Introduction
Endometrial cancer is among the most common
gynecologic malignancies affecting women worldwide, with ~417,000
new cases and 97,000 deaths recorded globally in 2020 alone, and
its incidence continues to rise steadily. Recent global cancer
statistics indicate that this increase is largely driven by aging
populations, rising obesity rates and lifestyle-associated risk
factors (1,2). Although prognosis is generally
favorable when endometrial cancer is diagnosed at an early stage,
the therapeutic landscape for advanced, recurrent, or
treatment-resistant disease remains limited, with poor long-term
outcomes despite multimodal treatment approaches (3). These challenges highlight the need to
identify novel agents capable of targeting key molecular
vulnerabilities in endometrial cancer cells.
Reactive oxygen species (ROS) have emerged as
central regulators of cancer cell biology, functioning as both
signaling mediators and inducers of cellular damage. Evidence
emphasizes the dual role of ROS: While moderate ROS levels may
support proliferation and adaptive signaling, excessive ROS
accumulation disrupts redox homeostasis, promotes mitochondrial
dysfunction and triggers apoptotic cell death (4,5).
Importantly, redox alterations are tightly interconnected with
mitogen-activated protein kinase (MAPK) signaling, particularly the
extracellular signal-regulated kinase (ERK1/2) pathway.
Dysregulated ERK1/2 activation contributes to enhanced
proliferation, survival signaling, and therapeutic resistance in
multiple malignancies, including endometrial carcinoma (6,7).
Accordingly, the ROS-ERK axis is increasingly recognized as a
critical determinant of cancer cell fate and a promising
therapeutic target.
Natural phenolic compounds have attracted sustained
interest in cancer research due to their capacity to modulate
oxidative stress and intracellular signaling networks across
multiple tumor types, including breast, colorectal, lung and
gynecologic cancers. Plant-derived phenolic compounds can influence
MAPK/ERK signaling, often through ROS-dependent mechanisms, thereby
suppressing tumor cell proliferation and promoting cell death
(8). Salicin, a phenolic glycoside
traditionally isolated from willow bark, has a long history of
pharmacological relevance and has recently gained renewed attention
for its potential anticancer properties. Modern formulation
strategies, including salicin-based nanocarriers and other
phytochemical nano-delivery systems, have demonstrated improved
cytotoxic and antiproliferative effects in cancer cell models
through enhanced bioavailability, targeted delivery, and
redox-mediated mechanisms (9). In
addition, structural derivatives of salicin, such as salicin
dimethyl ether, have been shown to inhibit tumor cell proliferation
and induce apoptotic signaling (10).
Importantly, mechanistic studies have provided
evidence that salicin and certain salicin derivatives can modulate
intracellular redox status and ERK-related signaling. For example,
salicin has been reported to inhibit angiogenesis via blockade of
the ROS-ERK pathway (11). In
addition, salicin dimethyl ether has been reported to exert
inhibitory effects in laryngeal cancer cells through activation of
apoptosis, supporting the plausibility that salicin-derived
structures can engage stress-survival signaling nodes relevant to
cancer cell fate (10). However,
despite these insights, the effects of D-salicin on ROS production
and ERK1/2 activity in gynecologic malignancies, especially
endometrial cancer, remain insufficiently explored.
To evaluate the causal involvement of oxidative
stress in D-salicin-induced cytotoxicity, antioxidant modulation
strategies were incorporated into the experimental design.
N-acetylcysteine (NAC), a well-established precursor of
intracellular glutathione (GSH), acts as a thiol-based antioxidant
and redox regulator (12,13). NAC is widely used in experimental
cancer models to test whether ROS generation directly contributes
to cytotoxic and apoptotic responses (14,15).
By replenishing intracellular thiol pools and restoring redox
homeostasis, NAC facilitates functional interrogation of
ROS-dependent signaling pathways, including MAPK/ERK modulation
(16). Accordingly, NAC
co-treatment provides a pathway-level approach to assess whether
D-salicin-induced ERK1/2 alterations and apoptotic responses are
mediated by oxidative stress-dependent processes (15).
The present study investigated the anticancer
effects of D-salicin in the endometrial adenocarcinoma-derived
Ishikawa cell line. Cell viability was assessed using the MTT
assay, apoptotic responses were quantified via caspase-3 ELISA,
intracellular oxidative stress was measured through ROS assays, and
changes in phospho-ERK1/2 expression were evaluated using
immunofluorescence. In addition, NAC co-treatment experiments were
conducted to examine whether restoration of intracellular redox
balance modifies D-salicin-induced cytotoxicity and ERK1/2
signaling alterations. To the best of our knowledge, this is among
the first studies of D-salicin-mediated ROS-ERK1/2 modulation in
endometrial cancer cells using antioxidant-based functional
validation. Through this integrated experimental approach, the
present study aimed to elucidate whether D-salicin exerts its
anticancer effects via activation of oxidative stress and
modulation of the ROS-ERK1/2 signaling axis in endometrial cancer
cells.
Materials and methods
Cell lines and culture conditions
The Ishikawa human endometrial adenocarcinoma cell
line and human dermal fibroblasts (HDF; PCS-201-012) were obtained
from the American Type Culture Collection (ATCC) and cultured
according to the supplier's recommendations. HDF cells are
non-immortalized normal human dermal fibroblasts and, under
standard culture conditions, typically exhibit lower baseline
proliferative activity and ERK1/2 signaling compared with actively
dividing cancer cell lines, consistent with their non-malignant
phenotype. All cells were maintained at 37°C in a humidified
atmosphere containing 5% CO2 under standard culture
conditions. Since commercially available ATCC cells were used, no
additional ethical approval was required.
