTis). This multifaceted approach
involves synergistic targeting of interconnected epigenetic
pathways driving tumorigenesis. Preclinical evidence demonstrates
that co-administration of HDACis with DNMTis achieves coordinated
reversal of aberrant epigenetic silencing, reactivating tumor
suppressor genes frequently muted in malignancies (36,148).
When these epigenetic modifiers are combinatorially inhibited, the
tumor microenvironment undergoes notable alterations. This not only
enhances the antitumor immune response of the body (preclinical
studies have confirmed that HDACis combined with immunotherapy can
increase the immunogenicity of tumor cells) but also further
improves the therapeutic efficacy of immune checkpoint inhibitors
(89,149,150). Mechanistically, this multi-drug
combination strategy can simultaneously block multiple aberrantly
activated oncogenic signaling pathways, among which the PI3K/AKT
and NF-κB pathways are the most representative targets (151). Incorporating HMTis into
combination frameworks amplifies therapeutic impact through layered
epigenetic reprogramming. Dual modulation of histone acetylation
and methylation status enables comprehensive restoration of
silenced tumor suppressor networks key to apoptosis induction
(35,152,153). This multitarget strategy overcomes
limitations inherent to monotherapy by exploiting synergistic
interactions across parallel epigenetic regulatory axes (154). Ongoing clinical trials
systematically evaluate diverse combinations with conventional
chemotherapy and novel agents, aiming to maximize efficacy while
minimizing chemotoxicities associated with traditional cytotoxic
regimens (155–159). The integration of multimodal
epigenetic therapies represents a paradigm shift towards precision
medicine in cancer treatment.
HDACs are key regulators of the TIME. By
epigenetically modifying immune regulatory genes, HDACs modulate
the dynamics of immune cells and facilitate tumor immune evasion.
Elevated HDAC activity reduces the infiltration of effector cells
such as natural killer and CD8+ T cells, thereby
establishing an immunosuppressive microenvironment (12,38,160).
Dysregulated HDAC function further exacerbates immune evasion by
inhibiting tumor suppressor pathways and upregulating molecules
such as programmed cell death-ligand 1 (143,161). For instance, in pancreatic
adenocarcinoma, upregulated HDACs impede T cell-mediated antitumor
immune responses and recruit MDSCs to form an immune barrier
(126).
HDAC-targeted therapy has notable immunomodulatory
potential. Combining HDACis with immune checkpoint blockade therapy
can reverse immunosuppression (151,162). At the molecular level, HDAC
inhibition increases the expression levels of major
histocompatibility complex class I molecules on tumor cells and
enhances the activity of key components in the antigen-processing
system, thus improving T-cell recognition efficiency (2,44). It
also reprograms tumor-associated macrophages from the
immunosuppressive M2 phenotype to the pro-inflammatory M1
phenotype, thus weakening the ability of the tumor to evade
immunity (149,163).
Additionally, HDACs regulate the function of
regulatory T cells (Tregs) by interacting with forkhead box protein
P3 (FOXP3) regulators. For example, HDAC3 forms a complex with
FOXP3 negative regulators to inhibit FOXP3 activity, while HDACis
disrupt this complex to relieve the inhibition, reducing
Treg-mediated immunosuppression and enhancing the cytotoxicity of
effector T cells. The identification of the HDAC-FOXP3-Treg axis
provides a theoretical basis for the combination of HDAC-targeted
therapy with immunotherapy (164).
Currently, the research and development of novel
HDACis focuses on enhancing subtype selectivity, reducing toxicity
and overcoming drug resistance, driven by interdisciplinary
technologies. By resolving the active pocket structures of HDAC
subtypes using X-ray crystallography and cryo-electron microscopy,
combined with high-throughput screening, candidate molecules can be
rapidly identified. For instance, tetrapheno(α)3-methyl derivatives
exhibit nanomolar-level inhibitory activity against HDAC1/6 and
demonstrate antiproliferative effects in various cancer cell lines
(165,166). Using molecular docking and
molecular dynamics simulations to optimize compound structures,
dual-target drugs such as 4-arylaminoquinoline derivatives have
also been developed, which can simultaneously target the HDAC and
EGFR pathways (124,167,168). Additionally, PROTACs technology
has emerged as a novel direction; for example, HDAC7 PROTAC
degraders can specifically degrade HDAC7 in DLBCL, with antitumor
activity >10-fold higher compared with that of traditional
inhibitors and low toxicity to normal cells (169).
Preclinical evaluation of novel HDACis requires
multi-dimensional validation across the molecular-cell-animal
spectrum. At the molecular level, the selectivity for target
subtypes is verified; at the cellular level, the effects on
drug-resistant strains and other cell types are assessed; and at
the animal level, in situ xenograft models are used to
monitor efficacy and drug distribution. Meanwhile, toxicity to
systems such as the hematological, digestive and nervous systems is
closely monitored as well. Pharmacokinetic-pharmacodynamic models
are employed to optimize dosing regimens or targeted delivery
technologies are used to increase drug concentrations in tumor
tissues and reduce systemic toxicity. For clinical trials, cancer
types and patient groups are selected based on the properties of
the drug. For example, HDAC11 inhibitors are first investigated in
HCC (113–116), while dual-target drugs are
administered to patients with pathway abnormalities (58,124,167,168). In combination therapy trials,
relevant molecular markers need to be monitored (37,56,57,100).
Although challenges exist, such as the lack of structural data for
certain HDAC subtypes and the difficulty of preclinical models in
simulating tumor heterogeneity, these can be addressed through
homology modeling and patient-derived xenograft models. In the
future, integrating biomarker stratification should facilitate the
advancement of more HDACis with high efficacy and low toxicity into
clinical practice. The key results of recent clinical trials of
therapeutic regimens based on histone deacetylase inhibitors are
summarized in Table I.
Mechanistic insights, therapeutic innovations and
unresolved questions in HDAC-targeted cancer therapy.
The present review systematically analyzed the role
of HDACs in cancer and their therapeutic prospects, demonstrating
notable complementarity to and expansion of previous studies
(170,171). In terms of research scope, it
collates the mechanisms of action of HDAC isoforms across five
cancer types, including ovarian cancer, while specifically
dissecting the functional differences and regulatory networks of
class I isoforms such as HDAC1 and HDAC3. Building on previous
studies focused on lung cancer (172), this review further extends the
analysis of common mechanisms across cancer types. For instance,
HDAC6 can enhance drug resistance in various solid tumors-including
lung, breast, hepatocellular, ovarian, pancreatic cancers and
CRC-via deacetylation of α-tubulin (173,174), thereby providing a solid
theoretical basis for the development of pan-cancer HDAC-targeted
therapeutic strategies (37).
Beyond the expansion of the research scope, the
present review also highlighted notable innovations in combination
therapy strategies: It proposes a novel HDACi + dual-target drug
regimen (for example, combining HDACis with EGFR/PI3K dual-target
inhibitors). This strategy achieves synergistic antitumor effects
by simultaneously blocking two critical oncogenic pathways: The
EGFR signaling pathway and the PI3K/Akt signaling pathway,
addressing a key limitation of previous studies that only focused
on the combination of HDAC7 PROTAC degraders with immunotherapy
(112,175). Notably, a preclinical study on AML
reported a synergistic mechanism between HDACs and DNMTs mediated
by an acetylation-demethylation crosstalk, further verifying the
scientific validity of the multi-targeted epigenetic regulator
targeting strategy proposed in the present review (36).
Although this review comprehensively summarizes the
research and therapeutic advances related to HDACs, it has certain
limitations. First, the tumor coverage is limited, as the
functional mechanisms of HDAC isoforms in high-incidence solid
tumors such as lung and liver cancer and more hematological
malignancies have not been systematically investigated, making it
difficult to reflect their pan-cancer regulatory characteristics.
