Introduction
According to the 2021 World Health Organization
(WHO) Classification of Tumors of the Central Nervous System,
gliosarcoma (GSM) is a distinct histopathological variant of
isocitrate dehydrogenase (IDH)-wild-type glioblastoma (GBM),
categorized as a WHO Grade IV neoplasm, the highest malignancy
grade for central nervous system tumors (1,2).
Clinically, GSM is exceptionally rare, accounting for only 1.5–8%
of all GBM cases, with a median onset age of ~52 years and a slight
male predominance (male-to-female ratio, 1.2:1-1.5:1) (3–5). The
prognosis is poor, with untreated patients having a median overall
survival (OS) time of only 4 months. Even with standard multimodal
therapy (following the Stupp protocol), the median OS time only
extends to 6.6–18.5 months (most commonly 12–15 months) (6–8).
Prognostic factors include tumor location (supratentorial lesions
fare better than infratentorial ones), extent of surgical resection
(gross-total resection improves survival) and molecular markers
such as O6-methylguanine-DNA methyltransferase promoter
methylation (4,6,9).
The preoperative diagnosis of GSM is challenging due
to non-specific clinical and imaging features. Clinically,
supratentorial GSM typically causes seizures, focal neurological
deficits or elevated intracranial pressure (headaches and nausea),
while infratentorial cases may present with ataxia, diplopia or
brainstem compression symptoms (10). Imaging often shows a heterogeneously
enhancing mass with peritumoral edema, overlapping with malignant
meningioma (distinguished by epithelial membrane antigen (EMA)
positivity and glial fibrillary acidic protein (GFAP) negativity),
anaplastic astrocytoma (no biphasic histology and lower Ki-67
index), or metastatic tumors (history of extracranial primary
tumors and lineage-specific markers) (11–13). A
definitive diagnosis relies on histopathology (biphasic
glial-sarcomatous components) and immunohistochemistry
(GFAP-positive glial cells, smooth muscle actin
(SMA)/vimentin-positive sarcomatous cells) (12,13).
Treatment primarily involves maximal safe surgical resection (often
subtotal for deep/infratentorial lesions to preserve neural
function), followed by adjuvant intensity-modulated radiotherapy
(IMRT; total, 60 Gy in 30 fractions) and concurrent temozolomide
chemotherapy (75 mg/m2 during radiotherapy, then 150–200
mg/m2 for 5 days every 28 days for 6 cycles) (8,14),16,17). Despite this, >70% of
patients experience recurrence within 12 months, with no
standardized salvage therapy (6,15).
GSM most commonly arises in supratentorial regions
(temporal, frontal and parietal lobes), with infratentorial
(cerebellar and brainstem) cases accounting for <5% (3,9). This
rarity, combined with the anatomical constraints of the posterior
fossa, makes cerebellar GSM particularly challenging to diagnose
and treat. The present study reports an unusual case of GSM
originating in the right cerebellar hemisphere with extension to
the parahippocampal region, detailing the patient's clinical
presentation, comprehensive imaging findings, pathological
characteristics, treatment course and outcomes. A review of the
relevant literature is also included to enhance understanding of
this rare tumor's behavior and management complexities.
Case report
Patient presentation
A 55-year-old woman presented to the General
Hospital of Western Theater Command (Chengdu, China) in April 2022
with a 2-month history of diplopia. At 1 month prior to admission,
the patient developed intermittent, dull headaches localized to the
right occipital region. The symptoms progressively worsened,
culminating in an unsteady gait with a tendency to tilt to one
side. A physical examination revealed instability upon standing
when the left eye was covered (positive Romberg sign), while
stability was maintained with the right eye covered. The right
finger-nose test was positive, indicating cerebellar dysfunction.
No facial numbness, dysphagia or limb motor deficits were observed.
Routine laboratory investigations were within normal limits. All
imaging, neurosurgical procedures and pathological examinations
were performed at the General Hospital of Western Theater Command.
The patient was first admitted to the Department of Neurosurgery in
April 2022. At 10 days post-admission, the patient underwent
resection of the right cerebellar space-occupying lesion,
craniotomy decompression and repair of cerebrospinal fluid leakage
via the infratentorial supracerebellar approach. The operation
lasted 6 h and 2 min, with a smooth procedure and satisfactory
anesthesia effect. Intraoperative blood loss was 200 ml, and no
blood transfusion was required. The patient regained spontaneous
breathing uneventfully postoperatively. In August 2022, the patient
subsequently received cranial radiotherapy combined with concurrent
chemotherapy. The chemotherapeutic regimen consisted of oral
temozolomide administered once daily, as later described. In March
2023, the patient returned to the Oncology Outpatient Department at
the General Hospital of Western Theater Command for a follow-up
magnetic resonance imaging (MRI) examination.
