Introduction
Esophageal squamous cell carcinoma (ESCC) continues
to impose a disproportionate global disease burden, with
incidence-to-mortality ratios approaching unity and 5-year survival
rates remaining <20% (1).
Contemporary therapeutic advances, including neoadjuvant
chemoradiotherapy and immune-checkpoint blockade, have not markedly
improved long-term outcomes. This is largely due to late-stage
presentation, early micrometastatic dissemination and primary or
acquired therapeutic refractoriness (2). At the molecular level, ESCC exhibits a
complex landscape dominated by tumor protein 53 mutations,
cell-cycle dysregulation and extensive epigenetic rewiring that
collectively orchestrate initiation, invasion and metastatic
colonization (3,4). Within this epigenetic circuitry,
dysregulation of long non-coding RNAs (lncRNAs) has emerged as a
key driver that modulates chromatin architecture, transcriptional
programs and post-transcriptional networks, thereby offering
tractable biomarkers and therapeutic vulnerabilities for precision
oncology (5).
lncRNAs are >200 nucleotides in length and lack
canonical open reading frames, yet they exert pleiotropic
regulatory functions by sculpting chromatin topology, orchestrating
transcription factor assemblies and modulating mRNA stability and
translation efficiency (6–8). Among these transcripts, LINC00184 has
been repeatedly implicated in oncogenesis, serving as an
independent predictor of poor prognosis in breast cancer (9) and associating with aggressive disease
courses in gastric cancer through its ability to accelerate
G1/S transition and metastatic dissemination (10). Nevertheless, its functional
contribution to ESCC and the molecular circuits it governs remain
to be elucidated.
CpG dinucleotide methylation, installed and
maintained by DNA methyltransferases (DNMT), constitutes a
principal epigenetic switch that silences gene expression through
the addition of methyl moieties to promoter-rich CpG islands
(11). Hypermethylation of
tumor-suppressor loci is a universal hallmark of malignancy
(12). As the principal maintenance
DNA methyltransferase, DNA methyltransferase 1 (DNMT1) perpetuates
pre-existing methylation marks through successive cell divisions,
thereby locking tumor-suppressor genes in a long-term silent state.
Consistent with this function, DNMT1 is frequently upregulated
across diverse cancer types, including prostate, pancreatic and
gastric cancer, and serves a key role in extinguishing
anti-oncogenic transcriptional programs (13–15).
This epigenetic silencing mechanism is exemplified in ESCC by the
tumor suppressor N-Myc downstream regulated gene 2 (NDRG2). While
NDRG2 is established to inhibit proliferation and promote apoptosis
in various cancer types, including lung, liver and pancreatic
cancer (16–18), its expression is consistently
downregulated in ESCC, a phenomenon strongly associated with
promoter hypermethylation (19,20).
Notably, NDRG2 functions as a key upstream inhibitor of the
oncogenic PI3K/AKT pathway; NDRG2 can suppress pathway activity by
enhancing PTEN or recruiting protein phosphatase 2A (PP2A) to
facilitate AKT dephosphorylation, thereby curbing tumor growth
(21). Since hyperactivation of the
PI3K/AKT axis is a major driver of ESCC progression (22), the epigenetic silencing of NDRG2
presents a plausible mechanism for its constitutive activation.
lncRNAs have emerged as key regulators that can guide epigenetic
modifiers like DNMT1 to specific genomic loci (23). Building on this paradigm and the
aforementioned established associations, the present study
hypothesized that the lncRNA LINC00184 functions as an oncogenic
driver in ESCC by recruiting DNMT1 to the NDRG2 promoter, inducing
its hypermethylation and transcriptional silencing. The consequent
loss of NDRG2 would then relieve the brake on the PI3K/AKT pathway,
leading to its sustained activation and ultimately promoting tumor
cell proliferation and survival.
The present study systematically dissected the
oncogenic circuitry of LINC00184 in ESCC by integrating gain- and
loss-of-function analyses with pharmacological inhibition of DNMT1.
Specifically, the present study investigated how
LINC00184-directed, DNMT1-dependent hypermethylation of the NDRG2
promoter governs proliferative capacity, apoptotic threshold and
migratory potential of esophageal squamous carcinoma cells.
Furthermore, the present study employed 5-azacytidine (5-AZA), a
clinically approved DNMT1 inhibitor-to evaluate the reversibility
of LINC00184-evoked epigenetic silencing and to validate the
therapeutic tractability of the LINC00184-DNMT1-NDRG2 axis. These
data will not only refine current mechanistic understanding of ESCC
biology but will also provide pre-clinical evidence for
repositioning DNMT1 inhibitors in precision oncology of esophageal
cancer.
Materials and methods
Cell lines and cell culture
ESCC lines KYSE-150 and TE-1 were obtained from the
Cell Bank of the Chinese Academy of Sciences. KYSE-150 is one of
the most classic and extensively used cell lines in ESCC research,
with a well-defined ESCC origin and characteristics. TE-1 is
another well-characterized and commonly utilized ESCC cell line.
KYSE-150 cultures were maintained in a 1:1 mixture of RPMI-1640 and
Ham's F-12 basal media supplemented with 10% (v/v) fetal bovine
serum (FBS) and 1% (v/v) penicillin-streptomycin. TE-1 cultures
were grown in RPMI-1640 containing identical supplements. Cells
were incubated at 37°C under a humidified atmosphere containing 5%
CO2 and routinely tested for mycoplasma contamination
(negative).
Plasmid construction and
transfection
For ectopic expression, the full-length LINC00184
sequence (gene ID, 100302691; National Center for Biotechnology
Information) was gene-synthesized and inserted into the
pLVX-mCMV-ZsGreen-IRES-Puro lentiviral backbone (Wuhan Viraltherapy
Technologies, Co., Ltd.) using conventional
restriction-enzyme-based cloning. Nucleic acid constructs were used
at a standardized concentration of 2 µg per well for 6-well plate
transfection. For transient transfection, logarithmically growing
cells were plated at 4×105 cells per well in 6-well
plates and transfected with either OE-LINC00184 or empty vector
controls (OE-NC) using Lipofectamine® 3000 (Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
Transfection was performed at 37°C for a continuous incubation
duration of 48 h under routine cell culture conditions. Total RNA
was isolated 48 h post-transfection and LINC00184 overexpression
was verified by reverse transcription-quantitative PCR (RT-qPCR)
using the primers detailed in Table
I. Specifically, total RNA was extracted from transfected cell
samples using RNAiso Plus (Takara Bio) as the RNA extraction
reagent. Reverse transcription was performed with the
PrimeScript™ RT Reagent Kit (Takara Bio), following the
manufacturer's recommended temperature protocols. The cDNA acquired
from reverse-transcribed total RNA served as the DNA template for
subsequent qPCR analysis. qPCR was conducted using
BeyoFast™ SYBR Green qPCR Mix (Beyotime Biotechnology).
