Introduction
Breast cancer (BRCA) remains one of the most
prevalent and fatal malignancies affecting women worldwide, with
>2.3 million new cases and 685,000 associated deaths every year
worldwide, imposing notable psychological and economic burdens on
both patients and their families (1,2).
Despite advances in surgical techniques, radiotherapy, chemotherapy
and targeted therapies, the heterogeneity of BRCA results in
variable treatment responses and outcomes among patients (1). This clinical variability underscores
the urgent need to identify novel biomarkers that can improve
prognostic accuracy and guide personalized therapeutic strategies.
In addition, the high incidence and mortality rates of BRCA
continue to pose a notable challenge to global public health,
highlighting the importance of refining prognostic tools to
optimize patient management.
Programmed cell death (PCD) encompasses a spectrum
of tightly regulated cellular processes, including apoptosis,
necroptosis, pyroptosis, ferroptosis and autophagy, all of which
collectively maintain tissue homeostasis and modulate immune
responses (3,4). Emerging evidence has implicated PCD
pathways as key modulators of tumor initiation, progression and
response to therapy (5,6). However, the specific roles and
regulatory mechanisms of PCD-related genes (CRGs) in BRCA remain
poorly understood. Previous studies have demonstrated that
alterations in PCD can contribute to tumor immune evasion and
therapeutic resistance (6).
However, to the best of our knowledge, comprehensive analyses
integrating PCD gene signatures with BRCA prognosis and
immunological context remain lacking. Therefore, elucidating the
landscape of CRGs in BRCA may provide key insights into tumor
biology and reveal novel prognostic biomarkers and therapeutic
targets.
Advances in high-throughput sequencing technologies
and bioinformatics have facilitated the exploration of
transcriptomic alterations and their clinical relevance in
oncology. In particular, the integration of RNA sequencing
(RNA-seq) data with machine learning approaches has emerged as a
powerful strategy to construct robust prognostic models, by
capturing complex gene expression patterns. Whilst previous
prognostic models in BRCA have mainly focused on limited gene sets
or clinical parameters, targeting multi-cohort transcriptomic
datasets and sophisticated computational algorithms can enhance
predictive performance and clinical applicability. Huang et
al (7) developed a nomogram
using survival predictors (tumor grade, T-stage, N-stage, LNR, ER
status, PR status, HER2 status) for predicting the overall survival
of patients with breast cancer. Another palmitoylation-related gene
model could be used for predicting the prognosis and treatment
response in breast cancer (8).
Multi-cohort and single-cell profiling of aging genes showed a good
performance in predicting the prognosis and therapeutic response in
breast cancer (9). Zeng et
al (10) developed a lipid
metabolism and ferroptosis-associated index for prognosis and
immunotherapy response prediction in hormone receptor-positive
breast cancer using multi-cohort data. Despite these methodological
progresses, to the best of our knowledge, few studies have
systematically developed and validated PCD gene-based signatures
for BRCA prognosis (11,12). Furthermore, their potential
association with the tumor immune microenvironment (TIME) and drug
sensitivity remain unknown.
In the present study, multiple publicly available
RNA-seq datasets encompassing BRCA tissues and normal controls were
applied to identify differentially expressed genes (DEGs)
associated with PCD. By integrating these data with curated CRG
lists, the present study employed univariate Cox regression
analysis to screen for prognostically significant genes.
Subsequently, a comprehensive machine learning framework was
implemented to evaluate the numerous algorithmic combinations,
culminating in an optimal gene signature model with high
concordance index (C-index) for survival prediction. This
integrative approach enables the capture of key PCD-related
molecular features underpinning BRCA heterogeneity.
Furthermore, the constructed PCD-based gene
signature (CDS) was investigated for its association with immune
cell infiltration and immune-related pathways within the tumor
microenvironment. Understanding the interplay between tumor cell
death programs and immune contexture is key to predicting
immunotherapy responsiveness and developing combination treatment
strategies (13). Additionally, the
association between CDS and chemotherapeutic drug sensitivity was
assessed using pharmacogenomic data, aiming to provide actionable
information for individualized treatment selection. Through this
multidimensional analysis, the present study aimed to establish a
novel prognostic biomarker that can not only stratify patient risk
but can also inform immunological and pharmacological aspects of
BRCA management. Collectively, the present study aimed to fill the
existing gap in BRCA prognostication by focusing on PCD gene
networks and their clinical implications.
Materials and methods
Data acquisition
To validate the present findings, data were
collected from various publicly available datasets. RNA-seq and
clinical data were sourced from The Cancer Genome Atlas (TCGA)
database (https://portal.gdc.cancer.gov/repository), which
offers a comprehensive molecular data repository for various cancer
types, including BRCA. The present study specifically selected
RNA-seq data for patients with BRCA, alongside corresponding
clinical information, such as survival rates, tumor staging and
other clinical variables.
Additionally, external validation cohorts were
incorporated to strengthen the reliability of the present study
model. These cohorts included the Molecular Taxonomy of Breast
Cancer International Consortium (METABRIC; http://ega-archive.org/studies/EGAS00000000083)
dataset and datasets from the Gene Expression Omnibus (GEO)
database (https://www.ncbi.nlm.nih.gov/geo), namely GSE20685
(14), GSE20711 (15), GSE42568 (16), GSE58812 (17) and GSE96058 (18).
Inclusion criteria for the present study were as
follows: i) A confirmed pathological diagnosis of invasive BRCA;
and ii) complete clinical and survival data. Patients with <6
months of follow-up, previous malignancies or metastatic BRCA were
excluded. This multi-cohort approach allowed for validation across
different populations, enhancing the generalizability of the
present study predictive signature. In addition, three
immunotherapy-related datasets, namely, IMvigor210 (19), GSE91061 (20) and GSE78220 (21), were utilized to assess the ability
of CDS in predicting the benefits of immunotherapy.
The PCD patterns included in the present study were
pyroptosis, ferroptosis, necroptosis, autophagy, immunological cell
death, entotic cell death genes, cuproptosis, parthanatos,
lysosome-dependent cell death, intrinsic apoptosis, extrinsic
apoptosis, necrosis, anoikis, apoptosis-like and necrosis-like
morphology. The present study obtained a set of genes associated
with PCD from multiple resources, including the Molecular
Signatures Database (22), Kyoto
Encyclopedia of Genes and Genomes database (23), articles (24,25)
and the GeneCards (26) database
(Table SI) (26,27).
Preprocessing of RNA-seq data
RNA-seq data from TCGA and GEO were preprocessed to
standardize and ensure consistency across datasets. Key
preprocessing steps included: i) Normalization, where RNA-seq data
was normalized using ‘DESeq2’ to adjust for sequencing depth
differences (27); ii) batch effect
correction, where the ‘ComBat’ method from the ‘sva’ R package
(version 3.46.0; R Development Core Team) was applied to correct
for batch effects (28); and iii)
log2 transformation, which was used on the normalized
counts to stabilize variance and prepare the data for subsequent
analysis.
Construction of CDS
A CDS was developed using a multi-step machine
learning framework. First, univariate Cox regression analysis
identified CRGs significantly associated with overall survival (OS)
in BRCA. Subsequently, 101 distinct model combinations derived from
10 machine learning algorithms were evaluated, including Random
Survival Forests (29), least
absolute shrinkage and selection operator (30), Ridge (31), Elastic Net (Enet) (32), CoxBoost (33), stepwise Cox regression (34), partial least squares Cox models
(35), supervised principal
component analysis (36),
generalized boosted regression (37) and survival support vector machines
(38). Detailed information about
the operation of these machine learning programs is presented in
Data S1.
Leave-one-out cross-validation was applied to TCGA
cohort to determine the optimal model. The accuracy of its
prediction of the overall survival rate of patients for each model
was assessed using Harrell's C-index (39). The combination designated as
Stepwise Cox (StepCox) (both) + Enet (α=0.9) demonstrated the
highest average C-index and was therefore selected as the final CDS
model. This model incorporated five genes and the risk score for
each patient was computed as follows: CDS score=[0.0135× anoctamin
6 (ANO6)level] + [0.079× polo-like kinase 1 (PLK1)level] + [0.1241×
solute carrier family 7 member 5 (SLC7A5)level] + [0.0615× tubulin
α-1C chain (TUBA1C)level] + [-0.165× transcobalamin 1
(TCN1)level].
Patients were then stratified into high- and
low-risk subgroups based on the median CDS score.
Nomogram construction
To further improve the clinical value, a nomogram
was constructed based on age, sex, T stage, N stage, clinical
stage, estrogen receptor, progesterone receptor and CDS-based risk
score using ‘nomogramEx’ R package (version 3.0; R Development Core
Team) (40). A calibration curve
was constructed to show the relationship between the actual and
predicted probabilities for the 1-, 3- and 5-year OS (41).
Immune microenvironment analysis
The relationship between the CDS-based risk score
(ANO6, PLK1, SLC7A5, TUBA1C and TCN1) and the TIME was investigated
using several computational approaches. The ‘immunedeconv’ R
package (version 2.0.3; R Development Core Team), which
incorporated multiple deconvolution algorithms, including Cell-type
Identification By Estimating Relative Subsets Of RNA Transcripts
(CIBERSORT), Estimating the Proportions of Immune and Cancer Cells
(42), xCell (43), Microenvironment Cell
Populations-counter (44) and Tumor
Immune Estimation Resource (45),
was used to estimate the abundance of various immune cell types
(46). CIBERSORT is a versatile
computational method for quantifying cell fractions from bulk
tissue gene expression profiles (47). The Estimation of Stromal and Immune
cells in Malignant Tumor tissues using Expression (ESTIMATE)
algorithm was used to compute stromal, immune and ESTIMATE scores,
reflecting the non-tumor cellular components in the tumor
microenvironment (48).