Imaging findings and follow-up
Preoperative brain MRI demonstrated a large
(4.0×2.8×2.7 cm), lobulated, mixed-signal-intensity mass occupying
the right cerebellar hemisphere and parahippocampal region. The
lesion exhibited partial diffusion restriction on
diffusion-weighted imaging/apparent diffusion coefficient
sequences. Susceptibility-weighted imaging revealed intralesional
punctate hypointensities suggestive of microhemorrhages or
calcifications. Post-contrast T1-weighted imaging revealed marked
heterogeneous enhancement. Significant perilesional edema,
manifesting as T1 hypointensity and T2 hyperintensity, was noted in
the adjacent cerebellar parenchyma, extending to involve the right
brainstem and cerebellar vermis. Mass effect resulted in
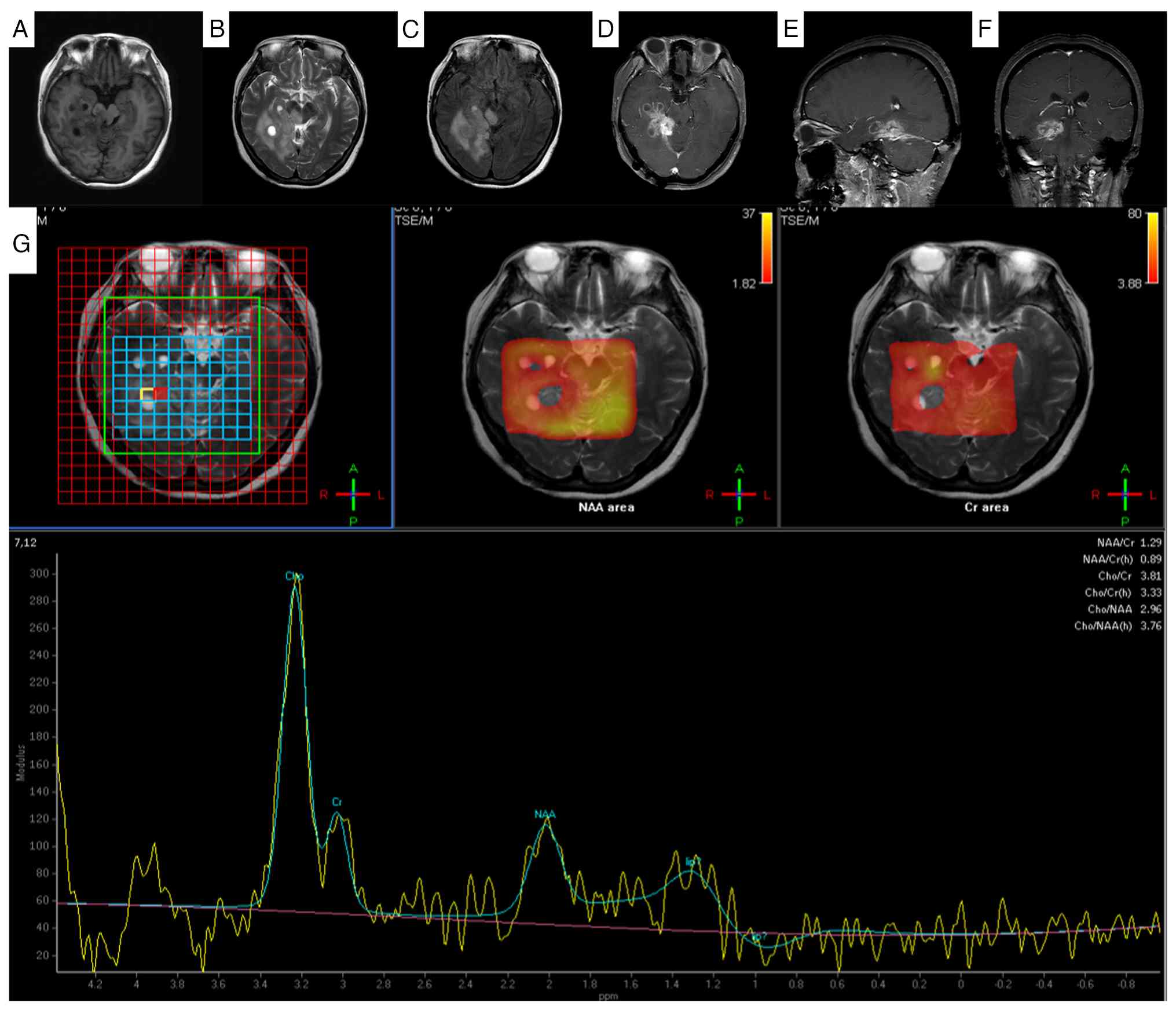
compression of the fourth ventricle. Magnetic resonance
spectroscopy (MRS) within the lesion demonstrated a
characteristically elevated choline (Cho) peak and a markedly
reduced N-acetylaspartate (NAA) peak, consistent with high cellular
turnover and neuronal loss. Patchy slightly prolonged T1 and
isointense T1 signals were noted in the right cerebellar hemisphere
and right parahippocampal region (Fig.
1A). On T2-weighted imaging, the same region exhibited slightly
prolonged T2 signals interspersed with small patchy slightly
shortened T2 signals (Fig. 1B),
with a lobulated mixed-signal mass identified in this area.
Susceptibility-weighted imaging revealed punctate hypointense foci
within the lesion (Fig. 1C).
Fluid-attenuated inversion recovery imaging showed hyperintense
signals in the lesion (Fig. 1D).
Marked heterogeneous enhancement of the lesion was seen on axial
(Fig. 1E) and coronal (Fig. 1F) post-contrast images. The lesion
involved the right brainstem and cerebellar vermis, with
compression of the adjacent fourth ventricle and brainstem. MRS of
the lesion area revealed a decreased NAA peak, a significant
elevation of the Cho peak and a nearly unchanged creatine (Cr) peak
(Fig. 1G).
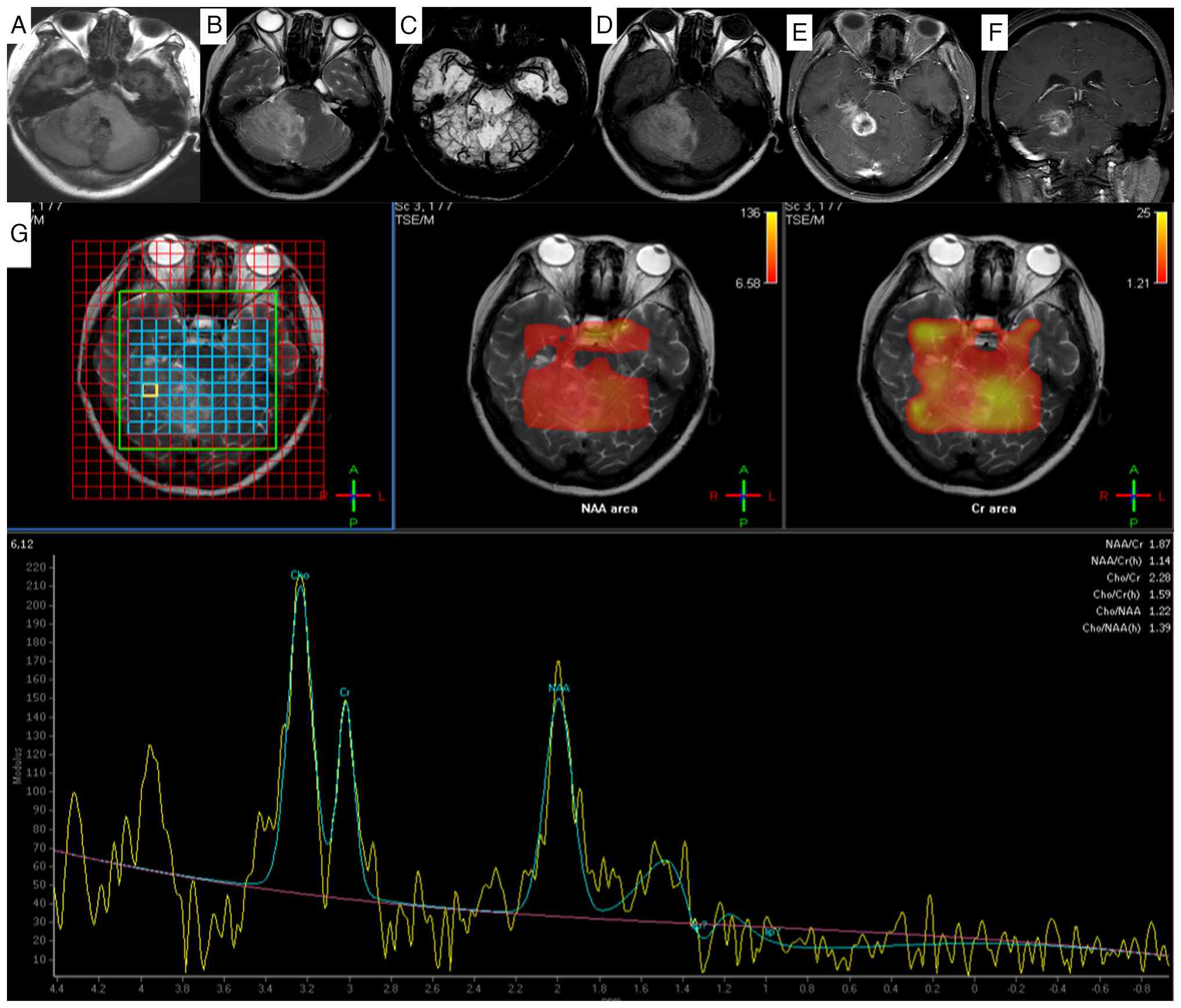 | Figure 1.Magnetic resonance imaging findings of
gliosarcoma in the right cerebellar hemisphere and right
parahippocampal region. (A) In the right cerebellar hemisphere and
right parahippocampal region, patchy slightly hyperintense and
isointense signals on T1-weighted imaging were observed. (B) On
T2-weighted imaging, slightly hyperintense signals were noted,
mixed with small patchy slightly hypointense signals. (C)