GAPDH was applied as the internal reference gene. Standard
thermocycling conditions were set as initial denaturation at 95°C
for 30 sec followed by 40 cycles of 95°C for 5 sec and 60°C for 30
sec. Relative gene expression levels were calculated using the
2−ΔΔCq method (24). For
ectopic expression of NDRG2, the full-length human NDRG2 coding
sequence (gene ID, 57447) was synthesized and cloned into the
identical lentiviral backbone, generating the overexpression
plasmid designated OE-NDRG2. For transient rescue experiments,
cells were co-transfected with the specified combinations of
OE-LINC00184, OE-NDRG2 or their respective OE-NC using
Lipofectamine® 3000. Successful ectopic expression of
NDRG2 was validated at translational levels by western blotting
analysis.
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Gene | Primer sequence
(5′-3′) | Size, bp |
|---|
| Homo GAPDH | F:
TCAAGAAGGTGGTGAAGCAGG | 115 |
|
| R:
TCAAAGGTGGAGGAGTGGGT |
|
| Homo LINC00184 | F:
CCAATGAGCAGGGACTATGAT | 145 |
|
| R:
GCAGAGAGGCAGGAAGGTTTA |
|
Knockdown of LINC00184 and DNMT1 by
small interfering RNA (siRNA) transfection
In total, three siRNA duplexes targeting distinct
regions of LINC00184 (sequences listed in Table II) were chemically synthesized by
Sangon Biotech Co., Ltd. KYSE-150 and TE-1 cells were transfected
in 6-well plates using Lipofectamine® RNAiMAX (Thermo
Fisher Scientific, Inc.). All siRNA transfections were performed at
37°C following the manufacturer's standard protocol, with a final
siRNA concentration of 50 nM. The random non-coding siRNA sequence
described below served as the negative control for mouse DNMT1
siRNA interference. Gene silencing efficiency was detected 48 h
after transfection, and all subsequent functional experiments were
performed at the same 48 h post-transfection time point. The
detection of knockdown efficiency by RT-qPCR adopted the identical
reagent system, thermal cycling parameters and calculation method
as aforementioned in the RT-qPCR section. siRNA #2, which produced
the highest knock-down (>70% reduction) was selected for all
subsequent loss-of-function assays. siRNA duplexes specifically
targeting mouse DNMT1 transcript: Sense (S),
5′-GCUGGGAGAUGGCGUCAUA-3′; antisense (AS),
5′-CAGGGAGAUACCGCAGUAU-3′; or a random non-coding mRNA sequence: S,
5′-UUCUCCGAACGUGUCACGUTT-3′; AS, 5′-ACGUGACACGUUCGGAGAATT-3′ were
synthesized and reconstituted in 1X siRNA buffer (Sangon Biotech
Co., Ltd.). The results of the knock-down efficiency are shown in
Fig. S1.
| Table II.Sequences of siRNA for LINC00184. |
Table II.
Sequences of siRNA for LINC00184.
| siRNA | Sequences
(5′-3′) |
|---|
| siLINC00184-1 | S:
GCAAGGCAUCACACAGAAUTT |
|
| AS:
AUUCUGUGUGAUGCCUUGCTT |
| siLINC00184-2 | S:
GGAAGAGACAUAAGGAGAATT |
|
| AS:
UUCUCCUUAUGUCUCUUCCTT |
| siLINC00184-3 | S:
GCCGUCAUCUACAAAGGAATT |
|
| AS:
UUCCUUUGUAGAUGACGGCTT |
Cell Counting Kit-8 (CCK-8) assay
For viability assessment, KYSE-150 and TE-1 cells
were seeded at 5×103 cells per well in 96-well plates,
allowed to attach overnight (37°C and 5% CO2) and then
subjected to plasmid transfection or 5-AZA treatment for 48 h.
Subsequently, 10 µl of CCK-8 reagent (Dojindo Laboratories, Inc.)
were added per well, incubated for 2 h at 37°C, and absorbance at
450 nm was recorded on a SpectraMax® i3× microplate
reader (Molecular Devices, LLC). To determine a functional and
non-cytotoxic concentration, the present study performed a
dose-optimization experiment using CCK-8 assays across a range of
concentrations (0, 0.2, 2, 5, 10 and 20 µM). Based on these results
(Fig. S2), the present study
selected 2 µM for subsequent functional rescue experiments, as it
effectively modulated the phenotype while keeping non-specific
cytotoxicity low (cell viability reduction <15%).
Transwell migration assay
KYSE-150 and TE-1 cells were seeded at
5×105 cells per well in 6-well plates and allowed to
attach overnight (37°C and 5% CO2). Following plasmid
transfection or 5-AZA exposure (48 h), cells were trypsinized,
washed twice with PBS and resuspended in serum-free RPMI-1640 to a
density of 3×105 cells/ml. A 200 µl aliquot was plated
into the upper compartment of 8.0 µm pore Transwell®
inserts (Corning, Inc.); the lower compartment contained 600 µl
complete medium (10% FBS) serving as chemoattractant. After 24 h
incubation (37°C and 5% CO2), non-invading cells on the
upper surface were gently removed with cotton swabs. Migratory
cells on the underside were fixed in 4% paraformaldehyde for 15
min, stained with 0.5% (w/v) crystal violet for 20 min at room
temperature, washed three times with PBS and captured under a
phase-contrast microscope. Five random fields per insert were
counted by two blinded investigators.
Apoptosis assay
After 48 h post-transfection or 5-AZA exposure,
KYSE-150 and TE-1 cells were collected, washed three times with PBS
and centrifuged. Apoptosis was then quantified using the Annexin
V-FITC/propidium iodide (PI) Apoptosis Detection Kit (cat. no.
C1062S; Beyotime Biotechnology). Briefly, cells were resuspended in
1X binding buffer and stained with 5 µl of FITC-conjugated Annexin
V and 10 µl of PI for 15 min in the dark at room temperature,
followed by the addition of 2 ml PBS. Apoptosis was evaluated using
flow cytometry with a FACScan instrument (BD Biosciences) and
analyzed using CellQuest™ software (version number
5.2.1; BD Biosciences). Unstained control and single-color
compensation controls (FITC single-stained and PI single-stained
cells) were applied to define negative thresholds and adjust
spectral compensation. Firstly, intact single-cell populations were
gated according to forward scatter and side scatter parameters to
exclude cellular debris and aggregates. Subsequently, quadrant
gating was performed on the Annexin V-FITC vs. PI dot plot to
distinguish viable cells, early apoptotic cells, late apoptotic
cells and necrotic cells for statistical analysis.
Methylation-specific PCR (MSP)
To evaluate the methylation status of the NDRG2
promoter, the EpiTect Plus DNA Bisulfite Kit (cat. no. 59124;
Qiagen GmbH) was employed. Genomic DNA was extracted from KYSE-150
and TE-1 cells using a conventional DNA isolation method (DP-34;
Tiangen Biotech Co., Ltd.). The concentration and purity of
extracted genomic DNA were determined via a Nanodrop
spectrophotometer. After quantifying the DNA concentration, 1 µg of
genomic DNA was subjected to bisulfite treatment following the
manufacturer's protocol. Subsequently, the bisulfite-treated DNA
was used as a template for PCR amplification, utilizing the primers
provided in Table III. The PCR
was performed under the following conditions: Initial denaturation
at 95°C for 10 min, followed by 35 cycles consisting of
denaturation at 95°C for 30 sec, annealing at 60°C for 30 sec and
extension at 72°C for 10 sec, with a final extension step at 72°C
for 10 min. The PCR products were analyzed via agarose gel
electrophoresis and visualized using an image analysis system. The
PCR amplicons were then analyzed by 1% agarose gel electrophoresis
and visualized using a Bio-Rad ChemiDoc Imaging System (Bio-Rad
Laboratories, Inc.) to assess the methylation status.
| Table III.Primer sequences for NDRG2
promoter. |
Table III.