Assessment of cell viability by
MTT
Cytotoxic responses to D-salicin were quantified
using the MTT colorimetric assay. Ishikawa and HDF cells were
seeded into 96-well plates and treated with escalating
concentrations of D-salicin (0–80 µM) for 72 h. D-salicin stock
solutions were prepared directly in complete culture medium, and no
additional organic solvent was used. After incubation, MTT was
added and allowed to metabolize for 4 h, generating formazan
crystals, which were subsequently dissolved with dimethyl sulfoxide
and quantified at 570 nm by a Multiskan FC Microplate Photometer
(Thermo Fisher Scientific, Inc.). Each condition was assayed in
technical triplicate, and experiments were independently repeated
at least three times. Cell viability was expressed as a percentage
of untreated control cells and dose-response curves were analyzed
using nonlinear regression to calculate IC25,
IC50, and IC75 values. To probe the
contribution of oxidative stress to D-salicin-induced cytotoxicity,
N-acetylcysteine (NAC) was included as an antioxidant co-treatment.
To determine an NAC concentration suitable for use in both
malignant and non-malignant cells without causing appreciable
cytotoxicity, Ishikawa and HDF cells were exposed to NAC (0.5–10
mM) for 72 h and viability was assessed by MTT under the same
conditions. The concentration used in subsequent co-treatment
experiments was chosen based on this preliminary NAC screening. For
NAC co-treatment experiments, cells were treated with D-salicin in
the presence or absence of NAC for 72 h, after which MTT was
performed as aforementioned. Untreated (vehicle control),
D-salicin-treated, NAC-only and D-salicin + NAC co-treated wells
were included to enable appropriate normalization and comparison of
treatment effects.
Intracellular ROS measurement
Intracellular ROS levels were quantified using the
DCFDA/H2DCFDA Cellular ROS Assay Kit (Abcam; cat. no.
ab113851) according to the manufacturer's guidelines, with minor
adjustments for adherent cell cultures. Ishikawa and HDF cells were
seeded into black-walled 96-well plates and allowed to attach
overnight. Cells were then exposed to D-salicin at the previously
determined IC25, IC50 and IC75
concentrations for 72 h. In addition, cells were treated with 0.5
mM NAC concomitantly with D-salicin and incubated for 72 h.
Untreated and NAC-only (0.5 mM) wells were included as negative
controls, and a 200 µM H2O2 treatment for 30
min was used as a positive control.
Following treatment, the wells were washed once with
Hank's Balanced Salt Solution (HBSS) and incubated with 20–25 µM
DCFDA in HBSS for 30 min at 37°C in the dark. After dye loading,
the plates were washed again with HBSS, and fluorescence was
recorded using a Varioskan (Varioskan Lux; Thermo Fisher
Scientific, Inc.) microplate reader (Ex/Em=485/535 nm). Background
fluorescence from blank wells was subtracted, and ROS levels were
expressed as relative fluorescence units normalized to total
protein content per well, determined using the BCA assay. Each
condition was assayed in technical triplicate, and experiments were
independently repeated at least three times.
Lipid peroxidation (MDA) and GSH
quantification by ELISA
To further characterize D-salicin-associated
oxidative stress at the levels of lipid peroxidation and
antioxidant capacity, malondialdehyde (MDA) and GSH levels were
quantified by ELISA. Ishikawa and HDF cells were treated with
D-salicin at the previously determined IC25,
IC50 and IC75 concentrations for 72 h,
alongside control wells. Following treatment, cells were washed
with PBS, harvested, and lysed according to the manufacturers'
sample preparation recommendations. Cell lysates underwent
centrifugation at 10,000 × g for 10 min at 4°C, and the resulting
supernatants were used for analysis. MDA and GSH were measured
using commercial ELISA kits (Elabscience; MDA ELISA Kit, cat. no.
E-EL-0060; GSH ELISA Kit, cat. no. E-EL-0026) according to the
manufacturers' instructions. Optical density was measured at 450 nm
using a microplate reader (Varioskan Lux; Thermo Fisher Scientific,
Inc.), and concentrations were calculated from the corresponding
standard curves. MDA levels were reported as ng/ml and GSH levels
as µg/ml. To allow comparability across wells, MDA and GSH values
were normalized to total protein content determined by the BCA
assay (reported as ng/mg protein and µg/mg protein, respectively).
Each condition was assayed in triplicate, and the experiments were
independently repeated at least three times (n=3 biological
replicates).
Caspase-3 protein quantification by
ELISA
Caspase-3 protein levels, used as an indicator of
apoptosis, were quantified using the Human Caspase-3 ELISA Kit (BT
Lab; cat. no. E4804Hu) according to the manufacturer's
instructions. All reagents and samples were stored at 2–8°C and
equilibrated to room temperature before use. Cell lysates underwent
centrifugation at 10,000 × g for 10 min at 4°C, and the resulting
supernatants were used for analysis. Standards and samples were
added to 96-well plates, and the sandwich ELISA procedure,
including antibody binding, incubation, and washing steps, was
performed as recommended. Optical density was measured at 450 nm
using a microplate reader (Varioskan Lux, Thermo Fisher Scientific,
Inc.), and caspase-3 concentrations were calculated from the
standard curve. Each condition was analyzed in triplicate, and the
experiment was independently repeated at least three times.
Immunofluorescence staining of
phospho-ERK1/2
Ishikawa and HDF cells were seeded onto sterile
glass coverslips placed in 24-well plates at a density of 2×104
cells/well and treated with D-salicin at IC25,
IC50, and IC75 concentrations previously
determined for Ishikawa cells for 72 h. For NAC co-treatment
experiments, cells were treated concomitantly with 0.5 mM
N-acetylcysteine (NAC) and D-salicin for 72 h. Following treatment,
cells were washed twice with phosphate-buffered saline (PBS) and
fixed with 4% paraformaldehyde for 15 min at room temperature.
Fixed cells were permeabilized with 0.1% Triton X-100 in PBS for 10
min and blocked with 3% BSA; Sigma-Aldrich; cat. no. A9647) in PBS
for 1 h at room temperature.. Cells were then incubated overnight
at 4°C with a rabbit polyclonal phospho-ERK1/2 (Thr202/Tyr204;
Affinity Biosciences; cat. no. Ab-AF1015; 1:200) primary antibody.
After three washes with PBS, cells were incubated with Alexa Fluor
594-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch
Laboratories, Inc.; cat. no. 111-585-003; 1:200) for 1 h at room
temperature in the dark. Following the final washes, coverslips
were mounted at room temperature using a DAPI-containing mounting
medium (MilliporeSigma; cat. no. F6057), enabling simultaneous
nuclear counterstaining.