Additionally, single-sample gene set enrichment analysis (ssGSEA)
through the ‘Gene Set Variation Analysis’ R package (version
1.46.0; R Development Core Team) was performed to quantify the
activity of immune-related pathways and functions. Several
computational tools were used to evaluate the role of CDS in
predicting immunotherapy benefit. Intratumor heterogeneity (ITH)
scores for BRCA were calculated using the ‘DEPTH2’ algorithm
(49). Tumor Immune Dysfunction and
Exclusion (TIDE) scores were obtained from the TIDE database
(http://tide.dfci.harvard.edu; version
1.0) (50). Immunophenoscores (IPS)
for patients with breast cancer were retrieved from The Cancer
Immunome Atlas database (https://tcia.at/home; version 1.0) (51). Tumor mutation burden (TMB) scores
for patients with BRCA were collected from TCGA database (52).
Drug sensitivity prediction
Drug sensitivity predictions were generated using
the ‘OncoPredict’ R package (R Development Core Team, version
no.0.2) (53), which leverages gene
expression profiles and pharmacogenomic data from the Genomics of
Drug Sensitivity in Cancer database (54) to estimate the half-maximal
inhibitory concentration (IC50) values. The
‘OncoPredict’ R package was utilized to predict IC50 of
various chemotherapeutic and targeted agents based on gene
expression profiles. Differences in drug sensitivity between the
high and low CDS score groups were compared to assess the potential
of the signature for the prediction of therapeutic response.
Protein expression data from the Human
Protein Atlas (HPA)
Protein expression levels of the CDS genes were
examined using immunohistochemistry data from the HPA (https://www.proteinatlas.org/) (55), which provides protein localization
and expression information in both normal and cancerous tissues.
Representative immunohistochemical images of CDS genes of in normal
and cancerous tissues was obtained from the ‘TISSUE’ resource and
‘CANCER’ resource of HPA, respectively.
Cell culture
Human BRCA cell lines (T47D, BT549, SKBR3, MCF-7 and
MDA-MB-468) and a normal breast epithelial cell line (Bst578Bst)
were acquired from the Shanghai Institute of Biochemistry and Cell
Biology. These cells were cultured in RPMI-1640 medium (Procell
Life Science & Technology Co., Ltd.) enriched with 10% FBS
(Procell Life Science & Technology Co., Ltd.) and 1% antibiotic
solution (Procell Life Science & Technology Co., Ltd.). Cells
were maintained at 37°C in an atmosphere containing 5%
CO2. The use of these cell lines follow the guidelines
of Shanghai Institute of Biochemistry and Cell Biology.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the aforementioned
cells (T47D, BT549, SKBR3, MCF-7, MDA-MB-468 and Bst578Bst) using
Triquick® Reagent (Beijing Solarbio Science &
Technology Co., Ltd.). According to the manufacturer's protocol,
complementary DNA (cDNA) was synthesized from the isolated RNA
employing the SweScript RT I FTTSt Strand cDNA Synthesis Kit (cat.
no. G3330-100; Wuhan Servicebio Technology Co., Ltd.). The
resulting cDNA was then used as a template for qPCR analysis
performed on a PCR system using 2X Fast SYBR Green qPCR Master Mix
(Low ROX) (cat. no. G3321-05; Wuhan Servicebio Technology Co.,
Ltd.). The thermocycling conditions were as follows: Initial
denaturation at 95°C for 5 min; followed by 35 cycles at 95°C for
30 sec, 55°C for 30 sec and 70°C for 30 sec. Gene expression levels
were quantified using the 2−ΔΔCq method (56) and normalized to GAPDH expression.
Primer sequences used were as follows: GAPDH forward (F),
5′-GTCTCCTCTGACTTCAACAGCG-3′ and reverse (R),
5′-ACCACCCTGTTGCTGTAGCCAA-3′; anoctamin 6 (ANO6) F,
5′-ATGAAAAAGATGAGCAGGAATG-3′ and R, 5′-TTATTCTGATTTTGGCCGTA-3′;
Polo-like kinase 1 (PLK1) F, 5′-GGCAACCTTTTCCTGAATGA-3′ and PLK1 R,
5′-AATGGACCACACATCCACCT-3′; solute carrier family 7 member 5
(SLC7A5) F, 5′-GTCCAATCTAGATCCCAACTTCTC-3′ and R,
5′-ATTCCATCCTCCATAGGCAAAG-3′; tubulin α 1C (TUBA1C) F,
5′-GCTTCAAGGTTGGCATTAA-3′ and R, 5′-GAGCAATACCACAGCTGTT-3′;
transcobalamin (TCN) F, 5′-CATCCGCCTAAAACCTCTGTT'-3′ and TCN R,
5′-CCGAGCTTACATCTGACAATCTG-3′.
Statistical analysis
Statistical analyses were conducted in R software
(version 4.2.1; R Development Core Team). Survival differences
between risk groups were assessed using Kaplan-Meier analysis and
the log-rank test. For survival analysis, right-censoring was
handled using the Cox Proportional Hazards Model, accounting for
censored observations. The proportional hazards assumption was
evaluated using Schoenfeld residuals. The independent prognostic
value of the CDS was evaluated using univariate and multivariate
Cox proportional hazards models. Multicollinearity was assessed
using correlation matrices and variance inflation factor (VIF).
Genes with high pairwise correlations (r>0.8) or VIF values
>10 were excluded to prevent redundancy in the multivariate
analysis. Time-dependent receiver operating characteristic (ROC)
curves were plotted to determine the predictive accuracy of the
signature at 1, 3 and 5 years. GSEA was performed to identify
biological pathways associated with CDS risk groups. Comparisons
between two groups for continuous variables were conducted using
the unpaired Student's t-test or Wilcoxon rank-sum test. One-way
ANOVA (parametric) followed by a post-hoc test (Bonferroni) was
performed to compare differences between three or more groups,
whilst correlations were analyzed using Pearson's or Spearman's
rank correlation analysis. The data were presented as mean ±
standard deviation. P<0.05 was considered to indicate a
statistically significant difference.
Results
Identification of prognostic CRGs
Comparative transcriptomic analysis between BRCA and
normal breast tissues identified 2,404 DEGs in the TCGA dataset
(Fig. S1A and C). Intersection
with a curated list of CRGs yielded 373 candidate genes implicated
in PCD (Fig. S1C). Univariate Cox
regression analysis further refined this set to 40 CRGs by being
significantly associated with OS, representing potential prognostic
biomarkers for BRCA (Fig.
S1B).
Development and validation of a
machine learning-based CDS
The present study constructed a prognostic CDS using
a comprehensive machine learning framework. Among the 101 algorithm
combinations evaluated, the StepCox (both) + Enet (α=0.9) model
achieved the highest average C-index of 0.79 (Fig. 1A) and was selected for subsequent
analysis. This model incorporated five genes to calculate a CDS
risk score for each patient.
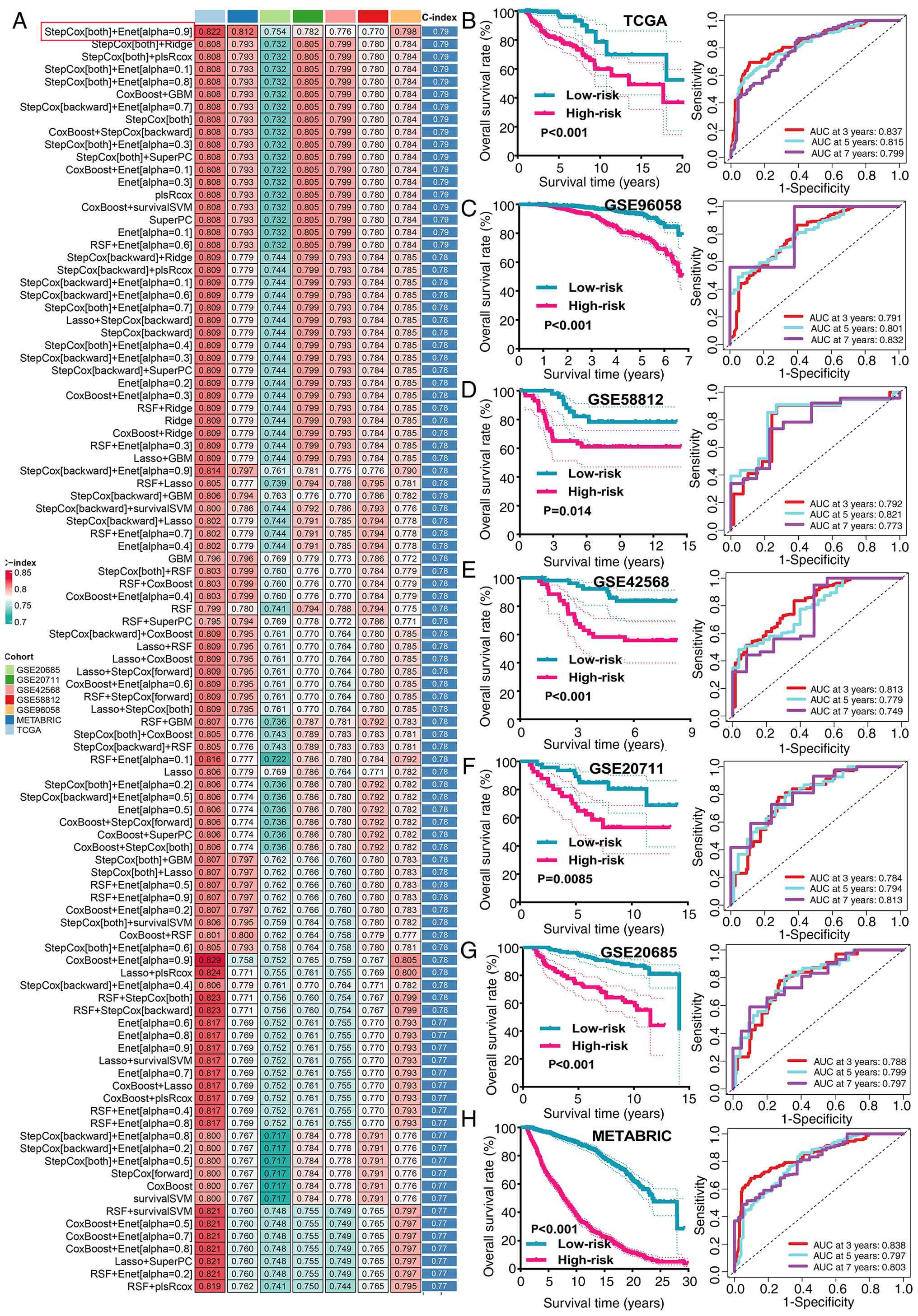 | Figure 1.Machine learning develops a
prognostic CDS. (A) C-index of 101 prognostic models of TCGA and
all included GEO datasets. Kaplan-Meier survival analysis for
patients stratified by the median CDS risk score and their
corresponding receiver operating characteristic curve in (B) TCGA,
(C) GSE96058, (D) GSE58812, (E) GSE42568, (F) GSE20711, (G)
GSE20685 and (H) METABRIC datasets. The darker the red, the larger
the C-index; the darker the green, the smaller the C-index. CDS,
programmed cell death-based gene signature; TCGA, The Cancer Genome
Atlas; GEO, Gene Expression Omnibus; AUC, area under the curve;
METABRIC, Molecular Taxonomy of Breast Cancer International
Consortium; C-index, concordance index. |
Stratification of patients into the high- and
low-risk groups based on the median CDS score revealed significant
survival disparities. Survival analysis pertaining to the composite
signature risk groups demonstrated that patients with a high CDS
score exhibited significantly shorter OS across all datasets tested
(Fig. 1B-H). The time-dependent ROC
analysis confirmed the robust predictive power of the model, with
area under the curve (AUC) values for 3-, 5- and 7-year survival
reaching 0.837, 0.815 and 0.799, respectively, in TCGA cohort
(Fig. 1B). Consistent performance
was observed in the METABRIC and GEO validation cohorts, where AUC
values ranged from 0.749 to 0.838 across various time-points
(Fig. 1C-H).