Susceptibility-weighted imaging demonstrates punctate hypointense
foci within the lesion. (D) Fluid-attenuated inversion recovery
imaging shows hyperintensity. The lesions exhibited obvious
inhomogeneous enhancement on (E) axial and (F) coronal images. The
lesion involves the right portion of the brainstem and cerebellar
vermis, with compression of the adjacent fourth ventricle and
brainstem. (G) The peak value of NAA in the lesion area was
decreased, the peak value of Cho was significantly increased, and
the peak value of Cr remained basically unchanged. MRI, magnetic
resonance imaging; NAA, N-acetylaspartate; Cr, creatine; TSE/M,
turbo spin echo/magnetization. |
At the 2-month postoperative follow-up, MRI
confirmed tumor recurrence in the primary site. The recurrent
lesion exhibited trans-tentorial extension, involving the right
cerebellar tentorium and right brainstem, with persistent
heterogeneous contrast enhancement (Fig. 2).
 | Figure 2.Recurrence of gliosarcoma on 2-month
follow-up magnetic resonance imaging. A lobulated mixed-signal mass
predominantly with (A) slightly long T1 and (B) slightly long T2
signals is present in the right cerebellar hemisphere and right
parahippocampal region. (C) Fluid-attenuated inversion recovery
imaging shows hyperintensity. After enhancement, the (D) axial, (E)
sagittal and (F) coronal images of the lesion exhibited obvious
inhomogeneous enhancement and annular enhancement. The lesion is
growing across the tentorium, with thickening and significant
enhancement of the adjacent right cerebellar tentorium; it involves
the right portion of the brainstem, causing compression of the
adjacent fourth ventricle and brainstem. (G) Magnetic resonance
spectroscopy of the recurrent lesion reveals a decreased NAA peak
and an increased Cho peak in the lesion area. The left panel shows
the voxel placement (yellow grid). The middle and right panels
display the spectrum without pseudo-color mapping, likely due to
extremely low metabolite concentrations, incomplete water
suppression (note the prominent water peak on the right), and/or
post-processing thresholds. NAA, N-acetylaspartate; Cr, creatine;
TSE/M, turbo spin echo/magnetization. |
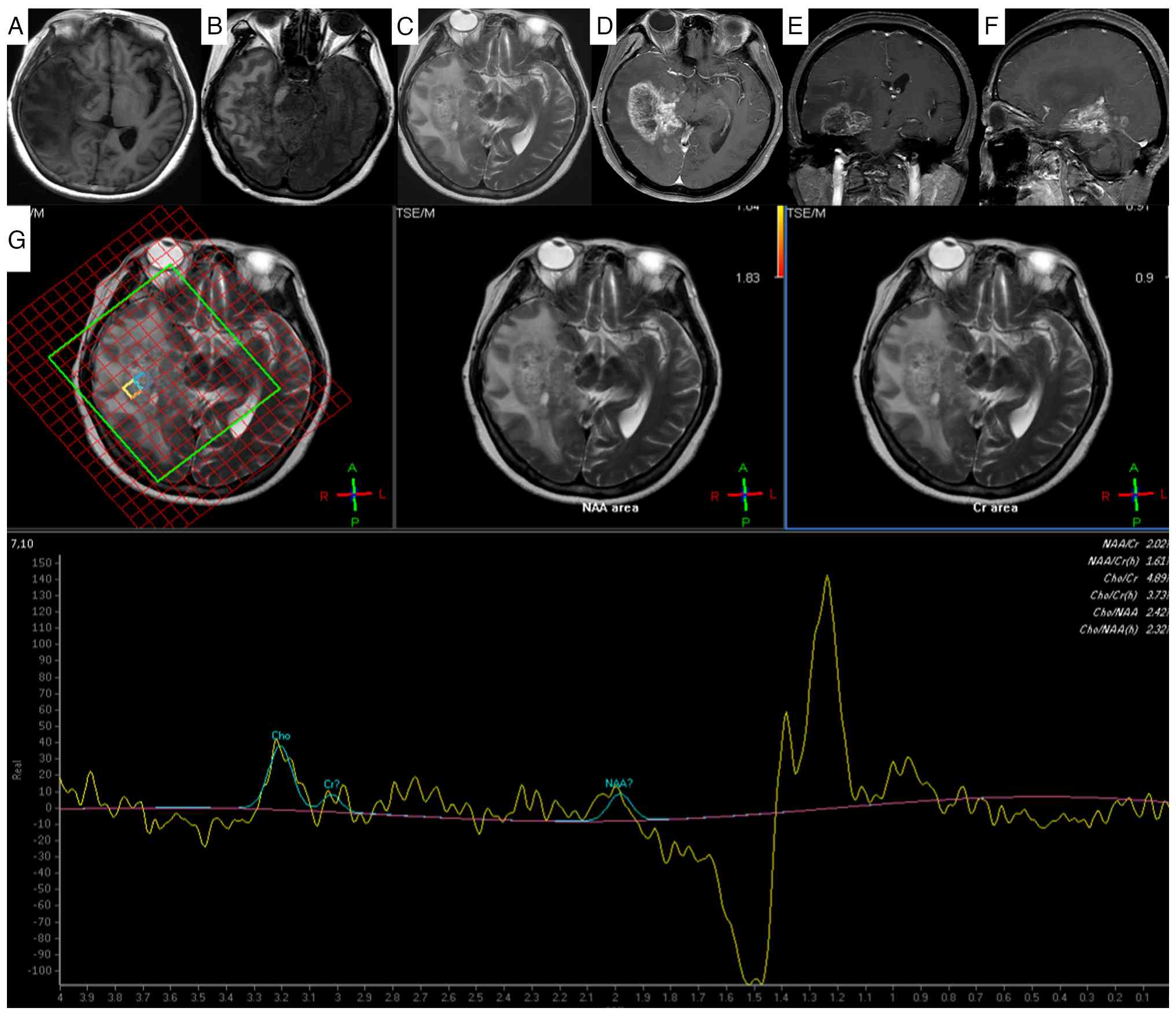
By the 12-month follow-up, the recurrent tumor had
enlarged considerably, with further involvement of the right
temporal lobe. The imaging revealed extensive perilesional edema, a
significant mass effect with midline shift and evidence of brain
herniation (Fig. 3).