Fluorescence images were acquired using a
fluorescence microscope (Zeiss Calibri 7; Zeiss AG) under identical
imaging parameters for all experimental groups. For quantitative
analysis, fluorescence intensity measurements were performed using
ImageJ software (version 2.16.0; National Institutes of Health).
For each treatment concentration, at least three randomly selected
microscopic fields were analyzed, and a minimum of 50 cells per
field were evaluated to determine the mean fluorescence intensity
of phospho-ERK1/2. Corrected total cell fluorescence (CTCF) values
were calculated for each cell using the standard formula
(CTCF=Integrated Density-[Area × Mean Background Fluorescence]) and
analyzed as absolute fluorescence intensity measurements without
normalization to a fixed reference value.
Statistical analysis
All quantitative data are presented as the mean ±
standard deviation (SD) of at least three independent experiments
(n≥3). Statistical analyses were performed using GraphPad Prism
8.4.2 (Dotmatics). Differences between groups were evaluated using
one-way ANOVA) followed by Tukey's multiple comparisons test when
more than two groups were compared. All graphs and statistical
outputs were generated using GraphPad Prism. P<0.05 was
considered to indicate a statistically significant difference.
Results
Cell viability
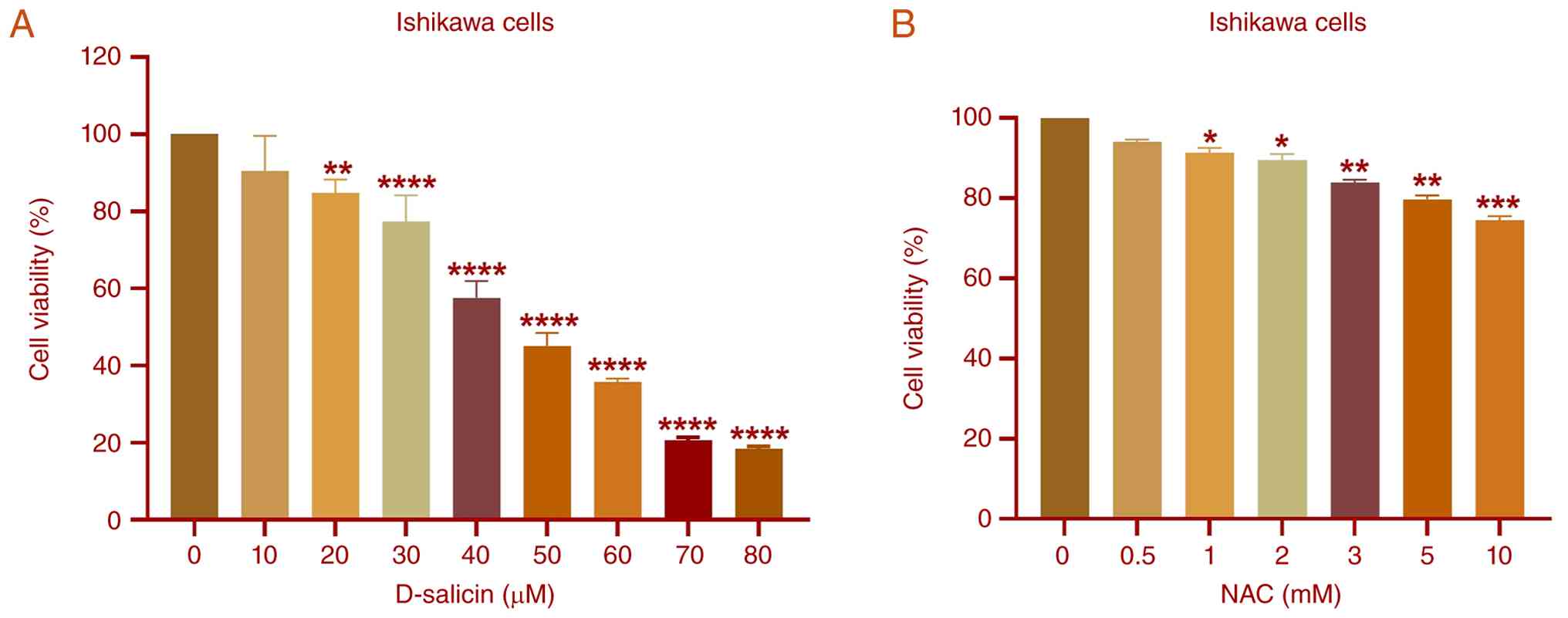
Treatment of Ishikawa cells with D-salicin for 72 h
induced a concentration-dependent reduction in cell viability
(Fig. 1A). While exposure to 10 µM
D-salicin did not result in a statistically significant decrease
compared with untreated cells, significant reductions in cell
viability were observed starting from 20 µM. These reductions
became progressively more marked at higher concentrations. The mean
viability decreased from 100% in the control cells to ~16.9% at 80
µM D-salicin. Nonlinear regression analysis yielded an
IC50 value of ~48.3 µM, with corresponding
IC25 and IC75 values of 26.7 and 69.9 µM,
respectively, indicating a marked cytotoxic effect of D-salicin on
Ishikawa cells.
To support selection of a sub-cytotoxic NAC
concentration for subsequent co-treatment experiments, a
preliminary NAC screening was performed in Ishikawa cells (Fig. 1B). At 0.5 mM NAC, viability did not
markedly differ from control, whereas higher concentrations
produced modest but significant reductions. Accordingly, 0.5 mM NAC
was selected as a sub-cytotoxic concentration for subsequent
co-treatment experiments. The slight (5–10%) reduction in cell
viability observed at this concentration was not statistically
significant and was therefore considered negligible; all subsequent
experimental results were normalized to their respective control
groups.