Previous studies also explored the functional roles
of ANO6, PLK1, SLC7A5, TUBA1C and TCN1 within cellular and
molecular processes associated with PCD and tumor-immune
interactions. PLK1 is a key regulator of the cell cycle,
particularly during mitosis. PLK1 governs various stages of cell
division and has been implicated in apoptosis resistance. PLK1
upregulation has been associated with decreased apoptosis and
increased tumor progression, suggesting a role in immune evasion
(57). SLC7A5 is a transporter of
essential amino acids, such as leucine, serving a key role in
metabolic reprogramming. The transporter is key to nutrient sensing
and mTOR activation, processes that are closely associated with
cell survival. Furthermore, recent studies have suggested that
SLC7A5 may modulate ferroptosis (58,59).
ANO6 is involved in phospholipid scrambling and membrane dynamics.
This process is central to the execution of apoptosis and
ferroptosis, since the exposure of phosphatidylserine on the cell
surface is a hallmark of early apoptotic events (60,61).
TUBA1C is a structural component of the microtubule network that is
key to mitosis, cell division and apoptosis pathways (62). TCN1 is a vitamin B12-binding
protein, which facilitates cellular uptake of cobalamin. Previous
studies have suggested that TCN1 is involved in regulating immune
responses and may influence cell survival under metabolic stress
conditions. TCN1 has been implicated in tumor progression and
metastasis, potentially through its effects on cellular metabolism
and DNA synthesis, both of which can influence cell death pathways,
including apoptosis (63,64).
CDS as an independent risk factor for
BRCA
The CDS demonstrated notably increased prognostic
performance compared with conventional clinical variables,
including age, tumor grade, stage and estrogen
receptor/progesterone receptor status, as indicated by a higher
C-index (Fig. 2A). Both univariate
and multivariate Cox regression analyses confirmed that the CDS
score served as an independent prognostic factor in TCGA, METABRIC
and all GEO datasets (Fig. 2B and
C). This independent predictive capacity was consistently
observed across diverse clinical subgroups.
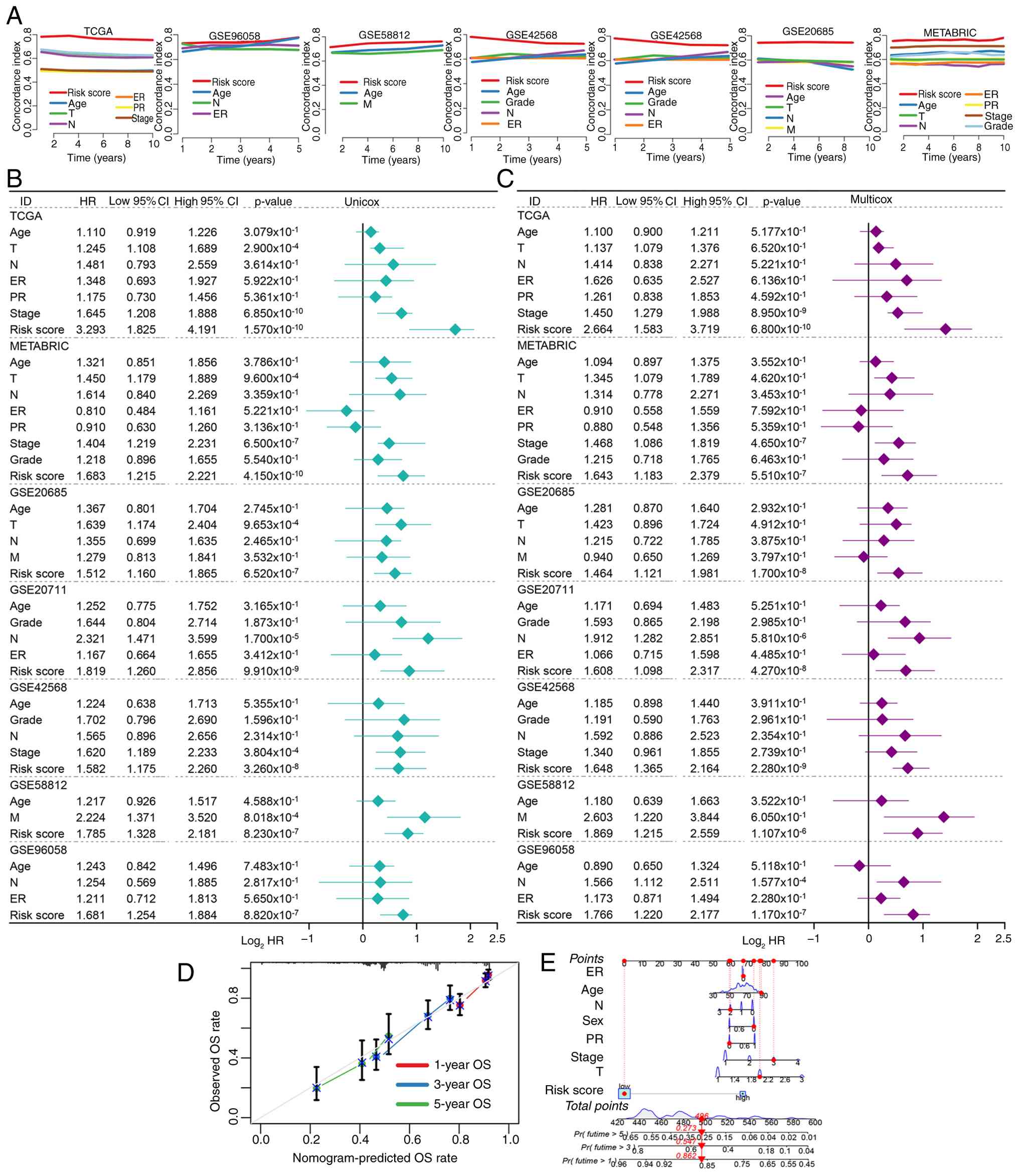 | Figure 2.Evaluation of the predictive
performance of CDS in patients with BRCA. (A) Concordance-index of
CDS and clinical characteristics in TCGA, METABRIC and the 5
included GEO datasets. Prognostic risk factors identified by (B)
univariate and (C) multivariate Cox regression analysis. (D)
Nomogram developed based on CDS and clinical characters. (E)
Calibration evaluated the role of the nomogram in evaluating the
prognosis of patients with BRCA. CDS, programmed cell death-based
gene signature; BRCA, breast cancer; TCGA, The Cancer Genome Atlas;
GEO, Gene Expression Omnibus; METABRIC, Molecular Taxonomy of
Breast Cancer International Consortium; ER, estrogen receptor; PR,
progesterone receptor; OS, overall survival; HR, hazard ratio. |
Construction of a clinical
nomogram
To facilitate clinical translation, the present
study developed a nomogram integrating the CDS score with a number
of key clinical parameters, such as age, tumor grade and clinical
stage (Fig. 2D). Calibration curves
demonstrated notable agreement between predicted and observed
survival probabilities in both training and validation sets
(Fig. 2E), supporting the utility
of the nomogram for individualized risk assessment and treatment
planning.