 | Figure 3.Enlargement of the recurrent
gliosarcoma with brain herniation formation on 1-year follow-up
magnetic resonance imaging. The dura mater in the right occipital
region was thickened with obvious enhancement. The right cerebellar
hemisphere, right parahippocampal region and right temporal lobe
exhibited patchy mixed signal shadows. (A) On the T1WI sequence,
slightly longer T1 signals were observed, and (B) on the T2WI
sequence, slightly longer T2 signals were observed. (C)
Fluid-attenuated inversion recovery imaging shows isointense and
slightly hyperintense signals. After enhancement, the (D) axial,
(E) sagittal and (F) coronal images of the lesion exhibited obvious
inhomogeneous enhancement and annular enhancement. The lesion grows
across the tentorium, with thickening and significant enhancement
of the adjacent right cerebellar tentorium; it involves the right
portion of the brainstem, causing compression of the adjacent
fourth ventricle, right lateral ventricle and brainstem, with
leftward shift of the midline structures. (G) Magnetic resonance
spectroscopy reveals a decreased NAA peak and an increased Cho peak
in the lesion area. NAA, N-acetylaspartate; Cr, creatine; TSE/M,
turbo spin echo/magnetization. |
Operative record
Under general anesthesia, the patient was positioned
in a left lateral prone position with Mayfield head frame fixation.
A right paramedian occipital straight incision was made. A
craniotomy was performed, exposing the transverse sinus. Upon dural
opening, significant cerebellar swelling and elevated intracranial
pressure were noted. A subtentorial supracerebellar approach was
utilized. After arachnoid dissection and mobilization of venous
structures, a firm, poorly demarcated tumor was identified in the
tentorial region. Due to persistent tension in the posterior fossa
despite cerebrospinal fluid (CSF) release and dehydration, a
complete dissection was deemed unsafe. An internal decompression
was performed via piecemeal resection of the tumor and a portion of
the cerebellar tissue. Meticulous dissection preserved the superior
cerebellar artery and key venous structures. An attempt to access
the supratentorial component via tentorial incision was limited by
anatomical constraints, resulting in residual supratentorial tumor.
Hemostasis was achieved, the dural defect was repaired with an
artificial graft and the bone flap was secured. A surgical drain
was placed before layered closure.
Immunohistochemistry
The postoperative tumor tissue was fixed in 10%
neutral buffered formalin at 25°C for 24 h, routinely processed,
paraffin-embedded and sectioned at a 4-µm thickness.
Immunohistochemical staining was performed using the EnVision
two-step method. Endogenous peroxidase activity was blocked with 3%
hydrogen peroxide for 10 min at room temperature. Antigen retrieval
was performed with heat induction using EDTA buffer (pH 9.0) at a
constant temperature of 100°C in a microwave, followed by graded
alcohol rehydration. Sections were then blocked with normal goat
serum (OriGene Technologies, Inc.) for 30 min at room temperature.
Primary antibodies (all Fuzhou Maixin Biotechnology Development
Co., Ltd.) were applied and incubated overnight at 4°C in a
humidified chamber. A detailed list of the antibodies, including
the clones, dilutions and catalog numbers, is shown in Table I. Subsequently, the HRP-labeled
secondary antibody from the EnVision kit (catalog no. KIT-5001;
Fuzhou Maixin Biotechnology Development Co., Ltd.) was applied and
incubated for 30 min at room temperature. Hematoxylin and eosin
(H&E) staining was performed on the gliosarcoma tissues
following a standardized protocol, with all procedures conducted at
25°C unless otherwise specified. Freshly resected gliosarcoma
tissues (≤3 mm in thickness) were fixed in 10% neutral buffered
formalin (pH 7.4) at 25°C for 24 h (a maximum fixation duration of
72 h was applied to avoid over-fixation), then processed for
routine paraffin embedding. Serial 4-µm thick sections were cut
using a rotary microtome, mounted on positively charged glass
slides and baked at 60°C for 30 min for adhesion. H&E staining
was implemented using the Hematoxylin-Eosin Staining Solution
(Shaanxi Medical Device Preparation No. 20180101, Xi'an Meijiajia,
China) with the following steps: Xylene deparaffinization (10, 10
and 5 min); graded alcohol rehydration (100% ethanol, 2×5 min; 95%
ethanol, 3 min; 85% ethanol, 3 min; and 75% ethanol, 3 min); Harris
hematoxylin staining (5–8 min); differentiation in 1% acid alcohol
(3–5 sec); bluing in 0.5% lithium carbonate solution (10–20 sec);
alcoholic Eosin Y staining (1–3 min); graded alcohol dehydration
(95% ethanol, 3 min; 100% ethanol, 2×3 min); xylene clearing (2×5
min); and permanent mounting with neutral balsam. For
immunohistochemical staining, the chromogenic reaction was
visualized using a DAB substrate kit (cat. no. ab64238; Abcam),
followed by hematoxylin counterstaining. All stained slides were
examined under an Olympus BX53 bright-field light microscope
(equipped with a DP74 digital camera; Olympus Corporation), and
representative images were captured at magnifications of ×100
(scale bar, 200 µm), ×200 (scale bar, 100 µm) and ×400 (scale bar,
50 µm; for identifying the biphasic glial and sarcomatous
components) for documentation.
 | Table I.Antibodies for
immunohistochemistry. |
Table I.