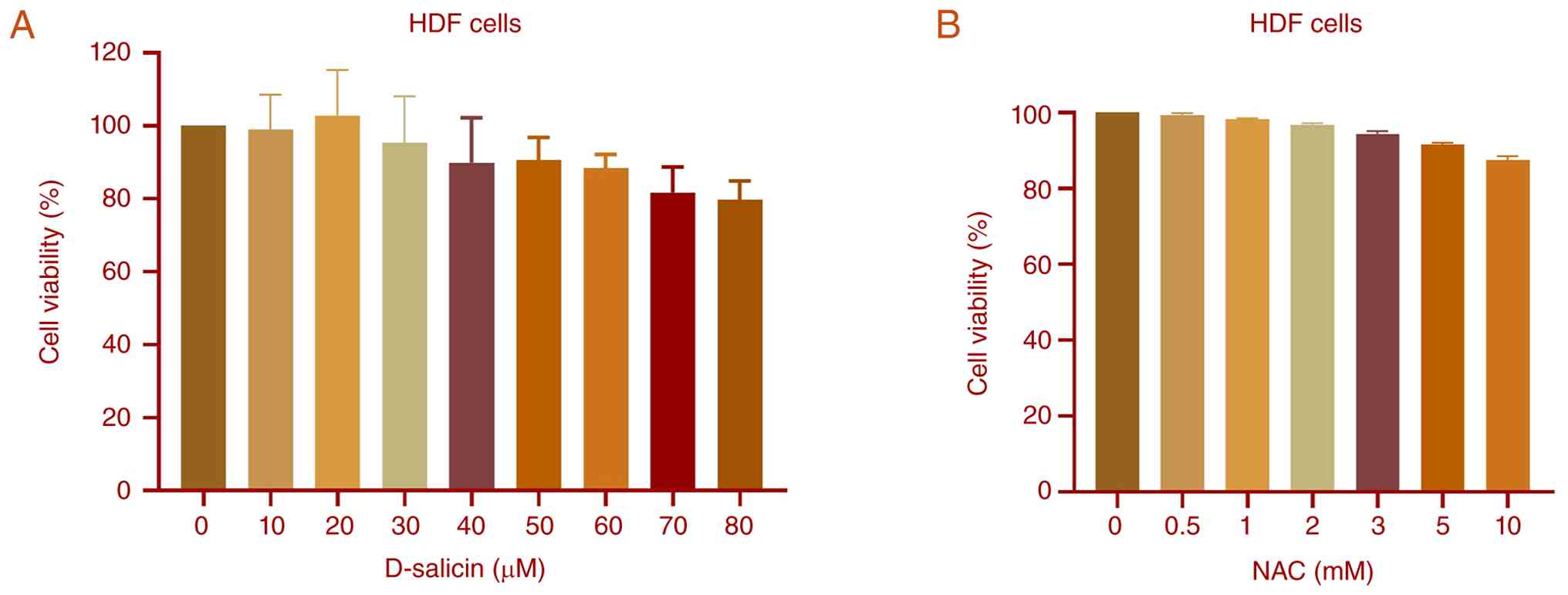
By contrast, HDF cells maintained relatively high
viability across the same concentration range (0–80 µM) following
72 h of D-salicin treatment (Fig.
2A). Consistent with this, Shapiro-Wilk normality testing
confirmed that all datasets followed a normal distribution. There
were no significant differences between control and treatment
groups, indicating that D-salicin did not significantly affect the
viability of HDF cells under the experimental conditions. In
addition, HDF cells exhibited minimal sensitivity to NAC across the
tested range (Fig. 2B), with no
statistically significant differences observed at any
concentration, suggesting that NAC does not exert cytotoxic effects
on HDF cells within the tested dose range. These findings suggested
that D-salicin selectively reduces viability in Ishikawa
endometrial cancer cells while exerting minimal cytotoxic effects
on normal HDF cells.
Intracellular ROS levels
D-salicin treatment resulted in a significant and
dose-dependent increase in intracellular ROS levels in Ishikawa
endometrial cancer cells (Fig. 3A).
When normalized to the untreated control, ROS levels increased to
1.26–1.33-fold at 26.7 µM, 1.61–1.75-fold at 48.8 µM, and
2.00–2.28-fold at 69.9 µM D-salicin. All tested concentrations
induced significant elevations compared with the control (Fig. 3A).
![Effects of D-salicin and NAC
co-treatment on intracellular ROS levels in Ishikawa and HDF cells.
(A) Ishikawa cells and (B) HDF cells were treated with D-salicin at
IC25 (26.7 µM), IC50 (48.8 µM), and
IC75 (69.9 µM) for 72 h in the absence (−NAC) or
presence (+NAC) of N-acetylcysteine (NAC, 0.5 mM). Control
represents untreated cells, and NAC represents cells treated with
NAC alone (0.5 mM). For D-salicin-treated groups, ‘-NAC’ and ‘+NAC’
indicate the absence or presence of NAC at each corresponding
D-salicin concentration. Intracellular ROS levels were measured
using the DCFDA assay and expressed as RFU, normalized to the
untreated control. In Ishikawa cells, D-salicin induced a
dose-dependent increase in ROS levels, which was markedly
attenuated by NAC co-treatment. In HDF cells, D-salicin induced
only modest changes in ROS levels, and NAC co-treatment did not
markedly alter this response. Data are presented as mean ± SD (n≥3
independent experiments). Statistical analysis was performed using
one-way ANOVA followed by Tukey's multiple comparisons test. Hash
symbols (#) indicate comparisons between matched
D-salicin groups in the absence and presence of NAC at the
corresponding concentration [e.g., 26.7 (−NAC) vs. 26.7 (+NAC)].
*P<0.05, **P<0.01, ****P<0.0001 vs.
untreated control; #P<0.05,
####P<0.0001. NAC, N-acetylcysteine; ROS,
reactive oxygen species; HDF, human dermal fibroblasts; RFU,
relative fluorescence units.](/article_images/ol/32/1/ol-32-01-15666-g02.jpg) | Figure 3.Effects of D-salicin and NAC
co-treatment on intracellular ROS levels in Ishikawa and HDF cells.