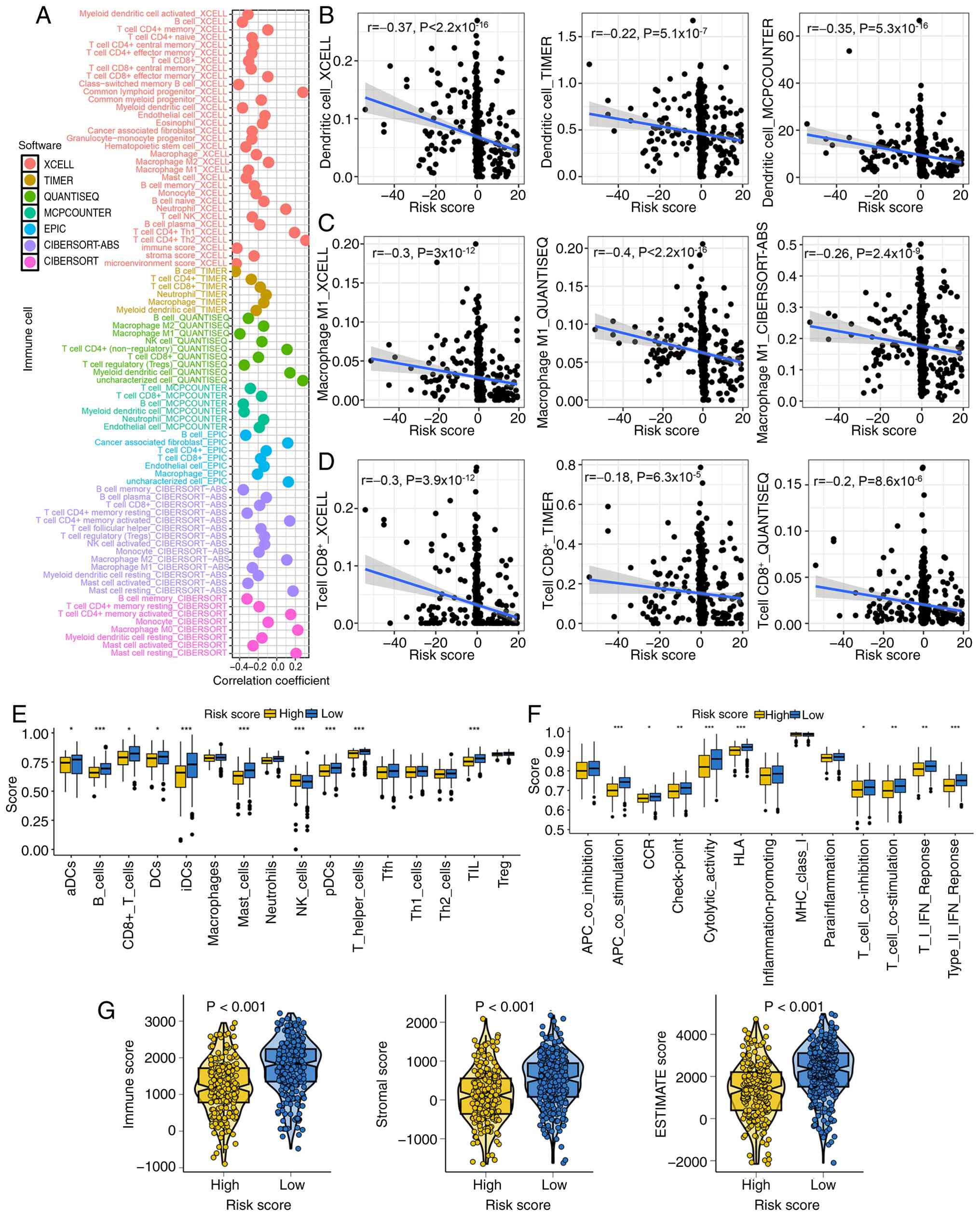
Correlation between CDS score and
immune microenvironment
Further analysis revealed a significant negative
correlation between the CDS and immune contexture. Immune
deconvolution indicated that high CDS scores were weekly associated
with reduced infiltration of dendritic cells, macrophage M1 and
CD8+ T cells (Fig.
3A-D). Based on the results of the CIBERSORT method, CDS score
indicated significant week negative correlations with the abundance
of CD4+ memory resting and CD4+ memory
activated T cells, memory B cells, macrophages M1, monocytes,
resting myeloid dendritic cells, and activated and resting mast
cells (Fig. S2A-H). Furthermore,
these CDS genes (ANO6, PLK1, SLC7A5, TUBA1C and TCN1) also
demonstrated significant correlations with CD8+ T-cell
infiltration, M1 macrophage abundance and immune score (Table SII). ssGSEA further confirmed that
the high-risk group exhibited multiple immune cell subsets,
including B and mast cells, and higher enrichment scores for
natural killer cells (Fig. 3E). By
contrast, low CDS scores were associated with enhanced activity of
immune activation pathways, such as antigen-presenting cell and
T-cell co-stimulation, cytolytic activity and type II IFN response
(Fig. 3F). Consistently, ESTIMATE,
immune and stromal scores were significantly lower in the high CDS
score group, indicating an immunologically ‘cold’ tumor
microenvironment (Fig. 3G).
CDS acts as a biomarker in predicting
immunotherapy response
Increased expression levels of human leukocyte
antigen (HLA)-related genes suggest a broader range of antigen
presentation, which could lead to the presentation of more
immunogenic antigens and potentially enhance the success of
immunotherapy (65,66). A low TIDE score and ITH score
indicate a reduced likelihood of immune escape and an improved
response to immunotherapy (67).
The present study next explored the potential of the CDS to predict
response to immune checkpoint inhibition. Patients with low CDS
scores exhibited elevated expression of most of the immune
checkpoint molecules (Fig. 4A) and
HLA-related genes (Fig. 4B). This
group also demonstrated markedly higher TMB and IPS (Fig. 4C and D), alongside lower T-cell
dysfunction (TIDE), immune escape and ITH scores (Fig. 4E-G), all indicators of favorable
immunotherapy response. Furthermore, patients with BRCA with a low
CDS score indicated higher antigen presentation gene set score
(Fig. S3A) and inflammatory
response gene set score (Fig.
S3B), and lower T-cell exhaustion gene set score (Fig. S3C).
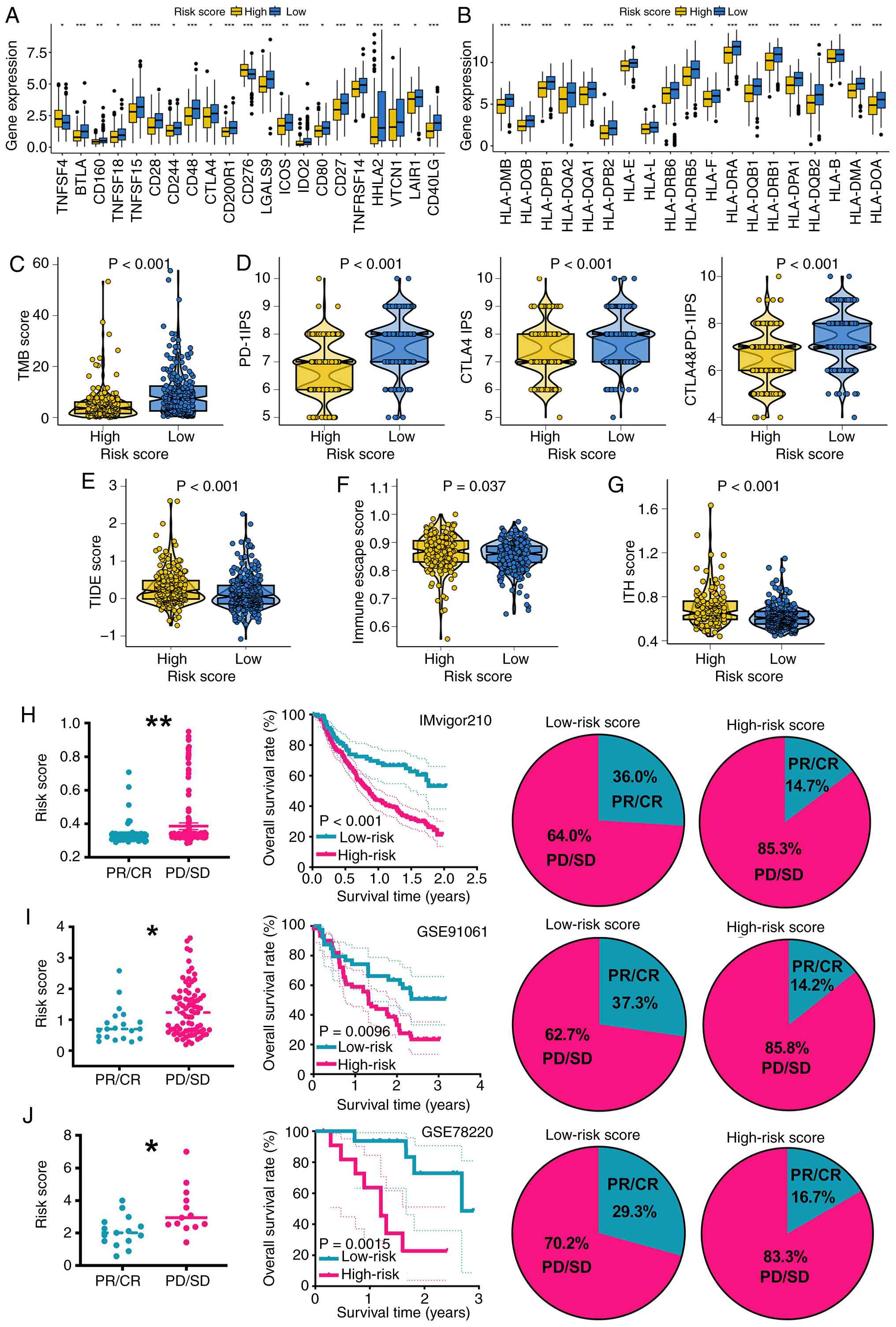 | Figure 4.CDS score acts as an indicator for
immunotherapy benefits in BRCA. Level of (A) immune checkpoint
genes and (B) HLA-related genes, (C) TMB score, (D) PD-1 and CTLA4
immunophenoscore, (E) TIDE, (F) immune escape and (G) ITH scores in
the different CDS score group. Overall rate and immunotherapy
response rate in the different CDS score groups in (H) IMvigor210,
(I) GSE91061 and (J) GSE78220 cohort. (A-G) Analyses are performed
in The Cancer Genome Atlas-BRCA cohorts to predict potential
immunotherapy response, which is then validated in (H-J)
independent immunotherapy cohorts. *P<0.05, **P<0.01 and
***P<0.001. CDS, programmed cell death-based gene signature;
BRCA, breast cancer; HLA, human leukocyte antigen; TMB, tumor
mutational burden; TIDE, Tumor Immune Dysfunction and Exclusion;
ITH, intratumor heterogeneity; PR, partial response; CR, complete
response; PD, progressive disease; SD, stable disease; PD-1
programmed cell death protein 1; CTLA-1, cytotoxic
T-lymphocyte-associated antigen 4. |
Validation in three independent immunotherapy
cohorts (IMvigor210, GSE91061 and GSE78220) confirmed that patients
with low CDS scores experienced significantly improved OS and
higher response rates following anti-PCD protein-1/PCD-ligand 1
treatment (Fig. 4H-J).