Primer sequences for NDRG2
promoter.
| Promoter | Primer sequences
(5′-3′) | Size, bp |
|---|
| Homo NDRG2 M | F:
AAGTTTATAGTGGTAAATTTATTCGG | 103 |
|
| R:
ATAAAAAACCAACTCAAACCCG |
|
| Homo NDRG2 U | F:
TGTGTAAAGTTTATAGTGGTAAATTTATTT | 109 |
|
| R:
ATAAAAAACCAACTCAAACCCACT |
|
Chromatin immunoprecipitation
(ChIP)
When the KYSE-150 cells reached 70–80% confluence,
the ChIP assay was performed using a commercial ChIP Kit (cat. no.
P2078; Beyotime Biotechnology). The cells were fixed with 1%
formaldehyde for 10 min at ambient temperature to cross-link the
DNA-protein complexes. Subsequently, the chromatin was fragmented
by sonication to generate DNA fragments of 200–1,000 base pairs.
The sheared chromatin was then subjected to immunoprecipitation
overnight at 4°C using specific antibodies: Mouse anti-DNMT1 (cat.
no. ab13537; 1:100; Abcam) and a negative control mouse anti-IgG
(cat. no. ab109489; 1:100; Abcam). Protein A/G agarose beads were
employed to capture the antibody-bound chromatin complexes. After
extensive washing to remove non-specific binding, the cross-links
were reversed by incubating the samples at 65°C overnight. The DNA
was subsequently purified using phenol/chloroform extraction and
ethanol precipitation. To verify the binding of DNMT1 to the NDRG2
promoter, PCR amplification was performed using primers specific to
the NDRG2 promoter region. The sequences of these primers are
detailed in Table IV. Fluorescent
quantitative PCR was performed with the 2−ΔΔCq
calculation. The presence of PCR products indicated the successful
enrichment of the NDRG2 promoter region in the immunoprecipitated
samples, thereby confirming the interaction between DNMT1 and the
NDRG2 promoter.
| Table IV.NDRG2 promoter-specific primers. |
Table IV.
NDRG2 promoter-specific primers.
| NDRG2-promoter | Primer sequences
(5′-3′) | Size, bp |
|---|
| 1 | F:
CAGACAGACCCCCAGTGTTC | 204 |
|
| R:
CGTTTCCTGTGGCTGAGACT |
|
| 2 | F:
AGTCTCAGCCACAGGAAACG | 173 |
|
| R:
GGAGGCTGGAGGAAAAAGAA |
|
| 3 | F:
CCAGAGACGGGACATTCAGT | 197 |
|
| R:
GCTGCTCAAGCCCTAGCTC |
|
RNA-binding protein
immunoprecipitation (RIP) assay
To elucidate the interaction between LINC00184 and
DNMT1, RIP assay was performed in the present study using the RIP
kit from EMD Millipore, following the manufacturer's instructions.
KYSE-150 cells were disrupted with RIPA lysis buffer (cat. no.
P0013B; Beyotime Biotechnology) for 5 min at 4°C. After lysis, the
cell lysate was centrifuged at 12,000 × g for 10 min at 4°C to
remove cell debris and insoluble impurities. A volume of 100 µl of
the lysate was incubated with 50 µl protein A/G magnetic beads
(MedChemExpress) conjugated with anti-DNMT1 antibody (cat. no.
ab13537; 1:100; Abcam) or control IgG (cat. no. ab109489; 1:100;
Abcam) for an extended period at 4°C. The magnetic bead-protein
complexes were subsequently isolated using a magnetic separator.
The complexes were then treated with proteinase K to release the
associated RNA. The purified RNA was analyzed via RT-qPCR. Total
RNA extracted from the RIP samples was reverse-transcribed into
cDNA using a reverse transcription kit, which was then used as the
template for qPCR amplification. The qPCR system included BeyoFast™
SYBR Green qPCR Mix, specific primers targeting the target genes
and cDNA template. The thermal cycling conditions were as follows:
Pre-denaturation at 95°C for 30 sec, followed by 40 cycles of
denaturation at 95°C for 5 sec and annealing/extension at 60°C for
30 sec. Relative gene expression levels were calculated using the
2−ΔΔCq method. The specific primers utilized in this
experiment are detailed in Table
I.
Western blotting
Protein lysates were obtained from KYSE-150 and TE-1
cells using a cell lysis buffer (cat. no. P0013; Beyotime
Biotechnology). Protein concentrations were determined using the
Bradford method and subsequently normalized. Equivalent quantities
ranging from 20–40 µg of protein were resolved on 12–15% gels using
SDS-PAGE and electrotransferred to PVDF membranes (MilliporeSigma).
After blocking with 5% BSA for 1 h at ambient temperature, the
membranes were probed with primary antibodies overnight at 4°C. The
primary antibodies utilized were: Mouse anti-GAPDH (cat. no.
60004-1-Ig; 1:1,000; Proteintech Group, Inc.), rabbit anti-NDRG2
(cat. no. Ab174850; 1:1,000; Abcam), mouse anti-AKT (cat. no.
60203-2-Ig; 1:1,000; Proteintech Group, Inc.), rabbit
anti-phosphorylated (p)-AKT (cat. no. 80455-1-RR; 1:1,000;
Proteintech Group, Inc.), rabbit anti-PI3K (cat. no. 4292; 1:1,000;
CST), and rabbit anti-p-PI3K (cat. no. 17366; 1:1,000; CST),
anti-DNMT1 (cat. no. 5032; 1:1,000; CST). The blots were washed
with TBST containing 0.1% Tween-20 at room temperature three times
for 15 min per wash. The membranes were then incubated with
HRP-conjugated secondary antibodies, goat anti-rabbit IgG (cat. no.
A0208; 1:5,000; Proteintech Group, Inc.) and goat anti-mouse IgG
(cat. no. SA00001-1; 1:5,000; Proteintech Group, Inc.) for 1 h at
room temperature. Protein bands were detected using a
Clarity™ Western ECL Substrate (Bio-Rad Laboratories,
Inc.) and their relative intensities were assessed by Quantity One
analysis tool (Bio-Rad Laboratories, Inc.).
Statistical analysis
Statistical analyses were conducted using GraphPad
Prism software (version 5; Dotmatics). For pairwise comparisons, an
unpaired Student's t-test was employed. In cases involving multiple
treatment groups, one-way ANOVA was performed, followed by Tukey's
post hoc test for multiple comparisons. All experiments in this
study were performed with no fewer than three independent
biological replicates and the number of experimental repeats varied
appropriately according to different experimental designs. Data are
presented as mean ± SEM. P<0.05 was considered to indicate a
statistically significant difference.
