Metabolic reprogramming has become the main topic of
a growing number of investigations examining the fundamental
origins of cancer. A artefact of this reprogramming is the Warburg
effect (1), a metabolic shift
defined by elevated glycolysis that leads to the excessive
accumulation of lactate (2). Long
dismissed as a simple metabolic ‘waste product’, lactate is now
understood to be a pivotal effector molecule; it functions as a key
energy substrate, a potent regulator of the tumor microenvironment
(TME) and an essential signaling molecule (3–5). The
recent identification of lysine lactylation (Kla) (6), a novel post-translational modification
(PTM) directly originating from lactate, has enhanced the new
understanding of the role of lactate. This finding has established
a direct and critical link between the metabolic state of a cell
and its epigenetic regulation. Lactylation represents a core
metabolic-epigenetic axis, translating the accumulation of lactate
into durable transcriptional programs. This axis is considered a
key driver of tumor cell proliferation, invasion, migration and,
most critically, therapeutic resistance (3,7,8). Given
its central role, understanding the enzymatic machinery that
governs this axis is paramount. The present review focuses on the
regulatory enzymes (‘writers’, ‘erasers’ and ‘readers’) that
control protein lactylation, discusses their mechanisms in cancer
and explores their therapeutic potential.
Recent tumor biology has shown that lactate is a key
source of energy for tumor cells. The ‘lactate shuttle’ enables
aerobic and hypoxic tumor cells to sustain a metabolic symbiosis,
in which glycolysis-derived lactate is continuously produced,
exchanged and reutilized. This coordinated circulation of lactate
fuels tumor proliferation, invasion and adaptation to an
oxygen-limited microenvironment (9,10).
Neurons can also uptake lactate through monocarboxylate transporter
(MCT)2 to use it as a fuel source (11). Research has revealed that lactate
can buffer dietary glucose and be utilized for energy almost as
efficiently as glucose (12). This
dual function of lactate connects tumor metabolism with systemic
energy balance.
Lactate is known to be a key regulator of cellular
metabolism and phenotype; it modulates metabolic reprogramming,
inflammatory responses and immune regulation. Lactate signaling
through G protein-coupled receptor (GPR)81 directly promotes energy
metabolism (13) or lactate itself
enters mitochondria to be incorporated into the TCA cycle,
activates the respiratory chain and increases ATP synthesis
(14). Most critically, lactate
serves as a central driver of immunosuppression within the TME.
Lactate activates GPR81, inhibiting MHCII presentation on the cell
surface, while reducing IL-12 secretion. This further weakens
antigen presentation capacity, preventing effective T cell
activation (15). In melanoma,
lactate is taken up by tumor-associated macrophages (TAMs),
reprogramming them toward a pro-tumor phenotype (16). While promoting tissue homeostasis,
lactate functions as both an immunosuppressant and a pro-carcinogen
(16). Concurrently, lactate
induces polarization of helper T (Th) and CD4+ T cells,
fostering an immunosuppressive environment that suppresses Th1
subsets critical for antitumor activity (17). Lactate inhibits the upregulation of
nuclear factor of activated T cells in T cells and natural killer
(NK) cells, leading to reduced interferon γ production. This
suppresses the tumor immune surveillance and antitumor functions of
T cells and NK cells, ultimately resulting in tumor immune escape
(18).
In conclusion, the function of lactate in tumor
biology has been entirely redefined. Large production of lactate
implies that metabolic reprogramming can be linked to epigenetic
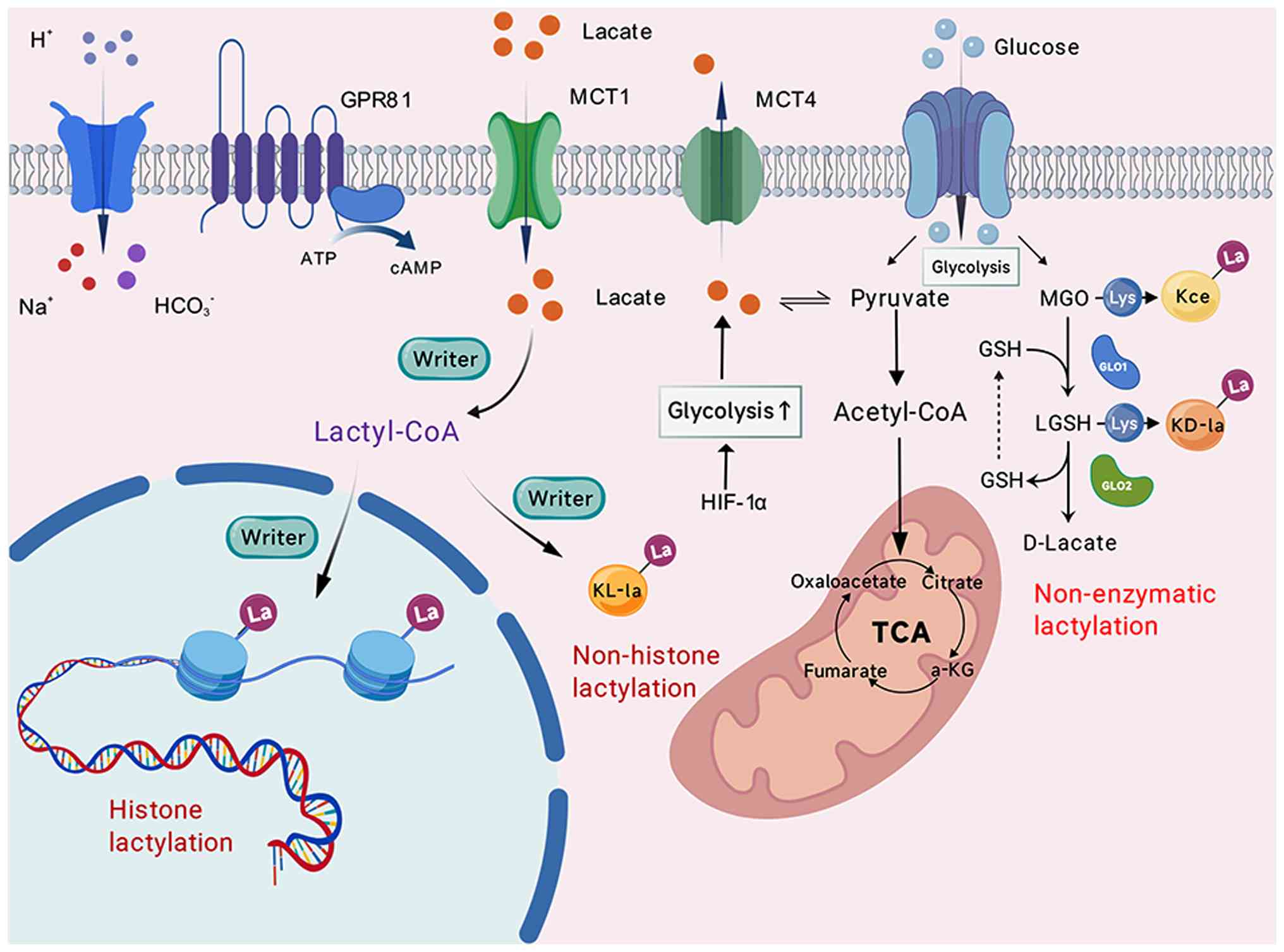
regulation through lactylation (19). Fig.
1 demonstrates the direct conversion of the lactate metabolic
into durable transcriptional programs that drive tumor
malignancy.
Kla provided the first evidence that lactate
directly modifies histones, leading to transcriptional changes in
macrophages and regulating the cellular transcriptome. This finding
has expanded the current understanding of the functions of lactate
(6). Subsequent studies (20–24)
revealed Kla as a versatile regulatory mechanism influencing gene
expression, metabolism and tumor progression. Tables I (writers) and II (erasers) (3,7,20–62)
collectively provide an enzymatic perspective on the functional
consequences of Kla, offering a comprehensive summary of specific
lactylation sites and their associated biological functions.
Lactate accumulation produced by glycolysis in tumor
cells is converted into lactyl-CoA, which serves as the donor of
the lactyl group. This lactyl group is subsequently transferred to
lysine residues on histone tails. This binding weakens the
attraction between histones and DNA, leading to a relaxed chromatin
structure and a more open state that is conducive to transcription,
thereby creating favorable conditions for gene transcription
initiation (63,64). Hla modifications, including at sites
such as H3K18 and H4K12, have been shown to play a key role in the
initiation, development, immune evasion and therapeutic resistance
of various cancer types. For example, H3K18la promotes cell cycle
and proliferation by specifically enriching at and directly
activating the promoters of key oncogenes, such as TTK protein
kinase, BUB1 mitotic checkpoint serine/threonine kinase B in
pancreatic ductal adenocarcinoma (PDAC) and c-Myc in colorectal
cancer (CRC) (20,65); it also drives cell invasion and
migration by activating the AKT-mTOR signaling pathway in gastric
cancer (GC) or enhancing epithelial-mesenchymal transition (EMT)
(66,67). Furthermore, H3K18la facilitates
immune evasion by promoting programmed death-ligand 1 (PD-L1)
expression and inhibiting CD8+ T cell function in GC and
lung adenocarcinoma (LUAD) (68,69),
and contributes to chemotherapy and radiotherapy resistance by
activating protective autophagy or ferroptosis resistance in CRC
and prostate cancer (70,71). Similar findings have been reported
for H4K12la (52,72). Recently, newly identified sites such
as H4K79la and H4K91la have also been shown to be involved in
breast cancer (BC) progression (73), indicating that, despite substantial
existing research, Hla continues to offer valuable potential for
the identification of new targets.
Throughout these tumor progression processes, the
persistently high lactate levels resulting from the Warburg effect
disrupt normal lactate metabolism. Since H3K18la affects DNA
transcriptional activity by enriching at the promoters of target
genes, such as the oncogene c-Myc (20), this lactylation modification
consequently confers therapy resistance by activating
survival-related genes, including glutamate-cysteine ligase
catalytic subunit (52).
Concurrently, some of those modifications (such as H3K18la and
H4K12la) further promote the expression of lactate metabolism
enzymes, leading to even higher intracellular lactate levels. This
creates a vicious positive feedback loop where lactate produced by
glycolysis reinforces glycolysis and lactate production through Hla
(74,75). In this manner, lactate is
transformed into a pivotal signaling molecule that governs
epigenetics and affects various processes, emerging as a critical
factor for the persistent malignant progression and therapeutic
suppression of cancer cells.
Unlike Hla, n-Hla occurs widely in the cell due to
the abundance of its substrate; it occurs in several cellular
compartments, including the nucleus, cytoplasm and mitochondria
(22,29,55),
and has broader biological consequences than Hla. First, n-Hla can
enhance protein stability, thereby amplifying oncogenic signaling
or promoting metabolic reprogramming. By competitively occupying
these sites, n-Hla inhibits proteasomal degradation and stabilizes
critical oncoproteins such as β-catenin (76), transcription factor EB (48), polypyrimidine tract binding protein
1 (38) and YTH domain containing 1
(YTHDC1) (3), thereby sustaining
oncogenic signaling and immune evasion. Second, n-Hla dynamically
reshapes the assembly, localization and function of signaling
complexes by regulating the conformation of proteins, thereby
enhancing or weakening their interactions with other proteins
(77). In addition, n-Hla can
enhance protein-nucleic acid interactions, which is evident in its
crosstalk with other modifications (such as m6A methylation and m5C
RNA modification), facilitating downstream signal transduction and
RNA processing (78–80). n-Hla can regulate energy metabolism
to facilitate signal transduction and the activation of oncogenic
pathways, including the AMP-activated protein kinase (21) and pentose phosphate (81) pathways. These effects not only alter
cellular energy metabolism but also expedite the malignant
development of tumor cells by supplying substrates for processes
such as nucleic acid synthesis. More notably, n-Hla is a mechanism
by which tumors utilize metabolic products to enhance the function
of immunosuppressive cells (32,82)
and inhibit tumor-killing cells (83), thereby shaping an inhibitory immune
microenvironment. By suppressing various immune cells, favorable
conditions are created for diminished tumor-inhibitory functions
and the onset of immune evasion, ultimately leading to a series of
immunotherapy resistance or suboptimal efficacy outcomes.
In summary, both Hla and n-Hla form a
lactate-derived network that reshapes gene expression and signaling
to drive tumor progression. Governed by specific writers, erasers
and readers, these regulatory nodes are fundamental for
understanding the lactylation landscape. Consequently, elucidating
their functions is crucial for translating this metabolic
modification into precise therapeutic targets and clinical
applications.
p300 [also known as EP300/lysine acetyltransferase
(KAT)3B] and its homologue CBP are well-studied histone
acetyltransferases (HATs) in acetylation modification (Kac)
(84,85). p300 is a prominent member of the
MYST family of acetyltransferases. Recent research indicates that
they also have lactyltransferase activity, as the HAT domain of
p300 contains a hydrophobic pocket that can accommodate a variety
of short-chain acyl-CoA molecules; it can recognize both acetyl-CoA
and structurally similar lactyl-CoA (26,48,49).
As the initially identified and most thoroughly
examined lactyl transferase, p300 significantly alters numerous
essential substrates. Thus, p300 is widely recognized as the
principal catalytic enzyme of Kla (20,24,29,33,53).
Lactylation of transcription factors regulated by p300/CBP
activates downstream signaling pathways, participating in tumor
growth and other biological processes including angiogenesis, cell
invasion and migration. For instance, under hypoxic conditions,
lactylation of the transcription factor Yin Yang 1 at its K183 site
by p300 directly activates the fibroblast growth factor 2 signaling
pathway, leading to tumor angiogenesis (86). Moreover, in several malignancies,
p300-mediated lactylation of ATP-binding cassette subfamily F
member 1 at K430 (22) and Snail1
(87) facilitates tumor cell
invasion and migration by advancing downstream processes such as
EMT.
The targeting specificity of p300 determines its
precise modifying activity. Recent findings have indicated that
p300, in combination with the histone chaperone anti-silencing
function protein 1 homolog A (ASF1A), directly regulates H3K18la
accumulation in the Snail1 promoter region, hence specifically
activating transcription and inducing atherosclerosis via
endothelial-to-mesenchymal transition (EndMT) (88). This shows that p300 may be led to
specific gene loci by different types of cofactors, triggering
changes at distinct places. Thus, examining these histone
chaperones could aid in understanding the mechanisms by which
writers precisely detect and bind targets, deciphering the
targeting specificity of lactylation changes and enabling the
development of novel intervention strategies.
Similar to p300/CBP, HATs of the MYST family have
also been found to function as lactyltransferases, likewise
depending on lactyl-CoA. Members of this family include KAT2A (also
known as GCN5), KAT8 (MOF), KAT5 (Tip60) and KAT7 (also known as
HBO1). For instance, KAT8-mediated lactylation of elongation factor
1 α2 at its K408 site enhances its GTPase activity, thereby
promoting protein synthesis as well as the proliferation and
migration of tumor cells (62).
Notably, the lactylation catalytic activity of KAT8 is dependent on
intracellular lactate levels, specifically requiring the
recognition of lactyl-CoA as a donor. However, knocking down KAT8
significantly reduces the modification level at lactylation sites,
without affecting lactate concentration, indicating its direct
involvement in the modification process rather than in regulating
lactate metabolism. That is, its mechanism of action is to transfer
the lactyl group from lactyl-CoA without affecting the metabolism
of lactate itself (62). In
addition, KAT8-mediated lactylation is also closely associated with
collagen synthesis (89) and
ferroptosis resistance (90).
Meanwhile, KAT2A and KAT5, which are involved in lactylation at
various sites, are closely related to processes such as promoting
DNA double-strand breaks and chemotherapy resistance (34,37,57).