Second, research on isoforms such as HDAC8 and HDAC10 is
superficial, and the regulatory networks among isoforms remain
unclear, precluding a complete understanding of their overall
regulatory patterns. Third, clinical data are mostly from
traditional regimens, with insufficient data on emerging therapies
and Phase III trial evidence for solid tumors. Fourth, tumor
microenvironment analysis only focuses on core immune cells,
ignoring other components and their cross-regulation with
microenvironmental characteristics. Fifth, the clinical validation
limitations of potential biomarkers have not been thoroughly
analyzed, nor has the establishment of combined detection systems
been explored, which fails to meet the needs of precise
treatment.
Due to the complexity of HDAC-related regulatory
networks and the multi-faceted challenges in clinical translation,
the present review concluded by clarifying the core mechanisms and
therapeutic potential of HDACs, while putting forward innovative
therapeutic strategies. It also suggested that interdisciplinary
collaboration, integrating insights from molecular biology,
pharmacology and clinical oncology, is expected to overcome key
hurdles in HDAC-targeted therapy, including off-target toxicity,
acquired drug resistance and the lack of reliable biomarkers.
Therefore, such efforts will accelerate the translation of
HDAC-targeted research from basic science to clinical practice and
potentially improve the prognosis of patients with cancer in the
future.
HDACs, as core factors in epigenetic regulation,
serve a key role in the oncogenic mechanisms, disease progression
and therapeutic resistance of various malignant tumors. Their
ability to reshape chromatin structure and regulate gene expression
programs makes them highly promising therapeutic targets, a finding
that has been validated by the clinical application of HDACis in
hematological malignancies; however, translating these advantages
into solid tumor therapy remains challenging due to tumor
heterogeneity, complex microenvironmental interactions and
differences in cellular responses, a discrepancy that underscores
the urgency of dissecting subtype-specific functions in different
cancer contexts. A key to future progress lies in the development
of subtype-selective HDACis that can precisely modulate oncogenic
pathways while minimizing off-target effects, as well as rational
combination therapy regimens (such as pairing HDACis with
immunotherapies, targeted agents or chemotherapy) that hold notable
potential through synergistic mechanisms (for example, combination
therapy with immune checkpoint blockade can activate antitumor
immune responses). Elucidating resistance mechanisms, including
compensatory signal activation, altered drug metabolism and
microenvironmental adaptation, is equally key to overcoming
treatment failure and the identification of biomarkers that predict
treatment response or resistance is expected to enable personalized
dosing strategies and stratified patient care. Emerging research
paradigms advocate for multidisciplinary combination therapies,
integrating HDAC-targeted approaches with innovative modalities
such as PROTAC degradation systems and dual epigenetic inhibitors.
By integrating cutting-edge insights from tumor biology, precision
medicine and clinical pharmacology, well-validated HDAC-targeted
strategies (supported by rigorous mechanistic studies and clinical
trials) have the potential to transform cancer treatment outcomes,
with the convergence of these elements expected to shift HDACis
from promising experimental tools to core components of modern
cancer treatment regimens.
Not applicable.
The present study was supported by the Natural Science
Foundation of Shandong (grant no. ZR2023QH453) and the National
Natural Science Foundation of China (grant no. 82403344).
Not applicable.
RL designed the review article, researched
references and wrote the majority of the manuscript. YZ researched
references and wrote the manuscript. HL revised and edited the
manuscript. FL revised the manuscript and acquired funding. Data
authentication is not applicable. All authors read and approved the
final version of the manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Liu YL, Yang PM, Shun CT, Wu MS, Weng JR
and Chen CC: Autophagy potentiates the anti-cancer effects of the
histone deacetylase inhibitors in hepatocellular carcinoma.
Autophagy. 6:1057–1065. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Maciejewski K, Giers M, Oleksiewicz U and
Czerwinska P: The epigenetic modifiers HDAC2 and HDAC7 inversely
associate with cancer stemness and immunity in solid tumors. Int J
Mol Sci. 25:78412024. View Article : Google Scholar
|
|
3
|
Bao Q, Li Y, Chen Y, Zheng J, Zhao J and
Hu T: Transcriptome-based network analysis related to histone
deacetylase genes and identified EMP1 as a potential biomarker for
prognosis in bladder cancer. Clin Genitourin Cancer. 23:1022622025.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liang T, Wang F, Elhassan RM, Cheng Y,
Tang X, Chen W, Fang H and Hou X: Targeting histone deacetylases
for cancer therapy: Trends and challenges. Acta Pharm Sin B.
13:2425–2463. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Klieser E, Neumayer B, Di Fazio P, Mayr C,
Neureiter D and Kiesslich T: HDACs as an emerging target in
endocrine tumors: A comprehensive review. Expert Rev Endocrinol
Metab. 18:143–154. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Contreras-Sanzón E, Prado-Garcia H,
Romero-Garcia S, Nuñez-Corona D, Ortiz-Quintero B, Luna-Rivero C,
Martínez-Cruz V and Carlos-Reyes Á: Histone deacetylases modulate
resistance to the therapy in lung cancer. Front Genet.
13:9602632022. View Article : Google Scholar
|
|
7
|
Liang R, Zhang J, Liu Z, Liu Z, Li Q, Luo
X, Li Y, Ye J and Lin Y: Mechanism and molecular network of
RBM8A-mediated regulation of oxaliplatin resistance in
hepatocellular carcinoma. Front Oncol. 10:5854522021. View Article : Google Scholar
|
|
8
|
Giordano F, Forestiero M, Leonetti AE,
Naimo GD, Marrone A, De Amicis F, Marsico S, Mauro L and Panno ML:
Valproic acid reduces invasiveness and cellular growth in 2D and 3D
glioblastoma cell lines. Int J Mol Sci. 26:66002025. View Article : Google Scholar
|
|
9
|
Zheng B, Jiang X, Liu Y, Cheng F, Zhang Y,
Niu C, Cong Z, Niu Z and He W: Elevated histone deacetylase 10
expression promotes the progression of clear cell renal cell
carcinoma by notch-1-PTEN signaling axis. Discov Oncol. 15:1562024.
View Article : Google Scholar
|
|
10
|
Jungwirth G, Yu T, Liu F, Cao J, Alaa
Eddine M, Moustafa M, Abdollahi A, Warta R, Unterberg A and
Herold-Mende C: Pharmacological landscape of FDA-approved
anticancer drugs reveals sensitivities to ixabepilone, romidepsin,
omacetaxine, and carfilzomib in aggressive meningiomas. Clin Cancer
Res. 29:233–243. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee H, Jung TY, Lim SH, Choi EJ, Lee J and
Min DS: Phospholipase D2 is a positive regulator of sirtuin 1 and
modulates p53-mediated apoptosis via sirtuin 1. Exp Mol Med.
53:1287–1297. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lin Y, Jing X, Chen Z, Pan X, Xu D, Yu X,
Zhong F, Zhao L, Yang C, Wang B, et al: Histone
deacetylase-mediated tumor microenvironment characteristics and
synergistic immunotherapy in gastric cancer. Theranostics.
13:4574–4600. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Haberland M, Montgomery RL and Olson EN:
The many roles of histone deacetylases in development and
physiology: Implications for disease and therapy. Nat Rev Genet.