Antibodies for
immunohistochemistry.
| Antibody | Clone number | Recommended
dilution | Catalog number | Supplier |
|---|
| Actin | MX083 | 1:100 | MAB-0871 | Fuzhou Maixin Biotech
Co., Ltd. |
| CD34 | QBEnd/10 | Ready-to-use | Kit-0004 | Fuzhou Maixin Biotech
Co., Ltd. |
| Pan-CK | AE1/AE3 | Ready-to-use | 0349 | Shanghai Long Island
Antibody Diagnostica Inc. |
| Caldesmon | MX077 | 1:50 | MAB-0865 | Fuzhou Maixin Biotech
Co., Ltd. |
| Calponin | MX023 | 1:100 | MAB-0712 | Fuzhou Maixin Biotech
Co., Ltd. |
| Desmin | MX046 | 1:50 | MAB-0766 | Fuzhou Maixin Biotech
Co., Ltd. |
| EMA | MX132E29 | Ready-to-use | MAB-1101 | Fuzhou Maixin Biotech
Co., Ltd. |
| ER | MXR030 | 1:50 | RMA-1065 | Fuzhou Maixin Biotech
Co., Ltd. |
| GFAP | MX-047 |
Ready-to-use/1:100 | MAB-0769 | Fuzhou Maixin Biotech
Co., Ltd. |
| Ki-67 | MX-006 |
Ready-to-use/1:50 | MAB-0672 | Fuzhou Maixin Biotech
Co., Ltd. |
| PR | MXR008 | 1:50 | RMA-0895 | Fuzhou Maixin Biotech
Co., Ltd. |
| S-100 | rMM-S1 |
Ready-to-use/1:100 | 0466 | Shanghai Long Island
Antibody Diagnostica Inc. |
| SMA | 1D6 |
Ready-to-use/1:100 | MAB-0575 | Fuzhou Maixin Biotech
Co., Ltd. |
| SOX-10 | MXR026 | 1:100 | RMA-1058 | Fuzhou Maixin Biotech
Co., Ltd. |
| STAT-6 | YE361 | 1:100 | YE361 | Abcam Shanghai
Trading Co., Ltd. |
| Vim | MX034 |
Ready-to-use/1:200 | MAB-0735 | Fuzhou Maixin Biotech
Co., Ltd. |
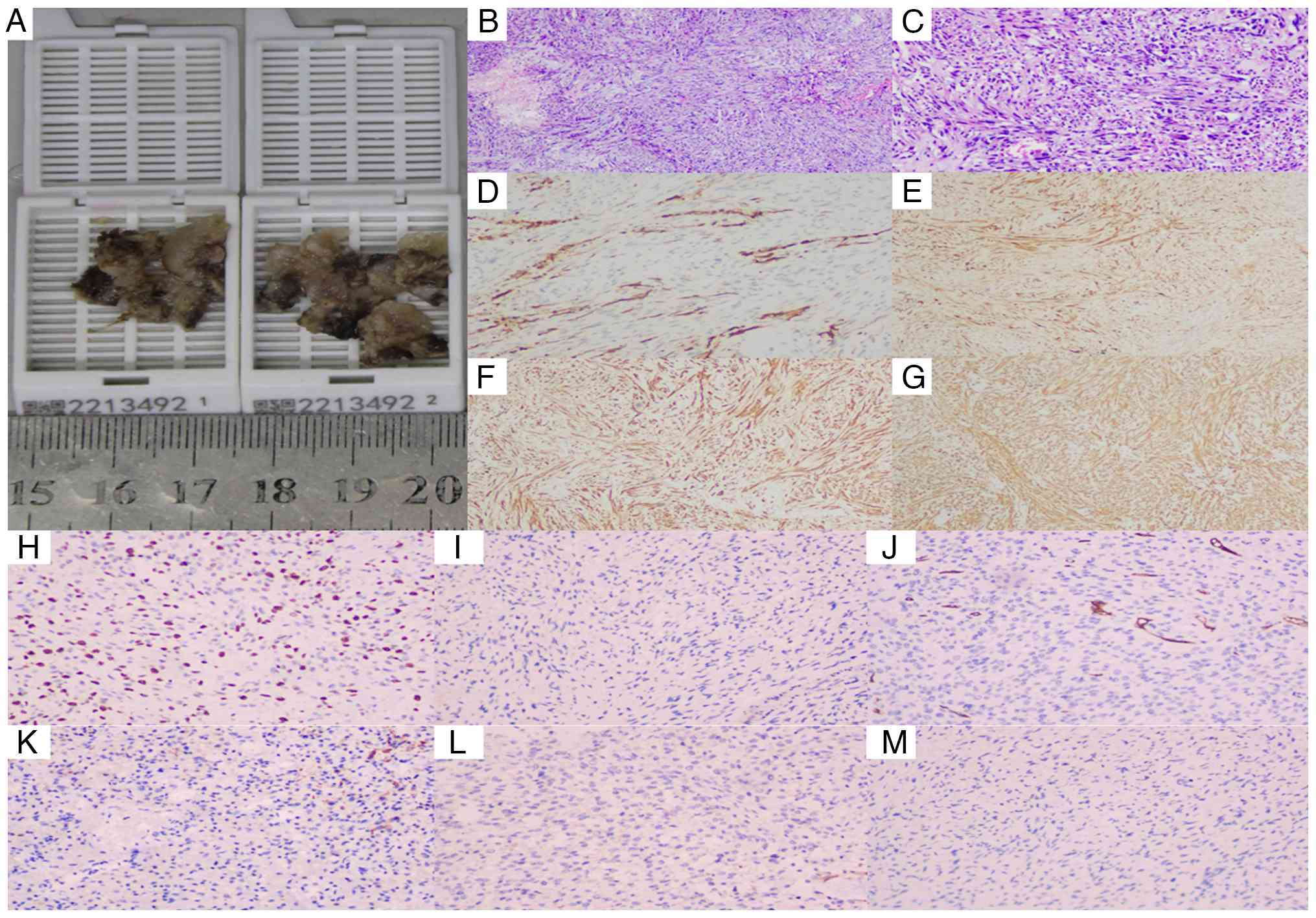
Pathological results
Histopathological examination confirmed the
diagnosis of GSM (WHO Grade IV) (2). Macroscopically, the tumor was
irregular with a firm, grey-brown cut surface. Microscopically, a
biphasic pattern was evident, comprising both glial and sarcomatous
components. The sarcomatous areas consisted of densely packed,
fascicularly arranged spindle cells with abundant cytoplasm and
prominent stromal vascular proliferation. The glial component was
composed of fibrillary and gemistocytic astrocytoma cells, arranged
in nests and diffuse sheets, with frequent mitotic figures
(Fig. 4B and C).