Although MYST family enzymes, including p300, are
widely regarded as major contributors to Kla, their involvement
largely reflects a functional extension of classical
acetyltransferases rather than a dedicated lactylation system. This
repurposed activity exposes several limitations of the
CoA-dependent model. First, the intracellular source and abundance
of lactyl-CoA remain insufficiently defined, and, even under
optimized conditions, its level is markedly lower than that of
acetyl-CoA (~1/1,000) (91),
suggesting that CoA-dependent lactylation may occur only within
restricted metabolic niches. Second, since these enzymes use both
acetyl-CoA and lactyl-CoA, Kla inevitably competes (92) or synergizes (75) with Kac at shared lysine residues,
raising unresolved questions regarding how these PTMs are
selectively regulated. These constraints indicate that
CoA-dependent acyltransferases may not fully account for the
efficiency or specificity of Kla, suggesting the potential
existence of non-enzymatic pathways that influence the progression
of this modification.
In contrast to lactyl-CoA-dependent HATs, AARS1/2
represent a mechanistically distinct class of lactylation writers
and their identification has resolved the dilemma of lactyl-CoA
scarcity. AARS1 functions as a direct lactate sensor; it binds to
l-lactate and utilizes ATP to catalyze the formation of a
lactyl-AMP intermediate, subsequently transferring the lactyl group
covalently to the lysine residues of target proteins and releasing
AMP (93). Previous molecular
docking research has shown that conserved residues within AARS1
(M46, R77, N216 and D239) directly bind lactate in a reaction that
requires only l-lactate and ATP (23). This mechanism is considerably more
direct than that proposed for p300 and explains the presence of
active lactylation in tumor tissues where a lactyl-CoA-producing
enzyme had not been previously identified.
AARS1 and AARS2 have different intracellular
localizations and thus regulate distinct biological processes.
AARS1 is primarily distributed in the cytoplasm and nucleus. In the
nucleus, it can lactylate key oncoproteins, such as yes-associated
protein (YAP) and TEA domain transcription factor 1, thereby
activating the Hippo signaling pathway and promoting GC cell
proliferation (36). Concurrently,
AARS1 can also lactylate the tumor suppressor p53, which inhibits
its DNA-binding ability and transcriptional activity, thereby
impairing its tumor-suppressive function (23). By contrast, AARS2 is localized to
the mitochondria and it regulates key enzymes involved in
mitochondrial aerobic metabolism, such as pyruvate dehydrogenase
E1α subunit 1 and carnitine palmitoyltransferase 2, through
lactylation, thereby reducing their activity and suppressing
oxidative phosphorylation (OXPHOS). In a high-lactate environment,
this inhibition of OXPHOS decreases cellular oxygen consumption and
enhances resistance to mitochondrial oxidative stress, ultimately
facilitating tumor cell adaptation to metabolic stress conditions
(94).
AARS1-mediated lactylation is also closely linked to
therapeutic resistance. In bladder cancer cells under high-glucose
conditions, lactylation of YTHDC1 downregulates JunD
proto-oncogene, AP-1 transcription factor subunit and reduces
Nectin4 expression, thereby diminishing the sensitivity of the
cells to the Nectin4-targeted antibody-drug conjugate, enfortumab
vedotin (55). Lactylation of the
key homologous recombination (HR) helicase BLM RecQ like helicase
(BLM) at K24 prevents its ubiquitin-dependent degradation, thereby
stabilizing the protein and promoting DNA end resection and HR
repair, ultimately leading to chemoresistance to anthracyclines
(56). Furthermore, lactylation of
nudix hydrolase 21 at K23 induces 3′-untranslated region
lengthening of ferredoxin 1 (FDX1) mRNA, resulting in reduced
protein expression. The decreased FDX1 levels weaken the capacity
of FDX1 to reduce Cu2+ to the more cytotoxic
Cu+, thereby conferring resistance to cuproptosis in
esophageal squamous cell carcinoma (ESCC) (30).
These findings indicate that AARS1 functions far
beyond a simple writer enzyme; it serves as a central hub
integrating metabolic signaling, protein synthesis and oncogenic
pathways. Canonically, AARS1 catalyzes the attachment of alanine to
its cognate tRNA, initiating protein synthesis. However, under
high-lactate conditions, lactate competes with the traditional
substrate L-alanine for binding to the active site of AARS1,
allowing it to operate as a lactyltransferase (23,95).
This functional shift effectively translates metabolic signals into
long-term activation of oncogenic pathways and malignant
phenotypes. Mechanistically, the lactyltransferase activity of
AARS1 depends on its nuclear translocation, which requires a
C-terminal, evolutionarily conserved nuclear localization signal
(NLS) motif. The importin protein karyopherin subunit alpha 4 binds
to this NLS, allowing AARS1 to enter the nucleus (36). Therefore, blocking AARS1 nuclear
translocation can selectively disrupt tumor cell signaling without
affecting normal cellular lactate metabolism, thus representing a
promising therapeutic strategy.
However, AARS1 appears to lack a highly specific
recognition mechanism for lactate and may not distinguish it from
homologous amino acids such as serine and glycine, potentially
leading to confounding effects from other amino acid modifications
(93). Furthermore, the lactylation
of targets by AARS1 may not be precise, as its substrate scope
appears to be broad, possibly leading to the simultaneous
lactylation of numerous other proteins that could also promote
tumor progression (23).
Recent research has identified several enzymes
responsible for synthesizing lactyl-CoA, thereby confirming the
physiological relevance and research potential of the CoA-dependent
pathway.
GTPSCS has also been identified as a lactyl-CoA
synthetase in glioma. After entering the nucleus, it binds with
p300 to form a lactyltransferase complex. A hydrogen bond at the
N308 site binds to lactate to generate lactyl-CoA, specifically
enhancing Hla (such as H3K18la), thereby upregulating the
expression of growth differentiation factor 15 and driving glioma
proliferation and radiotherapy resistance (47).
The identification of these enzymes provides a solid
biochemical foundation for the lactyl-CoA-dependent lactylation
pathway, resolving the issue of lactyl-CoA production; it indicates
that cells have evolved specialized mechanisms to ensure that, when
needed, these regulatory enzymes can undergo nuclear translocation,
provide an adequate supply of lactyl-CoA substrate and act in
concert with downstream lactyl-CoA transferases (such as p300) to
achieve precise epigenetic regulation of specific genes.
Histone deacetylases (HDACs) comprise a family of 18
enzymes. Notably, specific members, particularly the class I HDACs
(HDAC1-3) and the class III sirtuins (SIRT1-3), have been confirmed
to possess robust delactylase activity.
Members of the HDAC family are the primary erasers
of lactylation. Specifically, class I HDACs (HDAC1, HDAC2 and
HDAC3) have been confirmed to possess strong delactylase activity.
The catalytic activity of classical HDACs (classes I, II, and IV)
is dependent on a Zn2+ located at the bottom of the
active site pocket. This ion activates the carbonyl bond of the
lactyl group, making it easier for water molecules to hydrolyze it
through nucleophilic attack. This mechanism is structurally
comparable to their conventional deacetylation action; both
processes restore chromatin to its compact shape and repress gene
transcription (97,98). Among these enzymes, HDAC3 exhibits
the strongest activity and can remove both L- and D-isomers of
lactylation (98). HDAC1-3 can
reduce the Kla levels of Hla (20,24,33)
and n-Hla (26,30,34),
acting as negative regulators in this process. For instance, HDAC3
associates with p300 and Brahma-related gene 1 (Brg1), which is a
key chromatin remodeling factor, to control the expression of
H3K18la. This activates ETS-related gene and MMP9, which leads to
liver metastasis in CRC (33).
In addition, deacetylation modifications can also
promote malignant cancer progression. HDAC2 is deeply involved in
regulating multiple Kla-mediated mechanisms of therapeutic
resistance. Specifically, HDAC2-driven delactylation of
methyltransferase-like 3 (METTL3), a key m6A RNA methyltransferase,
enhances m6A methylation associated with DNA damage
repair, contributing to cisplatin resistance in triple-negative
breast cancer (31). Furthermore,
HDAC2 decreases p300-mediated PD-L1 K189la, thereby promoting PD-L1
nuclear translocation and cholesterol biosynthesis, which in turn
drives hepatocellular carcinoma (HCC) growth (29). Moreover, HDACs display broad
substrate adaptability, showing robust activity not only against
l-lactylation but also against D-lactylation and various other
short-chain acyl modifications (97).
SIRT6 has been reported to be involved in the
delactylation of H3K9 and H3K18, demonstrating a complementary role
with class I HDACs in deacylation. While single knockout of SIRT6
does not significantly elevate global histone acetylation levels,
H3K9 acetylation markedly increases when SIRT6 is knocked out
concurrently with HDAC inhibition. This indicates a functional
compensation or overlap between SIRT6 and HDACs in deacetylation
(103). Furthermore, previous
research indicates that NAM has no significant effect on either the
delactylation activity in cell lysates or on intracellular Kla
levels, which suggests that HDAC1-3 may function as the primary
intracellular erasers for lactylation, rather than SIRTs (98).
Reader proteins of protein lactylation specifically
recognize and bind to lactyl groups, thereby influencing gene
expression and cellular activities by modulating chromatin
structure or protein function. Research on Kla reader proteins
remains limited. Drawing parallels from Kac studies, readers
primarily include proteins containing bromodomains [BRDs; such as
BRD4 and tripartite motif containing 33 (TRIM33)], plant
homeodomain (PHD) zinc finger domains (such as bromodomain PHD
finger transcription factor) and YEATS domains.
Brg1 is a core subunit of the BAF
(Brg1/Brm-associated factor) chromatin remodeling complex and
specifically recognizes and marks H3K18la during the reprogramming
of induced pluripotent stem cells (33,104).
Brg1 collaborates with Dux proteins to promote metabolic
reprogramming and mesenchymal-to-epithelial transition; it also
recruits writer enzymes such as p300/CBP, forming a positive
feedback loop that further upregulates H3K18la levels, thus
enhancing chromatin accessibility and transcriptional activity
(104). A previous study has shown
that the long non-coding RNA (lncRNA) STEAP3-AS1 may regulate Brg1
in H3K18la, leading to liver metastasis in CRC (33). This suggests that readers do not
control the Kla process independently; instead, the lactylation
signal needs to be recognized and marked by a reader, which then
recruits powerful chromatin remodeling machinery to execute
subsequent functions.
DPF2 was one of the first highly specific
lactylation reader proteins to be identified (105). DPF2 is a member of the BAF
chromatin regulatory complex and has been identified as a specific
reader that regulates H3K14 lactylation in cervical cancer (CC);
its double PHD finger domain directly binds to H3K14la through
hydrogen bonds and hydrophobic interactions, showing no affinity
for the structurally similar H3K14 acetylation, thus exhibiting
high specificity. Functionally, DPF2 is recruited by H3K14la to the
promoter regions of genes, where it recruits the BAF chromatin
remodeling complex to activate the transcription of the downstream
target oncogenes MYC and cyclin cyclin D1, thereby driving cell
proliferation and progression in cancer types such as CC (105). However, a limitation of the
aforementioned study is that it did not clarify whether DPF2
recognizes and binds H3K14la independently or relies on the BAF
complex, with DPF2 merely serving as a key binding site. An
important future direction of research will be to determine whether
DPF2 recognizes H3K14la independently or as part of the BAF
complex, and to what extent this specific recognition mechanism is
conserved across different cancer types
BRDs are a classic reader module for acetylated
lysine, present in >40 human proteins, which plays a core role
in transcriptional regulation (106,107). The BRD of TRIM33 achieves
high-affinity binding with Kla by forming a hydrogen bond between a
unique glutamate residue (E1041) in its binding pocket and the
hydroxyl group of Kla. The PHD domain of TRIM33 recognizes H3K9me3,
while its BRD binds H3K18ac or Kla. This ‘multi-recognition’
further enhances its ability to recognize and bind chromatin,
enabling it to translate metabolic signals from lactate into
epigenetic regulation at the chromatin level, making it a potential
therapeutic target for diseases associated with
metabolic-epigenetic dysregulation (108). Since BRDs can recognize both
acetylation and lactylation sites (for example, BRD4 is involved in
the malignant progression caused by both Kla and Kac) (109,110), BRD4 inhibitors such as JQ1
(111) are likely to inhibit both
signals simultaneously, achieving a multi-inhibitory effect. This
could play a broad role in various cancer types caused by metabolic
reprogramming.
Although research on the readers of Kla is still in
its early stages, these proteins are becoming recognized as the
main reason for the specific recognition and downstream signaling
of this modification. Reader proteins have the unique ability to
bind to the lactyl group and send its signal to downstream gene
targets. This is different from the less specific writer and eraser
enzymes, which often control multiple types of acyl modifications.
This intrinsic specificity indicates that targeting the distinct
binding sites of reader proteins constitutes a highly attractive
therapeutic approach, potentially providing a more accurate
intervention than the inhibition of the generally active writers
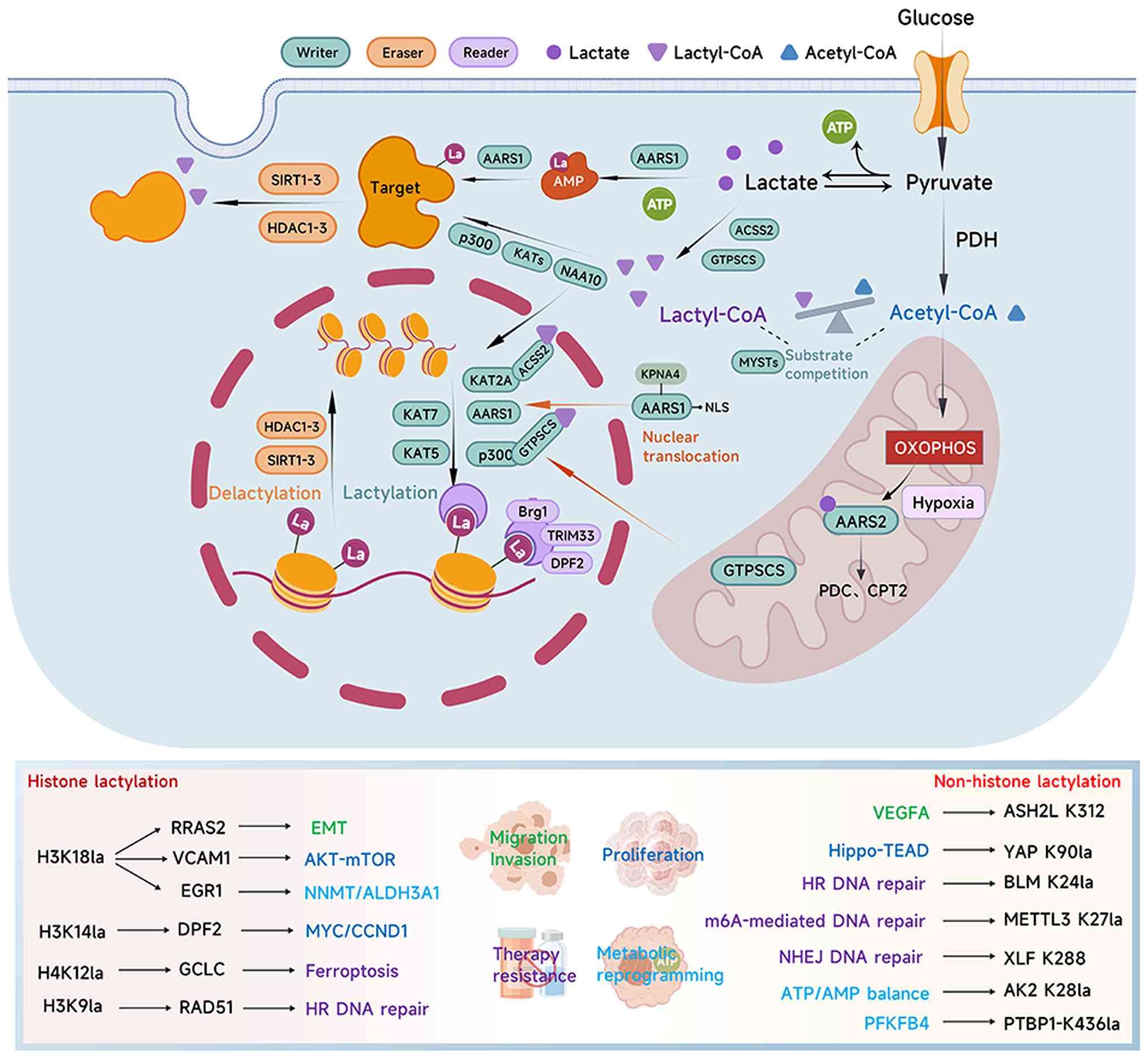
and erasers. Fig. 2 depicts the
structural interplay of the ‘writer-eraser-reader’ system,
demonstrating how these enzymes coordinate to translate
intracellular lactate levels into precise gene expression
outcomes.