10:32–42. 2009. View Article : Google Scholar
|
|
14
|
Kundu R, Banerjee S, Baidya SK, Adhikari N
and Jha T: A quantitative structural analysis of AR-42 derivatives
as HDAC1 inhibitors for the identification of promising structural
contributors. SAR QSAR Environ Res. 33:861–883. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Guo K, Ma Z, Zhang Y, Han L, Shao C, Feng
Y, Gao F, Di S, Zhang Z, Zhang J, et al: HDAC7 promotes NSCLC
proliferation and metastasis via stabilization by deubiquitinase
USP10 and activation of β-catenin-FGF18 pathway. J Exp Clin Cancer
Res. 41:912022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu L, Cao H, Yang JW, Meng WX, Yang C,
Wang JT, Yu MM and Wang BS: HDAC5-mediated PRAME regulates the
proliferation, migration, invasion, and EMT of laryngeal squamous
cell carcinoma via the PI3K/AKT/mTOR signaling pathway. Open Med
(Wars). 18:202306652023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang Y, Ding P, Wang Y, Shao C, Guo K,
Yang H, Feng Y, Ning J, Pan M, Wang P, et al: HDAC7/c-Myc signaling
pathway promotes the proliferation and metastasis of choroidal
melanoma cells. Cell Death Dis. 14:382023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lu B, Qiu R, Wei J, Wang L, Zhang Q, Li M,
Zhan X, Chen J, Hsieh IY, Yang C, et al: Phase separation of
phospho-HDAC6 drives aberrant chromatin architecture in
triple-negative breast cancer. Nat Cancer. 5:1622–1640. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jin J, Meng T, Yu Y, Wu S, Jiao CC, Song
S, Li YX, Zhang Y, Zhao YY, Li X, et al: Human HDAC6 senses valine
abundancy to regulate DNA damage. Nature. 637:215–223. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pandey UB, Batlevi Y, Baehrecke EH and
Taylor JP: HDAC6 at the intersection of autophagy, the
ubiquitin-proteasome system and neurodegeneration. Autophagy.
3:643–645. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bhagat RP, Jyotisha Dasgupta I, Amin SA,
Jakkula P, Bhattacharya A, Qureshi IA and Gayen S: First report on
analysis of chemical space, scaffold diversity, critical structural
features of HDAC11 inhibitors. Mol Divers. 29:3679–3702. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sriramareddy SN, Faião-Flores F, Emmons
MF, Saha B, Chellappan S, Wyatt C, Smalley I, Licht JD, Durante MA,
Harbour JW and Smalley KSM: HDAC11 activity contributes to MEK
inhibitor escape in uveal melanoma. Cancer Gene Ther. 29:1840–1846.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Buglio D, Khaskhely NM, Voo KS,
Martinez-Valdez H, Liu YJ and Younes A: HDAC11 plays an essential
role in regulating OX40 ligand expression in Hodgkin lymphoma.
Blood. 117:2910–2917. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Das T, Bhattacharya A, Jha T and Gayen S:
Exploration of fingerprints and data mining-based prediction of
some bioactive compounds from Allium sativum as histone deacetylase
9 (HDAC9) inhibitors. Curr Comput Aided Drug Des. 21:270–284. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
West AC and Johnstone RW: New and emerging
HDAC inhibitors for cancer treatment. J Clin Invest. 124:30–39.
2014. View Article : Google Scholar
|
|
26
|
Debbarma M, Sarkar K and Sil SK:
Dissecting the epigenetic orchestra of HDAC isoforms in breast
cancer development: A review. Med Oncol. 42:12024. View Article : Google Scholar
|
|
27
|
Sanaei M and Kavoosi F: Histone
deacetylase inhibitors, intrinsic and extrinsic apoptotic pathways,
and epigenetic alterations of histone deacetylases (HDACs) in
hepatocellular carcinoma. Iran J Pharm Res. 20:324–336.
2021.PubMed/NCBI
|
|
28
|
Han S, Fan H, Zhong G, Ni L, Shi W, Fang
Y, Wang C, Wang L, Song L, Zhao J, et al: Nuclear KRT19 is a
transcriptional corepressor promoting histone deacetylation and
liver tumorigenesis. Hepatology. 81:808–822. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jasim SA, Altalbawy FMA, Abohassan M,
Oghenemaro EF, Bishoyi AK, Singh RP, Kaur P, Sivaprasad GV,
Mohammed JS and Hulail HM: Histone deacetylases (HDACs) roles in
inflammation-mediated diseases; current knowledge. Cell Biochem
Biophys. 83:1375–1386. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang K, Peng X, Zhou P, Li X, Shen L,
Yang L and Zhou Q: HDAC1/2-mediated deacetylation of KLF9 promotes
the malignant progression of nasopharyngeal carcinoma via CDH17.
Oncogene. 44:3183–3198. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sahu RK, Dhakshnamoorthy J, Jain S, Folco
HD, Wheeler D and Grewal SIS: Nucleosome remodeler exclusion by
histone deacetylation enforces heterochromatic silencing and
epigenetic inheritance. Mol Cell. 84:3175–3191.e8. 2024. View Article : Google Scholar
|
|
32
|
Talom A, Barhoi A, Jirpu T, Dawn B and
Ghosh A: Clinical progress and functional modalities of HDAC
inhibitor-based combination therapies in cancer treatment. Clin
Transl Oncol. 28:71–85. 2026. View Article : Google Scholar
|
|
33
|
Sandonà M, Cavioli G, Renzini A, Cedola A,
Gigli G, Coletti D, McKinsey TA, Moresi V and Saccone V: Histone
deacetylases: Molecular mechanisms and therapeutic implications for
muscular dystrophies. Int J Mol Sci. 24:43062023. View Article : Google Scholar
|
|
34
|
Lu-Culligan WJ, Connor LJ, Xie Y, Ekundayo
BE, Rose BT, Machyna M, Pintado-Urbanc AP, Zimmer JT, Vock IW,
Bhanu NV, et al: Acetyl-methyllysine marks chromatin at active
transcription start sites. Nature. 622:173–179. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Joseph R, Dasari SK, Umamaheswaran S,
Mangala LS, Bayraktar E, Rodriguez-Aguayo C, Wu Y, Nguyen N, Powell
RT, Sobieski M, et al: EphA2- and HDAC-targeted combination therapy
in endometrial cancer. Int J Mol Sci. 25:12782024. View Article : Google Scholar
|
|
36
|
Blagitko-Dorfs N, Schlosser P, Greve G,
Pfeifer D, Meier R, Baude A, Brocks D, Plass C and Lübbert M:
Combination treatment of acute myeloid leukemia cells with DNMT and
HDAC inhibitors: Predominant synergistic gene downregulation
associated with gene body demethylation. Leukemia. 33:945–956.
2019. View Article : Google Scholar
|
|
37
|
McGuire CK, Meehan AS, Couser E, Bull L,
Minor AC, Kuhlmann-Hogan A, Kaech SM, Shaw RJ and Eichner LJ:
Transcriptional repression by HDAC3 mediates T cell exclusion from
Kras mutant lung tumors. Proc Natl Acad Sci USA.
121:e23176941212024. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yan M, Cao H, Tao K, Xiao B, Chu Y, Ma D,
Huang X, Han Y and Ji T: HDACs alters negatively to the tumor
immune microenvironment in gynecologic cancers. Gene.
885:1477042023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim YK, Lee EK, Kang JK, Kim JA, You JS,
Park JH, Seo DW, Hwang JW, Kim SN, Lee HY, et al: Activation of
NF-kappaB by HDAC inhibitor apicidin through Sp1-dependent de novo
protein synthesis: Its implication for resistance to apoptosis.