Results
Overexpression of LINC00184 promotes
proliferation and migration while inhibiting apoptosis in ESCC
cells
In order to explore the role of LINC00184 in ESCC
cell proliferation, migration and apoptosis, KYSE-150 and TE-1
cells were transfected with the OE-LINC00184 plasmid to overexpress
LINC00184. The expression level of LINC00184 in KYSE-150 and TE-1
cells following transfection was verified using RT-qPCR. The
results demonstrated that the expression level of LINC00184 was
significantly increased after transfection with the OE-LINC00184
plasmid in KYSE-150 (Fig. 1A) and
TE-1 cells (Fig. 1B). In addition,
the biological behaviors of cells were assessed using CCK-8
staining, Transwell and apoptosis assays. As presented in Fig. 1C and D, overexpression of LINC00184
markedly enhanced the cell viability of these two cells. The
Transwell assay results revealed the increased number of migrated
cells in the OE-LINC00184 groups compared with the control groups
which means that overexpression of LINC00184 significantly promoted
the migration of KYSE-150 cells (Fig.
1E and F) and TE-1 cells (Fig. 1G
and H). Additionally, flow cytometry analysis of apoptosis
indicated that the apoptotic rates in the OE-LINC00184 groups were
significantly lower compared with those in the control groups which
means that overexpression of LINC00184 reduced the apoptosis rate
in both KYSE-150 cells (Fig. 1I and
J) and TE-1 cells (Fig. 1K and
L). In summary, these findings suggested that overexpression of
LINC00184 serves a key role in promoting the proliferation,
migration, and inhibiting apoptosis of ESCC cells.
LINC00184 negatively regulates NDRG2
and activates the PI3K/AKT pathway in ESCC cells
To elucidate the molecular mechanisms underlying the
effects of LINC00184 in ESCC cells, the present study focused on
its impact on NDRG2 expression and the PI3K/AKT signaling pathway.
NDRG2 is a tumor suppressor protein that inhibits cell
proliferation and promotes apoptosis in various cancer types,
including lung, liver and pancreatic cancer (16–18).
NDRG2 can inhibit the PI3K/AKT pathway by acting as a bridge
between PP2A and PTEN, leading to the dephosphorylation of
phosphatidylinositol-3,4,5-triphosphate and subsequent inhibition
of AKT activity (21). Due to this
background, the present study hypothesized that LINC00184 might
influence ESCC cell behavior by regulating NDRG2, thereby affecting
the PI3K/AKT pathway. Western blotting analysis in KYSE-150 and
TE-1 cells revealed that overexpression of LINC00184 significantly
decreased NDRG2 protein levels (Fig.
2A, B, F and G). Consistent with this, RT-qPCR analysis
confirmed that LINC00184 overexpression also led to a significant
reduction in NDRG2 mRNA levels in both cell lines (Fig. 2E and J), demonstrating
transcriptional downregulation. Concurrently, the overexpression
increased the phosphorylation of AKT (Fig. 2A, C, F and H) and PI3K (Fig. 2A, D, F and I), indicative of
PI3K/AKT pathway activation. These results suggested that LINC00184
may promote ESCC cell proliferation and survival by
transcriptionally repressing NDRG2 and leading to reduced NDRG2
protein expression, thereby influencing the PI3K/AKT pathway.
 | Figure 2.LINC00184 negatively regulates NDRG2
and activate the PI3K/AKT pathway in esophageal squamous cell
carcinoma cells. (A) Western blotting analysis reveals the protein
expression levels of NDRG2, p-AKT (Ser473), AKT, p-PI3K (Tyr458)
and PI3K in KYSE-150 transfected with OE-NC or OE-LINC00184
plasmid. (B) Quantitative analysis of immunoblots of NDRG2
presented in (A). (C) Quantitative analysis of immunoblots of
pAKT/AKT presented in (A). (D) Quantitative analysis of immunoblots
of pPI3K/PI3K presented in (A). (E) RT-qPCR analysis of NDRG2 mRNA
levels in KYSE-150 cells under control conditions and following
overexpression of LINC00184. (F) Western blotting analysis
demonstrating the protein expression levels of NDRG2, p-AKT
(Ser473), AKT, p-PI3K (Tyr458) and PI3K in KYSE-150 and TE-1 cells
transfected with OE-NC or OE-LINC00184 plasmid. (G) Quantitative
analysis of immunoblots of NDRG2 presented in (F). (H) Quantitative
analysis of immunoblots of pAKT/AKT presented in (F). (I)
Quantitative analysis of immunoblots of pPI3K/PI3K presented in
(F). (J) RT-qPCR analysis of NDRG2 mRNA levels in TE-1 cells under
control conditions and following overexpression of LINC00184. Data
are presented as mean ± SEM (n=3). Comparisons between two groups
were performed using the Student's t-test. Statistical significance
is indicated as *P<0.05, **P<0.01 and ***P<0.001. OE,
overexpression; NC, negative control; RT-qPCR, reverse
transcription-quantitative PCR; NDRG2, N-Myc downstream regulated
gene. |
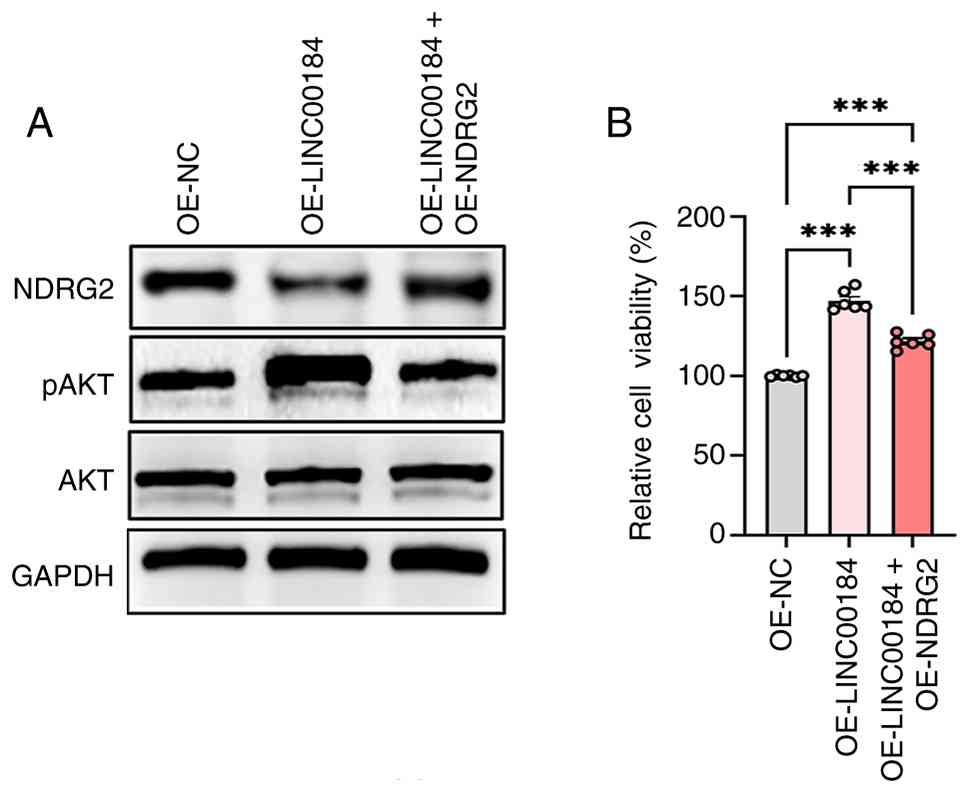
Restoration of NDRG2 attenuates
LINC00184-induced oncogenic phenotypes
To directly establish the causal role of NDRG2
downregulation in mediating the effects of LINC00184, the present
study performed a rescue experiment by re-expressing NDRG2 in TE-1
cells stably overexpressing LINC00184. Western blotting analysis
confirmed the successful restoration of NDRG2 protein (Figs. 3A and S3A). Notably, this restoration
significantly reversed the LINC00184-induced hyperphosphorylation
of AKT (Figs. 3A and S3B), indicating that NDRG2 reconstitution
effectively suppressed the activation of the PI3K/AKT pathway.