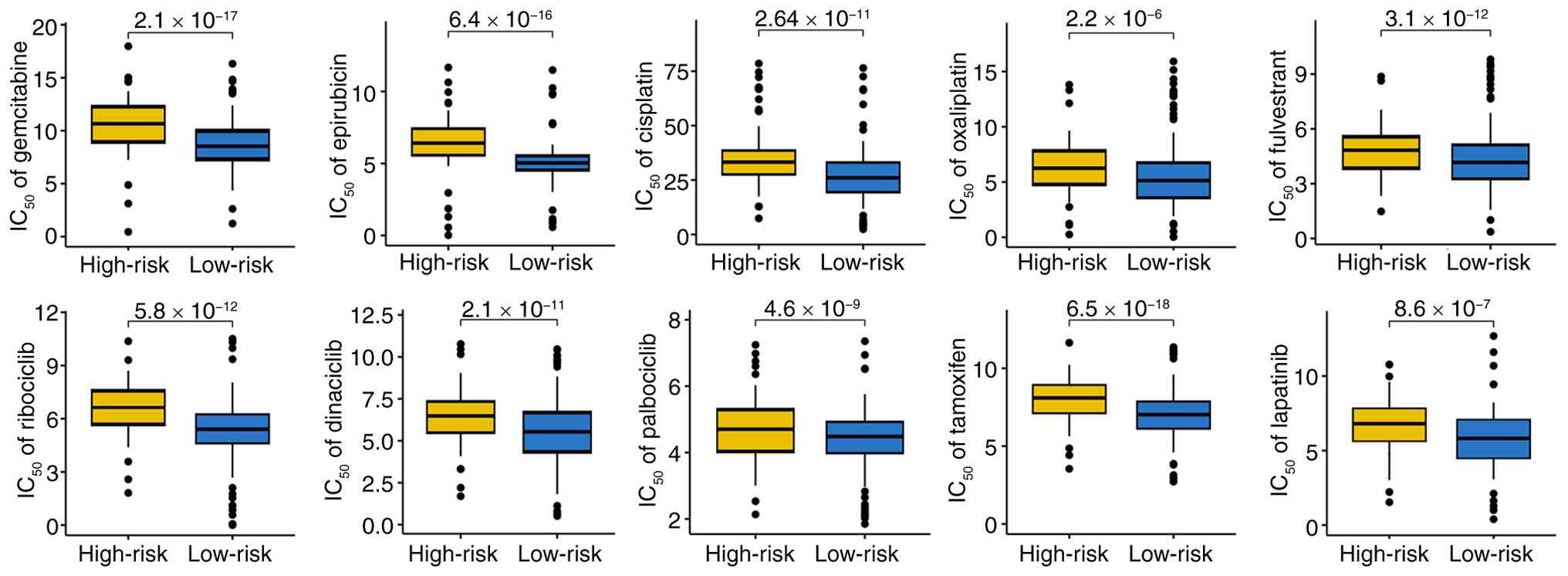
CDS and drug sensitivity
Drug sensitivity analysis revealed that patients in
the low CDS score group were significantly more sensitive to a
range of chemotherapeutic, endocrine and targeted agents, including
fulvestrant, oxaliplatin, gemcitabine, epirubicin, cisplatin,
lapatinib, ribociclib, dinaciclib, palbociclib and tamoxifen
(Fig. 5A and B). These findings
position the CDS as a potential biomarker in guiding personalized
therapy selection beyond immunotherapy.
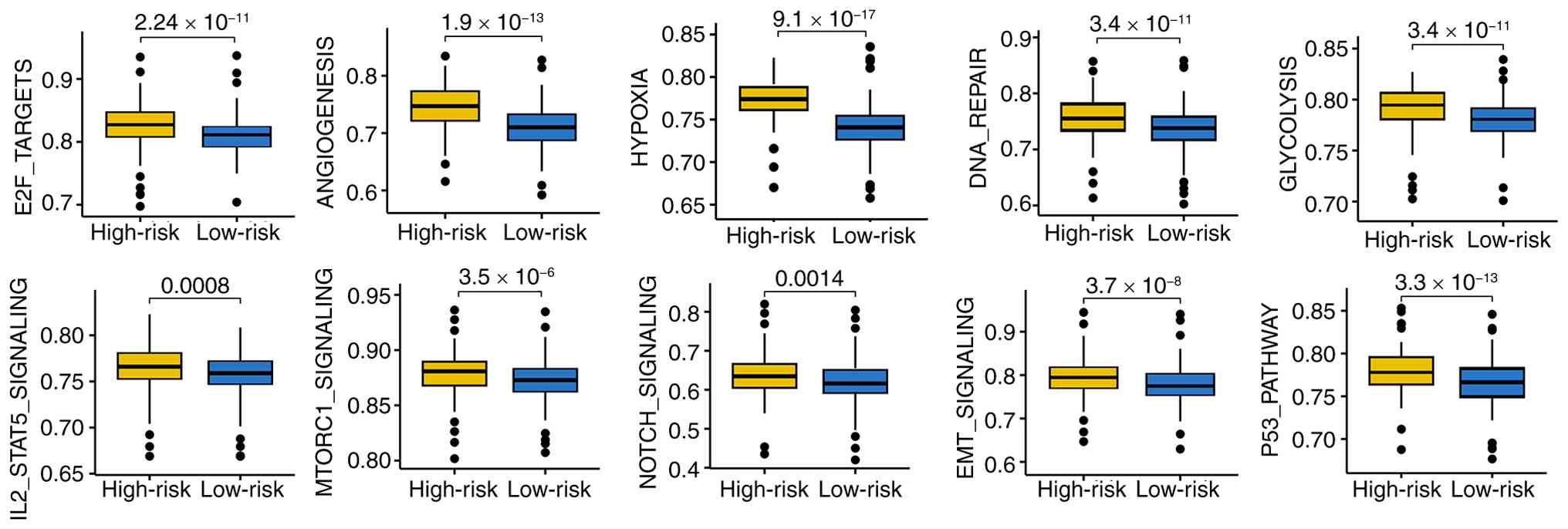
Biological pathways associated with
CDS
ssGSEA revealed that high CDS scores were
significantly enriched in oncogenic and metabolic pathways,
including ‘p53 pathway’, ‘EMT signaling’, ‘NOTCH signaling’,
‘mTORC1 signaling’, ‘IL2-STAT5 signaling’, ‘hypoxia’, ‘glycolysis’,
‘DNA repair’, ‘angiogenesis’ and E2F targets (Fig. 6). These pathways are collectively
associated with tumor progression, treatment resistance and immune
suppression.
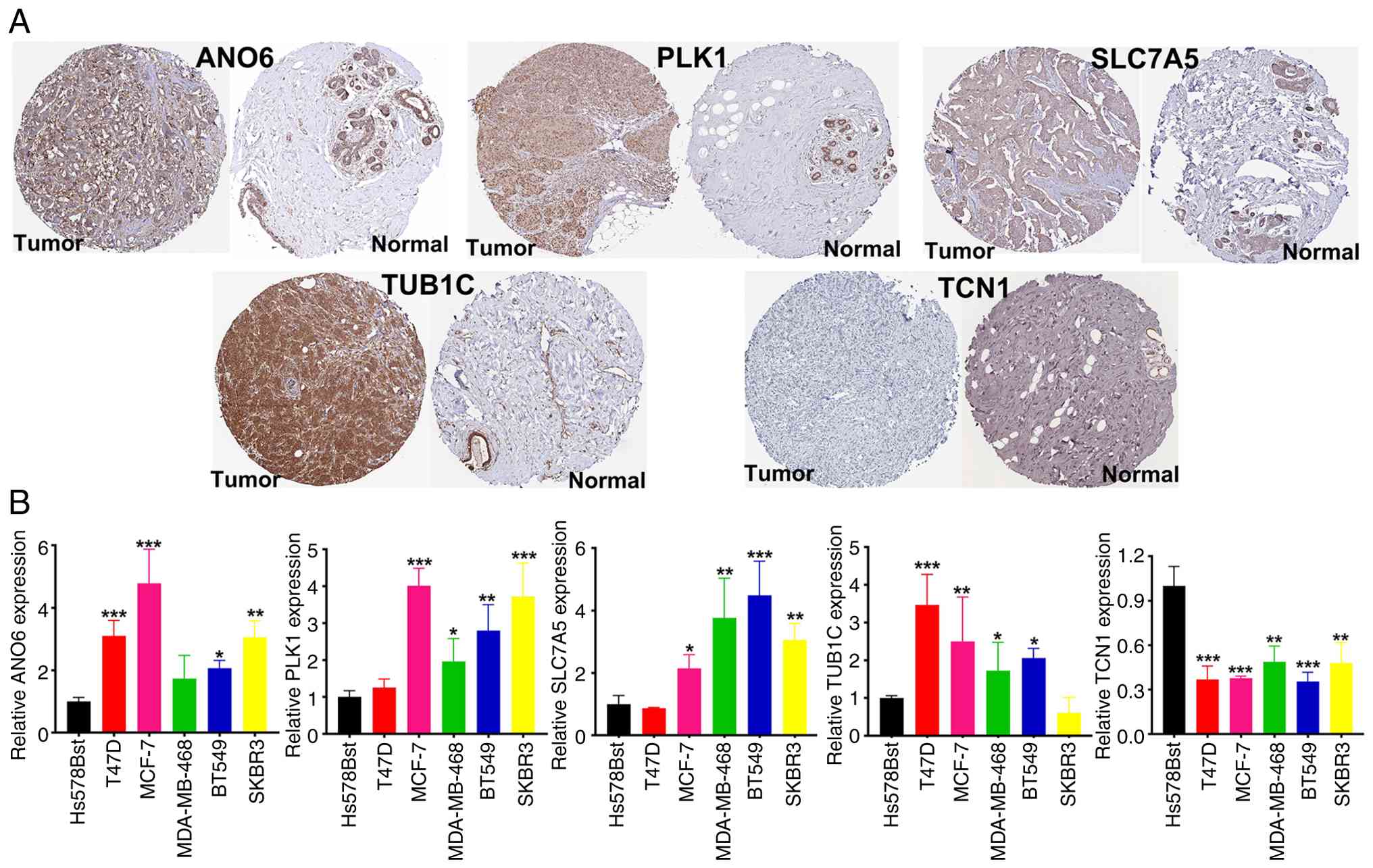
Verification of the expression of CDS
genes
mRNA and protein expression analyses corroborated
the present bioinformatic findings. Expression levels of ANO6,
PLK1, SLC7A5, and TUB1C were found to be significantly upregulated
in BRCA tissues and cell lines, whereas TCN1 was downregulated
compared with those in normal breast tissues and normal human
breast cells (Fig. 7A and B).