(A) Ishikawa cells and (B) HDF cells were treated with D-salicin at
IC25 (26.7 µM), IC50 (48.8 µM), and
IC75 (69.9 µM) for 72 h in the absence (−NAC) or
presence (+NAC) of N-acetylcysteine (NAC, 0.5 mM). Control
represents untreated cells, and NAC represents cells treated with
NAC alone (0.5 mM). For D-salicin-treated groups, ‘-NAC’ and ‘+NAC’
indicate the absence or presence of NAC at each corresponding
D-salicin concentration. Intracellular ROS levels were measured
using the DCFDA assay and expressed as RFU, normalized to the
untreated control. In Ishikawa cells, D-salicin induced a
dose-dependent increase in ROS levels, which was markedly
attenuated by NAC co-treatment. In HDF cells, D-salicin induced
only modest changes in ROS levels, and NAC co-treatment did not
markedly alter this response. Data are presented as mean ± SD (n≥3
independent experiments). Statistical analysis was performed using
one-way ANOVA followed by Tukey's multiple comparisons test. Hash
symbols (#) indicate comparisons between matched
D-salicin groups in the absence and presence of NAC at the
corresponding concentration [e.g., 26.7 (−NAC) vs. 26.7 (+NAC)].
*P<0.05, **P<0.01, ****P<0.0001 vs.
untreated control; #P<0.05,
####P<0.0001. NAC, N-acetylcysteine; ROS,
reactive oxygen species; HDF, human dermal fibroblasts; RFU,
relative fluorescence units. |
In the presence of NAC (+NAC), ROS levels at each
D-salicin concentration remained significantly higher than in the
control, although the magnitude of increase was consistently
reduced compared with D-salicin treatment alone.
Direct comparisons between matched D-salicin
concentrations in the absence and presence of NAC (26.7 vs. 26.7 +
NAC; 48.8 vs. 48.8 + NAC; 69.9 vs. 69.9 + NAC) demonstrated a
significant attenuation of ROS levels following NAC co-treatment.
Despite this reduction, ROS levels in the NAC co-treatment groups
remained moderately elevated relative to baseline at higher
concentrations, indicating partial but not complete reversal of
oxidative stress. NAC alone did not markedly alter ROS levels
compared with the untreated control.
By contrast, HDF normal fibroblasts exhibited
minimal changes in intracellular ROS levels following D-salicin
exposure (Fig. 3B). Only the
highest concentration (69.9 µM) induced a modest but statistically
significant increase compared with control, while no significant
differences were observed among lower concentrations. In the
presence of NAC, ROS levels were modestly elevated at 48.8 and 69.9
µM compared with control. However, direct comparisons between
D-salicin-treated groups in the absence and presence of NAC
revealed no significant differences at any concentration.
These results indicated that D-salicin
preferentially induces oxidative stress in Ishikawa cancer cells,
with limited effects on normal fibroblasts.
Lipid peroxidation and GSH levels
D-salicin-associated oxidative stress was further
evaluated by measuring lipid peroxidation (MDA) and GSH) levels in
cell lysate after 72 h exposure to IC25 (26.7 µM),
IC50 (48.8 µM), and IC75 (69.9 µM)
concentrations (Fig. 4A-D).
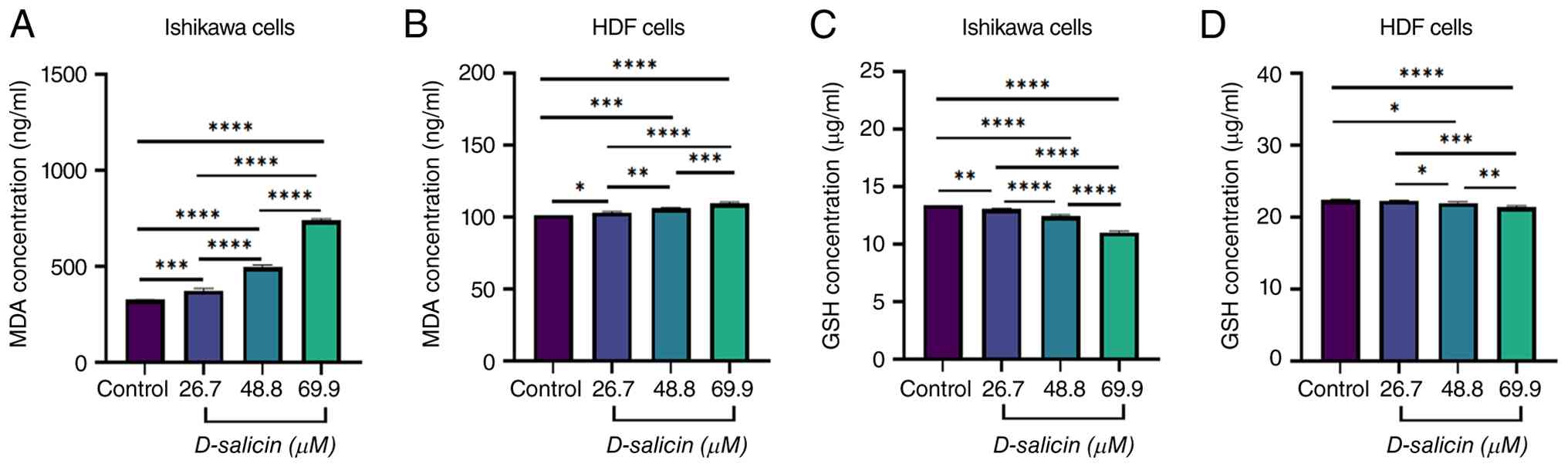 | Figure 4.Effects of D-salicin on lipid
peroxidation (MDA) and GSH levels in Ishikawa and HDF cells. (A)
MDA concentrations in Ishikawa cells following 72 h exposure to
D-salicin at IC25 (26.7 µM), IC50 (48.8 µM),
and IC75 (69.9 µM). (B) MDA concentrations (ng/ml) in
HDF cells under identical treatment conditions. (C) GSH
concentrations (µg/ml) in Ishikawa cells following 72 h D-salicin
exposure at IC25, IC50, and IC75.
(D) GSH concentrations (µg/ml) in HDF cells under identical
conditions. MDA and GSH were quantified in cell lysates by ELISA,
and concentrations were calculated from standard curves. Data are
presented as mean ± SD from three independent experiments (n=3).