Three isomers of Kla have been reported:
l-lactyl-lysine (Kl-la), d-lactyl-lysine (Kd-la) and
N-ε-(carboxyethyl)lysine (Kce). While Kl-la is enzymatically
regulated and serves as the dominant form due to the abundance of
l-lactate in mammalian cells (112,113), Kd-la and Kce are generated through
non-enzymatic mechanisms central to the metabolism of the
glycolysis byproduct methylglyoxal (MGO). Kce is formed through the
direct binding of MGO to lysine residues. Concurrently, MGO is
catalyzed by glyoxalase 1 (GLO1) to conjugate with glutathione,
forming S-d-lactoylglutathione (LGSH), which is subsequently
hydrolyzed by GLO2 to produce d-lactate. The generation of the
reactive intermediate LGSH can lead to the non-enzymatic
lactylation of lysine residues, forming Kd-la (114,115). This process is driven directly by
LGSH and does not depend on traditional lactyltransferases. Since
mammalian LDH and glycolysis almost exclusively produce l-lactate
(113), the physiological pool of
d-lactate is generally limited. Apart from the MGO pathway,
d-lactate is primarily derived from carbohydrate fermentation by
gut microbiota such as Lactobacilli (116).
Current evidence indicates that, in HCC, d-lactate
activates macrophages to induce TNF-α and IL-6 production,
remodeling the TME by regulating M2 TAMs into an immunosuppressive
state (117,118). In ESCC, cancer cells upregulate
LDHD to metabolize d-lactate into pyruvate. This metabolic
adaptation meets energy demands and facilitates the evasion of
d-lactate-induced ferroptosis, highlighting LDHD as a viable
therapeutic target (119).
Furthermore, both l- and d-lactate have been shown to facilitate
DNA repair and confer resistance to anticancer therapy in CC cells
via HDAC inhibition and hydroxycarboxylic acid receptor 1
activation (120). Crucially, the
structural similarity between l- and d-lactate suggests a potential
for crosstalk and competition. d-lactate has been observed to
promote H3K18la levels (121),
raising the possibility that d-lactate or its derivatives could
competitively bind to the active pockets of writers, erasers or
readers, thereby interfering with the regulation of l-lactylation.
Whether this interaction represents a synergistic amplification of
lactylation signaling or an antagonistic blockade remains to be
further investigated. Given that d-lactate participates primarily
via non-enzymatic routes while l-lactate relies on enzymatic
transfer (4,112), precise experimental designs are
required to dissect the specific contributions of
lactyl-CoA-dependent vs. -independent pathways.
As lactate can be converted to lactyl-CoA, the
direct substrate for lactylation, the most effective intervention
strategy is to inhibit its production and transport at the source
with the aim of reducing lactate accumulation in the TME, thereby
diminishing the substrate supply for downstream Kla.
Firstly, the glycolytic inhibitor 2-deoxy-d-glucose
(2-DG) suppresses hexokinase activity and blocks the glycolytic
flux, leading to a reduction in site-specific lactylation. 2-DG
suppresses LUAD progression (125)
and restores lenvatinib sensitivity by disrupting the insulin-like
growth factor 2 mRNA-binding protein 3 - phosphoenolpyruvate
carboxykinase 2 - S-adenosylmethionine - m6A feedback
loop in HCC (80). Secondly,
targeting LDH directly limits the conversion of pyruvate to
lactate. LDH inhibition suppresses the lactylation of DNA repair
proteins such as meiotic recombination 11-K673la (126) and Nijmegen breakage syndrome
1-K388la (34), impairing HR and
enhancing chemosensitivity. The LDH inhibitor GSK2837808A blocks
mitochondrial Mic60-K282la, disrupts cristae remodeling and
oxidative metabolism and restores sensitivity to EGFR-tyrosine
kinase inhibitors (127). Oxamate,
another LDH inhibitor, reduces lactate accumulation and alleviates
immunosuppression within the TME by blocking H3K18la, thereby
enhancing chimeric antigen receptor T cell activity in glioblastoma
(128). Combined treatment with
2-DG and oxamate further suppresses H3K18la, augments
CD8+ T cell cytotoxicity and prevents immune evasion in
non-small cell lung cancer (NSCLC) (129). Finally, blocking lactate shuttling
across the plasma membrane disrupts metabolic symbiosis between
tumor and stromal cells, reversing the acidic TME. The MCT4
inhibitor VB124 (130) and the
glucose transporter 1 inhibitor BAY-876 (131) have shown promising prospects. The
dual MCT1/4 inhibitor 7ACC1 suppresses oxaliplatin resistance by
preventing lactate transfer from cancer-associated fibroblasts and
inhibiting anthrax toxin receptor 1-K453la (132).
Despite encouraging preclinical evidence, the safety
and efficacy of these inhibitors require validation through
large-scale animal models and clinical trials. Notably, a phase I
clinical trial of the MCT1 inhibitor AZD3965 for advanced solid
tumors (NCT01791595) has been completed, laying the groundwork for
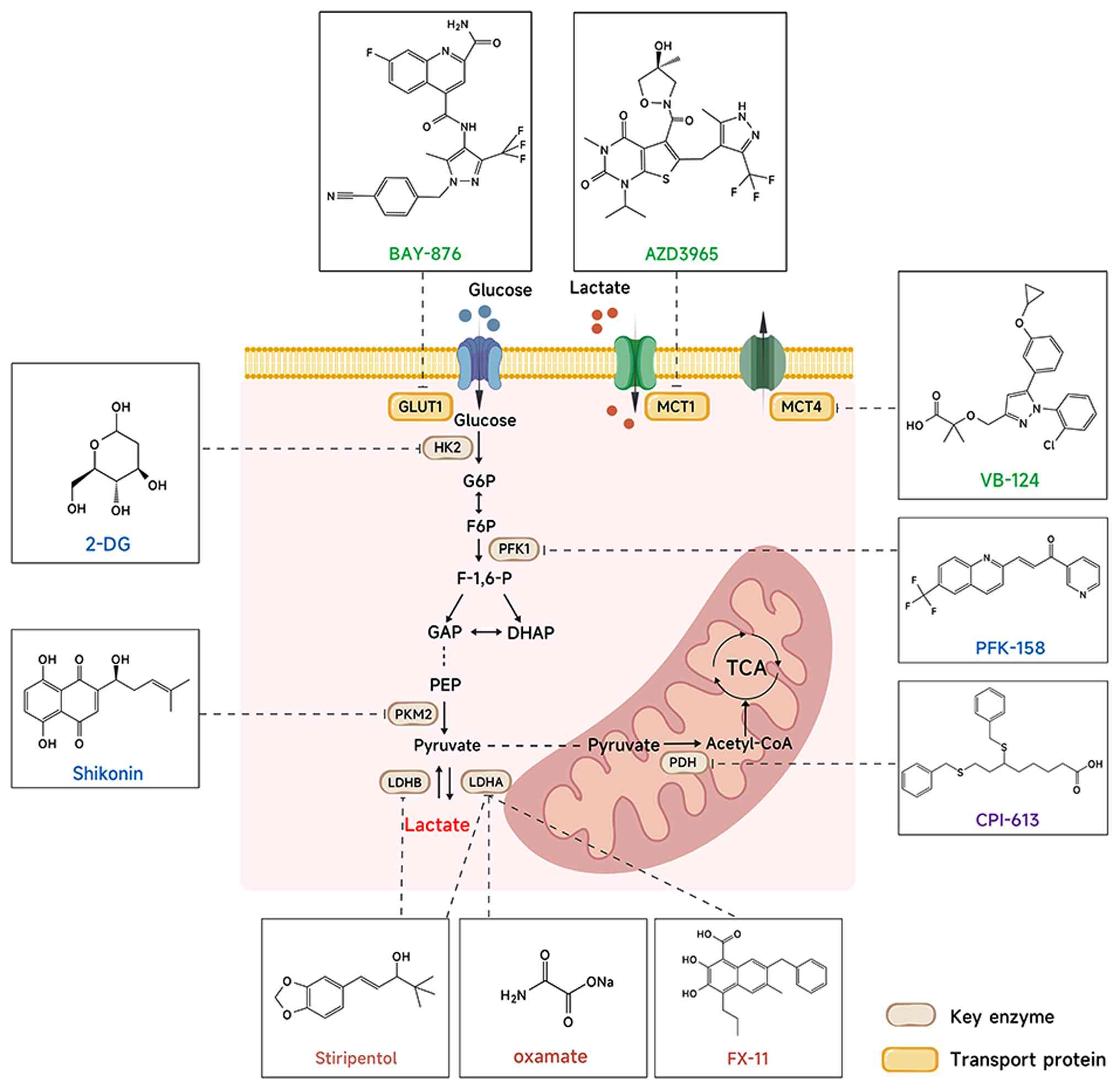
future clinical development. Fig. 3
summarizes representative inhibitors acting on key metabolic nodes,
while Table III summarizes
representative inhibitors targeting enzymes involved in lactate
metabolism and protein lactylation, along with their inhibitory
potency and the cancer models evaluated (133–156).
At present, commonly used small molecule inhibitors
for p300/CBP are primarily A-485 (67,86)
and C646 (51). These highly
selective and cell-permeable catalytic inhibitors of p300/CBP
competitively bind to their HAT domain thereby inhibiting acyl
transfer, and have shown good efficacy in various cancer types,
including HCC, PDAC, CRC and BC (49,51,52,73).
Due to the challenges posed by the functional
overlap and off-target effects of conventional small-molecule
inhibitors, developing novel therapeutic modalities that afford
high-specificity regulation has become a key research priority. In
this context, targeted protein degradation technologies such as
PROTACs have emerged as particularly promising strategies. PROTACs
are hetero-bifunctional molecules that link a target protein to an
E3 ubiquitin ligase, inducing the ubiquitination of the target
protein and its subsequent degradation by the proteasome. With
their potential for lower toxicity and the ability to overcome drug
resistance, PROTACs may represent a superior alternative to
conventional small-molecule inhibitors (145,161). For instance, the PROTAC hexokinase
2 (HK2) degrader-1 has been shown to overcome the limited efficacy
of the traditional glycolysis inhibitor 2-DG, which arises from
substrate competition. This approach effectively inhibits
glycolysis and reduces lactate levels, causing a significant
decrease in downstream H3K18la levels, reversing the EndMT process
and markedly alleviating the progression of atherosclerosis in a
mouse model (88).
Some progress has already been made in developing
PROTACs that target specific lactylated sites (54,162).
For instance, the PROTAC BP3 targets tumor necrosis factor
receptor-associated protein 1 degradation, thereby alleviating
vascular smooth muscle cell senescence and atherosclerosis induced
by H4K12la (162). In
lactylation-driven tumors, this strategy focuses on degrading key
downstream effector proteins to halt malignant progression. For
example, H3K18la is known to upregulate aurora kinase B (65) and acetyl-CoA acetyltransferase 2
(ACAT2) (54). Consequently,
PROTACs targeting these proteins could potentially enhance
antitumor efficacy. Specifically, in PDAC, the ACAT2-targeting
degrader AP1 reverses cholesterol-linked immunosuppression driven
by the H3K18la/ACAT2/mitochondrial carrier 2 axis. Furthermore,
combining AP1 with anti-programmed cell death protein 1 (anti-PD-1)
therapy was shown to significantly inhibit tumor growth and restore
antitumor immunity in a previous study (54).
However, extending this concept to the lactylation
regulatory enzymes themselves offers a more upstream intervention
strategy. This approach allows for the elimination of the entire
protein, including its non-catalytic scaffolding functions and
protein-protein interaction capabilities. This is particularly
important for multi-domain proteins such as p300/CBP and HDACs,
achieving results that traditional small-molecule inhibitors cannot
match. Notably, PROTACs targeting these enzymes have already
demonstrated significant translational potential in Kac. For
instance, the p300/CBP degrader dCBP-1 (163) exhibited significantly superior
anti-proliferative activity in multiple myeloma cells compared with
catalytic or BRD inhibitors (A-485 and GNE-781). Similarly,
previous research on HDACs indicates that optimizing linkers and
selecting appropriate E3 ligases can yield highly specific
degraders, thus underscoring the design flexibility of this
technology (161). A recent study
reported the development of a series of SIRT2-directed PROTAC
degraders derived from the conventional inhibitor Tenovin-6. The
lead compound, W10, catalyzed target protein degradation and
demonstrated potent anti-proliferative activity across multiple
ovarian cancer cell lines. Notably, this approach enhanced the
anticancer activity of W10 by ~53-fold compared with its parent
inhibitor Tenovin-6 and no pathological organ damage was observed
in mouse models (145).
Since PROTACs target the entire protein for
degradation, they offer a more precise intervention than
traditional inhibitors, but this approach is not without
limitations. Complete removal of multifunctional enzymes that
regulate multiple PTMs may result in new toxicities or off-target
effects. Therefore, precisely blocking the interactions or
functions necessary for the enzyme's role within the lactylation
pathway may be a more sophisticated approach than completely
eliminating the enzyme.
Due to the extensive overlap between the regulatory
machinery of lactylation and multiple PTMs, existing inhibitors and
drugs are often non-selective. Directly regulating these regulatory
enzymes can lead to broad, unpredictable changes in gene expression
and potential toxic side effects. Current strategies primarily
focus on targeting upstream metabolic enzymes or lactate
transporters as well as on lactylation inhibitors and
small-molecule peptides designed for specific modification sites
(mainly n-Hla). While these methods can effectively reduce lactate
and modification levels, they still lack precision or are difficult
to develop (164,165). A more promising alternative may be
the development of agents that can selectively inhibit the
generation of key substrates and enzyme binding. For instance, by
blocking the interaction between GTPSCS and p300, it is possible to
inhibit local lactyl-CoA synthesis without affecting succinyl-CoA
synthesis or the acetylation function of p300 (47). This has been demonstrated through
site-directed mutagenesis, which can inhibit the nuclear
translocation of GTPSCS and its subsequent catalytic activity for
lactyl-CoA synthesis. Similarly, the nuclear translocation of ACSS2
and its function as a lactyl-CoA synthetase are regulated by
phosphorylation at S267. Disrupting the ACSS2-KAT2A interaction has
been shown to effectively inhibit tumor growth without altering
histone acetylation levels (96).