Correspondingly, functional assays demonstrated that re-expression
of NDRG2 significantly attenuated the enhanced cell viability
promoted by LINC00184 overexpression (Fig. 3B). Collectively, these data provided
direct genetic evidence that the downregulation of NDRG2 is a
pivotal and causative event through which LINC00184 activates the
PI3K/AKT pathway and drives proliferative advantage in ESCC
cells.
LINC00184 regulates NDRG2 expression
through DNMT1-mediated methylation of the NDRG2 promoter
To elucidate the regulatory mechanisms of LINC00184
on NDRG2 expression in ESCC cells, the present study first
validated the effects of both LINC00184 overexpression and
knockdown using siRNA (Fig. 4A and
B). The present study identified that overexpression of
LINC00184 markedly increased the methylation level of the NDRG2
promoter, as confirmed by MSP assay (Fig. 4C and D). By contrast, silencing
LINC00184 using siRNA markedly reduced the methylation level of the
NDRG2 promoter (Fig. 4C and D).
These initial findings prompted further investigation into the role
of DNA methylation in LINC00184-mediated NDRG2 regulation. DNMT1 is
a key enzyme responsible for maintaining DNA methylation patterns,
particularly during DNA replication (13). Due to its key function in epigenetic
regulation, the present study hypothesized that LINC00184 might
influence NDRG2 expression through DNMT1-mediated methylation of
the NDRG2 promoter. To investigate this hypothesis, the present
study conducted ChIP assays to examine the enrichment of DNMT1 at
the NDRG2 promoter region. The results indicated that
overexpression of LINC00184 led to significantly increased
enrichment of DNMT1 at the NDRG2 promoter (Fig. 4E), suggesting that LINC00184
recruits DNMT1 to the NDRG2 promoter. By contrast, silencing
LINC00184 using siRNA significantly decreased the enrichment of
DNMT1 at the NDRG2 promoter (Fig.
4E), further supporting the role of LINC00184 in DNMT1
recruitment. To confirm the direct interaction between LINC00184
and DNMT1, the present study performed RIP assays. The results
revealed that overexpression of LINC00184 significantly increased
the enrichment of LINC00184 by DNMT1 (Fig. 4F and G), indicating a direct
interaction between LINC00184 and DNMT1. By contrast, silencing
LINC00184 significantly decreased this enrichment (Fig. 4F and G), reinforcing the notion that
LINC00184 directly interacts with DNMT1. To further confirm the
role of DNMT1 in LINC00184-mediated NDRG2 suppression, ESCC cells
overexpressing LINC00184 were treated with the DNMT1 inhibitor
5-AZA. The results demonstrated that 5-AZA treatment effectively
reversed the LINC00184-induced methylation of the NDRG2 promoter
(Fig. 4H and I) and restored NDRG2
expression (Fig. 4J-M). This
indicated that the methylation changes induced by LINC00184 are
reversible and that DNMT1 activity is a key factor in this process.
To establish a more specific causal association between DNMT1
activity and this regulatory axis, the present study employed siRNA
to directly knock down DNMT1 in LINC00184-overexpressing cells.
This genetic intervention successfully reversed the
LINC00184-induced downregulation of NDRG2 protein (Figs. 4N and S4A), mirroring the effect observed with
the pharmacological inhibitor 5-AZA. Notably, to definitively rule
out the alternative possibility that LINC00184 suppresses NDRG2 by
first upregulating DNMT1 expression, the present study assessed
DNMT1 levels following LINC00184 manipulation. Neither DNMT1 mRNA
(Fig. 4O) nor total protein levels
(Figs. 4P and S4B) were significantly altered by
LINC00184 overexpression. This key control experiment confirmed
that LINC00184 does not affect DNMT1 abundance but instead acts by
modulating the functional recruitment and activity of pre-existing
DNMT1 to epigenetically silence the NDRG2 promoter. Therefore, the
convergent evidence from pharmacological (5-AZA) and genetic
(siDNMT1) inhibition, coupled with the unchanged DNMT1 expression
profile, solidifies DNMT1 as the key epigenetic effector downstream
of LINC00184.