Each condition was assayed in technical triplicate, and experiments
were independently repeated at least three times (n≥3 biological
replicates). Statistical analysis was performed using one-way ANOVA
followed by Tukey's multiple comparisons test. *P<0.05,
**P<0.01, ***P<0.001, ****P<0.0001.
MDA, malondialdehyde; GSH, glutathione; HDF, human dermal
fibroblasts. |
In Ishikawa cells, D-salicin induced a marked,
concentration-dependent increase in MDA concentrations (Fig. 4A). Compared with control, MDA levels
were significantly elevated at 26.7 µM and increased further at
48.8 and 69.9 µM. Pairwise comparisons confirmed a progressive rise
in lipid peroxidation between successive doses.
In HDF cells, MDA concentrations also increased
following D-salicin exposure, although the magnitude of change was
more modest than in Ishikawa cells (Fig. 4B). Relative to control, significant
increases were observed at 26.7 48.8 and 69.9 µM, indicating that
D-salicin can induce lipid peroxidation in normal fibroblasts,
albeit to a lesser extent.
Conversely, GSH levels in Ishikawa cells decreased
markedly and in a dose-dependent manner following D-salicin
treatment (Fig. 4C). Compared with
control, GSH concentrations were reduced at 26.7 µM and decreased
further at 48.8 and 69.9 µM, with pairwise comparisons indicating
greater depletion at higher concentrations.
In HDF cells, GSH levels showed a small but
statistically significant reduction following D-salicin exposure
(Fig. 4D). Compared with control,
significant decreases were detected at 48.8 and 69.9 µM, whereas no
significant change was observed at 26.7 µM. However, the absolute
magnitude of GSH depletion was limited compared with Ishikawa
cells, consistent with a comparatively attenuated redox disruption
in normal fibroblasts.
Collectively, these results indicated that D-salicin
promotes oxidative stress characterized by increased lipid
peroxidation (MDA) and depletion of the antioxidant pool (GSH). The
overall redox imbalance was more pronounced in Ishikawa cancer
cells, supporting preferential disruption of redox homeostasis in
the malignant cell model under identical exposure conditions.
Although D-salicin also induced modest but statistically
significant changes in MDA and GSH levels in HDF cells (Fig. 4B and D), these oxidative changes in
HDF cells were not accompanied by significant reductions in cell
viability or caspase-3 activation. This suggests that normal
fibroblasts retain sufficient redox buffering capacity to tolerate
D-salicin-induced oxidative perturbations without engaging
cytotoxic or apoptotic programs, supporting a degree of functional
selectivity even in the presence of partial oxidative stress
induction in non-malignant cells.
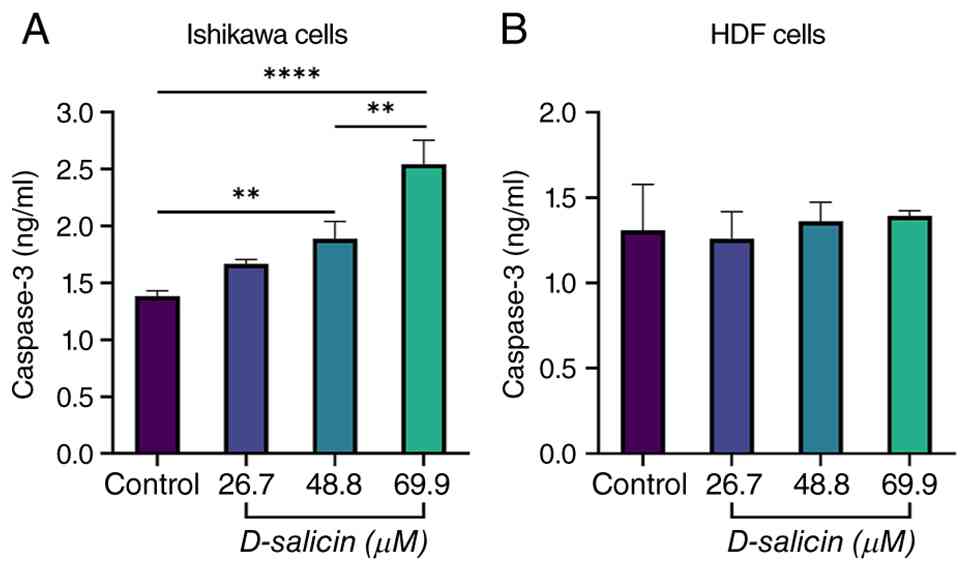
Caspase-3 response to D-salicin
exposure
In Ishikawa endometrial cancer cells, D-salicin
treatment induced a concentration-dependent increase in caspase-3
protein levels (Fig. 5A). Compared
with control cells, exposure to 26.7 µM D-salicin did not result in
a statistically significant change. However, treatment with 48.8 µM
led to a significant elevation in caspase-3, while the highest
concentration (69.9 µM) produced a pronounced and highly
significant increase. Pairwise comparisons among D-salicin-treated
groups further demonstrated that caspase-3 levels at 69.9 µM were
significantly higher than those observed at 26.7 and 48.8 µM. No
significant difference was detected between the 26.7 and 48.8 µM
treatment groups, indicating that apoptotic activation became more
prominent at higher D-salicin concentrations. By contrast, HDF
cells exhibited no statistically significant changes in caspase-3
protein levels following D-salicin treatment at any concentration
tested (Fig. 5B). These findings
suggested that D-salicin selectively activates caspase-3-associated
apoptotic signaling in Ishikawa cancer cells while sparing normal
fibroblasts.