Another strategy may involve targeting the reader proteins. Once
the specific recognition and binding domains of readers such as
TRIM33 or DPF2 are clearly identified, drugs that block their
lactylated lysine-binding pockets could be developed, offering
higher specificity and drugs that are more therapeutically
actionable. This highlights the need for a thorough mechanistic
understanding of lactylation regulatory enzymes to rationally
develop more efficient lactylation-targeted therapies (115).
The substrate-based technique is a more advanced
treatment approach. Rather than blocking or degrading the entire
enzyme, this method achieves targeted intervention by disrupting
key molecular interactions required for lactylation. Its main
advantage is its ability to selectively inhibit the lactylation
pathway while keeping the other regulatory functions of the enzyme,
reducing off-target consequences. However, this strategy is still
mostly conceptual. To apply it effectively, the current
understanding of the structural and molecular mechanisms involved
needs to be enhanced.
Kla promotes tumor adaptation and survival against
numerous treatments by regulating DNA repair, protein stability,
metabolic pathways and immune evasion (32,166–168). Table
IV illustrates the role of lactylation modification in therapy
resistance and the current interventions targeting it. Notably, in
addition to developing highly specific tools, synergistic
combination therapies could be designed (7,8,30,32,34,37,49,55–57,80,82,126,127,132,166–177).
By simultaneously targeting lactate metabolism enzymes and
lactylation regulatory enzymes, dual control can be achieved at
both the source and the modification pathway, thus enhancing the
effectiveness of tumor treatment. Blocking lactate metabolism
reduces the substrate supply for Kla at its source, thereby
reversing resistance to various anticancer treatments, including
chemotherapy (cisplatin) resistance (34,126),
oxaliplatin resistance (132) and
targeted therapy (80).
As a key signaling molecule, lactate is the
substrate that drives multiple immunosuppressive processes. For
instance, targeting key metabolic enzymes or using glycolysis
inhibitors can reduce lactate production and lactylation levels,
thereby enhancing the efficacy of immune checkpoint blockade
therapy, improving NK and CD8+ T cell infiltration and
counteracting the regulatory T cell expansion caused by
MOESIN-K72la (82). Through dietary
interventions or metabolic inhibitors combined with anti-PD-1/PD-L1
antibodies, the immune evasion in CRC caused by KAT2A-mediated
PD-L1 lactylation can be reversed (59).
Furthermore, since lactylation is intricately
linked with resistance to numerous anticancer drug therapies,
inhibitors targeting lactylation regulatory enzymes can restore
sensitivity in therapy-resistant cases. This approach achieves an
additive therapeutic effect when combined with treatments such as
radiotherapy and chemotherapy. For example, using the
KAT2A-targeting inhibitor MB-3 (57) and the CBP-targeting inhibitor
SGC-CBP30 (126) restores
sensitivity to cisplatin. The AARS1 inhibitor β-alanine reverses
the poor efficacy of enfortumab vedotin therapy caused by YTHDC1
(55). Meanwhile, the HDAC2
inhibitor tucidinostat can reverse cisplatin resistance by
impairing DNA damage repair (31).
Another HDAC2 inhibitor, CAY-10683, in combination with
terbinafine, enhances the reduction of cholesterol levels and
suppresses HCC proliferation by reversing PD-L1-K189la (29).
These interventions, aimed at altering enzymes,
have reinstated resistance induced by lactylation, resulting in a
more extensive antitumor effect. A dual-targeting strategy focused
on lactate metabolism and lactylation regulatory enzymes offers the
potential to simultaneously address immunosuppression and drug
resistance, establishing a novel method for precision lactylation
intervention.
Beyond its therapeutic implications, Kla is
emerging as a critical diagnostic and prognostic biomarker.
Specific Kla modifications are associated with distinct
pathological states; for instance, adenylate kinase 2-K28la signals
reduce p53 activity in HCC (21),
while FASN-K673la reflects lipid metabolic imbalance in NSCLC
(178). Similarly, elevated
H3K18la levels are clinically associated with advanced tumor stages
and poor prognosis in NSCLC. Mechanistically, H3K18la drives immune
evasion by activating the POM121 transmembrane nucleoporin/MYC axis
to directly upregulate PD-L1 expression. Consequently, monitoring
H3K18la could serve as a vital stratification tool to identify
patients likely to benefit from combined metabolic and anti-PD-1
immunotherapies (129).
Crucially, integrating Hla (specifically H3K18la)
with existing clinical indicators addresses current limitations in
predicting immunotherapy responses. For instance, in head and neck
squamous cell carcinoma, H3K18la-mediated upregulation of Related
RAS viral (r-ras) oncogene homolog 2 is positively correlated with
both chemoresistance and high tumor mutational burden (179). Beyond tissue analysis, the
relative stability of histone modifications compared with unstable
metabolites supports their utility in liquid biopsy (180). In pancreatic cancer (PC) and CRC,
serum H3K18la levels show strong correlations with tumor
progression and traditional markers such as CEA and CA19-9,
highlighting its potential for dynamic, non-invasive monitoring of
tumor metabolism (180,181). Furthermore, H3K18la serves as an
independent prognostic biomarker for PC severity (181). Combined assessment of AARS2 and
its catalyzed product, H3K18la, constitutes a composite biomarker
for evaluating intestinal ischemia-reperfusion injury and
associated ferroptosis. This offers a novel molecular perspective
for understanding and monitoring such tissue damage (182).
With the advancement of multi-omics and machine
learning, lactylation-related genes and lncRNAs are being utilized
to construct sophisticated prognostic models. These models
demonstrate promising potential across various cancer types by
linking lactylation to immune infiltration, immunotherapy response
and drug sensitivity (183,184).
Such approaches efficiently narrow the scope of potential target
screening, providing an effective reference framework for the
precise identification of therapeutic targets and biomarkers.
The regulatory network of Kla is not an isolated
system. It has complex and diverse interactions with other PTMs,
including Kac, succinylation and crotonylation. These interactions,
which use various substrates along the same metabolic pathway and
are coordinated by similar regulatory enzymes, turn metabolic
activity into long-term and widespread cellular functional outputs.
In this process, extensive crosstalk between distinct PTMs creates
a highly tuned network defined by substrate competition and
overlapping regulatory mechanisms.
The competition for acyl-CoA precursors produced by
various metabolic pathways is the main reason of this crosstalk.
Pyruvate, a byproduct of glycolysis, is converted into lactyl-CoA
and acetyl-CoA, the substrates of Kla and Kac. The Warburg effect
causes the metabolic flux to be switched towards lactate, which
results in a decrease in acetyl-CoA and an excess of lactoyl-CoA.
The competitive and synergistic interaction between these two
modifications is fueled by the difference in local substrate
concentrations caused by this shift (115,185).
Notably, the role of lactate is not exclusive to
Kla. Previous research has shown that lactate can also promote
histone H3K27 acetylation (H3K27ac), which transcriptionally
suppresses the pro-inflammatory functions of macrophages while
enhancing the expression of immunosuppressive factors (186). At the same time, lactate can also
mediate increases in the stemness of CD8+ T cells
through H3K27ac (187). Since the
substrates for all these modifications are tightly interconnected
through glucose metabolism, a crosstalk of modification signals is
inevitable.
In M1 macrophages, Hla exhibits different temporal
dynamics than Kac. During the later stages of M1 polarization,
driven by lactate produced during early inflammation, Kla appears
to replace Kac, activating genes involved in inflammation
resolution and homeostatic repair (6). In addition, Kla and Kac can act
synergistically on the same protein. For example, p300/CBP promotes
high mobility group box 1 lactylation while simultaneously
inhibiting SIRT1 deacetylase activity via the Hippo/YAP pathway,
thereby activating acetylation and ultimately exacerbating sepsis
progression (75). A competitive
association exists between Kla and Kac at H3K18. HK2 expression
enhances glycolytic activity and lactate production, influencing
gene expression by affecting H3K18la. However, H3K18ac is
suppressed during in vitro hepatic stellate cell (HSC)
activation. Exogenous lactate supplementation improves lactylation
while decreasing acetylation. Concurrently, class I HDAC inhibitors
enhance H3K18ac while inhibiting H3K18la, thus reducing HSC
activation (92). Due to the
intricate interaction between PTMs, regulating a single modifying
site cannot provide an optimal therapeutic intervention.
Kla and Kac share the same writers and erasers,
which creates a complex regulatory landscape involving multiple
PTMs. This is also reflected in other emerging modifications
(188–190). For instance, lactylation shares
regulatory systems with other modifications such as succinylation
and crotonylation, including enzymes such as p300/CBP, KAT2A, HDACs
and SIRTs (188,189). Furthermore, these PTMs often need
to share coenzyme synthetases (such as ACSS2) to provide the
substrate acyl-CoAs, leading to substrate competition. The
modification type catalyzed by p300/CBP is directly dependent on
the relative concentration of available acyl-CoA substrates at its
active site. Under a high lactate environment, the p300 active site
is more likely to encounter lactyl-CoA, causing its catalytic
activity to switch from writing Kac to writing Kla. Secondly,
lactyl-CoA synthetases produce large quantities of lactyl-CoA,
creating a local environment where lactyl-CoA is dominant, and
synergize with lactyl-CoA transferases to promote lactylation, such
as the synergistic effect of ACSS2 and KAT2A (96). Similarly, GTPSCS translocates into
the nucleus and forms a complex with p300, generating a high local
concentration of lactyl-CoA, which in turn enhances H3K18la
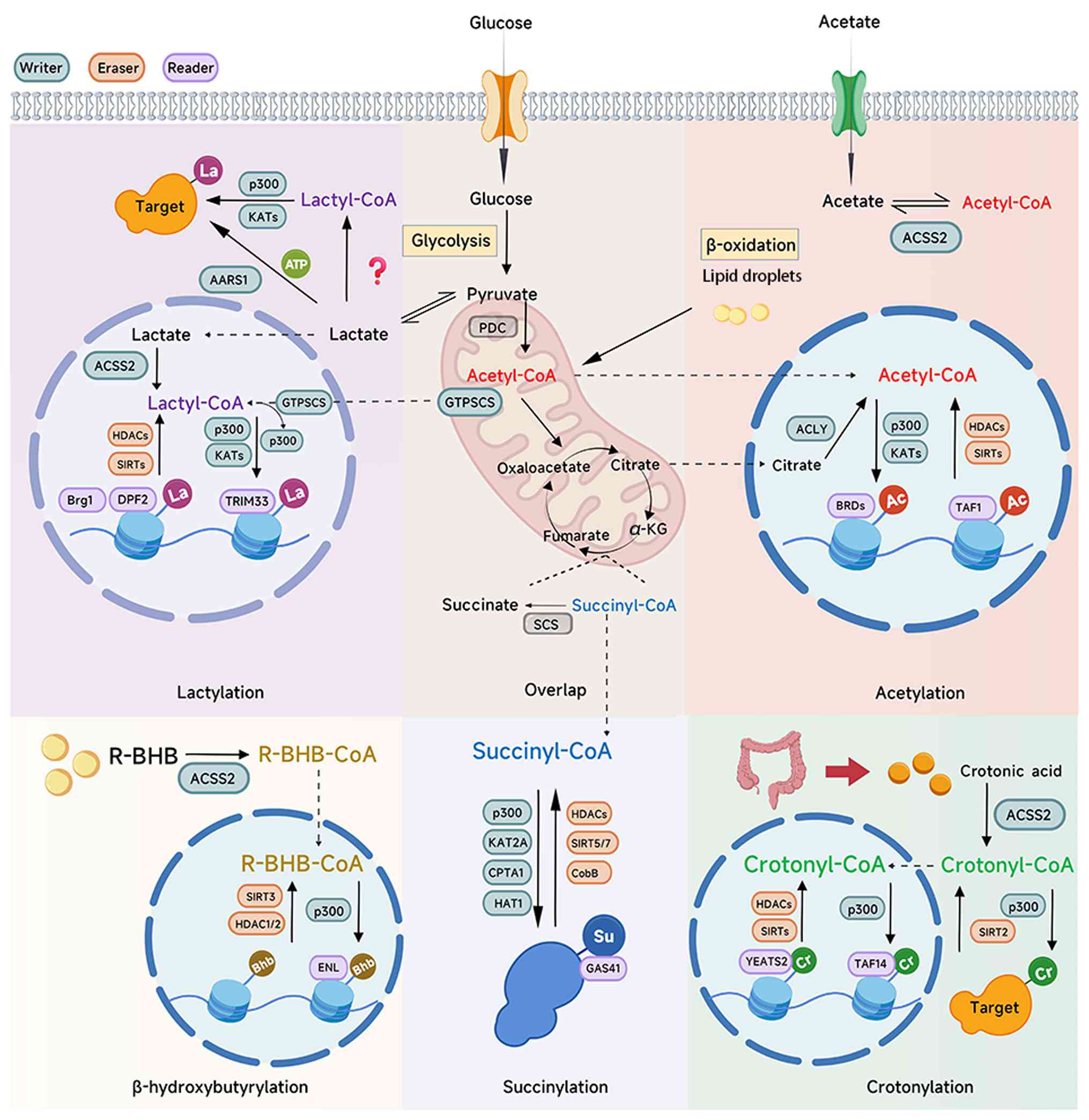
(47). Fig. 4 illustrates this integrated
regulatory network, highlighting how shared enzymatic machinery and
substrate competition facilitate the crosstalk between diverse
PTMs.
Previous structural studies have revealed that the
HAT domain of p300 contains a well-defined acyl-CoA-binding channel
formed by key residues such as Y1397, W1436, Y1446 and C1438
(191,192). This hydrophobic tunnel confers
selectivity toward different acyl-CoA species, enabling p300 to
discriminate among acetyl-CoA, crotonyl-CoA, butyryl-CoA and other
acyl donors. Due to substrate-assisted chain repositioning and the
limited volume of the aliphatic back pocket, the catalytic
efficiency of p300 decreases progressively as the acyl-chain length
increases (191). Short and
hydrophobic groups such as the acetyl moiety fit optimally into the
narrow pocket, supporting highly efficient catalysis. By contrast,
the lactyl group is not only bulkier, but also contains a polar
hydroxyl group; its additional steric and electrostatic constraints
likely require subtle conformational adjustments of the HAT domain
to accommodate lactyl-CoA, ultimately reducing catalytic efficiency
compared with acetyl-CoA. These structural features provide a
mechanistic explanation for why p300 exhibits differential
reactivity towards Kac and Kla. More notably, they highlight that
competition between lactylation and acetylation may arise directly
from shared catalytic machinery: When multiple acyl-CoA species
coexist in the nucleus, they may compete for occupancy of the same
acyl-CoA channel within p300, thereby influencing the balance
between distinct PTMs. However, simulating the competitive
association between the PTMs under physiological conditions remains
challenging. It is difficult to replicate the complex environment
faced by tumors and to exclude interference from multiple processes
to precisely determine which modification is specifically
regulated. At present, lysine-to-threonine site mutations are
commonly used to mimic lactylation (22,74),
but they still cannot fully replicate the effects of lactylation in
the body. Recently developed bio-orthogonal systems based on
genetic code expansion may provide improved tools for authentic
simulation by directly inserting the lactyl group at target lysine
sites (23,64,93).