 | Figure 4.LINC00184 regulates NDRG2 expression
through DNMT1-mediated methylation of the NDRG2 promoter. (A)
Expression level of LINC00184 in KYSE-150 cells following treatment
with siRNA targeting LINC00184 (n=3). (B) Expression level of
LINC00184 in TE-1 cells following treatment with siRNA targeting
LINC00184 (n=3). (C) Methylation level of the NDRG2 promoter in
KYSE-150 cells detected via MSP assay after overexpression or
silencing of LINC00184. (D) Methylation level of the NDRG2 promoter
in TE-1 cells detected via MSP assay after overexpression or
silencing of LINC00184. (E) Enrichment of DNMT1 at the NDRG2
promoter region detected by chromatin immunoprecipitation assay and
quantified using RT-qPCR in KYSE-150 cells with overexpression or
silencing of LINC00184 (n=3). (F) Enrichment of LINC00184 bound to
DNMT1 detected by RNA immunoprecipitation assay and quantified
using RT-qPCR in KYSE-150 cells after overexpression or silencing
of LINC00184 (n=3). (G) Enrichment of LINC00184 bound to DNMT1
detected by RNA immunoprecipitation assay and quantified using
RT-qPCR in TE-1 cells after overexpression or silencing of
LINC00184 (n=3). (H) Methylation level of the NDRG2 promoter in
KYSE-150 cells measured by MSP assay following LINC00184
overexpression combined with 5-AZA treatment. (I) Methylation level
of the NDRG2 promoter in TE-1 cells measured by MSP assay following
LINC00184 overexpression combined with 5-AZA treatment. (J) Western
blotting analysis of NDRG2 protein expression in KYSE-150 cells
after LINC00184 overexpression and 5-AZA intervention. (K)
Quantitative analysis of NDRG2 protein grayscale values obtained
from the western blotting results in (J) (n=3). (L) Western
blotting analysis of NDRG2 protein expression in TE-150 cells after
LINC00184 overexpression and 5-AZA intervention. (M) Quantitative
analysis of NDRG2 protein grayscale values obtained from the
western blotting results in (L) (n=3). (N) Western blotting
detection of NDRG2 protein levels under control conditions, single
overexpression of LINC00184, and combined treatment with
OE-LINC00184 + si-DNMT1. (O) RT-qPCR detection of relative DNMT1
mRNA levels in control cells and LINC00184-overexpressing cells
(n=6). (P) Western blotting analysis of total DNMT1 protein levels
in control cells and LINC00184-overexpressing cells; GAPDH was used
as the loading control (n=3). Data are presented as mean ± SEM
(n=3). Comparisons between two groups were performed using the
unpaired Student's t-test. Comparisons among multiple groups were
analyzed by one-way analysis of variance. Statistical significance
is indicated as *P<0.05 and ***P<0.001. OE, overexpression;
NC, negative control; NDRG2, N-Myc downstream regulated gene;
RT-qPCR, reverse transcription-quantitative PCR; DNMT1, DNA
methyltransferase 1; si/siRNA, small interfering RNA; lnc/lncRNA,
long non-coding RNA; 5-AZA, 5-azacytidine; MSP,
methylation-specific PCR. M, methylation; U, unmethylation. |
Inhibition of DNMT1 abrogates
LINC00184-induced PI3K/AKT pathway activation
Having established that LINC00184 recruits DNMT1 to
silence NDRG2, the present study then investigated whether this
epigenetic mechanism is responsible for the downstream activation
of the oncogenic PI3K/AKT pathway. The present study first
confirmed that pharmacological inhibition of DNMT1 with 5-AZA in
LINC00184-overexpressing cells effectively reversed the
hyperphosphorylation of both PI3K and AKT (Fig. 5A-F). To establish a specific genetic
association, siRNA-mediated knockdown of DNMT1 was performed.
Consistent with the pharmacological data, siDNMT1 significantly
attenuated the LINC00184-induced increase in AKT phosphorylation
(Fig. 5G and H). This convergence
of evidence from independent inhibitory strategies definitively
confirmed that the DNMT1-mediated epigenetic silencing of NDRG2 is
the key mechanistic event through which LINC00184 activates the
PI3K/AKT pathway in ESCC cells.
 | Figure 5.Inhibition of DNMT1 abrogates
LINC00184-induced PI3K/AKT pathway activation and functional
phenotypes. (A) Western blotting analysis showing the protein
expression level of pAKT, AKT, pPI3K and PI3K in KYSE-150 cells
following overexpression of LINC00184 and treatment with 5-AZA. (B)
Quantitative analysis of the pAKT/AKT protein expression level
derived from the immunoblots in (A). (C) Quantitative analysis of
the pPI3K/PI3K protein expression level derived from the
immunoblots in (A). (D) Western blotting analysis showing the
protein expression level of pAKT, AKT, pPI3K and PI3K in TE-1 cells
following overexpression of LINC00184 and treatment with 5-AZA. (E)
Quantitative analysis of the pAKT/AKT protein expression level
derived from the immunoblots in (D). (F) Quantitative analysis of
the pPI3K/PI3K protein expression level derived from the
immunoblots in (D). (G) Western blotting analysis of p-AKT and
total AKT levels in esophageal squamous cell carcinoma cells under
the following conditions: Control, OE-LINC00184 and OE-LINC00184 +
si-DNMT1. (H) Quantitative analysis of the pAKT/AKT protein
expression level corresponding to the immunoblots presented in (G).
Data are presented as mean ± SEM (n=3). Comparisons among multiple
groups were analyzed by one-way analysis of variance. Statistical
significance is indicated as *P<0.05, **P<0.01 and
***P<0.001. OE, overexpression; NC, negative control; NDRG2,
N-Myc downstream regulated gene; RT-qPCR, reverse
transcription-quantitative PCR; DNMT1, DNA methyltransferase 1;
si/siRNA, small interfering RNA; 5-AZA, 5-azacytidine. |
DNMT1 inhibition reverses
LINC00184-induced malignant phenotypes in ESCC cells
To determine whether the DNMT1-mediated epigenetic
axis is functionally required for the oncogenic phenotypes driven
by LINC00184, the present study inhibited DNMT1 activity using both
pharmacological and genetic approaches. Treatment of
LINC00184-overexpressing cells with the DNMT1 inhibitor 5-AZA
significantly reversed the pro-tumorigenic phenotypes: It
significantly reduced cell viability (Fig. 6A and F), significantly attenuated
enhanced cell migration (Fig. 6B, D, G
and I) and significantly increased the apoptotic rate (Fig. 6C, E, H and J) back towards control
levels. To provide specific genetic confirmation, siRNA-mediated
knockdown of DNMT1 (siDNMT1) was performed in the same context.
Consistent with the pharmacological inhibition, genetic depletion
of DNMT1 also significantly reduced the enhanced cell viability
induced by LINC00184 overexpression (Fig. 6K).
Collectively, these results demonstrated that DNMT1
activity is key to executing the full spectrum of the oncogenic
functions of LINC00184, including promoting proliferation,
migration and suppressing apoptosis. The concordance between
chemical and genetic inhibition solidifies DNMT1 as a key
downstream effector and a potential therapeutic target within this
pathway.
Discussion
The present study revealed the significant role of
LINC00184 in ESCC progression, highlighting its oncogenic function
through DNMT1-mediated methylation of the NDRG2 promoter and
subsequent activation of the PI3K/AKT signaling pathway. The
present study results indicated that LINC00184 promotes ESCC cell
proliferation and migration while inhibiting apoptosis by
recruiting DNMT1 to the NDRG2 promoter, causing hypermethylation
and silencing of NDRG2. This epigenetic modification activates the
PI3K/AKT pathway, driving the malignant phenotype of ESCC cells.
Notably, treatment with the DNMT1 inhibitor 5-AZA effectively
reversed these effects, suggesting potential therapeutic strategies
targeting the LINC00184/DNMT1/NDRG2 axis in ESCC.
Beyond the classical competing endogeneous RNA
(ceRNA) network, LINC00184 orchestrates oncogenic signaling through
multiple, context-dependent modalities. In ovarian carcinoma, it
sequesters miR-1305 to relieve contactin-1 suppression and enhance
tumor cell fitness (25); in
gastric cancer, it competes with miR-145 for ANGPT2 mRNA, thereby
driving angiogenesis and metastasis (9). Prostate cancer studies revealed that
LINC00184 reduces miR-105-5p activity, leading to PD-L1
upregulation and docetaxel refractoriness (26), whereas in cholangiocarcinoma it
suppresses miR-23b-3p to elevate ANXA2 and accelerate proliferation
(27). In non-small cell lung
cancer, the lncRNA enhances aggressiveness via the
miR-524-5p/high-mobility group box 2 axis (28). The present study expands the known
functional repertoire of LINC00184 by demonstrating its capacity to
directly recruit the epigenetic regulator DNMT1 and facilitate DNA
methylation, thereby revealing a previously unrecognized
chromatin-based mechanism of action in ESCC, to the best of our
knowledge.