Cell Death Differ. 13:2033–2041. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Luo J, Su F, Chen D, Shiloh A and Gu W:
Deacetylation of p53 modulates its effect on cell growth and
apoptosis. Nature. 408:377–381. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Singh T, Kaur P, Singh P, Singh S and
Munshi A: Differential molecular mechanistic behavior of HDACs in
cancer progression. Med Oncol. 39:1712022. View Article : Google Scholar
|
|
42
|
Sharma S, Tyagi W, Tamang R and Das S:
HDAC5 modulates SATB1 transcriptional activity to promote lung
adenocarcinoma. Br J Cancer. 129:586–600. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ling R, Wang J, Fang Y, Yu Y, Su Y, Sun W,
Li X and Tang X: HDAC-an important target for improving tumor
radiotherapy resistance. Front Oncol. 13:11936372023. View Article : Google Scholar
|
|
44
|
Mao C, Fan W, Liu J, Yang F, Li W, Li L,
Shi Z, Li Q, Yuan Z, Jiang Y and Chu B: Targeting HDAC and PARP
enhances STING-dependent antitumor immunity in STING-deficient
tumor. Adv Sci (Weinh). 12:e079042025. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Rodrigues Moita AJ, Bandolik JJ, Hansen
FK, Kurz T, Hamacher A and Kassack MU: Priming with HDAC inhibitors
sensitizes ovarian cancer cells to treatment with cisplatin and
HSP90 inhibitors. Int J Mol Sci. 21:83002020. View Article : Google Scholar
|
|
46
|
Li Z, Wu YH, Guo YQ, Min XJ and Lin Y:
Tasquinimod promotes the sensitivity of ovarian cancer cells to
cisplatin by down-regulating the HDAC4/p21 pathway. Korean J
Physiol Pharmacol. 29:191–204. 2025. View Article : Google Scholar
|
|
47
|
Kim M, Lu F and Zhang Y: Loss of
HDAC-mediated repression and gain of NF-κB activation underlie
cytokine induction in ARID1A- and PIK3CA-mutation-driven ovarian
cancer. Cell Rep. 17:275–288. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Feng Q, Hao S, Liu X, Yan Z, Sheng K, Li
Y, Zhang P and Sheng X: HDAC7 promotes ovarian cancer malignancy
via AKT/mTOR signalling pathway. J Cell Mol Med. 28:e701202024.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Xu L, Wang J, Liu B, Fu J, Zhao Y, Yu S,
Shen L, Yan X and Su J: HDAC9 contributes to serous ovarian cancer
progression through regulating epithelial-mesenchymal transition.
Biomedicines. 10:3742022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xu L, Yan X, Wang J, Zhao Y, Liu Q, Fu J,
Shi X and Su J: The roles of histone deacetylases in the regulation
of ovarian cancer metastasis. Int J Mol Sci. 24:150662023.
View Article : Google Scholar
|
|
51
|
Chi AJ, Hsu JL, Xiao YX, Chern JW, Guh JH,
Yu CW and Hsu LC: A novel HDAC6 inhibitor enhances the efficacy of
paclitaxel against ovarian cancer cells. Molecules. 30:27932025.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Thomine C, Guillemot S, Weiswald LB,
Florent R, Abeilard E, Giffard F, Brotin E, Briand M, Dolivet E,
Poulain L and Villedieu M: The anticancer effect of the HDAC
inhibitor belinostat is enhanced by inhibitors of Bcl-xL
or Mcl-1 in ovarian cancer. Mol Oncol. 19:3325–3341. 2025.
View Article : Google Scholar
|
|
53
|
Begum S, Irvin SD, Cox CK, Huang Z, Wilson
JJ, Monroe JD and Gibert Y: Anti-ovarian cancer migration and
toxicity characteristics of a platinum(IV) pro-drug with axial HDAC
inhibitor ligands in zebrafish models. Invest New Drugs.
42:644–654. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Natarajan U, Venkatesan T and Rathinavelu
A: Effect of the HDAC inhibitor on histone acetylation and
methyltransferases in A2780 ovarian cancer cells. Medicina
(Kaunas). 57:4562021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Si L, Lai T, Zhao J, Jin Y, Qi M, Li M, Fu
H, Shi X, Ma L and Guo R: Identification of a novel pyridine
derivative with inhibitory activity against ovarian cancer
progression in vivo and in vitro. Front Pharmacol. 13:10644852022.
View Article : Google Scholar
|
|
56
|
Valdez BC, Tsimberidou AM, Yuan B, Baysal
MA, Chakraborty A, Andersen CR and Andersson BS: Synergistic
cytotoxicity of histone deacetylase and poly-ADP ribose polymerase
inhibitors and decitabine in breast and ovarian cancer cells:
Implications for novel therapeutic combinations. Int J Mol Sci.
25:92412024. View Article : Google Scholar
|
|
57
|
Qiu J, Ren T, Liu Q, Jiang Q, Wu T, Cheng
LC, Yan W, Qu X, Han X and Hua K: Dissecting the distinct tumor
microenvironments of HRD and HRP ovarian cancer: Implications for
targeted therapies to overcome PARPi resistance in HRD tumors and
refractoriness in HRP tumors. Adv Sci (Weinh). 11:e23097552024.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wang Y, Wen J, Sun X, Sun Y, Liu Y, Cheng
X, Wu W, Liu Q and Ren F: CUDC-907 exhibits potent antitumor
effects against ovarian cancer through multiple in vivo and in
vitro mechanisms. Cancer Chemother Pharmacol. 93:295–306. 2024.
View Article : Google Scholar
|
|
59
|
Zheng Y and Yang X, Wang C, Zhang S, Wang
Z, Li M, Wang Y, Wang X and Yang X: HDAC6, modulated by miR-206,
promotes endometrial cancer progression through the PTEN/AKT/mTOR
pathway. Sci Rep. 10:35762020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Li Y, Zhou W, Li L, Li JW, Li T, Huang C,
Lazaro-Camp VJ, Kavlashvili T, Zhang Y, Reyes H, et al: Enhancing
progestin therapy via HDAC inhibitors in endometrial cancer. Am J
Cancer Res. 12:5029–5048. 2022.PubMed/NCBI
|
|
61
|
Nahar S, Yu J, Lee H, Tran DN, Li R, Kim
TH, Jung JS, Kim K, Yoo JY and Jeong JW: MIG-6 regulates
HDAC1-mediated angiogenesis and tumorigenesis in PTEN-deficient
endometrioid endometrial cancer. Mol Cancer Res. Jan 21–2026.(Epub
ahead of print). View Article : Google Scholar
|
|
62
|
Psilopatis I, Pergaris A, Giaginis C and
Theocharis S: Histone deacetylase inhibitors: A promising
therapeutic alternative for endometrial carcinoma. Dis Markers.
2021:78506882021. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Edwards K, Yao S, Pisano S, Feltracco V,
Brusehafer K, Samanta S, Oommen OP, Gazze SA, Paravati R, Maddison
H, et al: Hyaluronic acid-functionalized nanomicelles enhance SAHA
efficacy in 3D endometrial cancer models. Cancers (Basel).
13:40322021. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Pothuri B, Sawaged Z, Karpel HC, Li X, Lee
J, Musa F, Lutz K, Reese E, Blank SV, Boyd LR, et al: A phase 2
feasibility study of nab-paclitaxel and carboplatin in epithelial
carcinoma of the uterus. Gynecol Oncol. 190:209–214. 2024.
View Article : Google Scholar
|
|
65
|
Lin CK, Liu ST, Wu ZS, Wang YC and Huang
SM: Mechanisms of cisplatin in combination with repurposed drugs
against human endometrial carcinoma cells. Life (Basel).
11:1602021.PubMed/NCBI
|
|
66
|
Wu Q, Zhang W, Liu Y, Huang Y, Wu H and Ma
C: Histone deacetylase 1 facilitates aerobic glycolysis and growth
of endometrial cancer. Oncol Lett. 22:7217212021. View Article : Google Scholar
|
|
67
|
Ma M, Fei X, Jiang D, Chen H, Xie X, Wang
Z and Huang Q: Research progress on the mechanism of histone
deacetylases in ferroptosis of glioma. Oncol Rev. 18:14321312024.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Chen X, Wang Z, Li C, Zhang Z, Lu S, Wang
X, Liang Q, Zhu X, Pan C, Wang Q, et al: SIRT1 activated by AROS
sensitizes glioma cells to ferroptosis via induction of NAD+
depletion-dependent activation of ATF3. Redox Biol. 69:1030302024.
View Article : Google Scholar
|
|
69
|
Pai P, Das I, Reddy Y, Venkidesh BS,
Bhandari P, Madalageri M, Sadashivanavar V, Pai KSR, Rao P,
Oruganti S, et al: Targeting glioblastoma with HDAC inhibitors:
Insights into hydroxamic acid-based therapeutic strategies. Acta
Neuropathol Commun. 14:92025. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Fan F, Liu P, Bao R, Chen J, Zhou M, Mo Z,
Ma Y, Liu H, Zhou Y, Cai X, et al: A dual PI3K/HDAC inhibitor
induces immunogenic ferroptosis to potentiate cancer immune
checkpoint therapy. Cancer Res. 81:6233–6245. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Mormino A, Cocozza G, Fontemaggi G,
Valente S, Esposito V, Santoro A, Bernardini G, Santoni A, Fazi F,
Mai A, et al: Histone-deacetylase 8 drives the immune response and
the growth of glioma. Glia. 69:2682–2698. 2021. View Article : Google Scholar
|
|
72
|
Han W, Yu F, Cao J, Dong B, Guan W and Shi
J: Valproic acid enhanced apoptosis by promoting autophagy via
Akt/mTOR signaling in glioma. Cell Transplant.