From this perspective, compared with the MYST
family, which is involved in crosstalk with multiple modifications,
AARS1 appears to be a more precise therapeutic target. However, the
crucial role of AARS1 in protein translation means that targeting
it for inhibition would lead to notable toxicity. Indeed, mutations
in AARS1 itself are associated with various diseases, such as
Charcot-Marie-Tooth disease type 2N and infantile epileptic
encephalopathy (193,194). β-alanine, which has a similar
structure to lactate, may be a theoretical candidate for developing
specific interventions as it has been shown to inhibit lactylation
in molecular experiments (23,55);
however, further research is needed before it can become a clinical
therapeutic drug. A potential therapeutic strategy could involve
structural studies of AARS1 to decouple its tRNA synthetase
function from its lactyltransferase function. This would enable the
development of specific inhibitors that selectively block the
lactate-binding activity of AARS1 without compromising its
canonical role in protein synthesis.
Current enzymological research on lactylation in
tumors primarily focuses on a limited number of known regulatory
enzymes, such as p300, HDACs or SIRTs, and often relies on chemical
inhibitors or protein-protein interaction analyses to infer their
roles. This strategy is reasonable, given the availability of
established inhibitors and the mechanistic overlap with Kac, but it
has several limitations and risks. First, inhibitor-based studies
often suffer from off-target effects, raising doubts about whether
the observed changes in lactylation are direct or secondary to
overall metabolic shifts. Second, protein interaction assays can
reveal correlations but not causality, leaving the true enzymatic
drivers uncertain. Third, focusing only on enzymes already
implicated in other PTMs may overlook other writers or erasers that
may be specific to lactylation (23,115).
It has been shown that supplementing with exogenous sodium lactate
in combination with p300 knockdown paradoxically increases Hla
compared with p300 knockdown alone. This suggests that high levels
of upstream lactate can override the limiting effect of p300, and
it is more likely that p300 is not the sole writer of Hla and that
other enzymes may be involved in this process, or even that p300 is
not the primary writer for Kla (49). Finally, systemic interference with
histone-modifying factors can have broad cellular effects, which
can complicate the interpretation of mechanisms specific to
lactylation. In conclusion, while this strategy is a necessary
starting point, it needs to be combined with more precise methods
such as proteomics and CRISPR screening to broaden the scope of
consideration. Utilizing structural biology and the analysis of
substrate-binding pockets, and possibly simulating substrates under
physiological conditions (165),
may be necessary to obtain more exclusive evidence to enhance the
scientific rigor and reliability of enzymological research. For
example, innovative methods have been successfully applied to the
study of Kla erasers. Fan et al (45) introduced two innovative,
function-oriented chemical probes. The first, an affinity-based
probe named p-H4K16laAlk, allows for the UV-induced covalent
capture of transiently interacting proteins directly from cell
lysates. This facilitates the unbiased proteomic identification of
endogenous erasers, as demonstrated by its successful capture of
SIRT3. The second, a fluorogenic probe termed p-H4K16laNBD,
provides a real-time readout upon the enzymatic removal of the
lactyl group. Together, these tools advance the detection of
lactylation regulatory enzymes from indirect inference toward
direct identification and dynamic functional characterization.
Crucially, species differences significantly impact
AARS1-mediated lactylation. Although the lactate-dependent exposure
of the NLS is a conserved mechanism, variations in the
lactate-sensing and catalytic domains suggest that sensitivity may
differ between rodents and humans (36). Furthermore, downstream targets such
as p53 operate within species-specific networks, implying that
murine findings may not fully mirror human tumor biology (23). Therefore, rigorous validation using
human-derived organoids or patient-derived xenograft models is
essential to ensure preclinical findings translate effectively to
clinical contexts.
Future studies should incorporate models with
greater human relevance, including humanized mouse systems and
patient-derived organoids, to more reliably evaluate drug exposure,
toxicity and lactylation-dependent signaling.
Future intervention strategies for lactylation
regulatory enzymes must extend beyond traditional small-molecule
inhibitors to incorporate more precise approaches. First, by
integrating metabolic monitoring technology, precise monitoring of
lactate production, transport and the Kla process can be achieved,
such as the lactate sensor FiLa (198), which can provide real-time dynamic
data, thus helping to understand the role of lactylation in tumors
and, in turn, guide the optimization and evaluation of subsequent
intervention strategies. This, in turn, facilitates the
optimization of therapeutic timing and the evaluation of
intervention strategies based on real-time metabolic responses.
Furthermore, by establishing biomarkers based on lactylated
molecules and by developing and validating detection methods for
clinical samples to assess the lactylation levels of specific sites
or proteins, it may be possible to predict sensitivity to targeted
metabolic or epigenetic therapies or to monitor
pharmacodynamics.
Second, developing highly specific intervention
tools is key to overcoming off-target effects. The vast diversity
of lactylated protein substrates makes site-specific intervention
complex. Although targeting and inhibiting lactylated proteins can
reduce toxic side effects, the screening and validation processes
still rely on high-throughput proteomics, CRISPR-based methods and
custom antibodies, which are both time-consuming and technically
demanding. This suggests that intervening with lactylation
regulatory enzymes would be more efficient and meaningful. In
addition to continuing the search for regulatory enzymes, it is
important to fully elucidate the regulatory mechanisms of
lactylation and, on that basis, develop inhibitors that are
specific to lactylation without affecting other PTMs. Before that
is achieved, another direction to consider is shifting from the
traditional approach of inhibiting enzymatic catalytic activity to
blocking the binding ability between regulatory enzymes and
specific sites, such as the interaction between lactyl-CoA
synthetase and writers. The use of potent and highly selective
inhibitors, coupled with robust drug delivery systems to achieve
sustained interference with regulatory enzymes, offers a promising
strategy to overcome the off-target effects of conventional
inhibitors and demonstrates significant translational
potential.
Lactylation functions as a pivotal bridge
connecting the metabolic Warburg effect to epigenetic
reprogramming. The present review summarizes the core enzymatic
machinery governing this process, including writers, erasers and
readers. Collectively, this regulatory network orchestrates a
malignancy-associated transcriptomic profile; it drives metastasis,
reshapes the immune microenvironment toward an immunosuppressive
state and actively promotes resistance to chemotherapy and
immunotherapy.
Despite these advances in uncovering the
lactylation regulatory enzymes and identifying its diverse roles in
tumor progression and therapy resistance, translating these
findings into clinical therapies faces critical hurdles. First,
targeting upstream metabolic enzymes often causes systemic toxicity
due to off-target effects on normal tissues. Second,
species-specific differences in enzyme activity limit the
predictive value of conventional animal models. Third, the
extensive crosstalk between lactylation and other acyl
modifications creates a complex competitive landscape, complicating
the identification of specific therapeutic targets. To overcome
these barriers, future research must shift toward precision
intervention that bypasses broad metabolic disruption in favor of
selective molecular targeting. The present review highlights the
need to move beyond non-selective catalytic inhibitors to advanced
modalities, such as PROTACs that degrade specific enzymes or
inhibitors that disrupt specific writer-reader interactions.
Additionally, validating lactylation signatures as liquid biopsy
biomarkers is essential for patient stratification. By resolving
these structural and translational challenges, targeting the
lactylation regulatory network offers a transformative strategy to
reprogram tumors and overcome therapeutic resistance.
This research was funded by the Key Research Program from the
4310 Program of Hengyang Medical College, University of South China
(grant no. 20224310NHYCG12) and the National Natural Science
Foundation of China (grant no. 82173008).
Not applicable.
GT conceived and designed the study. GT and HZ
drafted the manuscript and provided overall supervision of the
project. RL, CZ, YL, XL and JQ conducted the literature search and
study selection, performed data collation from the included
references, and contributed to specific sections of the manuscript.
JZ contributed to the study design, provided funding support,
supervised the project and offered critical revision suggestions.
All authors participated in the revision of the manuscript and have
read and approved the final version. Data authentication is not
applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Koppenol WH, Bounds PL and Dang CV: Otto
warburg's contributions to current concepts of cancer metabolism.
Nat Rev Cancer. 11:325–337. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Alberghina L: The warburg effect
explained: Integration of enhanced glycolysis with heterogeneous
mitochondria to promote cancer cell proliferation. Int J Mol Sci.
24:157872023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dai C, Tang Y, Yang H and Zheng J: YTHDC1
lactylation regulates its phase separation to enhance target mRNA
stability and promote RCC progression. Mol Cell. 85:2733–2748.e7.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Iozzo M, Pardella E, Giannoni E and
Chiarugi P: The role of protein lactylation: A kaleidoscopic
post-translational modification in cancer. Mol Cell. 85:1263–1279.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu Z, Peng Y, Chen W, Xia F, Song T and Ke
Q: Lactylation-driven transcriptional activation of FBXO33 promotes
gallbladder cancer metastasis by regulating p53 polyubiquitination.
Cell Death Dis. 16:1442025. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang D, Tang Z, Huang H, Zhou G, Cui C,
Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, et al: Metabolic
regulation of gene expression by histone lactylation. Nature.
574:575–580. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Niu K, Chen Z, Li M, Ma G, Deng Y, Zhang
J, Wei D, Wang J and Zhao Y: NSUN2 lactylation drives cancer cell
resistance to ferroptosis through enhancing GCLC-dependent
glutathione synthesis. Redox Biol. 79:1034792025. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang T, Zhang S, Nie K, Cheng C, Peng X,
Huo J and Zhang Y: ZNF207-driven PRDX1 lactylation and NRF2
activation in regorafenib resistance and ferroptosis evasion. Drug
Resist Updat. 82:1012742025. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Allen E, Miéville P, Warren CM, Saghafinia
S, Li L, Peng MW and Hanahan D: Metabolic symbiosis enables
adaptive resistance to anti-angiogenic therapy that is dependent on
mTOR signaling. Cell Rep. 15:1144–1160. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Végran F, Boidot R, Michiels C, Sonveaux P
and Feron O: Lactate influx through the endothelial cell
monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway
that drives tumor angiogenesis. Cancer Res. 71:2550–2560. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Leija RG, Curl CC, Arevalo JA, Osmond AD,
Duong JJ, Huie MJ, Masharani U and Brooks GA: Enteric and systemic
postprandial lactate shuttle phases and dietary carbohydrate carbon
flow in humans. Nat Metab. 6:670–677. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Netzahualcoyotzi C and Pellerin L:
Neuronal and astroglial monocarboxylate transporters play key but
distinct roles in hippocampus-dependent learning and memory
formation. Prog Neurobiol. 194:1018882020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yan K, He Q, Lin D, Liang J, Chen J, Xie Z
and Chen Z: Promotion of NAD+ recycling by the hypoxia-induced
shift in the lactate dehydrogenase isozyme profile reduces the
senescence of human bone marrow-derived endothelial progenitor
cells. Free Radical Biol Med. 208:88–102. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Roland CL, Arumugam T, Deng D, Liu SH,
Philip B, Gomez S, Burns WR, Ramachandran V, Wang H,
Cruz-Monserrate Z and Logsdon CD: Cell surface lactate receptor
GPR81 is crucial for cancer cell survival. Cancer Res.
74:5301–5310. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gottfried E, Kunz-Schughart LA, Ebner S,
Mueller-Klieser W, Hoves S, Andreesen R, Mackensen A and Kreutz M:
Tumor-derived lactic acid modulates dendritic cell activation and
antigen expression. Blood. 107:2013–2021. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Colegio OR, Chu NQ, Szabo AL, Chu T,
Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC,
Phillips GM, et al: Functional polarization of tumour-associated
macrophages by tumour-derived lactic acid. Nature. 513:559–563.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Comito G, Iscaro A, Bacci M, Morandi A,
Ippolito L, Parri M, Montagnani I, Raspollini MR, Serni S, Simeoni
L, et al: Lactate modulates CD4+ T-cell polarization and induces an
immunosuppressive environment, which sustains prostate carcinoma
progression via TLR8/miR21 axis. Oncogene. 38:3681–3695. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Brand A, Singer K, Koehl GE, Kolitzus M,
Schoenhammer G, Thiel A, Matos C, Bruss C, Klobuch S, Peter K, et
al: LDHA-associated lactic acid production blunts tumor
immunosurveillance by T and NK cells. Cell Metab. 24:657–671. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jiang S, Li H, Zhang L, Mu W, Zhang Y,
Chen T, Wu J, Tang H, Zheng S, Liu Y, et al: Generic diagramming
platform (GDP): A comprehensive database of high-quality biomedical
graphics. Nucleic Acids Res. 53:D1670–D1676. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li F, Si W, Xia L, Yin D, Wei T, Tao M,
Cui X, Yang J, Hong T and Wei R: Positive feedback regulation
between glycolysis and histone lactylation drives oncogenesis in
pancreatic ductal adenocarcinoma. Mol Cancer. 23:902024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang Z, Yan C, Ma J, Peng P, Ren X, Cai S,
Shen X, Wu Y, Zhang S, Wang X, et al: Lactylome analysis suggests
lactylation-dependent mechanisms of metabolic adaptation in
hepatocellular carcinoma. Nat Metab. 5:61–79. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hong H, Han H, Wang L, Cao W, Hu M, Li J,
Wang J, Yang Y, Xu X, Li G, et al: ABCF1-K430-lactylation promotes
HCC malignant progression via transcriptional activation of HIF1
signaling pathway. Cell Death Differ. 32:613–631. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zong Z, Xie F, Wang S, Wu X, Zhang Z, Yang
B and Zhou F: Alanyl-tRNA synthetase, AARS1, is a lactate sensor
and lactyltransferase that lactylates p53 and contributes to
tumorigenesis. Cell. 187:2375–2392.e33. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dai J, Lu X, Zhang C, Qu T, Li W, Su J,
Guo R, Yin D, Wu P, Han L and Zhang E: NNMT promotes acquired
EGFR-TKI resistance by forming EGR1 and lactate-mediated double
positive feedback loops in non-small cell lung cancer. Mol Cancer.
24:792025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zou L, Liu Z, Peng L, Wu B, Huang Y, Wen
X, Han D, Tan W and Zhou Y: Delactylation of H3K9 by Sirtuin6
inhibits MGMT transcription and reverses temozolomide resistance in
glioblastoma. Int J Biol Macromol. 327((Pt 1)): 1473322025.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu S, Cai J, Qian X, Zhang J, Zhang Y,
Meng X, Wang M, Gao P and Zhong X: TPX2 lactylation is required for
the cell cycle regulation and hepatocellular carcinoma progression.
Life Sci Alliance. 8:e2024029782025. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shan G, Bian Y, Sui Q, Liang J, Ren S, Pan
B, Shi H, Zheng Z, Zeng D, Zhu J, et al: Ferroptosis-induced SUMO2
lactylation counteracts ferroptosis by enhancing ACSL4 degradation
in lung adenocarcinoma. Cell Discov. 11:812025. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Han H, Wang S, Ma L, Yin H, Cheng X, Wang
Y, Xia S, Zhang Y, Zhang Y, Zhu R, et al: ASH2L-K312-lac stimulates
angiogenesis in tumors to expedite the malignant progression of
hepatocellular carcinoma. Adv Sci (Weinh). 12:e094772025.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang X, Li Y, Tang Y, Liu Z, Liu Y, Fu X,
Guo S, Ma J, Ma F, Zhu Z, et al: PD-L1 delactylation-promoted
nuclear translocation accelerates liver cancer growth through
elevating SQLE transcription activity. Cancer Lett. 630:2179012025.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lin J, Yin Y, Cao J, Zhang Y, Chen J, Chen
R, Zou B, Huang C, Lv Y, Xu S, et al: NUDT21 lactylation reprograms
alternative polyadenylation to promote cuproptosis resistance. Cell
Discov. 11:522025. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
He X, Li Y, Li J, Li Y, Chen S, Yan X, Xie
Z, Du J, Chen G, Song J and Mei Q: HDAC2-mediated METTL3
delactylation promotes DNA damage repair and chemotherapy
resistance in triple-negative breast cancer. Adv Sci (Weinh).