To the best of our knowledge, the present study
identified NDRG2 as a previously unrecognized downstream target
whose expression is stringently controlled by LINC00184-mediated
epigenetic rewriting. Although NDRG2 has been reported to be
transcriptionally repressed by Myc (29), silenced by specific miRNAs (30) or modified post-translationally
(31) in various malignancies.
While previous studies have established the importance of NDRG2 in
inhibiting tumor progression in lung (32), liver (33) and breast cancer (34), the regulatory mechanisms controlling
its expression in ESCC remain to be elucidated. The present study
findings revealed that LINC00184 recruits DNMT1 to install CpG
island hypermethylation on the NDRG2 promoter, functionally
extinguishing this tumor suppressor in esophageal squamous cells.
Reversal of this silencing by 5-AZA not only validates the
methylation-dependent mechanism but also highlights a clinically
actionable route to reactivate NDRG2 in patients with ESCC.
Beyond chromatin-level regulation, the present study
revealed a functional crosstalk between LINC00184 and the PI3K/AKT
cascade. NDRG2 loss-of-function triggered by LINC00184-directed
methylation relieves the brake on PI3K and AKT phosphorylation,
thereby amplifying pro-survival signaling in ESCC cells. Due to the
well-documented role of hyperactive PI3K/AKT in driving esophageal
tumorigenesis and chemoresistance (22,35,36),
pharmacological restoration of NDRG2 via DNMT1 blockade offers a
rational strategy to dampen this oncogenic axis. The observed
attenuation of PI3K and AKT phosphorylation following 5-AZA
treatment substantiates DNMT1 as a tractable target to counteract
LINC00184-mediated pathway hyperactivation.
The functional reversal achieved with 5-AZA
underscores the therapeutic value of intercepting the
LINC00184-DNMT1 interface. While DNMT1 inhibitors have demonstrated
clinical efficacy in myeloid neoplasms (37), the present study pre-clinical data
now extend their utility to solid tumors, specifically ESCC. Beyond
classical DNMT1 blockade, rational design of AS oligonucleotides or
small-molecule disruptors that specifically interfere with
LINC00184-DNMT1 complex formation could provide a
precision-medicine avenue in reactivating the NDRG2-PI3K/AKT brake
in ESCC.
Despite these advances, the present study had
certain limitations. The experiments were conducted primarily in
cell lines, and further validation in primary cells, animal models
and clinical samples is warranted to confirm the relevance of the
present study findings in vivo. Furthermore, the precise
molecular mechanisms by which LINC00184 recruits DNMT1 to the NDRG2
promoter remain to be fully elucidated. Future studies are
warranted to explore the broader network interactions of LINC00184
with other known tumor suppressors or oncogenic pathways, which
will help to contextualize its role in cancer beyond the NDRG2
axis. Future studies could also explore whether other epigenetic
modifiers or co-factors are involved in the LINC00184-DNMT1-NDRG2
regulatory axis. Additionally, while the CCK-8 and Transwell assays
were performed with three independent biological replicates at
standardized time points to confirm the qualitative effect of
LINC00184 on ESCC cell proliferation and migration, performing
multi-time-point kinetics would offer further insights into the
dynamics of these processes and thus, represent a beneficial
direction for future studies. Notably, the lack of formal power
calculations prior to conducting statistical analyses with
Student's t-test and ANOVA also represents a limitation of the
present study. Furthermore, the present study utilized MSP to
assess the methylation status of the NDRG2 promoter CpG islands, a
standard approach in detecting promoter hypermethylation; however,
the absence of bisulfite sequencing constitutes a limitation of the
present study, as this method would have provided single-base
resolution to characterize the methylation landscape in further
detail. This higher-resolution analysis represents a valuable
avenue for follow-up research to refine current understanding of
NDRG2 promoter methylation patterns.
In conclusion, the present study revealed a novel
oncogenic function of LINC00184 in ESCC: LINC00184 acts as a
molecular guide that recruits DNMT1 to epigenetically silence the
tumor suppressor NDRG2. This specific silencing event, distinct
from the known ceRNA role of LINC00184 in other cancer types,
subsequently relieves the brake on the PI3K/AKT pathway, a key
driver of ESCC progression. Thus, to the best of our knowledge, the
LINC00184/DNMT1/NDRG2 axis represents a previously unrecognized
epigenetic circuit integral to ESCC pathogenesis, offering both
mechanistic insight and a potential therapeutic target for this
aggressive cancer in the future.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Shaanxi Key Research and
Development Program (grant nos. 2022SF-495 and
2024SF-YBXM-121).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
JG conceptualized the study, analyzed experimental
data, conducted validation experiments, and drafted and revised the
manuscript critically. SF designed the core methodology,
participated in data interpretation and revised relevant manuscript
content. JL performed data analysis with software, summarized
research results, and revised the analytical sections of the paper.
LL optimized the experimental scheme and acquisition of data. JZ
verified data authenticity and supplemented results analysis. FW
was responsible for the acquisition of data, and the analysis and
interpretation of data. WW organized data sorting, optimized
research progress management, and contributed to data analysis,
interpretation, and manuscript preparation. XC improved the
methodology framework, and analysis and interpretation of data. EL
supervised the whole study, acquired funding, helped with
conception and design, revised the full manuscript, and took
overall responsibility for the research. JL and XC confirm the
authenticity of all the raw data. All authors have read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Abnet CC, Arnold M and Wei WQ:
Epidemiology of esophageal squamous cell carcinoma.
Gastroenterology. 154:360–373. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Uhlenhopp DJ, Then EO, Sunkara T and
Gaduputi V: Epidemiology of esophageal cancer: Update in global
trends, etiology and risk factors. Clin J Gastroenterol.
13:1010–1021. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chang J, Zhao X, Wang Y, Liu T, Zhong C,
Lao Y, Zhang S, Liao H, Bai F, Lin D and Wu C: Genomic alterations
driving precancerous to cancerous lesions in esophageal cancer
development. Cancer Cell. 41:2038–2050.e5. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu WJ, Zhao Y, Chen X, Miao ML and Zhang
RQ: Epigenetic modifications in esophageal cancer: An evolving
biomarker. Front Genet. 13:10874792023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang L, Wang Y, Gao J, Zhou X, Huang M,
Wang X and He Z: Non-coding RNA: A promising diagnostic biomarker
and therapeutic target for esophageal squamous cell carcinoma
(review). Oncol Lett. 27:2552024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Karakas D and Ozpolat B: The role of
LncRNAs in translation. Noncoding RNA. 7:162021.PubMed/NCBI
|
|
7
|
Herman AB, Tsitsipatis D and Gorospe M:
Integrated lncRNA function upon genomic and epigenomic regulation.