29:9636897209818782020. View Article : Google Scholar
|
|
73
|
Thotala D, Karvas RM, Engelbach JA, Garbow
JR, Hallahan AN, DeWees TA, Laszlo A and Hallahan DE: Valproic acid
enhances the efficacy of radiation therapy by protecting normal
hippocampal neurons and sensitizing malignant glioblastoma cells.
Oncotarget. 6:35004–35022. 2015. View Article : Google Scholar
|
|
74
|
Mehndiratta S, Qian B, Chuang JY, Liou JP
and Shih JC: N-methylpropargylamine-conjugated hydroxamic acids as
dual inhibitors of monoamine oxidase A and histone deacetylase for
glioma treatment. J Med Chem. 65:2208–2224. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Kunadis E, Lakiotaki E, Korkolopoulou P
and Piperi C: Targeting post-translational histone modifying
enzymes in glioblastoma. Pharmacol Ther. 220:1077212021. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Riyas Mohamed FR and Yaqinuddin A:
Epigenetic reprogramming and antitumor immune responses in gliomas:
A systematic review. Med Oncol. 42:2132025. View Article : Google Scholar
|
|
77
|
Ozair A, Bhat V, Alisch RS, Khosla AA,
Kotecha RR, Odia Y, McDermott MW and Ahluwalia MS: DNA methylation
and histone modification in low-grade gliomas: Current
understanding and potential clinical targets. Cancers (Basel).
15:13422023. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Khalid U, Simovic M, Hammann LA, Iskar M,
Wong JKL, Kumar R, Jugold M, Sill M, Bolkestein M, Kolb T, et al: A
synergistic interaction between HDAC- and PARP inhibitors in
childhood tumors with chromothripsis. Int J Cancer. 151:590–606.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
He Q, Yu C, Li Y, Hao P, Mai H, Guo R,
Zhong G, Zhang K, Wong C, Chen Q and Chen Y: ERRα contributes to
HDAC6-induced chemoresistance of osteosarcoma cells. Cell Biol
Toxicol. 39:813–825. 2023. View Article : Google Scholar
|
|
80
|
Wei LH, Torng PL, Hsiao SM, Jeng YM, Chen
MW and Chen CA: Histone deacetylase 6 regulates estrogen receptor
alpha in uterine leiomyoma. Reprod Sci. 18:755–762. 2011.
View Article : Google Scholar
|
|
81
|
Stopper D, Biermann L, Watson PR, Li J,
König B, Gaynes MN, Pessanha de Carvalho L, Klose J, Hanl M,
Hamacher A, et al: Exploring alternative zinc-binding groups in
histone deacetylase (HDAC) inhibitors uncovers DS-103 as a potent
ethylhydrazide-based HDAC inhibitor with chemosensitizing
properties. J Med Chem. 68:4426–4452. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Long J, Jia MY, Fang WY, Chen XJ, Mu LL,
Wang ZY, Shen Y, Xiang RF, Wang LN, Wang L, et al: FLT3 inhibition
upregulates HDAC8 via FOXO to inactivate p53 and promote
maintenance of FLT3-ITD+ acute myeloid leukemia. Blood.
135:1472–1483. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
McGuire JJ, Nerlakanti N, Lo CH, Tauro M,
Utset-Ward TJ, Reed DR and Lynch CC: Histone deacetylase inhibition
prevents the growth of primary and metastatic osteosarcoma. Int J
Cancer. 147:2811–2823. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Sawai T, Yamanegi K, Nishiura H, Futani H
and Tachibana T: Sodium valproate enhances semaphorin 3A-mediated
anti-angiogenesis and tumor growth inhibition in human osteosarcoma
cells. Anticancer Res. 43:2539–2550. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Rossi M, De Martino V, Di Giuseppe L,
Battafarano G, Di Gregorio J, Terreri S, Marampon F, Minisola S and
Del Fattore A: Anti-proliferative, pro-apototic and anti-migratory
properties of HDAC inhibitor PXD-101 on osteosarcoma cell lines.
Arch Biochem Biophys. 734:1094892023. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Magar AG, Morya VK, Koh YH and Noh KC:
Synergistic HDAC4/8 inhibition sensitizes osteosarcoma to
doxorubicin via pAKT/RUNX2 pathway modulation. Int J Mol Sci.
26:35742025. View Article : Google Scholar
|
|
87
|
Collier CD, Getty PJ and Greenfield EM:
Targeting the cancer epigenome with histone deacetylase inhibitors
in osteosarcoma. Adv Exp Med Biol. 1258:55–75. 2020. View Article : Google Scholar
|
|
88
|
Ding K, Liu H, Yang H, Zhu H, Ma J, Peng
H, Huang H, Shi W, Cao L, Wu W, et al: A prospective phase 2 study
of combination epigenetic therapy against relapsed/refractory
peripheral T cell lymphoma. Med. 5:1393–1401.e2. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Pu J, Liu T, Wang X, Sharma A,
Schmidt-Wolf IGH, Jiang L and Hou J: Exploring the role of histone
deacetylase and histone deacetylase inhibitors in the context of
multiple myeloma: Mechanisms, therapeutic implications, and future
perspectives. Exp Hematol Oncol. 13:452024. View Article : Google Scholar
|
|
90
|
Gössl FJ, Polo P, Helmprobst F, Menzenbach
A, Visekruna A, Gress TM, Adhikary T and Lauth M: ER-phagy mediates
the anti-tumoral synergism between HDAC inhibition and
chemotherapy. Cell Commun Signal. 23:2022025. View Article : Google Scholar
|
|
91
|
Xu T, Fang Y, Gu Y, Xu D, Hu T, Yu T, Xu
YY, Shen HY, Ma P and Shu Y: HDAC inhibitor SAHA enhances antitumor
immunity via the HDAC1/JAK1/FGL1 axis in lung adenocarcinoma. J
Immunother Cancer. 12:e0100772024. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Ferro A, Graikioti D, Gezer E,
Athanassopoulos CM and Cuendet M: Entinostat-bortezomib hybrids
against multiple myeloma. Molecules. 28:14562023. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Hirano M, Imai Y, Kaito Y, Murayama T,
Sato K, Ishida T, Yamamoto J, Ito T, Futami M, Ri M, et al:
Small-molecule HDAC and Akt inhibitors suppress tumor growth and
enhance immunotherapy in multiple myeloma. J Exp Clin Cancer Res.
40:1102021. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Florean C, Lernoux M, Lorant A, Losson H,
Bormans G, Schnekenburger M and Diederich M: HDAC6 inhibitors
sensitize resistant t(11;14) multiple myeloma cells to a
combination of bortezomib and BH3 mimetics. Haematologica.
110:784–790. 2025.
|
|
95
|
García-Guerrero E, Götz R, Doose S, Sauer
M, Rodríguez-Gil A, Nerreter T, Kortüm KM, Pérez-Simón JA, Einsele
H, Hudecek M and Danhof S: Upregulation of CD38 expression on
multiple myeloma cells by novel HDAC6 inhibitors is a class effect
and augments the efficacy of daratumumab. Leukemia. 35:201–214.