Immunohistochemical staining was critical for
establishing the definitive diagnosis. The staining profile
revealed a distinct immunophenotype: The neoplastic cell exhibited
focal positivity for calponin and GFAP (Fig. 4D), weak positivity for S-100
(Fig. 4E) and strong, diffuse
positivity for smooth muscle actin (SMA) and vimentin (Fig. 4F and G, respectively). A high
proliferative index was indicated by a Ki-67 labeling index of 30%
(Fig. 4H). Furthermore, the tumor
cells were consistently negative for a panel of markers including
caldesmon, CD34, cytokeratin (CK), EMA and progesterone receptor,
as illustrated in Fig. 4I-M. This
combined pattern of strong co-expression of SMA and vimentin with a
high Ki-67 index, alongside the absence of specific lineage
markers, was instrumental in narrowing the differential
diagnosis.
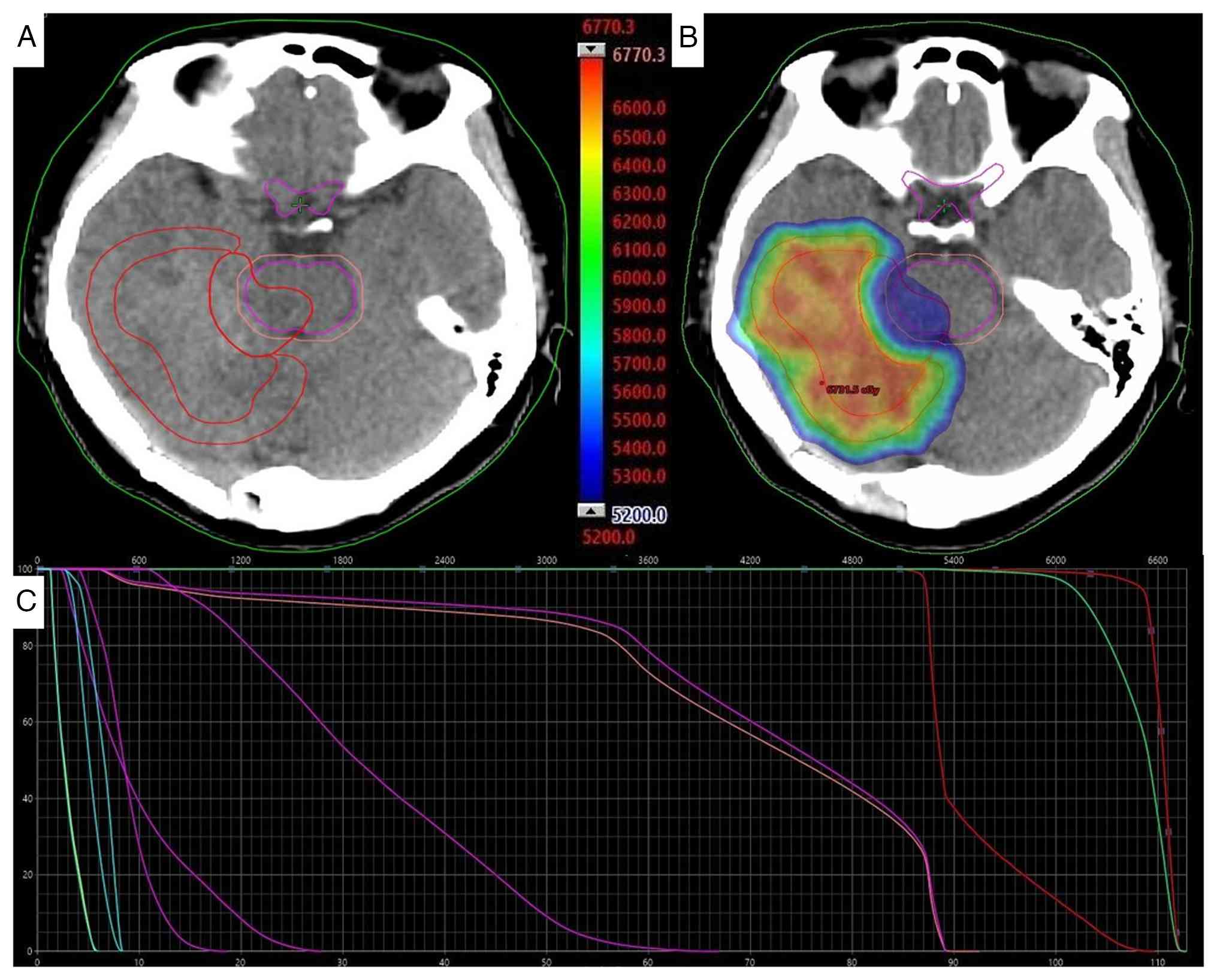
Postoperative treatment
Before radiotherapy, a head CT scan was performed
using a helical tomotherapy system (Accuray, Inc.). The scanning
parameters were as follows: The patient was positioned in a supine
position, utilizing helical scanning with a tube voltage of 120 kV,
tube current ranging from 200 to 250 mAsec and a slice thickness of
1 mm. The scanning range extended 2 cm from the upper edge of the
parietal scalp to the lower part of the skull base. This scan was
conducted for radiotherapy localization. The patient subsequently
received adjuvant IMRT concurrent with daily oral temozolomide at a
dose of 100 mg for a total of 38 days. The planning target volume
(PTV) for the postoperative bed (PTV1) received 6,500 cGy in 30
fractions, while the adjacent brainstem region (PTV2) received
5,200 cGy (max dose, 5,358.8 cGy) (Fig.