Mol Cell. 82:2252–2266. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ferrer J and Dimitrova N: Transcription
regulation by long non-coding RNAs: Mechanisms and disease
relevance. Nat Rev Mol Cell Biol. 25:396–415. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Piao HY, Guo S, Jin H, Wang Y and Zhang J:
LINC00184 involved in the regulatory network of ANGPT2 via ceRNA
mediated miR-145 inhibition in gastric cancer. J Cancer.
12:2336–2350. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yao Y, Zhang T, Qi L, Zhou C, Wei J, Feng
F, Liu R and Sun C: Integrated analysis of co-expression and ceRNA
network identifies five lncRNAs as prognostic markers for breast
cancer. J Cell Mol Med. 23:8410–8419. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Weisenberger DJ, Lakshminarasimhan R and
Liang G: The role of DNA methylation and DNA methyltransferases in
cancer. Adv Exp Med Biol. 1389:317–348. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhao N, Lai C, Wang Y, Dai S and Gu H:
Understanding the role of DNA methylation in colorectal cancer:
Mechanisms, detection, and clinical significance. Biochim Biophys
Acta Rev Cancer. 1879:1890962024. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li Z, Li B, Yu H, Wang P, Wang W, Hou P,
Li M, Chu S, Zheng J, Mao L and Bai J: DNMT1-mediated epigenetic
silencing of TRAF6 promotes prostate cancer tumorigenesis and
metastasis by enhancing EZH2 stability. Oncogene. 41:3991–4002.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li A, Omura N, Hong SM and Goggins M:
Pancreatic cancer DNMT1 expression and sensitivity to DNMT1
inhibitors. Cancer Biol Ther. 9:321–329. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Etoh T, Kanai Y, Ushijima S, Nakagawa T,
Nakanishi Y, Sasako M, Kitano S and Hirohashi S: Increased DNA
methyltransferase 1 (DNMT1) protein expression correlates
significantly with poorer tumor differentiation and frequent DNA
hypermethylation of multiple CpG islands in gastric cancers. Am J
Pathol. 164:689–699. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee KW, Lim S and Kim KD: The function of
N-Myc downstream-regulated gene 2 (NDRG2) as a negative regulator
in tumor cell metastasis. Int J Mol Sci. 23:93652022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li SJ, Wang WY, Li B, Chen B, Zhang B,
Wang X, Chen CS, Zhao QC, Shi H and Yao L: Expression of NDRG2 in
human lung cancer and its correlation with prognosis. Med Oncol.
30:4212013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu XL, Liu XP, Lin SX, Deng YC, Liu N, Li
X and Yao LB: NDRG2 expression and mutation in human liver and
pancreatic cancers. World J Gastroenterol. 10:3518–3521. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Feng RB, Zhou QZ, Cheng R, Li P, Zhu ST,
Min L and Zhang ST: Expression and significance of N-myc downstream
regulated gene 2 in the process of esophageal squamous cell
carcinogenesis. Bioengineered. 13:3275–3283. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ahn CH, Kim JH, Shim HW, Shin WJ, Cho YA
and Yoon HJ: Biological and prognostic significance of NDRG2
downregulation in oral squamous cell carcinoma. Oral Dis.
30:4287–4302. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Morishita K, Nakahata S and Ichikawa T:
Pathophysiological significance of N-myc downstream-regulated gene
2 in cancer development through protein phosphatase 2A
phosphorylation regulation. Cancer Sci. 112:22–30. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Luo Q, Du R, Liu W, Huang G, Dong Z and Li
X: PI3K/Akt/mTOR signaling pathway: Role in esophageal squamous
cell carcinoma, regulatory mechanisms and opportunities for
targeted therapy. Front Oncol. 12:8523832022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huang W, Li H, Yu Q, Xiao W and Wang DO:
LncRNA-mediated DNA methylation: An emerging mechanism in cancer
and beyond. J Exp Clin Cancer Res. 41:1002022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Han Y, You J, Han Y, Liu Y, Huang M, Lu X,
Chen J and Zheng Y: LINC00184 promotes ovarian cancer cells
proliferation and cisplatin resistance by elevating CNTN1
expression via sponging miR-1305. Onco Targets Ther. 14:2711–2726.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang W, Xin J, Lai J and Zhang W: LncRNA
LINC00184 promotes docetaxel resistance and immune escape via
miR-105-5p/PD-L1 axis in prostate cancer. Immunobiology.
227:1521632022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sun HB, Zhang GC, Liu J and Nie CS: Long
noncoding RNA LINC00184 facilitates the proliferation, metastasis,
and adenine metabolism of cholangiocarcinoma via modulating
hsa-miR-23b-3p/ANXA2 axis. Environ Toxicol. 36:1576–1590. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang W, Li L and Zhao L: LINC00184 plays
an oncogenic role in non-small cell lung cancer via regulation of
the miR-524-5p/HMGB2 axis. J Cell Mol Med. 25:9927–9938. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yao L, Zhang J and Liu X: NDRG2: A
Myc-repressed gene involved in cancer and cell stress. Acta Biochim
Biophys Sin (Shanghai). 40:625–635. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Feng L, Xie Y, Zhang H and Wu Y:
Down-regulation of NDRG2 gene expression in human colorectal cancer
involves promoter methylation and microRNA-650. Biochem Biophys Res
Commun. 406:534–538. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tantai J, Pan X and Hu D: RNF4-mediated
SUMOylation is essential for NDRG2 suppression of lung
adenocarcinoma. Oncotarget. 7:26837–26843. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ma Z, Ma Y, Feng J, Xu Z, Cheng C, Qin J,
Li S, Jiang J and Kong R: NDRG2 acts as a negative regulator of the
progression of small-cell lung cancer through the modulation of the
PTEN-AKT-mTOR signalling cascade. Toxicol Appl Pharmacol.
485:1169152024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lee DC, Kang YK, Kim WH, Jang YJ, Kim DJ,
Park IY, Sohn BH, Sohn HA, Lee HG, Lim JS, et al: Functional and
clinical evidence for NDRG2 as a candidate suppressor of liver
cancer metastasis. Cancer Res. 68:4210–4220. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zheng J, Liu Q, Li Y, Yang J, Ma J, Yu F,
Shi H, Ren Q, Zhang R, Zhang J, et al: NDRG2 expression regulates
CD24 and metastatic potential of breast cancer cells. Asian Pac J
Cancer Prev. 11:1817–1821. 2020.PubMed/NCBI
|
|
35
|
Liu B, Wang C, Chen P, Cheng B and Cheng
Y: RACKI induces chemotherapy resistance in esophageal carcinoma by
upregulating the PI3K/AKT pathway and Bcl-2 expression. Onco
Targets Ther. 11:211–220. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shi N, Yu H and Chen T: Inhibition of
esophageal cancer growth through the suppression of PI3K/AKT/mTOR
signaling pathway. Onco Targets Ther. 12:7637–7647. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sigalotti L, Altomonte M, Colizzi F, Degan
M, Rupolo M, Zagonel V, Pinto A, Gattei V and Maio M:
5-Aza-2′-deoxycytidine (decitabine) treatment of hematopoietic
malignancies: A multimechanism therapeutic approach? Blood.
101:4644–4646. 2003. View Article : Google Scholar : PubMed/NCBI
|