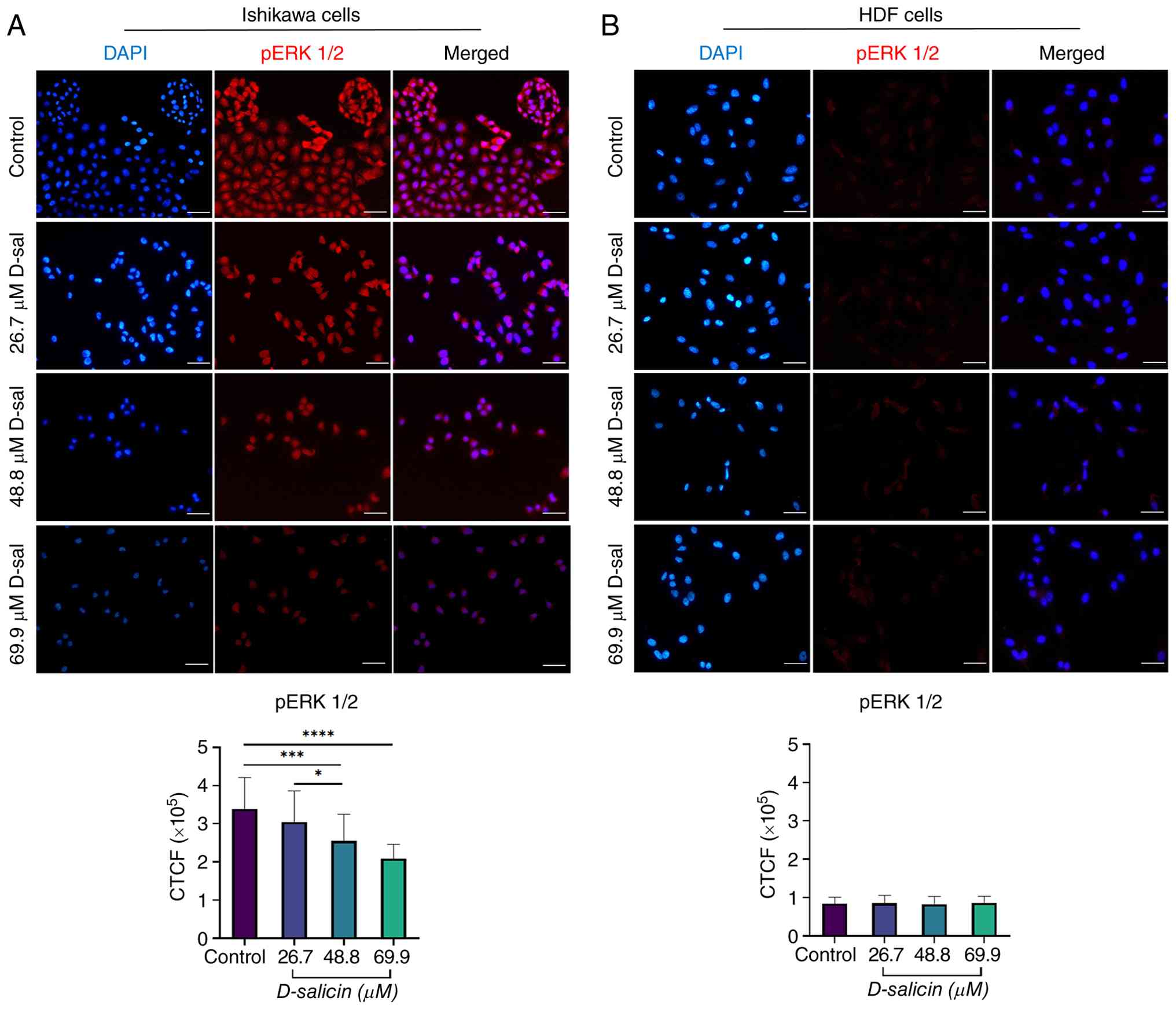
Effect of D-salicin on ERK1/2
phosphorylation in Ishikawa and HDF cells
Immunofluorescence analysis revealed a marked,
concentration-dependent suppression in ERK1/2 phosphorylation in
Ishikawa cells following D-salicin treatment (Fig. 6A). In control Ishikawa cells,
phosphorylated (p)-ERK1/2 immunoreactivity was prominently detected
throughout the cytoplasm and nucleus. Treatment with D-salicin
resulted in a progressive reduction in p-ERK1/2 fluorescence
intensity, particularly at higher concentrations.
p-ERK1/2 fluorescence intensity was not markedly
altered at 26.7 µM compared with control (Fig. 6A). However, treatment with 48.8 and
69.9 µM D-salicin led to a significant decrease in p-ERK1/2 levels
relative to control (Fig. 6A). In
addition, p-ERK1/2 intensity at 69.9 µM was significantly lower
than at 26.7 µM and a modest but significant difference was also
observed between 26.7 and 48.8 µM (Fig.
6A). No significant difference was detected between 48.8 µM and
69.9 µM (Fig. 6A), suggesting a
plateau effect at higher concentrations.
By contrast, D-salicin treatment did not induce
significant changes in ERK1/2 phosphorylation in HDF cells. Basal
p-ERK1/2 expression was detectable but relatively low in HDF cells;
however, it remained stable and did not show significant modulation
upon D-salicin treatment. Immunofluorescence images showed
consistently low p-ERK1/2 signal intensity across all treatment
groups, and quantitative analysis confirmed the absence of
statistically significant differences between control and
D-salicin-treated HDF cells at any concentration tested (Fig. 6B).
To further examine whether D-salicin-induced
suppression of ERK1/2 phosphorylation is linked to oxidative
stress, NAC co-treatment experiments were performed using D-salicin
at its IC50 concentration (48.8 µM) in Ishikawa cells
(Fig. 7A and B). In these
experiments, cells were treated with NAC alone (0.5 mM), D-salicin
alone (48.8 µM), or D-salicin + NAC (0.5 mM), and p-ERK1/2
immunofluorescence intensity was quantified by CTCF. NAC alone did
not significantly alter p-ERK1/2 levels compared with control. By
contrast, D-salicin significantly reduced p-ERK1/2 intensity
relative to control. Importantly, NAC co-treatment partially
restored p-ERK1/2 phosphorylation compared with D-salicin alone,
indicating that the ERK1/2 phosphorylation changes induced by
D-salicin are at least partly NAC-sensitive under these
conditions.