2021. View Article : Google Scholar
|
|
96
|
Zhan W, Liao X, Liu J, Tian T, Yu L and Li
R: USP38 regulates the stemness and chemoresistance of human
colorectal cancer via regulation of HDAC3. Oncogenesis. 9:482020.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Szigety KM, Liu F, Yuan CY, Moran DJ,
Horrell J, Gochnauer HR, Cohen RN, Katz JP, Kaestner KH, Seykora
JT, et al: HDAC3 ensures stepwise epidermal stratification via
NCoR/SMRT-reliant mechanisms independent of its histone deacetylase
activity. Genes Dev. 34:973–988. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Dai B, Wang F, Wang Y, Zhu J, Li Y, Zhang
T, Zhao L, Wang L, Gao W, Li J, et al: Targeting HDAC3 to overcome
the resistance to ATRA or arsenic in acute promyelocytic leukemia
through ubiquitination and degradation of PML-RARα. Cell Death
Differ. 30:1320–1333. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Li L, Hao S, Gao M, Liu J, Xu X, Huang J,
Cheng G and Yang H: HDAC3 inhibition promotes antitumor immunity by
enhancing CXCL10-mediated chemotaxis and recruiting of immune
cells. Cancer Immunol Res. 11:657–673. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Han R, Luo Y, Gao J, Zhou H, Wang Y, Chen
J, Zheng G and Ling C: HDAC3: A multifaceted modulator in
immunotherapy sensitization. Vaccines (Basel). 13:1822025.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Eichner LJ, Curtis SD, Brun SN, McGuire
CK, Gushterova I, Baumgart JT, Trefts E, Ross DS, Rymoff TJ and
Shaw RJ: HDAC3 is critical in tumor development and therapeutic
resistance in Kras-mutant non-small cell lung cancer. Sci Adv.
9:eadd32432023. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Wu X, Yang C, Sun F, Zhang Y, Wang Y, Li X
and Zheng F: Enterotoxigenic bacteroides fragilis (ETBF) enhances
colorectal cancer cell proliferation and metastasis through
HDAC3/miR-139-3p pathway. Biochem Genet. 62:3904–3919. 2024.
View Article : Google Scholar
|
|
103
|
Cassandri M, Porrazzo A, Pomella S, Noce
B, Zwergel C, Aiello FA, Vulcano F, Milazzo L, Camero S, Pajalunga
D, et al: HDAC3 genetic and pharmacologic inhibition
radiosensitizes fusion positive rhabdomyosarcoma by promoting DNA
double-strand breaks. Cell Death Discov. 10:3512024. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Zhou Y, Jin X, Ma J, Ding D, Huang Z,
Sheng H, Yan Y, Pan Y, Wei T, Wang L, et al: HDAC5 loss impairs RB
repression of pro-oncogenic genes and confers CDK4/6 inhibitor
resistance in cancer. Cancer Res. 81:1486–1499. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Fan J, Lou B, Chen W, Zhang J, Lin S, Lv
FF and Chen Y: Down-regulation of HDAC5 inhibits growth of human
hepatocellular carcinoma by induction of apoptosis and cell cycle
arrest. Tumour Biol. 35:11523–11532. 2014. View Article : Google Scholar
|
|
106
|
Lyu H, Ishimura A, Suzuki R, Buyanbat K,
Batbayar G, Meguro-Horike M, Horike SI, Yano S and Suzuki T: HDAC5,
an early osimertinib-responsive gene, is a novel therapeutic target
for the drug resistance in EGFR-mutant lung adenocarcinoma cells.
Biochem Biophys Rep. 42:1020162025.PubMed/NCBI
|
|
107
|
Pan P, Qin G, Wang B, Yu H, Chen J, Liu J,
Bing K, Shen J, Ren D, Zhao Y, et al: HDAC5 loss enhances
phospholipid-derived arachidonic acid generation and confers
sensitivity to cPLA2 inhibition in pancreatic cancer. Cancer Res.
82:4542–4554. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Liu C, Zheng D, Pu X and Li S: HDAC7: A
promising target in cancer. Front Oncol. 14:13279332024. View Article : Google Scholar
|
|
109
|
Lu P, Deng S, Liu J, Xiao Q, Zhou Z, Li S,
Xin J, Shu G, Yi B and Yin G: Tweety homolog 3 promotes colorectal
cancer progression through mutual regulation of histone deacetylase
7. MedComm (2020). 5:e5762024. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Gautam N, Chapagain PP, Adhikari NP and
Tiwari PB: Characterization of molecular interactions between HDAC7
and MEF2A. J Biomol Struct Dyn. 1–10. 2024.(Epub ahead of print).
View Article : Google Scholar
|
|
111
|
Lu W, Zhuang G, Guan Y, Li Y, Liu L and
Xiao M: Comprehensive analysis of HDAC7 expression and its
prognostic value in diffuse large B cell lymphoma: A review.
Medicine (Baltimore). 102:e345772023. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Jin Y, Qi X, Yu X, Cheng X, Chen B, Wu M,
Zhang J, Yin H, Lu Y, Zhou Y, et al: Discovery of a potential
hematologic malignancies therapy: Selective and potent HDAC7 PROTAC
degrader targeting non-enzymatic function. Acta Pharm Sin B.
15:1659–1679. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Bi L, Ren Y, Feng M, Meng P, Wang Q, Chen
W, Jiao Q, Wang Y, Du L, Zhou F, et al: HDAC11 regulates glycolysis
through the LKB1/AMPK signaling pathway to maintain hepatocellular
carcinoma stemness. Cancer Res. 81:2015–2028. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Wang W, Ding B, Lou W and Lin S: Promoter
hypomethylation and miR-145-5p downregulation-mediated HDAC11
overexpression promotes sorafenib resistance and metastasis of
hepatocellular carcinoma cells. Front Cell Dev Biol. 8:7242020.
View Article : Google Scholar
|
|
115
|
Liu Y, Tong X, Hu W and Chen D: HDAC11: A
novel target for improved cancer therapy. Biomed Pharmacother.
166:1154182023. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Chen J, Cheng F, Sahakian E, Powers J,
Wang Z, Tao J, Seto E, Pinilla-Ibarz J and Sotomayor EM: HDAC11
regulates expression of C/EBPβ and immunosuppressive molecules in
myeloid-derived suppressor cells. J Leukoc Biol. 109:891–900. 2021.
View Article : Google Scholar
|
|
117
|
Villagra A, Cheng F, Wang HW, Suarez I,
Glozak M, Maurin M, Nguyen D, Wright KL, Atadja PW, Bhalla K, et
al: The histone deacetylase HDAC11 regulates the expression of
interleukin 10 and immune tolerance. Nat Immunol. 10:92–100. 2009.
View Article : Google Scholar
|
|
118
|
Li R, Wu X, Zhao P, Xue K and Li J: A
pan-cancer analysis identifies HDAC11 as an immunological and
prognostic biomarker. FASEB J. 36:e223262022. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Zhao C, Dong H, Xu Q and Zhang Y: Histone
deacetylase (HDAC) inhibitors in cancer: A patent review
(2017-present). Expert Opin Ther Pat. 30:263–274. 2020. View Article : Google Scholar
|
|
120
|
Biersack B, Polat S and Höpfner M:
Anticancer properties of chimeric HDAC and kinase inhibitors. Semin
Cancer Biol. 83:472–486. 2022. View Article : Google Scholar
|
|
121
|
Zhou WH, Luo Y, Li RX, Degrace P, Jourdan
T, Qiao F, Chen LQ, Zhang ML and Du ZY: Inhibition of mitochondrial
fatty acid β-oxidation activates mTORC1 pathway and protein
synthesis via Gcn5-dependent acetylation of raptor in zebrafish. J
Biol Chem. 299:1052202023. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Mukherjee A, Zamani F and Suzuki T:
Evolution of slow-binding inhibitors targeting histone deacetylase
isoforms. J Med Chem. 66:11672–11700. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
San-Miguel JF, Hungria VTM, Yoon SS,
Beksac M, Dimopoulos MA, Elghandour A, Jedrzejczak WW, Günther A,
Nakorn TN, Siritanaratkul N, et al: Panobinostat plus bortezomib
and dexamethasone versus placebo plus bortezomib and dexamethasone
in patients with relapsed or relapsed and refractory multiple
myeloma: A multicentre, randomised, double-blind phase 3 trial.