Discussion
BRCA remains one of the most prevalent and lethal
malignancies affecting women worldwide, posing notable challenges
to public health due to its high incidence and mortality rates.
Despite advances in surgical interventions, radiotherapy,
chemotherapy and targeted therapies, treatment outcomes typically
vary markedly among patients, reflecting the heterogeneity of the
disease and its complex biological underpinnings. Furthermore,
conventional therapies are frequently limited by adverse effects
and the emergence of resistance, underscoring an urgent need for
more precise prognostic tools and therapeutic strategies to improve
patient management and survival.
In the present study, integrated transcriptomic
datasets and advanced machine learning techniques were applied to
develop a novel gene signature based on CRGs for prognostic
evaluation in BRCA. This signature demonstrated robust
discrimination between patient risk groups and revealed significant
associations with immune cell infiltration and drug sensitivity
profiles, suggesting its potential utility in guiding personalized
treatment decisions. The present study findings establish a
foundation for the further exploration of PCD pathways in BRCA
progression and therapeutic response, offering novel insights into
the molecular mechanisms that may underlie tumor immune evasion and
chemoresistance.
The five genes constituting the CDS, namely, ANO6,
PLK1, SLC7A5, TUBA1C and TCN1, serve distinct but interrelated
roles in tumor biology and immune regulation, offering biological
plausibility for the observed correlation between the CDS score and
immune infiltration patterns. ANO6 encodes a calcium-activated
phospholipid scramblase that mediates the externalization of
phosphatidylserine during apoptosis and other forms of PCD. This
process is key to the recognition and clearance of dying cells by
phagocytes and antigen-presenting cells (68). Aberrant ANO6 expression can impair
efferocytosis, leading to accumulation of apoptotic bodies and
secondary necrosis, which in turn promotes chronic inflammation and
immunosuppression within the tumor microenvironment. PLK1 is a
master regulator of mitotic progression and DNA damage repair.
Upregulation of PLK1 in BRCA promotes uncontrolled proliferation
and genomic instability, conditions that are frequently accompanied
by immune evasion (69).
Mechanistically, PLK1 can suppress type I IFN signaling and reduce
antigen presentation, thereby blunting cytotoxic T-cell activation
(70,71). SLC7A5 encodes a large neutral amino
acid transporter that facilitates leucine uptake, activating the
mTORC1 pathway (72).
Hyperactivation of mTOR signaling drives metabolic reprogramming
toward an immunosuppressive phenotype by increasing lactate
production and depleting nutrients in the tumor microenvironment
(73). Elevated SLC7A5 expression
has been associated with the reduced infiltration of CD8+ T cells
and impaired antitumor immunity due to competition for essential
amino acids between tumor and immune cells (74). The present study findings highlight
the importance of a biologically grounded approach to predictive
signature construction, wherein gene selection is informed by
established roles in PCD and immune modulation. The five-gene
signature identified in the present study, consisting of PLK1,
SLC7A5, ANO6, TUBA1C and TCN1, captures key aspects of cell cycle
regulation, metabolic reprogramming and membrane remodeling, all of
which are key determinants of cell survival and death under immune
pressure. Although the present study model demonstrated strong
predictive performance (C-index), it is key to emphasize that these
genes were not selected based purely on statistical associations,
but rather due to their biological relevance to PCD-related
processes. The ability of these genes to influence tumor cell
survival and immune modulation underscores the biologically
plausible nature of this signature. However, the present study
acknowledges that whilst the computational model demonstrates
promise, the exact molecular interactions between these genes and
their role in immunotherapy response should be further explored
using experimental and clinical validation.
PCD pathways, including apoptosis, necroptosis and
ferroptosis, intersect with both tumor immunity and metabolic
regulation, serving key roles in cancer progression, immune evasion
and therapy resistance. Apoptosis is a fundamental mechanism for
eliminating damaged cells, including tumor cells. Dysregulation of
apoptosis allows tumors to evade immune surveillance, as apoptotic
cells release signals that promote immune clearance. Genes, such as
PLK1 regulate mitosis and apoptosis, where its upregulation in
tumors leads to apoptosis resistance, promoting immune evasion and
chemotherapy resistance (75,76).
Necroptosis is triggered when apoptosis is inhibited, often due to
stress or inflammatory signals. Necroptotic cell death can release
damage-associated molecular patterns, which activate the immune
system (77). However, uncontrolled
necroptosis can also enhance tumor progression through inflammation
(78,79). Ferroptosis is an iron-dependent,
oxidative cell death pathway that is closely associated with
metabolic regulation. Ferroptosis is influenced by the availability
of amino acids and lipids, as well as the cellular redox state.
SLC7A5, by controlling nutrient uptake, serves a notable role in
modulating oxidative stress and ferroptosis susceptibility
(80). Ferroptotic cells can
influence the immune system by affecting cGAS-STING signals that
activate both innate and adaptive immune responses (80,81).
The inverse correlation between high CDS scores and
immune cell infiltration, particularly cytotoxic CD8+ T
cells and dendritic cells, underscores a potential
immunosuppressive tumor microenvironment. This observation
parallels findings that high-risk patients, as defined by PCD
signatures, exhibit diminished immune activation and increased
immune escape mechanisms. Mechanistically, PCD may influence
antigen presentation and immune surveillance through mitochondrial
dysfunction and altered metabolic states, leading to reduced
recruitment or function of effector immune cells (82). In the present study, the ssGSEA
analysis corroborates this by revealing decreased expression levels
of immune activation-related gene sets in high CDS groups,
indicating a suppression of key immune pathways, such as IFN
signaling and T-cell cytotoxicity. These results suggested that CDS
not only prognosticates survival but also reflects the
immunological landscape, providing a rationale for the integration
of CDS assessment in immunotherapy stratification. Targeting the
interplay between PCD and immune modulation may thus represent a
novel therapeutic avenue in BRCA.
The differential sensitivity to chemotherapeutic
agents observed between patients with high and low CDS scores
reveals a clinically relevant association between CRG expression
and drug response. The increased sensitivity of patients with low
CDS to agents such as oxaliplatin and gemcitabine, evidenced by
significantly lower IC50 values, aligns with previous
studies associating PCD pathways to chemosensitivity (83,84).
PCD may modulate drug efficacy by affecting mitochondrial
metabolism and reactive oxygen species generation, which are key
mediators of chemotherapy-induced cell death (85). Furthermore, machine learning-derived
prognostic models incorporating CRGs have demonstrated promise in
predicting chemotherapy response in breast cancer, suggesting that
CDS can serve as a biomarker for personalized chemotherapy regimens
(86). These findings advocate for
the prospective clinical validation of CDS-guided treatment
strategies and highlight the potential of integrating molecular
signatures with pharmacogenomics to optimize therapeutic outcomes
in BRCA.
The limitations of the present study primarily stem
from the lack of experimental validation, which raises concerns
about the robustness of the identified association between CRGs and
BRCA prognosis. Furthermore, the relatively small sample size may
hinder the generalizability of the results, potentially limiting
the applicability of the CDS model across diverse patient
populations. Additionally, the batch effects inherent in the
datasets utilized could introduce variability that may confound the
analysis, emphasizing the necessity of cautious interpretation of
the results in clinical contexts. Notably, the immunotherapy
response predictions are based on computational analyses derived
largely from cancer cell line-based datasets and public databases.
Therefore, these findings should be interpreted with caution and
require further validation in experimental models and well-designed
clinical cohorts.
Future studies should include experimental
validation of the core CDS genes (ANO6, PLK1, SLC7A5, TUBA1C and
TCN1) and signature scores using techniques such as western
blotting and reverse transcription-quantitative PCR in independent
collections of human BRCA tissues to confirm their protein and mRNA
expression patterns and solidify their clinical relevance.
Furthermore, the functions and potential molecular mechanism of CDS
genes in BRCA will be explored in future research.
In summary, the present study successfully
identified multiple CRGs associated with BRCA prognosis and
constructed a CDS model grounded in gene expression data,
demonstrating its potential in predicting patient outcomes and
treatment responses. This approach not only enhances prognostic
capabilities but also potentially lays the groundwork for
personalized treatment strategies. Future research should aim to
validate these findings through larger, multicentric studies and
investigate the clinical utility of the CDS model, ultimately
striving to integrate these biomarkers into routine clinical
practice to improve therapeutic decision-making.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
NW prepared the original draft and conducted the
bioinformatics and experimental investigation. ZF designed the
present study, provided supervision and reviewed the manuscript. NW
and ZF confirm the authenticity of all the raw data. Both authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Khan MM, Yalamarty SSK, Rajmalani BA,
Filipczak N and Torchilin VP: Recent strategies to overcome breast
cancer resistance. Crit Rev Oncol Hematol. 197:1043512024.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xiong X, Zheng LW, Ding Y, Chen YF, Cai
YW, Wang LP, Huang L, Liu CC, Shao ZM and Yu KD: Breast cancer:
Pathogenesis and treatments. Signal Transduct Target Ther.