12:e24131212025. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen J, Zhao D, Wang Y, Liu M, Zhang Y,
Feng T, Xiao C, Song H, Miao R, Xu L, et al: Lactylated
Apolipoprotein C-II induces immunotherapy resistance by promoting
extracellular lipolysis. Adv Sci (Weinh). e24063332024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lv J, Yu X, Liu X, Zhang Q, Zhang M, Gao
J, Sun Z, Zhang F, Zuo Y and Ren S: The LncRNA STEAP3-AS1 promotes
liver metastasis in colorectal cancer by regulating histone
lactylation through chromatin remodelling. J Exp Clin Cancer Res.
44:2052025. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen H, Li Y, Li H, Chen X, Fu H, Mao D,
Chen W, Lan L, Wang C, Hu K, et al: NBS1 lactylation is required
for efficient DNA repair and chemotherapy resistance. Nature.
631:663–669. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li J, Feng L, Zhang L, Zhang W, Zhang Y,
Liu X, Cai Q, Zhao W, Huang G and Lu C: Saikosaponin D mitigates
radioresistance in triple-negative breast cancer by inducing MRE11
de-lactylation via HIF1α/HDAC5 pathway. Theranostics. 15:8935–8951.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ju J, Zhang H, Lin M, Yan Z, An L, Cao Z,
Geng D, Yue J, Tang Y, Tian L, et al: The alanyl-tRNA synthetase
AARS1 moonlights as a lactyl-transferase to promote YAP signaling
in gastric cancer. J Clin Invest. 134:e1745872024. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jin M, Huang B, Yang X, Wang S, Wu J, He
Y, Ding X, Wang X, Wang Z, Yang J, et al: Lactylation of XLF
promotes non-homologous end-joining repair and chemoresistance in
cancer. Mol Cell. 85:2654–2672.e7. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhou Z, Yin X, Sun H, Lu J, Li Y, Fan Y,
Lv P, Han M, Wu J, Li S, et al: PTBP1 lactylation promotes glioma
stem cell maintenance through PFKFB4-driven glycolysis. Cancer Res.
85:739–757. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Du R, Gao Y, Yan C, Ren X, Qi S, Liu G,
Guo X, Song X, Wang H, Rao J, et al: Sirtuin 1/sirtuin 3 are robust
lysine delactylases and sirtuin 1-mediated delactylation regulates
glycolysis. iScience. 27:1109112024. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sun L, Zhang Y, Yang B, Sun S, Zhang P,
Luo Z, Feng T, Cui Z, Zhu T, Li Y, et al: Lactylation of METTL16
promotes cuproptosis via m6A-modification on FDX1 mRNA in gastric
cancer. Nat Commun. 14:65232023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen C, Zhang Y, Zang Y, Fan Z, Han Y, Bai
X, Wang A, Zhang J, Wang J and Zhang K: SIRT3 functions as an
eraser of histone H3K9 lactylation to modulate transcription for
inhibiting the progression of esophageal cancer. Mol Cell
Proteomics. 24:1009732025. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tang Y, Dai C, Yang H, Yang X, Chen Z, Dou
J, Zhang L, Bai T and Zheng J: KAT8-mediated MDH2 lactylation
promotes renal cancer progression by enhancing mitochondrial
function and stress resistance. Int J Biol Macromol.
322:1465712025. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jin J, Bai L, Wang D, Ding W, Cao Z, Yan
P, Li Y, Xi L, Wang Y, Zheng X, et al: SIRT3-dependent
delactylation of cyclin E2 prevents hepatocellular carcinoma
growth. EMBO Rep. 24:e560522023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li C, Ge C, Wang Q, Teng P, Jia H, Yao S
and Huang Z: Sirtuin 3-mediated delactylation of malic enzyme 2
disrupts redox balance and inhibits colorectal cancer growth. Cell
Oncol (Dordr). 48:979–990. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fan Z, Liu Z, Zhang N, Wei W, Cheng K, Sun
H and Hao Q: Identification of SIRT3 as an eraser of H4K16la.
iScience. 26:1077572023. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shi L, Zhang L, Ma Z, Hu Z, Geng Z, Liu H,
Wang J, Feng K, Geng Y, Liu Y, et al: H3K18 lactylation drives the
progression of silica nanoparticles-induced pulmonary fibrosis via
promoting macrophage M1 polarization. J Hazard Mater.
496:1392862025. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu R, Ren X, Park YE, Feng H, Sheng X,
Song X, AminiTabrizi R, Shah H, Li L, Zhang Y, et al: Nuclear
GTPSCS functions as a lactyl-CoA synthetase to promote histone
lactylation and gliomagenesis. Cell Metab. 37:377–394.e9. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Huang Y, Luo G, Peng K, Song Y, Wang Y,
Zhang H, Li J, Qiu X, Pu M, Liu X, et al: Lactylation stabilizes
TFEB to elevate autophagy and lysosomal activity. J Cell Biol.
223:e2023080992024. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sun K, Zhang X, Shi J, Huang J, Wang S, Li
X, Lin H, Zhao D, Ye M, Zhang S, et al: Elevated protein
lactylation promotes immunosuppressive microenvironment and
therapeutic resistance in pancreatic ductal adenocarcinoma. J Clin
Invest. 135:e1870242025. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yang L, Niu K, Wang J, Shen W, Jiang R,
Liu L, Song W, Wang X, Zhang X, Zhang R, et al: Nucleolin
lactylation contributes to intrahepatic cholangiocarcinoma
pathogenesis via RNA splicing regulation of MADD. J Hepatol.
81:651–666. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Jiang C, He X, Chen X, Huang J, Liu Y,
Zhang J, Chen H, Sui X, Lv X, Zhao X, et al: Lactate accumulation
drives hepatocellular carcinoma metastasis through facilitating
tumor-derived exosome biogenesis by Rab7A lactylation. Cancer Lett.
627:2176362025. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Deng J, Li Y, Yin L, Liu S, Li Y, Liao W,
Mu L, Luo X and Qin J: Histone lactylation enhances GCLC expression
and thus promotes chemoresistance of colorectal cancer stem cells
through inhibiting ferroptosis. Cell Death Dis. 16:1932025.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ji Y, Xu Z, Tang L, Huang T, Mu X, Ni C,
Tang B, Lu H, Zhang C, Yang S and Wang X: O-GlcNAcylation of YBX1
drives a glycolysis-histone lactylation feedback loop in
hepatocellular carcinoma. Cancer Lett. 631:2179572025. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yang J and Yu X, Xiao M, Xu H, Tan Z, Lei
Y, Guo Y, Wang W, Xu J, Shi S and Yu X: Histone lactylation-driven
feedback loop modulates cholesterol-linked immunosuppression in
pancreatic cancer. Gut. 74:1859–1872. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Xing Z, Yang T, Li X, Xu H, Hong Y, Shao
S, Li T, Ye L, Li Y, Jin X and Wei Y: High-glucose-associated
YTHDC1 lactylation reduces the sensitivity of bladder cancer to
enfortumab vedotin therapy. Cell Rep. 44:1155452025. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Li X, Zhang C, Mei Y, Zhong W, Fan W, Liu
L, Feng Z, Bai X, Liu C, Xiao M, et al: Irinotecan alleviates
chemoresistance to anthracyclines through the inhibition of
AARS1-mediated BLM lactylation and homologous recombination repair.
Signal Transduction Targeted Ther. 10:2142025. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sun C, Li X, Teng Q, Liu X, Song L,
Schiöth HB, Wu H, Ma X, Zhang Z, Qi C, et al: Targeting
platinum-resistant ovarian cancer by disrupting histone and RAD51
lactylation. Theranostics. 15:3055–3075. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zheng B, Pan Y, Qian F, Liu D, Ye D, Yu B,
Zhong S, Zheng W, Wang X, Zhou B, et al: High sugar induced RCC2
lactylation drives breast cancer tumorigenicity through
upregulating MAD2L1. Adv Sci (Weinh). 12:e24155302025. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Tong H, Jiang Z, Song L, Tan K, Yin X, He
C, Huang J, Li X, Jing X, Yun H, et al: Dual impacts of
serine/glycine-free diet in enhancing antitumor immunity and
promoting evasion via PD-L1 lactylation. Cell Metab.
36:2493–2510.e9. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Jia M, Yue X, Sun W, Zhou Q, Chang C, Gong
W, Feng J, Li X, Zhan R, Mo K, et al: ULK1-mediated metabolic
reprogramming regulates Vps34 lipid kinase activity by its
lactylation. Sci Adv. 9:eadg49932023. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Niu Z, Chen C, Wang S, Lu C, Wu Z, Wang A,
Mo J, Zhang J, Han Y, Yuan Y, et al: HBO1 catalyzes lysine
lactylation and mediates histone H3K9la to regulate gene
transcription. Nat Commun. 15:35612024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Xie B, Zhang M, Li J, Cui J, Zhang P, Liu
F, Wu Y, Deng W, Ma J, Li X, et al: KAT8-catalyzed lactylation
promotes eEF1A2-mediated protein synthesis and colorectal
carcinogenesis. Proc Natl Acad Sci USA. 121:e23141281212024.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Sui Y, Shen Z, Wang Z, Feng J and Zhou G:
Lactylation in cancer: Metabolic mechanism and therapeutic
strategies. Cell Death Discov. 11:682025. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zong Z, Zhang L and Zhou F: Lactylation in
cancer: Advances, challenges, and future perspectives. Cancer Res.
85:3192–3195. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Li Y, Peng J, Wu D, Xie Q, Hou Y, Li L,
Zhang X, Liang Y, Feng J, Chen J, et al: Histone
lactylation-boosted AURKB facilitates colorectal cancer progression
by inhibiting HNRNPM-mediated PSAT1 mRNA degradation. J Exp Clin
Cancer Res. 44:2332025. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhao Y, Jiang J, Zhou P, Deng K, Liu Z,
Yang M, Yang X, Li J, Li R and Xia J: H3K18 lactylation-mediated
VCAM1 expression promotes gastric cancer progression and metastasis
via AKT-mTOR-CXCL1 axis. Biochem Pharmacol. 222:1161202024.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Liu S, He Y, Jin L, Shi S, Zhang J, Xie W,
Yang M, Zhang Q and Kong H: H3K18 lactylation-mediated SIX1
upregulation contributes to silica-induced epithelial-mesenchymal
transition of airway epithelial cells. Toxicology. 514:1541092025.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Wu G, Cheng H, Yin J, Zheng Y, Shi H, Pan
B, Li M, Zhao M, Liang J, Bian Y, et al: NDRG1-driven lactate
accumulation promotes lung adenocarcinoma progression through the
induction of an immunosuppressive microenvironment. Adv Sci
(Weinh). 12:e012382025. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhou S, Xiao L, Hu L, Zuo F, Wang Y, Fei
B, Dai J and Zhou X: CAFs promote immune evasion in gastric cancer
through histone lactylation-mediated suppression of NCAPG
ubiquitination. J Transl Med. 23:9892025. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Ji FH, Qian YH, Guo XC, Liao HH, Huang JC,
Xu ZH, Yu MM, Wu YY, Bao JW, Chen HJ, et al: Targeting RPS6KC1 to
overcome enzalutamide resistance in prostate cancer. Biomark Res.
13:1092025. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Li W, Zhou C, Yu L, Hou Z, Liu H, Kong L,
Xu Y, He J, Lan J, Ou Q, et al: Tumor-derived lactate promotes
resistance to bevacizumab treatment by facilitating autophagy
enhancer protein RUBCNL expression through histone H3 lysine 18
lactylation (H3K18la) in colorectal cancer. Autophagy. 20:114–130.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Lu B, Chen S, Guan X, Chen X, Du Y, Yuan
J, Wang J, Wu Q, Zhou L, Huang X and Zhao Y: Lactate accumulation
induces H4K12la to activate super-enhancer-driven RAD23A expression
and promote niraparib resistance in ovarian cancer. Mol Cancer.
24:832025. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Liu J, Zhao L, Yan M, Jin S, Shang L, Wang
J, Wang Q, Zhao S, Shen Z, Liu T, et al: H4K79 and H4K91 histone
lactylation, newly identified lactylation sites enriched in breast
cancer. J Exp Clin Cancer Res. 44:2522025. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zhao Z, Zhang Z, Cai Q, Yang R, Liang H,
Qian B, Xiao B, Jiang Y, Wang L, Wang X and Cai J: Lactylation
increases the stability of RBM15 to drives m6A modification in
non-small-cell lung cancer cells. FASEB J. 39:e704932025.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Yang K, Fan M, Wang X, Xu J, Wang Y, Tu F,
Gill PS, Ha T, Liu L, Williams DL and Li C: Lactate promotes
macrophage HMGB1 lactylation, acetylation, and exosomal release in
polymicrobial sepsis. Cell Death Differ. 29:133–146. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Miao Z, Zhao X and Liu X: Hypoxia induced
β-catenin lactylation promotes the cell proliferation and stemness
of colorectal cancer through the wnt signaling pathway. Exp Cell
Res. 422:1134392023. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Liao J, Chen Z, Chang R, Yuan T, Li G, Zhu
C, Wen J, Wei Y, Huang Z, Ding Z, et al: CENPA functions as a
transcriptional regulator to promote hepatocellular carcinoma
progression via cooperating with YY1. Int J Biol Sci. 19:5218–5232.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Chen B, Deng Y, Hong Y, Fan L, Zhai X, Hu
H, Yin S, Chen Q, Xie X, Ren X, et al: Metabolic recoding of
NSUN2-mediated m5C modification promotes the progression of
colorectal cancer via the NSUN2/YBX1/m5C-ENO1 positive feedback
loop. Adv Sci (Weinh). 11:e23098402024. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Xiong J, He J, Zhu J, Pan J, Liao W, Ye H,
Wang H, Song Y, Du Y, Cui B, et al: Lactylation-driven
METTL3-mediated RNA m6A modification promotes immunosuppression of
tumor-infiltrating myeloid cells. Mol Cell. 82:1660–1677.e10. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Lu Y, Zhu J, Zhang Y, Li W, Xiong Y, Fan
Y, Wu Y, Zhao J, Shang C, Liang H and Zhang W: Lactylation-Driven
IGF2BP3-mediated serine metabolism reprogramming and RNA
m6A-modification promotes lenvatinib resistance in HCC. Adv Sci
(Weinh). 11:e24013992024. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Meng Q, Sun H, Zhang Y, Yang X, Hao S, Liu
B, Zhou H, Xu ZX and Wang Y: Lactylation stabilizes DCBLD1
activating the pentose phosphate pathway to promote cervical cancer
progression. J Exp Clin Cancer Res. 43:362024. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Gu J, Zhou J, Chen Q, Xu X, Gao J, Li X,
Shao Q, Zhou B, Zhou H, Wei S, et al: Tumor metabolite lactate
promotes tumorigenesis by modulating MOESIN lactylation and
enhancing TGF-β signaling in regulatory T cells. Cell Rep.