5). Supportive care included intravenous administration of 20%
mannitol (a routine dehydrating agent for cerebral edema) at a dose
of 250 ml per infusion, administered every 8 h (1–2 g/kg body
weight) via rapid intravenous drip (completed within 30–60 min),
for a total of 38 consecutive days.
Follow-up and outcome
The patient's most recent follow-up was in March
2023. At that time, a neurological examination revealed mild
limitations to the downward movement in the right eye, a positive
right finger-nose test and a positive Romberg sign, indicating
persistent cerebellar dysfunction. No significant abnormalities
were noted on routine laboratory tests. However, follow-up MRI
demonstrated significant enlargement and progression of the
recurrent lesion in the right cerebellar hemisphere and
parahippocampal region, with suspected brainstem invasion. This
rapid progression, observed despite combined modality therapy,
underscores the highly aggressive nature of cerebellar GSM.
Following this last evaluation, the patient's
condition continued to deteriorate due to disease progression, and
the patient succumbed to the disease in July 2023. This outcome
further underscores the dismal prognosis associated with this
aggressive tumor variant, particularly when located in the
posterior fossa.
Discussion
GSM has a poor prognosis with high recurrence rates
(15), as illustrated in the
present case, where the disease progressed rapidly leading to a
poor clinical status by the last follow-up in March 2023. Untreated
patients have a median overall survival time of 4 months, while
combined modality therapy extends this to 6.6–18.5 months (7). GSM is a highly aggressive tumor with a
median age of onset of ~52 years and a slight male predominance
(4). The clinical course is
typically rapid, characterized by significant mass effect and
peritumoral edema. Symptoms are directly related to the tumor's
location and invasion of adjacent structures, often leading to
elevated intracranial pressure (10). The current patient's presentation
with diplopia and occipital headaches is consistent with this
profile, exacerbated by the rare cerebellar location causing
brainstem compression. Beyond symptomatology, this anatomical
distinction fundamentally impacts therapeutic strategy. The compact
anatomy of the posterior fossa and proximity to critical brainstem
structures often preclude the aggressive surgical margins
achievable in non-eloquent supratentorial regions. Consequently,
the surgical goal in cerebellar GSM frequently shifts from a
gross-total resection to maximal safe debulking and cytoreduction,
inherently increasing the risk of local recurrence due to residual
disease burden.
MRI is the cornerstone of preoperative evaluation
for GSM. GSM typically presents as a heterogeneously enhancing mass
with ill-defined margins, marked edema and frequent dural
attachment, which can mimic a malignant meningioma (11). Signal characteristics often include
T1 hypointensity, T2 hyperintensity and heterogeneous post-contrast
enhancement, occasionally with a ‘reticular’ pattern (16). MRS consistently shows an elevated
Cho/NAA ratio, reflecting high cellularity and malignancy (17). The present case exhibited these
typical features, alongside the unusual finding of a large
cerebellar location and trans-tentorial spread.
The aggressive course observed in the present
patient, characterized by early recurrence and trans-tentorial
spread, can be critically contextualized within the broader
literature on GSM. While the biphasic histology and poor prognosis
are consistent with the defining features of GSM (1,12,13),
several aspects of the present case highlight its distinctiveness
and amplify its clinical challenge.
GSM most frequently arises in supratentorial
locations, particularly the temporal lobe, with cerebellar origins
being exceptionally rare (3,4). This
anatomical distinction is not merely incidental. Supratentorial
GSM, while aggressive, often permits a more extensive surgical
resection in non-eloquent areas. By contrast, in the present case,
the origin in the cerebellar hemisphere, with immediate proximity
to and compression of the brainstem, inherently limited the goal of
surgery to a subtotal resection for decompression. This unavoidable
surgical constraint likely contributed to the high residual tumor
burden, a factor strongly associated with early recurrence in GBM
and its variants (6,9). The presenting symptoms of ataxia and
brainstem compression are direct manifestations of this location,
differing from the more common seizures or focal neurological
deficits seen in temporal lobe GSM (10).
The Ki-67 proliferation index of 30% in the present
case is at the higher end of the spectrum reported for GSM
(typically ranging from 15 to 30%) (4,13).
This elevated proliferative activity provides a histopathological
link to the explosively aggressive behavior observed
radiologically, including the rapid trans-tentorial extension noted
at the 2-month follow-up. This pattern of spread is less commonly
detailed in reports of supratentorial GSM but may be facilitated in
cerebellar cases by anatomical routes along the tentorium.
Furthermore, the marked perilesional edema and heterogeneous
‘reticular’ enhancement pattern on MRI, as seen in the present
patient, are features described in GSM series (11,17).
However, when these features occur in the posterior fossa, the
resultant mass effect on the brainstem and fourth ventricle carries
more immediate life-threatening implications, such as herniation,
which ultimately occurred in the present patient at 12 months
postoperatively.
The median overall survival time for GSM treated
with multimodal therapy is 6.6–18.5 months (6,7). In
the present patient, the rapid progression leading to brain
herniation at 12 months aligns with the poorer end of this
spectrum. This outcome underscores that cerebellar GSM may confer
an even worse prognosis than its supratentorial counterparts, a
hypothesis supported by the compounded challenges of limited
resectability and critical adjacent neuroanatomy. This aligns with
findings of the study by Feng et al (9), which identified tumor location as a
significant prognostic factor in a nomogram analysis.
Pathologically, GSM is defined by its biphasic glial
(GFAP+) and sarcomatous
(vimentin+/SMA+) morphology (1,12,13).
There is no standardized treatment protocol for GSM.
Maximal safe surgical resection remains the primary treatment,
followed by adjuvant chemoradiotherapy, mirroring the Stupp
protocol for GBM (8,9). High-dose radiotherapy and combined
modality therapy have been identified as favorable prognostic
factors (14). Despite aggressive
therapy, the prognosis remains poor, with a median overall survival
time of 6.6–18.5 months and high recurrence rates (6). The early recurrence in the present
patient, despite combined-modality therapy, can be attributed to
the tumor's critical location limiting the extent of resection and
its inherent high malignancy, potentially with early CSF
dissemination.
In conclusion, the present study highlights a rare
case of cerebellar GSM with classic biphasic pathology and a highly
aggressive clinical course. Preoperative imaging, while suggestive
of a high-grade glioma, is non-specific, and a definitive diagnosis
relies on thorough histopathological and immunohistochemical
evaluation. The rapid recurrence and progression observed in the
present patient, despite multimodal therapy, underscore the
therapeutic challenges posed by GSM and emphasize the urgent need
for novel diagnostic and treatment strategies to improve patient
outcomes.