10:492025. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yuan J and Ofengeim D: A guide to cell
death pathways. Nat Rev Mol Cell Biol. 25:379–395. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ames EG and Thoene JG: Programmed cell
death in cystinosis. Cells. 11:6702022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vu A, Glassman I, Campbell G, Yeganyan S,
Nguyen J, Shin A and Venketaraman V: Host cell death and modulation
of immune response against mycobacterium tuberculosis infection.
Int J Mol Sci. 25:62552024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu J, Hong M, Li Y, Chen D, Wu Y and Hu
Y: Programmed cell death tunes tumor immunity. Front Immunol.
13:8473452022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang X, Luo Z, Liang W, Xie G, Lang X,
Gou J, Liu C, Xu X and Fu D: Survival nomogram for young breast
cancer patients based on the SEER database and an external
validation cohort. Ann Surg Oncol. 29:5772–5781. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhu H, Hu H, Hao B, Zhan W, Yan T, Zhang
J, Wang S, Hu H and Zhang T: Insights into a machine Learning-Based
Palmitoylation-Related gene model for predicting the prognosis and
treatment response of breast cancer patients. Technol Cancer Res
Treat. 23:153303382412634342024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang L, Zhang L, Shi X, Wang C, Chen X,
Li M, Ni N, Gao G, Wang T and Zhang X: Multi-cohort and single-cell
profiling of aging genes reveals prognostic and therapeutic targets
in breast cancer. iScience. 29:1148472026. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zeng C, Wang J, Zhao S, Wei Y, Qi Y, Liu
S, Wang Y, Ge H, Yang X, Tan Y, et al: Multi-cohort validation of a
lipid metabolism and ferroptosis-associated index for prognosis and
immunotherapy response prediction in hormone receptor-positive
breast cancer. Int J Biol Sci. 21:3968–3992. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang Z, Gao Z, Huang Q, Ling Z, Zhang L,
Li M, Xu Y and Liu M: Cuproptosis- and m6A-related lncRNA
prognostic signature for breast cancer, in which Z68871.1
contributes to triple-negative breast cancer progression. Int J
Biol Macromol. 321:1463212025. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jiang B, Zhu H, Feng W, Wan Z, Qi X, He R,
Xie L and Li Y: Database mining detected a Cuproptosis-related
prognostic signature and a related regulatory axis in breast
cancer. Dis Markers. 2022:90048302022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang L, Cui Y, Zhou G, Zhang Z and Zhang
P: Leveraging mitochondrial-programmed cell death dynamics to
enhance prognostic accuracy and immunotherapy efficacy in lung
adenocarcinoma. J Immunother Cancer. 12:e0100082024. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kao KJ, Chang KM, Hsu HC and Huang AT:
Correlation of microarray-based breast cancer molecular subtypes
and clinical outcomes: Implications for treatment optimization. BMC
Cancer. 11:1432011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dedeurwaerder S, Desmedt C, Calonne E,
Singhal SK, Haibe-Kains B, Defrance M, Michiels S, Volkmar M,
Deplus R, Luciani J, et al: DNA methylation profiling reveals a
predominant immune component in breast cancers. EMBO Mol Med.
3:726–741. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Clarke C, Madden SF, Doolan P, Aherne ST,
Joyce H, O'Driscoll L, Gallagher WM, Hennessy BT, Moriarty M, Crown
J, et al: Correlating transcriptional networks to breast cancer
survival: A large-scale coexpression analysis. Carcinogenesis.
34:2300–2308. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jézéquel P, Loussouarn D,
Guérin-Charbonnel C, Campion L, Vanier A, Gouraud W, Lasla H,
Guette C, Valo I, Verrièle V and Campone M: Gene-expression
molecular subtyping of triple-negative breast cancer tumours:
Importance of immune response. Breast cancer Res. 17:432015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Brueffer C, Vallon-Christersson J, Grabau
D, Ehinger A, Häkkinen J, Hegardt C, Malina J, Chen Y, Bendahl PO,
Manjer J, et al: Clinical value of RNA Sequencing-based classifiers
for prediction of the five conventional breast cancer biomarkers: A
report from the Population-Based multicenter Sweden Cancerome
analysis Network-breast initiative. JCO Precis Oncol. 22018.doi:
10.1200/PO.17.00135. PubMed/NCBI
|
|
19
|
Rosenberg JE, Galsky MD, Powles T,
Petrylak DP, Bellmunt J, Loriot Y, Necchi A, Hoffman-Censits J,
Perez-Gracia JL, van der Heijden MS, et al: Atezolizumab
monotherapy for metastatic urothelial carcinoma: Final analysis
from the phase II IMvigor210 trial. ESMO Open. 9:1039722024.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Riaz N, Havel JJ, Makarov V, Desrichard A,
Urba WJ, Sims JS, Hodi FS, Martín-Algarra S, Mandal R, Sharfman WH,
et al: Tumor and microenvironment evolution during immunotherapy
with nivolumab. Cell. 171:934–949.e16. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hugo W, Zaretsky JM, Sun L, Song C, Moreno
BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G,
et al: Genomic and transcriptomic features of response to Anti-PD-1
therapy in metastatic melanoma. Cell. 165:35–44. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liberzon A, Subramanian A, Pinchback R,
Thorvaldsdóttir H, Tamayo P and Mesirov JP: Molecular signatures
database (MSigDB) 3.0. Bioinformatics. 27:1739–1740. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kanehisa M, Furumichi M, Tanabe M, Sato Y
and Morishima K: KEGG: New perspectives on genomes, pathways,
diseases and drugs. Nucleic Acids Res. 45:D353–D361. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang Y and Zhang Q: Leveraging programmed
cell death signature to predict clinical outcome and immunotherapy
benefits in postoperative bladder cancer. Sci Rep. 14:229762024.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ding D, Wang L, Zhang Y, Shi K and Shen Y:
Machine learning developed a programmed cell death signature for
predicting prognosis and immunotherapy benefits in lung
adenocarcinoma. Transl Oncol. 38:1017842023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Stelzer G, Rosen N, Plaschkes I, Zimmerman
S, Twik M, Fishilevich S, Stein TI, Nudel R, Lieder I, Mazor Y, et
al: The GeneCards Suite: From gene data mining to disease genome
sequence analyses. Curr Protoc Bioinformatics. 54:1.30.1–1.30.33.
2016. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biol. 15:5502014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Leek JT, Johnson WE, Parker HS, Jaffe AE
and Storey JD: The sva package for removing batch effects and other
unwanted variation in high-throughput experiments. Bioinformatics.
28:882–883. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ishwaran H, Gerds TA, Kogalur UB, Moore
RD, Gange SJ and Lau BM: Random survival forests for competing
risks. Biostatistics. 15:757–773. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stocking JC, Taylor SL, Fan S, Wingert T,
Drake C, Aldrich JM, Ong MK, Amin AN, Marmor RA, Godat L, et al: A
least absolute shrinkage and selection Operator-derived predictive
model for postoperative respiratory failure in a heterogeneous
adult elective surgery patient population. CHEST Crit Care.
1:1000252023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Arashi M, Roozbeh M, Hamzah NA and
Gasparini M: Ridge regression and its applications in genetic
studies. PLoS One. 16:e02453762021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xu QF, Ding XH, Jiang CX, Yu KM and Shi L:
An elastic-net penalized expectile regression with applications. J
Appl Stat. 48:2205–2230. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu Y, Zhou C, Shen T, Yu X, Li Q, Jiang
T, Li W and Zhu Y: Identification of prognostic markers related to
homologous recombination deficiency in cholangiocarcinoma using
CoxBoost and LASSO machine learning techniques. Front Immunol.
17:16156572026. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li S, Xiao Y, Wang Y, Bai M, Du F and
Zhang H: Exploration of influencing factors for postoperative
recurrence in patients with Madelung's disease on the basis of
multivariate stepwise cox regression analysis. Clin Cosmet Investig
Dermatol. 16:103–110. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sadiq M, Alnagar DKF, Abdulrahman AT and
Alharbi R: The partial least squares spline model for public health
surveillance data. Comput Math Methods Med. 2022:87747422022.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pan PJ, Lee CH, Hsu NW and Sun TL:
Combining principal component analysis and logistic regression for
multifactorial fall risk prediction among community-dwelling older
adults. Geriatr Nurs. 57:208–216. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schmid M, Wickler F, Maloney KO, Mitchell
R, Fenske N and Mayr A: Boosted beta regression. PLoS One.
8:e616232013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Turki T and Wei Z: Boosting support vector
machines for cancer discrimination tasks. Comput Biol Med.
101:236–249. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Van Oirbeek R and Lesaffre E: An
application of Harrell's C-index to PH frailty models. Stat Med.
29:3160–3171. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Iasonos A, Schrag D, Raj GV and Panageas
KS: How to build and interpret a nomogram for cancer prognosis. J
Clin Oncol. 26:1364–1370. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Findlay JW and Dillard RF: Appropriate
calibration curve fitting in ligand binding assays. AAPS J.
9:E260–E267. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Racle J and Gfeller D: EPIC: A tool to
estimate the proportions of different cell types from bulk gene
expression data. Methods Mol Biol. 2120:233–248. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Aran D, Hu Z and Butte AJ: xCell:
Digitally portraying the tissue cellular heterogeneity landscape.