39:1109862022. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Gu J, Xu X, Li X, Yue L, Zhu X, Chen Q,
Gao J, Takashi M, Zhao W, Zhao B, et al: Tumor-resident microbiota
contributes to colorectal cancer liver metastasis by lactylation
and immune modulation. Oncogene. 43:2389–2404. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Son SM, Park SJ, Breusegem SY, Larrieu D
and Rubinsztein DC: p300 nucleocytoplasmic shuttling underlies
mTORC1 hyperactivation in hutchinson-gilford progeria syndrome. Nat
Cell Biol. 26:235–249. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Navarro-Corcuera A, Sehrawat TS,
Jalan-Sakrikar N, Gibbons HR, Pirius NE, Khanal S, Hamdan FH, Aseem
SO, Cao S, Banales JM, et al: Long non-coding RNA ACTA2-AS1
promotes ductular reaction by interacting with the p300/ELK1
complex. J Hepatol. 76:921–933. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wang X, Fan W, Li N, Ma Y, Yao M, Wang G,
He S, Li W, Tan J, Lu Q and Hou S: YY1 lactylation in microglia
promotes angiogenesis through transcription activation-mediated
upregulation of FGF2. Genome Biol. 24:872023. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Fan M, Yang K, Wang X, Chen L, Gill PS, Ha
T, Liu L, Lewis NH, Williams DL and Li C: Lactate promotes
endothelial-to-mesenchymal transition via Snail1 lactylation after
myocardial infarction. Sci Adv. 9:eadc94652023. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Dong M, Zhang Y, Chen M, Tan Y, Min J, He
X, Liu F, Gu J, Jiang H, Zheng L, et al: ASF1A-dependent
P300-mediated histone H3 lysine 18 lactylation promotes
atherosclerosis by regulating EndMT. Acta Pharm Sin B.
14:3027–3048. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Zou Y, Cao M, Tai M, Zhou H, Tao L, Wu S,
Yang K, Zhang Y, Ge Y, Wang H, et al: A feedback loop driven by
H4K12 lactylation and HDAC3 in macrophages regulates
lactate-induced collagen synthesis in fibroblasts via the TGF-β
signaling. Adv Sci (Weinh). 12:e24114082025. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Yuan J, Yang M, Wu Z, Wu J, Zheng K, Wang
J, Zeng Q, Chen M, Lv T, Shi Y, et al: The lactate-primed KAT8-PCK2
axis exacerbates hepatic ferroptosis during ischemia/reperfusion
injury by reprogramming OXSM-dependent mitochondrial fatty acid
synthesis. Adv Sci (Weinh). 12:e24141412025. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Varner EL, Trefely S, Bartee D, von
Krusenstiern E, Izzo L, Bekeova C, O'Connor RS, Seifert EL, Wellen
KE, Meier JL and Snyder NW: Quantification of lactoyl-CoA
(lactyl-CoA) by liquid chromatography mass spectrometry in
mammalian cells and tissues. Open Biol. 10:2001872020. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Rho H, Terry AR, Chronis C and Hay N:
Hexokinase 2-mediated gene expression via histone lactylation is
required for hepatic stellate cell activation and liver fibrosis.
Cell Metab. 35:1406–1423.e8. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Li H, Liu C, Li R, Zhou L, Ran Y, Yang Q,
Huang H, Lu H, Song H, Yang B, et al: AARS1 and AARS2 sense
L-lactate to regulate cGAS as global lysine lactyltransferases.
Nature. 634:1229–1237. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Mao Y, Zhang J, Zhou Q, He X, Zheng Z, Wei
Y, Zhou K, Lin Y, Yu H, Zhang H, et al: Hypoxia induces
mitochondrial protein lactylation to limit oxidative
phosphorylation. Cell Res. 34:13–30. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Gao L, Guo J and Jia R: AARS1 and AARS2:
From protein synthesis to lactylation-driven oncogenesis.
Biomolecules. 15:13232025. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Zhu R, Ye X, Lu X, Xiao L, Yuan M, Zhao H,
Guo D, Meng Y, Han H, Luo S, et al: ACSS2 acts as a lactyl-CoA
synthetase and couples KAT2A to function as a lactyltransferase for
histone lactylation and tumor immune evasion. Cell Metab.
37:361–376.e7. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Gonzatti MB, Hintzen JCJ, Sharma I, Najar
MA, Tsusaka T, Marcinkiewicz MM, Da Silva Crispim CV, Snyder NW,
Burslem GM and Goldberg EL: Class I histone deacetylases catalyze
lysine lactylation. J Biol Chem. 301:1106022025. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Moreno-Yruela C, Zhang D, Wei W, Bæk M,
Liu W, Gao J, Danková D, Nielsen AL, Bolding JE, Yang L, et al:
Class I histone deacetylases (HDAC1-3) are histone lysine
delactylases. Sci Adv. 8:eabi66962022. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Jennings EQ, Ray JD, Zerio CJ, Zerio CJ,
Trujillo MN, McDonald DM, Chapman E, Spiegel DA and Galligan JJ:
Sirtuin 2 regulates protein LactoylLys modifications. Chembiochem.
22:2102–2106. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Ramadori G and Coppari R: Pharmacological
manipulations of CNS sirtuins: Potential effects on metabolic
homeostasis. Pharmacol Res. 62:48–54. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Moreno-Yruela C, Bæk M, Monda F and Olsen
CA: Chiral posttranslational modification to lysine ε-amino groups.
Acc Chem Res. 55:1456–1466. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Zu H, Li C, Dai C, Pan Y, Ding C, Sun H,
Zhang X, Yao X, Zang J and Mo X: SIRT2 functions as a histone
delactylase and inhibits the proliferation and migration of
neuroblastoma cells. Cell Discov. 8:542022. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Nickel GA, Pederson NJ, Faheem Yang Z,
Bulf J and Diehl KL: Sirtuin 6 is a histone delactylase. J Biol
Chem. 301:1107952025. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Hu X, Huang X, Yang Y, Sun Y, Zhao Y,
Zhang Z, Qiu D, Wu Y, Wu G and Lei L: Dux activates
metabolism-lactylation-MET network during early iPSC reprogramming
with Brg1 as the histone lactylation reader. Nucleic Acids Res.
52:5529–5548. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Zhai G, Niu Z, Jiang Z, Zhao F, Wang S,
Chen C, Zheng W, Wang A, Zang Y, Han Y and Zhang K: DPF2 reads
histone lactylation to drive transcription and tumorigenesis. Proc
Natl Acad Sci USA. 121:e24214961212024. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Lin T, Hou Y, Liu X, Ullah I, Qiu S, Lu Z,
Qi J and Yuan Y: Fluorinated proteolysis targeting
chimeras-sorafenib nanoassembly for epigenetic remodeling to combat
multi-pathway drug resistance in hepatocellular carcinoma. ACS
Nano. 19:34659–34676. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Rigo V, Amaro A, Reggiani F, Fenoglio D,
Martini S, Altosole T, Petito M, Profumo C and Croce M: The
systematic comparison of enhancer of zeste homolog-2-,
bromodomain-containing proteins-, histone deacetylase-, and
DNA-methyltransferase 1-inhibitors in a syngeneic murine model of
melanoma reveals differential anti-tumoral and immunomodulatory
activities. MedComm (2020). 6:e703362025. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Nuñez R, Sidlowski PFW, Steen EA,
Wynia-Smith SL, Sprague DJ, Keyes RF and Smith BC: The TRIM33
bromodomain recognizes histone lysine lactylation. ACS Chem Biol.
19:2418–2428. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Zhang F, Zhou J, Lu P, Zhang X, Yang L, Wu
J, Zhang L, Zhang L, Pang J, Xie H, et al: Lactylation of histone
by BRD4 regulates astrocyte polarization after experimental
subarachnoid hemorrhage. J Neuroinflammation. 21:1862024.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Zhang YZ, Zhu HJ, Zhou XX, Li SJ, Han D,
Liu FX, Zhou HM, Jiang XY, Guan YY, Ren HR, et al: Discovery of
novel CBP/p300 and BRD4 dual-target PROTACs with potent antitumor
activity in prostate cancer. J Med Chem. Feb 6–2026.(Epub ahead of
print).
|
|
111
|
Wang L, Xu M, Kao CY, Tsai SY and Tsai MJ:
Small molecule JQ1 promotes prostate cancer invasion via
BET-independent inactivation of FOXA1. J Clin Invest.
130:1782–1792. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Zhang D, Gao J, Zhu Z, Mao Q, Xu Z, Singh
PK, Rimayi CC, Moreno-Yruela C, Xu S, Li G, et al: Lysine
l-lactylation is the dominant lactylation isomer induced by
glycolysis. Nat Chem Biol. 21:91–99. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Merkuri F, Rothstein M and Simoes-Costa M:
Histone lactylation couples cellular metabolism with developmental
gene regulatory networks. Nat Commun. 15:902024. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Gaffney DO, Jennings EQ, Anderson CC,
Marentette JO, Shi T, Schou Oxvig AM, Streeter MD, Johannsen M,
Spiegel DA, Chapman E, et al: Non-enzymatic lysine lactoylation of
glycolytic enzymes. Cell Chem Biol. 27:206–213.e6. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Sheng X, Lin H, Cole PA and Zhao Y:
Biochemistry and regulation of histone lysine L-lactylation. Nat
Rev Mol Cell Biol. 27:95–109. 2026. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Taylor SJ, Winter MG, Gillis CC, Silva
LAD, Dobbins AL, Muramatsu MK, Jimenez AG, Chanin RB, Spiga L,
Llano EM, et al: Colonocyte-derived lactate promotes E. coli
fitness in the context of inflammation-associated gut microbiota
dysbiosis. Microbiome. 10:2002022. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Han S, Bao X, Zou Y, Wang L, Li Y, Yang L,
Liao A, Zhang X, Jiang X, Liang D, et al: d-lactate modulates M2
tumor-associated macrophages and remodels immunosuppressive tumor
microenvironment for hepatocellular carcinoma. Sci Adv.
9:eadg26972023. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
McDonald B, Zucoloto AZ, Yu IL, Burkhard
R, Brown K, Geuking MB and McCoy KD: Programing of an intravascular
immune firewall by the gut microbiota protects against pathogen
dissemination during infection. Cell Host Microbe. 28:660–668.e4.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Pan L, Feng F, Wu J, Fan S, Han J, Wang S,
Yang L, Liu W, Wang C and Xu K: Demethylzeylasteral targets lactate
by inhibiting histone lactylation to suppress the tumorigenicity of
liver cancer stem cells. Pharmacol Res. 181:1062702022. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Wagner W, Ciszewski WM and Kania KD: L-
and D-lactate enhance DNA repair and modulate the resistance of
cervical carcinoma cells to anticancer drugs via histone
deacetylase inhibition and hydroxycarboxylic acid receptor 1
activation. Commun Signal. 13:362015. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Ma N, Wang L, Meng M, Wang Y, Huo R, Chang
G and Shen X: D-sodium lactate promotes the activation of NF-κB
signaling pathway induced by lipopolysaccharide via histone
lactylation in bovine mammary epithelial cells. Microb Pathog.
199:1071982025. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Rabbani N, Xue M and Thornalley PJ:
Activity, regulation, copy number and function in the glyoxalase
system. Biochem Soc Trans. 42:419–424. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Pantano F, Simonetti S, Iuliani M, Guillen
MJ, Cuevas C, Aviles P, Cavaliere S, Napolitano A, Cortellini A,
Mazzocca A, et al: S-p-bromobenzyl-glutathione cyclopentyl diester
(BBGC) as novel therapeutic strategy to enhance trabectedin
anti-tumor effect in soft tissue sarcoma preclinical models.
Oncogene. 43:2986–2994. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Tang J, Zhong J, Yang Z, Su Q and Mo W:
Glyoxalase 1 inhibitor BBGC suppresses the progression of chronic
lymphocytic leukemia and promotes the efficacy of palbociclib.
Biochem Biophys Res Commun. 650:96–102. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Wang M, He T, Meng D, Lv W, Ye J, Cheng L
and Hu J: BZW2 modulates lung adenocarcinoma progression through
glycolysis-mediated IDH3G lactylation modification. J Proteome Res.
22:3854–3865. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Chen Y, Wu J, Zhai L, Zhang T, Yin H, Gao
H, Zhao F, Wang Z, Yang X, Jin M, et al: Metabolic regulation of
homologous recombination repair by MRE11 lactylation. Cell.
187:294–311.e21. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Zheng J, She H, Han R, Tang J, Dou Y, Lu
C, Huang D, Lin C, Wu D, He C, et al: Dapk2 dysfunction leads to
Mic60 lactylation and mitochondrial metabolic reprogramming,
promoting lung cancer EGFR-TKI resistance and metastasis. Dev Cell.
60:3267–3284.e7. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Sun T, Liu B, Li Y, Wu J, Cao Y, Yang S,
Tan H, Cai L, Zhang S, Qi X, et al: Oxamate enhances the efficacy
of CAR-T therapy against glioblastoma via suppressing
ectonucleotidases and CCR8 lactylation. J Exp Clin Cancer Res.
42:2532023. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Zhang C, Zhou L, Zhang M, Du Y, Li C, Ren
H and Zheng L: H3K18 lactylation potentiates immune escape of
non-small cell lung cancer. Cancer Res. 84:3589–3601. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Ma XM, Geng K, Wang P, Jiang Z, Law BYK
and Xu Y: MCT4-dependent lactate transport: A novel mechanism for
cardiac energy metabolism injury and inflammation in type 2
diabetes mellitus. Cardiovasc Diabetol. 23:962024. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Li Y, Tang S, Shi X, Lv J, Wu X, Zhang Y,
Wang H, He J, Zhu Y, Ju Y, et al: Metabolic classification suggests
the GLUT1/ALDOB/G6PD axis as a therapeutic target in
chemotherapy-resistant pancreatic cancer. Cell Rep Med.
4:1011622023. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
He J, Li W, Wang S, Lan J, Hong X, Liao L,
Kang D, Wang W, Wang R, Zhang W, et al: Cancer associated
fibroblasts-derived lactate induces oxaliplatin treatment
resistance by promoting cancer stemness via ANTXR1 lactylation in
colorectal cancer. Cancer Lett. 631:2179172025. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Galli U, Mesenzani O, Coppo C, Sorba G,
Canonico PL, Tron GC and Genazzani AA: Identification of a sirtuin
3 inhibitor that displays selectivity over sirtuin 1 and 2. Eur J
Med Chem. 55:58–66. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Wang X, He H, Rui W, Zhang N, Zhu Y and
Xie X: TRIM38 triggers the uniquitination and degradation of
glucose transporter type 1 (GLUT1) to restrict tumor progression in
bladder cancer. J Transl Med. 19:5082021. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Sołtyka-Krajewska M, Ziemniak M,
Zawadzka-Kazimierczuk A, Skrzypczyk P, Siwiak-Niedbalska E,
Jaśkiewicz A, Zieliński R, Fokt I, Skóra S, Koźmiński W, et al:
Potent biological activity of fluorinated derivatives of
2-deoxy-d-glucose in a glioblastoma model. Biomedicines.
12:22402024. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Mondal S, Roy D, Sarkar Bhattacharya S,
Jin L, Jung D, Zhang S, Kalogera E, Staub J, Wang Y, Xuyang W, et
al: Therapeutic targeting of PFKFB3 with a novel glycolytic
inhibitor PFK158 promotes lipophagy and chemosensitivity in
gynecologic cancers. Int J Cancer. 144:178–189. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Biswal S, Panda M and Biswal BK: Shikonin
stimulates mitochondria-mediated apoptosis by enhancing
intracellular reactive oxygen species production and DNA damage in
oral cancer cells. J Cell Biochem. 126:e306712025. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Giraud C, Treluyer JM, Rey E, Chiron C,
Vincent J, Pons G and Tran A: In vitro and in vivo inhibitory
effect of stiripentol on clobazam metabolism. Drug Metab Dispos.
34:608–611. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Azizzadeh B, Majidinia M and Gheysarzadeh
A: The reciprocal effects of autophagy and the warburg effect in
pancreatic ductal adenocarcinoma: An in vitro study. Med Oncol.
42:862025. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Mohammad GH, Vassileva V, Acedo P, Olde
Damink SWM, Malago M, Dhar DK and Pereira SP: Targeting pyruvate
kinase M2 and lactate dehydrogenase a is an effective combination
strategy for the treatment of pancreatic cancer. Cancers (Basel).