Acknowledgements
The authors would like to thank Dr Yihao Peng
(Department of Radiology, The General Hospital of Western Theater
Command, Chengdu, China) and Dr Jie Wu (Department of Radiology,
The General Hospital of Western Theater Command) for the
collection, sorting and verification of radiological and
histopathological data.
Funding
This research was supported by the Foundation of General
Hospital of Western Theater Command (grant no. 2024-YGLC-B07).
Availability of data and materials
The data generated in the present study are included
in the figures and/or tables of this article.
Authors' contributions
FW, YY, ZW, RJ designed the study and conceptual
framework. FW performed clinical data collection and case
follow-up, PW advised on patient treatment and analyzed patient
data, and YY and ZW assisted with clinical data collection. FW, PW
and TY obtained clinical specimens, performed imaging and conducted
the pathological analysis. FW sorted and verified the case data,
and PW, YY, RJ and TW analyzed and organized the patient data. FW,
PW and RJ drafted the case description and manuscript composition.
PW, YY and TY revised the manuscript and provided academic
polishing. FW, YY, RJ and PW prepared imaging, pathological figures
and tables. FW, PW and TY provided study design oversight and
clinical guidance. Project administration was performed by PW.
Funding acquisition was the responsibility of FW. All authors have
read and approved the final manuscript. FW and TY confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Written informed consent for the publication of this
case report and accompanying images was obtained from the patient
prior to her death.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Louis DN, Perry A, Reifenberger G, von
Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD,
Kleihues P and Ellison DW: The 2016 world health organization
classification of tumors of the central nervous system: A summary.
Acta Neuropathol (Berl). 131:803–820. 2016. View Article : Google Scholar
|
|
2
|
Louis DN, Perry A, Wesseling P, Brat DJ,
Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM,
Reifenberger G, et al: The 2021 WHO classification of tumors of the
central nervous system: A summary. Neuro Oncol. 23:1231–1251. 2021.
View Article : Google Scholar
|
|
3
|
Gerges C, Elder T, Penuela M, Rossetti N,
Maynard M, Jeong S, Wright CH, Wright J, Zhou X, Burant C, et al:
Comparative epidemiology of gliosarcoma and glioblastoma and the
impact of Race on overall survival: A systematic literature review.
Clin Neurol Neurosurg. 195:1060542020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen L, Rizk E, Sherief M, Chang M, Lucas
CH, Bettegowda C, Croog V, Mukherjee D, Rincon-Torroella J, Kamson
DO, et al: Molecular characterization of gliosarcoma reveals
prognostic biomarkers and clinical parallels with glioblastoma. J
Neurooncol. 171:403–411. 2024. View Article : Google Scholar
|
|
5
|
Karasev S, Talybov R, Chertoyev S,
Trofimova T, Mochalov V, Kleshchevnikova T, Loginova N and Karaseva
I: Diagnostic challenges of gliosarcoma: Case report of a rare
glioblastoma histopathological variant. Front Radiol.
5:16874012025. View Article : Google Scholar
|
|
6
|
Wang X, Jiang J, Liu M and You C:
Treatments of gliosarcoma of the brain: A systematic review and
meta-analysis. Acta Neurol Belg. 121:1789–1797. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
La Torre D, Della Torre A, Lo Turco E,
Longo P, Pugliese D, Lacroce P, Raudino G, Romano A, Lavano A and
Tomasello F: Primary intracranial gliosarcoma: Is it really a
variant of glioblastoma? an update of the clinical, radiological,
and biomolecular characteristics. J Clin Med. 13:832024. View Article : Google Scholar
|
|
8
|
Hashmi FA, Salim A, Shamim MS and Bari ME:
Biological characteristics and outcomes of gliosarcoma. J Pak Med
Assoc. 68:1273–1275. 2018.
|
|
9
|
Feng SS, Li HB, Fan F, Li J, Cao H, Xia
ZW, Yang K, Zhu XS, Cheng TT and Cheng Q: Clinical characteristics
and disease-specific prognostic nomogram for primary gliosarcoma: A
SEER population-based analysis. Sci Rep. 9:107442019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Saki A, Faghihi U and Balde I:
Differentiating gliosarcoma from glioblastoma: A novel approach
using PEACE and XGBoost to deal with datasets with ultra-high
dimensional confounders. Life (Basel). 14:8822024.PubMed/NCBI
|
|
11
|
Wang Y and Zhang Z: A case report:
Gliosarcoma associated with a germline heterozygous mutation in
MSH2. Front Neurol. 15:13882632024. View Article : Google Scholar
|
|
12
|
Jiang NN, Larrazabal R, Alsunbul W and Lu
JQ: Multiple pathological components in gliosarcoma. J Biomed Res.
35:408–410. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Castelli J, Feuvret L, Haoming QC, Biau J,
Jouglar E, Berger A, Truc G, Gutierrez FL, Morandi X, Le Reste PJ,
et al: Prognostic and therapeutic factors of gliosarcoma from a
multi-institutional series. J Neurooncol. 129:85–92. 2016.
View Article : Google Scholar
|
|
14
|
Adeberg S, Bernhardt D, Harrabi SB, Diehl
C, Koelsche C, Rieken S, Unterberg A, von Deimling A and Debus J:
Radiotherapy plus concomitant temozolomide in primary gliosarcoma.
J Neurooncol. 128:341–348. 2016. View Article : Google Scholar
|
|
15
|
Xu L, Yang Z, Chen H, Sun C, Tu C, Gu Z
and Luo M: Conditional survival and changing risk profile in
patients with gliosarcoma. Front Med (Lausanne). 11:14431572024.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Martinez A, Binks S, Pumarola M, Hardas A,
Easton A, Campo L, Browne M, Martins S, Garosi LS, Di Dona F and
Tauro A: Gliosarcoma associated with bilateral hippocampal
sclerosis in a cat presenting complex partial seizures with
orofacial involvement: A case report. Clin Case Rep. 12:e91232024.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fukada A, de Souza Queiroz L and Reis F:
Gliosarcomas: Magnetic resonance imaging findings. Arq
Neuropsiquiatr. 78:112–120. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gondi V, Bauman G, Bradfield L, Burri SH,
Cabrera AR, Cunningham DA, Eaton BR, Hattangadi-Gluth JA, Kim MM,
Kotecha R, et al: Radiation therapy for brain metastases: An ASTRO
clinical practice guideline. Pract Radiat Oncol. 12:265–282. 2022.
View Article : Google Scholar
|