Genome Biol. 18:2202017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Becht E, Giraldo NA, Lacroix L, Buttard B,
Elarouci N, Petitprez F, Selves J, Laurent-Puig P, Sautès-Fridman
C, Fridman WH and de Reyniès A: Estimating the population abundance
of tissue-infiltrating immune and stromal cell populations using
gene expression. Genome Biol. 17:2182016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li T, Fan J, Wang B, Traugh N, Chen Q, Liu
JS, Li B and Liu XS: TIMER: A web server for comprehensive analysis
of Tumor-infiltrating immune cells. Cancer Res. 77:e108–e110. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sturm G, Finotello F and List M:
Immunedeconv: An R package for unified access to computational
methods for estimating immune cell fractions from bulk
RNA-Sequencing data. Methods Mol Biol. 2120:223–232. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chen B, Khodadoust MS, Liu CL, Newman AM
and Alizadeh AA: Profiling tumor infiltrating immune cells with
CIBERSORT. Methods Mol Biol. 1711:243–259. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yoshihara K, Shahmoradgoli M, Martínez E,
Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW,
Levine DA, et al: Inferring tumour purity and stromal and immune
cell admixture from expression data. Nat Commun. 4:26122013.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Song D and Wang X: DEPTH2: An mRNA-based
algorithm to evaluate intratumor heterogeneity without reference to
normal controls. J Transl Med. 20:1502022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Fu J, Li K, Zhang W, Wan C, Zhang J, Jiang
P and Liu XS: Large-scale public data reuse to model immunotherapy
response and resistance. Genome Med. 12:212020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Charoentong P, Finotello F, Angelova M,
Mayer C, Efremova M, Rieder D, Hackl H and Trajanoski Z: Pan-cancer
immunogenomic analyses reveal Genotype-immunophenotype
relationships and predictors of response to checkpoint blockade.
Cell Rep. 18:248–262. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Samstein RM, Lee CH, Shoushtari AN,
Hellmann MD, Shen R, Janjigian YY, Barron DA, Zehir A, Jordan EJ,
Omuro A, et al: Tumor mutational load predicts survival after
immunotherapy across multiple cancer types. Nat Genet. 51:202–206.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Maeser D, Gruener RF and Huang RS:
oncoPredict: An R package for predicting in vivo or cancer patient
drug response and biomarkers from cell line screening data. Brief
Bioinform. 22:bbab2602021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yang W, Soares J, Greninger P, Edelman EJ,
Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR, et
al: Genomics of drug sensitivity in cancer (GDSC): A resource for
therapeutic biomarker discovery in cancer cells. Nucleic Acids Res.
41:D955–D961. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Colwill K and Gräslund S: A roadmap to
generate renewable protein binders to the human proteome. Nat
Methods. 8:551–558. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Li X, Chen G, Liu B, Tao Z, Wu Y, Zhang K,
Feng Z, Huang Y and Wang H: PLK1 inhibition promotes apoptosis and
DNA damage in glioma stem cells by regulating the nuclear
translocation of YBX1. Cell Death Discov. 9:682023. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chen Z, Cai H, Ye W, Wu J, Liu J, Xie Y,
Feng S, Jin Y, Lv Y, Ye H, et al: TP63 transcriptionally regulates
SLC7A5 to suppress ferroptosis in head and neck squamous cell
carcinoma. Front Immunol. 15:14454722024. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Cai Z, Zhang R, Liu R, Zhao L and Zhou L:
Plumbagin ameliorates ferroptosis of ovarian granulosa cells in
polycystic ovary syndrome by down-regulating SLC7A5 m6A methylation
modification through inhibition of YTHDF1. J Ovarian Res.
18:1152025. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wang H, Zhao W, Wang D and Chen J: ANO6
(TMEM16F) inhibits gastrointestinal stromal tumor growth and
induces ferroptosis. Open Med (Wars). 19:202409412024. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Liu G, Liu G, Chen H, Borst O, Gawaz M,
Vortkamp A, Schreiber R, Kunzelmann K and Lang F: Involvement of
Ca2+ activated Cl-channel Ano6 in platelet activation and
apoptosis. Cell Physiol Biochem. 37:1934–1944. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Liu YB, Dai WH, Chang JJ and Wei K:
CircRNA TUBA1C promotes proliferation and glucose metabolism, and
blocks apoptosis of osteosarcoma cells through sponging miR-143-3p.
Pol J Pathol. 75:215–227. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Liu GJ, Wang YJ, Yue M, Zhao LM, Guo YD,
Liu YP, Yang HC, Liu F, Zhang X, Zhi LH, et al: High expression of
TCN1 is a negative prognostic biomarker and can predict neoadjuvant
chemosensitivity of colon cancer. Sci Rep. 10:119512020. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zhu X, Jiang X, Zhang Q, Huang H, Shi X,
Hou D and Xing C: TCN1 deficiency inhibits the malignancy of
colorectal cancer cells by regulating the ITGB4 pathway. Gut Liver.
17:412–429. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Jhunjhunwala S, Hammer C and Delamarre L:
Antigen presentation in cancer: Insights into tumour immunogenicity
and immune evasion. Nat Rev Cancer. 21:298–312. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Li Y, Li Z, Tang Y, Zhuang X, Feng W, Boor
PPC, Buschow S, Sprengers D and Zhou G: Unlocking the therapeutic
potential of the NKG2A-HLA-E immune checkpoint pathway in T cells
and NK cells for cancer immunotherapy. J Immunother Cancer.
12:e0099342024. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X,
Li Z, Traugh N, Bu X, Li B, et al: Signatures of T cell dysfunction
and exclusion predict cancer immunotherapy response. Nat Med.
24:1550–1558. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Chen A, Yang C and Wang J: Multiple roles
of ANO6 in tumors, molecular mechanism and its potential
therapeutic value. Biochem Biophys Rep. 44:1022302025.PubMed/NCBI
|
|
69
|
Kandala S, Ramos M, Voith von Voithenberg
L, Diaz-Jimenez A, Chocarro S, Keding J, Brors B, Imbusch CD and
Sotillo R: Chronic chromosome instability induced by Plk1 results
in immune suppression in breast cancer. Cell Rep. 42:1132662023.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Lavallée É, Roulet-Matton M, Giang V,
Cardona Hurtado R, Chaput D and Gravel SP: Mitochondrial signatures
shape phenotype switching and apoptosis in response to PLK1
inhibitors. Life Sci Alliance. 8:e2024029122024. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Kong Y, Li C, Liu J, Wu S, Zhang M,
Allison DB, Hassan F, He D, Wang X, Mao F, et al: Single-cell
analysis identifies PLK1 as a driver of immunosuppressive tumor
microenvironment in LUAD. PLoS Genet. 20:e10113092024. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Sokolov AM, Holmberg JC and Feliciano DM:
The amino acid transporter Slc7a5 regulates the mTOR pathway and is
required for granule cell development. Hum Mol Genet. 29:3003–3013.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zhang H, Su X, Burley SK and Zheng XFS:
mTOR regulates aerobic glycolysis through NEAT1 and nuclear
paraspeckle-mediated mechanism in hepatocellular carcinoma.
Theranostics. 12:3518–3533. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Huang R, Wang H, Hong J, Wu J, Huang O, He
J, Chen W, Li Y, Chen X, Shen K and Wang Z: Targeting glutamine
metabolic reprogramming of SLC7A5 enhances the efficacy of
anti-PD-1 in triple-negative breast cancer. Front Immunol.
14:12516432023. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Moore XTR, Gheghiani L and Fu Z: The role
of Polo-like kinase 1 in regulating the forkhead box family
transcription factors. Cells. 12:13442023. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhang Z, Cheng L, Li J, Qiao Q, Karki A,
Allison DB, Shaker N, Li K, Utturkar SM, Atallah Lanman NM, et al:
Targeting Plk1 sensitizes pancreatic cancer to immune checkpoint
therapy. Cancer Res. 82:3532–3548. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Lu JV, Chen HC and Walsh CM: Necroptotic
signaling in adaptive and innate immunity. Semin Cell Dev Biol.
35:33–39. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Wang M, Yu F, Zhang Y and Li P: Programmed
cell death in tumor immunity: Mechanistic insights and clinical
implications. Front Immunol. 14:13096352023. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Tong X, Tang R, Xiao M, Xu J, Wang W,
Zhang B, Liu J, Yu X and Shi S: Targeting cell death pathways for
cancer therapy: Recent developments in necroptosis, pyroptosis,
ferroptosis, and cuproptosis research. J Hematol Oncol. 15:1742022.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Chen X, Li J, Kang R, Klionsky DJ and Tang
D: Ferroptosis: Machinery and regulation. Autophagy. 17:2054–2081.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Efimova I, Catanzaro E, Van der Meeren L,
Turubanova VD, Hammad H, Mishchenko TA, Vedunova MV, Fimognari C,
Bachert C, Coppieters F, et al: Vaccination with early ferroptotic
cancer cells induces efficient antitumor immunity. J Immunother
Cancer. 8:e0013692020. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Liu WQ, Lin WR, Yan L, Xu WH and Yang J:
Copper homeostasis and cuproptosis in cancer immunity and therapy.
Immunol Rev. 321:211–227. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zhao P, Yin S, Qiu Y, Sun C and Yu H:
Ferroptosis and pyroptosis are connected through autophagy: A new
perspective of overcoming drug resistance. Mol Cancer. 24:232025.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Su L, Chen Y, Huang C, Wu S, Wang X, Zhao
X, Xu Q, Sun R, Kong X, Jiang X, et al: Targeting Src reactivates
pyroptosis to reverse chemoresistance in lung and pancreatic cancer
models. Sci Transl Med. 15:eabl78952023. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Wan H, Yang X, Sang G, Ruan Z, Ling Z,
Zhang M, Liu C, Hu X, Guo T, He J, et al: CDKN2A was a
cuproptosis-related gene in regulating chemotherapy resistance by
the MAGE-A family in breast cancer: Based on artificial
intelligence (AI)-constructed pan-cancer risk model. Aging (Albany
NY). 15:11244–11267. 2023.PubMed/NCBI
|
|
86
|
Li S, Liu S, Zheng Y, Hong W, Du Y, Liu X,
Tang H, Meng X and Zheng Q: Machine learning analysis of
coagulation-related genes for breast cancer diagnosis and prognosis
prediction. Sci Rep. 15:354292025. View Article : Google Scholar : PubMed/NCBI
|