11:13722019. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Aktepe N and Yukselten Y: Induction of
apoptosis in human hormone-refractory prostate cancer cell lines by
using resveratrol in combination with AT-101. Andrologia.
54:e142672022. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Gao L, Xu Z, Huang Z, Tang Y, Yang D,
Huang J, He L, Liu M, Chen Z and Teng Y: CPI-613 rewires lipid
metabolism to enhance pancreatic cancer apoptosis via the AMPK-ACC
signaling. J Exp Clin Cancer Res. 39:732020. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Guo Y, Liu B, Liu Y, Sun W, Gao W, Mao S
and Chen L: Oncogenic chromatin modifier KAT2A activates MCT1 to
drive the glycolytic process and tumor progression in renal cell
carcinoma. Front Cell Dev Biol. 9:6907962021. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Cluntun AA, Badolia R, Lettlova S, Parnell
KM, Shankar TS, Diakos NA, Olson KA, Taleb I, Tatum SM, Berg JA, et
al: The pyruvate-lactate axis modulates cardiac hypertrophy and
heart failure. Cell Metab. 33:629–648.e10. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Wang D, Wu Y, Zhao Y, Ma X, Shi J, Ni J,
Gao Y, Cai H, Dong C and Zhou HB: The novel SIRT2-targeted PROTAC
degraders as the efficient agents for the treatment of ovarian
cancer. Eur J Med Chem. 302((Pt 1)): 1182952026. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Michaelides MR, Kluge A, Patane M, Van
Drie JH, Wang C, Hansen TM, Risi RM, Mantei R, Hertel C,
Karukurichi K, et al: Discovery of spiro oxazolidinediones as
selective, orally bioavailable inhibitors of p300/CBP histone
acetyltransferases. ACS Med Chem Lett. 9:28–33. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Liu R, Zhang Z, Yang H, Zhou K, Geng M,
Zhou W, Zhang M, Huang X and Li Y: Design, synthesis, and
biological evaluation of a new class of histone acetyltransferase
p300 inhibitors. Eur J Med Chem. 180:171–190. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Xiang Q, Wang C, Zhang Y, Xue X, Song M,
Zhang C, Li C, Wu C, Li K, Hui X, et al: Discovery and optimization
of 1-(1 H -indol-1-yl)ethanone derivatives as CBP/EP300 bromodomain
inhibitors for the treatment of castration-resistant prostate
cancer. Eur J Med Chem. 147:238–252. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Biel M, Kretsovali A, Karatzali E,
Papamatheakis J and Giannis A: Design, synthesis, and biological
evaluation of a small-molecule inhibitor of the histone
acetyltransferase Gcn5. Angew Chem Int Ed Engl. 43:3974–3976. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Al-Qazzan M, Al-Balas Q, Alnajjar B,
Kasabri V, Al-Hiari Y, Zayed A, AlKhateeb R and Al-Akeedi M:
Cytotoxic, anti-inflammatory, antioxidant, and anti-glyoxalase-I
evaluation of chelating substances: In silico and in vitro study.
PLoS One. 20:e03334052025. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Pun MD, Wu HH, Olatunji FP, Kesic BN,
Peters JW and Berkman CE: Phosphorus containing analogues of SAHA
as inhibitors of HDACs. J Enzyme Inhib Med Chem. 37:1315–1319.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Ning ZQ, Li ZB, Newman MJ, Shan S, Wang
XH, Pan DS, Zhang J, Dong M, Du X and Lu XP: Chidamide
(CS055/HBI-8000): A new histone deacetylase inhibitor of the
benzamide class with antitumor activity and the ability to enhance
immune cell-mediated tumor cell cytotoxicity. Cancer Chemother
Pharmacol. 69:901–909. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Song X, Wang H, Gao Y, Zhang W and Lei X:
Synthesis and biological evaluation of the fluoro analog of
romidepsin with improved selectivity for class I histone
deacetylases (HDACs). Bioorg Chem. 159:1083482025. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Hwang ES and Song SB: Nicotinamide is an
inhibitor of SIRT1 in vitro, but can be a stimulator in cells. Cell
Mol Life Sci. 74:3347–3362. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Spinck M, Bischoff M, Lampe P, Meyer-Almes
FJ, Sievers S and Neumann H: Discovery of dihydro-1,4-benzoxazine
carboxamides as potent and highly selective inhibitors of
sirtuin-1. J Med Chem. 64:5838–5849. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Bai X, Yao L, Ma X and Xu X: Small
molecules as SIRT modulators. Mini Rev Med Chem. 18:1151–1157.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Duvic M, Talpur R, Ni X, Zhang C, Hazarika
P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM and Frankel
SR: Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic
acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood.
109:31–39. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Apuri S and Sokol L: An overview of
investigational histone deacetylase inhibitors (HDACis) for the
treatment of non-hodgkin's lymphoma. Expert Opin Invest Drugs.
25:687–696. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Tsukihara S, Akiyama Y, Shimada S, Hatano
M, Igarashi Y, Taniai T, Tanji Y, Kodera K, Yasukawa K, Umeura K,
et al: Delactylase effects of SIRT1 on a positive feedback loop
involving the H19-glycolysis-histone lactylation in gastric cancer.
Oncogene. 44:724–738. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Jin H, Luo R, Li J, Zhao H, Ouyang S, Yao
Y, Chen D, Ling Z, Zhu W, Chen M, et al: Inhaled platelet
vesicle-decoyed biomimetic nanoparticles attenuate inflammatory
lung injury. Front Pharmacol. 13:10502242022. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Rodrigues DA, Roe A, Griffith D and
Chonghaile TN: Advances in the design and development of
PROTAC-mediated HDAC degradation. Curr Top Med Chem. 22:408–424.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Li X, Chen M, Chen X, He X, Li X, Wei H,
Tan Y, Min J, Azam T, Xue M, et al: TRAP1 drives smooth muscle cell
senescence and promotes atherosclerosis via HDAC3-primed histone H4
lysine 12 lactylation. Eur Heart J. 45:4219–4235. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Vannam R, Sayilgan J, Ojeda S,
Karakyriakou B, Hu E, Kreuzer J, Morris R, Herrera Lopez XI, Rai S,
Haas W, et al: Targeted degradation of the enhancer lysine
acetyltransferases CBP and p300. Cell Chem Biol. 28:503–514.e12.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Chen J, Huang Z, Chen Y, Tian H, Chai P,
Shen Y, Yao Y, Xu S, Ge S and Jia R: Lactate and lactylation in
cancer. Signal Transduction Targeted Ther. 10:382025. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Zong Z, Ren J, Yang B, Zhang L and Zhou F:
Emerging roles of lysine lactyltransferases and lactylation. Nat
Cell Biol. 27:563–574. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Chu YD, Cheng LC, Lim SN, Lai MW, Yeh CT
and Lin WR: Aldolase B-driven lactagenesis and CEACAM6 activation
promote cell renewal and chemoresistance in colorectal cancer
through the warburg effect. Cell Death Dis. 14:6602023. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Li G, Wang D, Zhai Y, Pan C, Zhang J, Wang
C, Huang R, Yu M, Li Y, Liu X, et al: Glycometabolic
reprogramming-induced XRCC1 lactylation confers therapeutic
resistance in ALDH1A3-overexpressing glioblastoma. Cell Metab.
36:1696–1710.e10. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Wang L, Zeng T, Wang Y, Wang G, Yu W,
Zhang J, Shi Y, Li J and Ding J: K90 lactylation orchestrates YAP
nuclear sequestration by impairing binding with exportin CRM1 and
enhances HCC malignancy. Cancer Lett. 611:2173862024. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Yang Z, Su W, Zhang Q, Niu L, Feng B,
Zhang Y, Huang F, He J, Zhou Q, Zhou X, et al: Lactylation of HDAC1
confers resistance to ferroptosis in colorectal cancer. Adv Sci
(Weinh). 12:e24088452025. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Chen Y, Yan Q, Ruan S, Cui J, Li Z, Zhang
Z, Yang J, Fang J, Liu S, Huang S, et al: GCLM lactylation mediated
by ACAT2 promotes ferroptosis resistance in KRASG12D-mutant cancer.
Cell Rep. 44:1157742025. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Chen S, Xie DF, Li S, Luo J, Han Y, Guo H,
Gao S, Huang X, Guan H, Huang R and Zhou PK: TAB182 regulates
glycolytic metabolism by controlling LDHA transcription to impact
tumor radiosensitivity. Cell Death Dis. 15:2092024. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Cao Y, Gao A, Li X, Min H, He C, Sun X,
Ding WQ and Zhou J: Elevated TAB182 enhances the radioresistance of
esophageal squamous cell carcinoma through G2-M checkpoint
modulation. Cancer Med. 10:3101–3112. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Jin X, Kuang Y, Li L, Li H, Zhao T, He Y,
Di C, Kang J, Yuan L, Yu B and Li Q: A positive feedback circuit
comprising p21 and HIF-1α aggravates hypoxia-induced
radioresistance of glioblastoma by promoting Glut1/LDHA-mediated
glycolysis. FASEB J. 36:e222292022. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Chen HJ, Yu MM, Huang JC, Lan FY, Liao HH,
Xu ZH, Yu YJ, Huang YC and Chen F: SLC4A4 is a novel driver of
enzalutamide resistance in prostate cancer. Cancer Lett.
597:2170702024. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Li A, Gong Z, Long Y, Li Y, Liu C, Lu X,
Li Q, He X, Lu H, Wu K, et al: Lactylation of LSD1 is an acquired
epigenetic vulnerability of BRAFi/MEKi-resistant melanoma. Dev
Cell. 60:1974–1990.e11. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Liu Y, Lin J, Yu Z, Li X, Lv X, Liu P, Sun
X, Zhang Z, Gao X, Sun K, et al: Tumor-associated schwann cell
remodeling under metabolic stress via lactate sensing orchestrates
pancreatic ductal adenocarcinoma development. Cell Metab.
37:1907–1925.e14. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Liu Y, Chen J, Ma L, Zhao S, Hui X, Xiong
W, Cheng S and Zhang Y: ZMIZ1 lactylation induces tamoxifen
resistance in breast cancer through increasing transcriptional
activity of nanog to impact cell stemness and cholesterol uptake.
Cell Biol Toxicol. 41:1172025. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Gao R, Li Y, Xu Z, Zhang F, Xu J, Hu Y,
Yin J, Yang K, Sun L, Wang Q, et al: Mitochondrial pyruvate carrier
1 regulates fatty acid synthase lactylation and mediates treatment
of nonalcoholic fatty liver disease. Hepatology. 78:1800–1815.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Miao S, Lin L, Long M and Fu Y: H3 lysine
18 lactylation-mediated RRAS2 facilitates migration and invasion of
head and neck squamous cell carcinoma. Sci Rep. 15:212822025.
View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Li S, Shi X and Zhou X: Histone
lactylation promoted colorectal cancer progression by enhancing
tumor necrosis factor transcription and served as a diagnostic
biomarker. Anticancer Drugs. 37:245–251. 2026. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Hou J, Guo M, Li Y and Liao Y: Lactylated
histone H3K18 as a potential biomarker for the diagnosis and
prediction of the severity of pancreatic cancer. Clinics (Sao
Paulo). 80:1005442024. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Dong W, Huang SX, Qin ML and Pan Z:
Mitochondrial alanyl-tRNA synthetase 2 mediates histone lactylation
to promote ferroptosis in intestinal ischemia-reperfusion injury.
World J Gastrointest Surg. 17:1067772025. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Sun X, Dong H, Su R, Chen J, Li W, Yin S
and Zhang C: Lactylation-related gene signature accurately predicts
prognosis and immunotherapy response in gastric cancer. Front
Oncol. 14:14855802024. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Jiao Y, Ji F, Hou L, Lv Y and Zhang J:
Lactylation-related gene signature for prognostic prediction and
immune infiltration analysis in breast cancer. Heliyon.
10:e247772024. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Eftekhari A, Sabir U and Kasumov T: The
role of lysine acetylation in metabolic sensing and proteostasis.
Pharmacol Ther. 274:1089082025. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Shi W, Cassmann TJ, Bhagwate AV, Hitosugi
T and Ip WKE: Lactic acid induces transcriptional repression of
macrophage inflammatory response via histone acetylation. Cell Rep.
43:1137462024. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Feng Q, Liu Z, Yu X, Huang T, Chen J, Wang
J, Wilhelm J, Li S, Song J, Li W, et al: Lactate increases stemness
of CD8 + T cells to augment anti-tumor immunity. Nat Commun.
13:49812022. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Wu T, Zhao Y, Zhang X, Wang Y, Chen Q,
Zhang M, Sheng H, Zhang Y, Guo J, Li J, et al: Short-chain acyl
post-translational modifications in cancers: Mechanisms, roles, and
therapeutic implications. Cancer Commun (Lond). 45:1247–1284. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
189
|
He S, Wang C, Li R, Xu K, Zhang W, Feng B,
Cheng S, Ren J and Chen J: The role of succinylation-mediated
metabolic reprogramming in tumor progression. Mol Biol Rep.
52:9542025. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Yang P, Qin Y, Zeng L, He Y, Xie Y, Cheng
X, Huang W and Cao L: Crotonylation and disease: Current progress
and future perspectives. Biomed Pharmacother. 165:1151082023.
View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Kaczmarska Z, Ortega E, Goudarzi A, Huang
H, Kim S, Márquez JA, Zhao Y, Khochbin S and Panne D: Structure of
p300 in complex with acyl-CoA variants. Nat Chem Biol. 13:21–29.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Liu X, Wang L, Zhao K, Thompson PR, Hwang
Y, Marmorstein R and Cole PA: The structural basis of protein
acetylation by the p300/CBP transcriptional coactivator. Nature.
451:846–850. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Setlere S, Jurcenko M, Gailite L, Rots D
and Kenina V: Alanyl-tRNA synthetase 1 gene variants in hereditary
neuropathy: Genotype and phenotype overview. Neurol Genet.
8:e2000192022. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Helman G, Mendes MI, Nicita F, Darbelli L,
Sherbini O, Moore T, Derksen A, Amy Pizzino, Carrozzo R, Torraco A,
et al: Expanded phenotype of AARS1-related white matter disease.
Genet Med. 23:2352–2359. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Yeung C, Gibson AE, Issaq SH, Oshima N,
Baumgart JT, Edessa LD, Rai G, Urban DJ, Johnson MS, Benavides GA,
et al: Targeting glycolysis through inhibition of lactate
dehydrogenase impairs tumor growth in preclinical models of ewing
sarcoma. Cancer Res. 79:5060–5073. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Lai C, Pursell N, Gierut J, Saxena U, Zhou
W, Dills M, Diwanji R, Dutta C, Koser M, Nazef N, et al: Specific
inhibition of hepatic lactate dehydrogenase reduces oxalate
production in mouse models of primary hyperoxaluria. Mol Ther.
26:1983–1995. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Rai G, Brimacombe KR, Mott BT, Urban DJ,
Hu X, Yang SM, Lee TD, Cheff DM, Kouznetsova J, Benavides GA, et
al: Discovery and optimization of potent, cell-active
pyrazole-based inhibitors of lactate dehydrogenase (LDH). J Med
Chem. 60:9184–9204. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Li X, Zhang Y, Xu L, Wang A, Zou Y, Li T,
Huang L, Chen W, Liu S, Jiang K, et al: Ultrasensitive sensors
reveal the spatiotemporal landscape of lactate metabolism in
physiology and disease. Cell Metab. 35:200–211.e9. 2023. View Article : Google Scholar : PubMed/NCBI
|