Lancet Oncol. 15:1195–1206. 2014. View Article : Google Scholar
|
|
124
|
Zhang L, Zhang Y, Mehta A, Boufraqech M,
Davis S, Wang J, Tian Z, Yu Z, Boxer MB, Kiefer JA, et al: Dual
inhibition of HDAC and EGFR signaling with CUDC-101 induces potent
suppression of tumor growth and metastasis in anaplastic thyroid
cancer. Oncotarget. 6:9073–9085. 2015. View Article : Google Scholar
|
|
125
|
San José-Enériz E, Gimenez-Camino N, Rabal
O, Garate L, Miranda E, Gómez-Echarte N, García F, Charalampopoulou
S, Sáez E, Vilas-Zornoza A, et al: Epigenetic-based differentiation
therapy for acute myeloid leukemia. Nat Commun. 15:55702024.
View Article : Google Scholar
|
|
126
|
Sim W, Lim WM, Hii LW, Leong CO and Mai
CW: Targeting pancreatic cancer immune evasion by inhibiting
histone deacetylases. World J Gastroenterol. 28:1934–1945. 2022.
View Article : Google Scholar
|
|
127
|
Li X, Wang Y, Deng S, Zhu G, Wang C,
Johnson NA, Zhang Z, Tirado CR, Xu Y, Metang LA, et al: Loss of
SYNCRIP unleashes APOBEC-driven mutagenesis, tumor heterogeneity,
and AR-targeted therapy resistance in prostate cancer. Cancer Cell.
41:1427–1449.e12. 2023. View Article : Google Scholar
|
|
128
|
Yang L, Qiu Q, Wang J, Wen Y, Li H, Liang
R, Feng Y, Wang F, Lin X, Tang M, et al: Preclinical and
first-in-human of purinostat mesylate, a novel selective HDAC I/IIb
inhibitor, in relapsed/refractory multiple myeloma and lymphoma.
Signal Transduct Target Ther. 10:2012025. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Bishton MJ, Harrison SJ, Martin BP,
McLaughlin N, James C, Josefsson EC, Henley KJ, Kile BT, Prince HM
and Johnstone RW: Deciphering the molecular and biologic processes
that mediate histone deacetylase inhibitor-induced
thrombocytopenia. Blood. 117:3658–3668. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Liu YM and Liou JP: An updated patent
review of histone deacetylase (HDAC) inhibitors in cancer
(2020-present). Expert Opin Ther Pat. 33:349–369. 2023. View Article : Google Scholar
|
|
131
|
Song S, Wen Y, Tong H, Loro E, Gong Y, Liu
J, Hong S, Li L, Khurana TS, Chu M and Sun Z: The HDAC3 enzymatic
activity regulates skeletal muscle fuel metabolism. J Mol Cell
Biol. 11:133–143. 2019. View Article : Google Scholar
|
|
132
|
Bülbül EF, Robaa D, Sun P, Mahmoudi F,
Melesina J, Zessin M, Schutkowski M and Sippl W: Application of
ligand- and structure-based prediction models for the design of
alkylhydrazide-based HDAC3 inhibitors as novel anti-cancer
compounds. Pharmaceuticals (Basel). 16:9682023. View Article : Google Scholar
|
|
133
|
Ren Y, Li S, Zhu R, Wan C, Song D, Zhu J,
Cai G, Long S, Kong L and Yu W: Discovery of STAT3 and histone
deacetylase (HDAC) dual-pathway inhibitors for the treatment of
solid cancer. J Med Chem. 64:7468–7482. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Xie X, Zhang O, Yeo MJR, Lee C, Tao R,
Harry SA, Payne NC, Nam E, Paul L, Li Y, et al: Converging
mechanism of UM171 and KBTBD4 neomorphic cancer mutations. Nature.
639:241–249. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Gao J, Hou B, Zhu Q, Yang L, Jiang X, Zou
Z, Li X, Xu T, Zheng M, Chen YH, et al: Engineered bioorthogonal
POLY-PROTAC nanoparticles for tumour-specific protein degradation
and precise cancer therapy. Nat Commun. 13:43182022. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Sun J, Lian X, Lv C, Li H, Lin Z, Luo S,
Liu Y, Xu Y, Jiang X, Xu W, et al: Trps1 acts as a regulator of
Sf-1 transcription and testosterone synthesis in mouse Leydig
cells. Cell Biol Toxicol. 39:3141–3157. 2023. View Article : Google Scholar
|
|
137
|
Chen M, Wang C, Wang X, Tu Z, Ding Z and
Liu Z: An ‘AND’ logic-gated prodrug micelle locally stimulates
antitumor immunity. Adv Mater. 36:e23078182024. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Li S, Liu Y, Tian T, Zhang T, Lin S, Zhou
M, Zhang X, Lin Y and Cai X: Bioswitchable delivery of microRNA by
framework nucleic acids: Application to bone regeneration. Small.
17:e21043592021. View Article : Google Scholar
|
|
139
|
Xing X, Zhong W, Tang P, Tao Q, Lu X and
Zhong L: Tracking intracellular nuclear targeted-chemotherapy of
chidamide-loaded Prussian blue nanocarriers by SERS mapping.
Colloids Surf B Biointerfaces. 229:1134692023. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Hong L, Ni M, Xue F, Jiang T, Wu X, Li C,
Liang S, Chen T, Luo C and Wu Q: The role of HDAC3 in pulmonary
diseases. Lung. 203:472025. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Zhang F, Yue K, Sun S, Lu S, Jia G, Zha Y,
Zhang S, Chou CJ, Liao C, Li X and Duan Y: Targeting histone
deacetylase 11 with a highly selective inhibitor for the treatment
of MASLD. Adv Sci (Weinh). 12:e24129032025. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Pramanik SD, Kumar Halder A, Mukherjee U,
Kumar D, Dey YN and R M: Potential of histone deacetylase
inhibitors in the control and regulation of prostate, breast and
ovarian cancer. Front Chem. 10:9482172022. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Woods DM, Sodré AL, Villagra A, Sarnaik A,
Sotomayor EM and Weber J: HDAC inhibition upregulates PD-1 ligands
in melanoma and augments immunotherapy with PD-1 blockade. Cancer
Immunol Res. 3:1375–1385. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Baretti M and Yarchoan M: Epigenetic
modifiers synergize with immune-checkpoint blockade to enhance
long-lasting antitumor efficacy. J Clin Invest. 131:e1510022021.
View Article : Google Scholar
|
|
145
|
Qian C, Lai CJ, Bao R, Wang DG, Wang J, Xu
GX, Atoyan R, Qu H, Yin L, Samson M, et al: Cancer network
disruption by a single molecule inhibitor targeting both histone
deacetylase activity and phosphatidylinositol 3-kinase signaling.
Clin Cancer Res. 18:4104–4113. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Zhou L and Yu CW: Epigenetic modulations
in triple-negative breast cancer: Therapeutic implications for
tumor microenvironment. Pharmacol Res. 204:1072052024. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Venneker S, van Eenige R, Kruisselbrink
AB, Palubeckaitė I, Taliento AE, Briaire-de Bruijn IH, Hogendoorn
PCW, van de Sande MAJ, Gelderblom H, Mei H, et al: Histone
deacetylase inhibitors as a therapeutic strategy to eliminate
neoplastic ‘stromal’ cells from giant cell tumors of bone. Cancers
(Basel). 14:47082022. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Guo F and Wang H: Potential of histone
deacetylase inhibitors for the therapy of ovarian cancer. Front
Oncol. 12:10571862022. View Article : Google Scholar
|
|
149
|
Li X, Su X, Liu R, Pan Y, Fang J, Cao L,
Feng C, Shang Q, Chen Y, Shao C and Shi Y: HDAC inhibition
potentiates anti-tumor activity of macrophages and enhances
anti-PD-L1-mediated tumor suppression. Oncogene. 40:1836–1850.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Nguyen A, Ho L, Hogg R, Chen L, Walsh SR
and Wan Y: HDACi promotes inflammatory remodeling of the tumor
microenvironment to enhance epitope spreading and antitumor
immunity. J Clin Invest. 132:e1592832022. View Article : Google Scholar
|
|
151
|
Zhang W, Ge L, Zhang Y, Zhang Z, Zhang W,
Song F, Huang P and Xu T: Targeted intervention of tumor
microenvironment with HDAC inhibitors and their combination therapy
strategies. Eur J Med Res. 30:692025. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Jing L, Qian Z, Gao Q, Sun R, Zhen Z, Wang
G, Yang X, Li H, Guo T and Zhang W: Diffuse midline glioma treated
with epigenetic agent-based immunotherapy. Signal Transduct Target
Ther. 8:232023. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Fresquet V, Garcia-Barchino MJ, Larrayoz
M, Celay J, Vicente C, Fernandez-Galilea M, Larrayoz MJ, Calasanz
MJ, Panizo C, Junza A, et al: Endogenous retroelement activation by
epigenetic therapy reverses the warburg effect and elicits
mitochondrial-mediated cancer cell death. Cancer Discov.
11:1268–1285. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Jenke R, Reßing N, Hansen FK, Aigner A and
Büch T: Anticancer therapy with HDAC inhibitors: Mechanism-based
combination strategies and future perspectives. Cancers (Basel).
13:6342021. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Chen Y, Su J, Li S, Chen F, Zhang Y, Wang
X, Zhang Y, Wang X, Yuan Z, Ren S, et al: Structural modifications
and prospects of histone deacetylase (HDAC) inhibitors in cancer.
Curr Med Chem. 32:8530–8555. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Zou Q, Zhang Y, Zhou H, Lai Y, Cao Y, Li
Z, Su N, Li W, Huang H, Liu P, et al: Chidamide, a histone
deacetylase inhibitor, combined with R-GemOx in relapsed/refractory
diffuse large B-cell lymphoma (TRUST): A multicenter, single-arm,
phase 2 trial. Cancer Med. 14:e709192025. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Wang L, Gao M, Wang T, Cui P, Chen G, Han
X, Ma Z, Yang W, Jing F, Ma J, et al: First-line treatment with
HDACis plus tislelizumab combined with chemotherapy in advanced
NSCLC: A single-arm phase II study. Oncologist. 30:oyaf1552025.
View Article : Google Scholar
|
|
158
|
Wieduwilt MJ, Pawlowska N, Thomas S, Olin
R, Logan AC, Damon LE, Martin T, Kang M, Sayre PH, Boyer W, et al:
Histone deacetylase inhibition with panobinostat combined with
intensive induction chemotherapy in older patients with acute
myeloid leukemia: Phase I study results. Clin Cancer Res.
25:4917–4923. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Gu S, Hou Y, Dovat K, Dovat S, Song C and
Ge Z: Synergistic effect of HDAC inhibitor chidamide with
cladribine on cell cycle arrest and apoptosis by targeting
HDAC2/c-Myc/RCC1 axis in acute myeloid leukemia. Exp Hematol Oncol.
12:232023. View Article : Google Scholar
|
|
160
|
Liu S, Ma S, Liu G, Hou L, Guan Y, Liu L,
Meng Y, Yu W, Liu T, Zhou L, et al: CK2B induces CD8+
T-cell exhaustion through HDAC8-mediated epigenetic reprogramming
to limit the efficacy of anti-PD-1 therapy in non-small-cell lung
cancer. Adv Sci (Weinh). 12:e24110532025. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Chen C, Li X, Zhao H, Liu M, Du J, Zhang
J, Yang X, Hou X and Fang H: Discovery of DNA-targeting HDAC
inhibitors with potent antitumor efficacy in vivo that trigger
antitumor immunity. J Med Chem. 65:3667–3683. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Liang R, Ding D, Li Y, Lan T, Ryabtseva S,
Huang S, Ren J, Huang H and Wei B: HDACi combination therapy with
IDO1i remodels the tumor microenvironment and boosts antitumor
efficacy in colorectal cancer with microsatellite stability. J
Nanobiotechnology. 22:7532024. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Guerriero JL, Sotayo A, Ponichtera HE,
Castrillon JA, Pourzia AL, Schad S, Johnson SF, Carrasco RD, Lazo
S, Bronson RT, et al: Class IIa HDAC inhibition reduces breast
tumours and metastases through anti-tumour macrophages. Nature.
543:428–432. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Chen KY, Kibayashi T, Giguelay A, Hata M,
Nakajima S, Mikami N, Takeshima Y, Ichiyama K, Omiya R, Ludwig LS,
et al: Genome-wide CRISPR screen in human T cells reveals
regulators of FOXP3. Nature. 642:191–200. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Gediya P, Vyas VK, Carafa V, Sitwala N,
Della Torre L, Poziello A, Kurohara T, Suzuki T, Sanna V, Raguraman
V, et al: Discovery of novel
tetrahydrobenzo[b]thiophene-3-carbonitriles as histone deacetylase
inhibitors. Bioorg Chem. 110:1048012021. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Elmezayen AD, Al-Obaidi A and Yelekçi K:
Discovery of novel isoform-selective histone deacetylases 5 and 9
inhibitors through combined ligand-based pharmacophore modeling,
molecular mocking, and molecular dynamics simulations for cancer
treatment. J Mol Graph Model. 106:1079372021. View Article : Google Scholar
|
|
167
|
Yao K, Li Y, Wei W, Liu S, Wang X, Xu J,
Zhang R, Wu Z, Guo C, Yang L and Hu L: Synthesis and biological
evaluation of novel 4-Arylaminoquinolines derivatives as EGFR/HDAC
inhibitors. Bioorg Med Chem Lett. 122:1302142025. View Article : Google Scholar
|
|
168
|
Parag-Sharma K, Tasoulas J, Musicant AM,
do Nascimento-Filho CHV, Zhu Z, Twomey C, Liu P, Castilho RM and
Amelio AL: Synergistic efficacy of combined EGFR and HDAC
inhibitors overcomes tolerance to EGFR monotherapy in salivary
mucoepidermoid carcinoma. Oral Oncol. 115:1051662021. View Article : Google Scholar
|
|
169
|
Cao J, Zhao W, Zhao C, Liu Q, Li S, Zhang
G, Chou CJ and Zhang Y: Development of a bestatin-SAHA hybrid with
dual inhibitory activity against APN and HDAC. Molecules.
25:49912020. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Zhang Y, Wang H, Zhan Z, Gan L and Bai O:
Mechanisms of HDACs in cancer development. Front Immunol.
16:15292392025. View Article : Google Scholar
|
|
171
|
Hai R, Yang D, Zheng F, Wang W, Han X,
Bode AM and Luo X: The emerging roles of HDACs and their
therapeutic implications in cancer. Eur J Pharmacol.
931:1752162022. View Article : Google Scholar
|
|
172
|
Sanaei M and Kavoosi F: Histone
deacetylases and histone deacetylase inhibitors: Molecular
mechanisms of action in various cancers. Adv Biomed Res. 8:632019.
View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Oliveira-Silva JM, de Oliveira LS,
Tagliéri JVM, Lopes LB, de Souza CVE, Batistão HKA and
Castro-Gamero AM: HDAC6: Tumor progression and beyond. Curr Cancer
Drug Targets. Jan 7–2025.(Epub ahead of print).
|
|
174
|
Jo H, Shim K and Jeoung D: Targeting HDAC6
to overcome autophagy-promoted anti-cancer drug resistance. Int J
Mol Sci. 23:95922022. View Article : Google Scholar
|
|
175
|
Kadier K, Niu T, Ding B, Chen B, Qi X,
Chen D, Cheng X, Fang Y, Zhou J, Zhao W, et al: PROTAC-mediated
HDAC7 protein degradation unveils its deacetylase-independent
proinflammatory function in macrophages. Adv Sci (Weinh).
11:e23094592024. View Article : Google Scholar : PubMed/NCBI
|