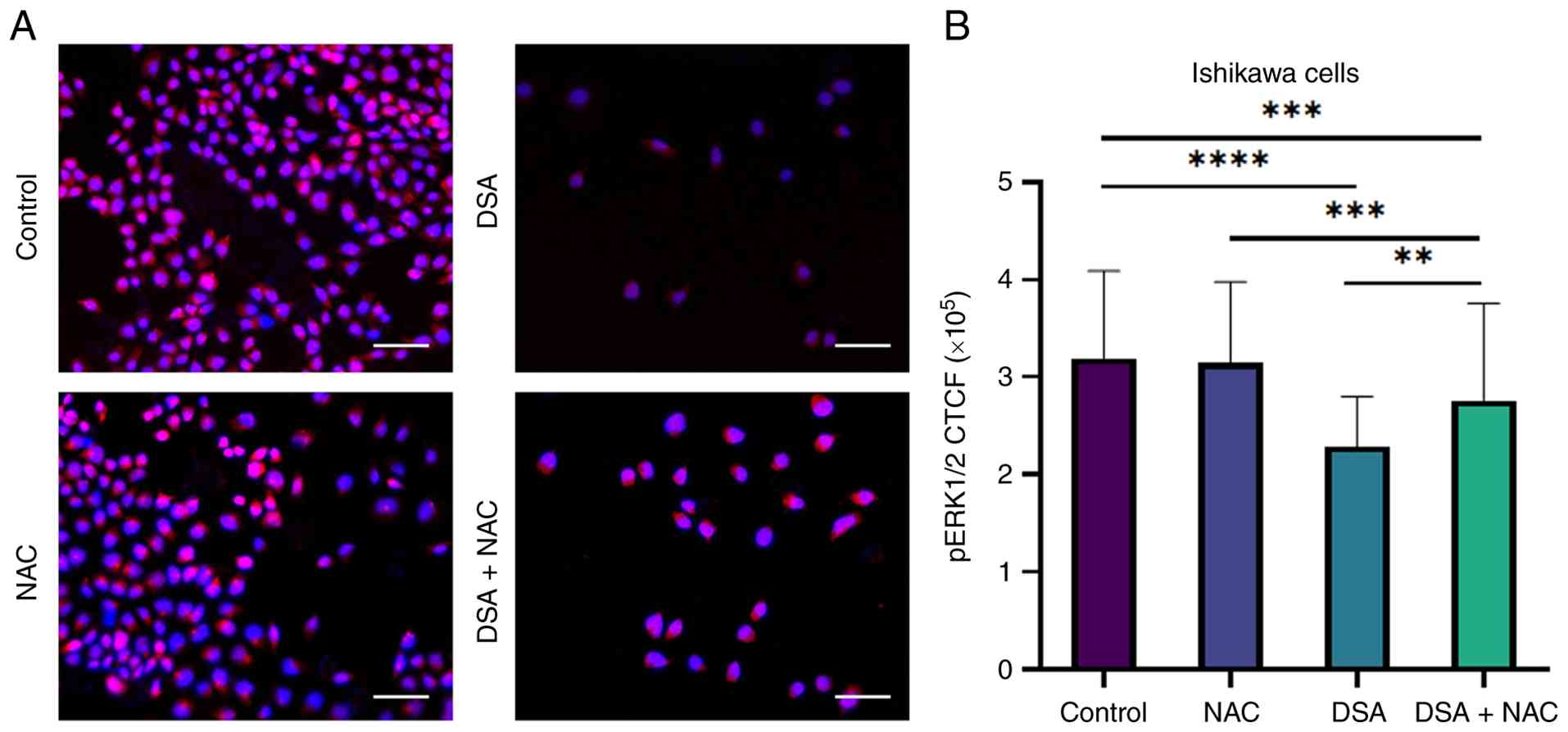 | Figure 7.Effect of NAC co-treatment on
D-salicin-induced suppression of ERK1/2 phosphorylation in Ishikawa
cells. (A) Representative immunofluorescence images showing
phospho-ERK1/2 (p-ERK1/2) expression in Ishikawa cells following 72
h treatment with control, NAC (0.5 mM), D-salicin
(IC50=48.8 µM), and D-salicin + NAC. p-ERK1/2
immunoreactivity is shown in red, and nuclei are counterstained
with DAPI (blue). D-salicin reduced p-ERK1/2 fluorescence intensity
compared with control, whereas NAC co-treatment partially restored
p-ERK1/2 signal. (B) Quantitative analysis of p-ERK1/2 fluorescence
intensity expressed as CTCF. Data are presented as mean ± SD from
three independent experiments. Statistical significance was
determined using one-way ANOVA followed by Tukey's multiple
comparisons test **P<0.01, ***P<0.001,
****P<0.0001). Scale bars, 100 µm. NAC, N-acetylcysteine;
p-, phosphorylated; CTCF, corrected total cell fluorescence; DSA,
D-salicin. |
Collectively, these findings indicated that
D-salicin selectively suppresses ERK1/2 phosphorylation in Ishikawa
endometrial cancer cells in a concentration-dependent manner, while
sparing ERK signaling in non-tumorigenic HDF cells. NAC
co-treatment attenuated D-salicin-associated p-ERK1/2 suppression
in Ishikawa cells, supporting a redox-linked (ROS-dependent)
contribution to ERK pathway modulation under these conditions.
Discussion
The observed concentration-dependent reduction in
Ishikawa cell viability, contrasted with the preserved viability of
HDF cells, suggests a degree of tumor selectivity. Such selective
cytotoxicity is a critical feature for candidate anticancer agents,
because clinical efficacy is frequently constrained by
dose-limiting toxicities in normal tissues and the need to widen
the therapeutic window (17,18).
This is particularly relevant in endometrial cancer, where advanced
or recurrent disease remains therapeutically challenging and
systemic treatment selection continues to evolve with histologic
and molecular stratification (7,19).
Although therapeutic options have expanded, improving response
depth and durability, especially in microsatellite-stable disease,
remains a major unmet need, supporting continued development of
agents with novel mechanisms and favorable safety profiles
(19,20). These challenges highlight the
rationale for exploring redox-modulating compounds that exploit
cancer-specific oxidative vulnerabilities to preferentially impact
malignant cells (18,21).
ROS analyses indicated that D-salicin induces a
robust and dose-dependent increase in intracellular oxidative
stress in Ishikawa cells, whereas normal fibroblasts exhibited
minimal ROS elevation. This finding aligns with the hypothesis that
numerous cancer cells operate close to a redox ‘tolerance limit,’
making them more susceptible to additional oxidative insults than
non-malignant cells (22).
Salicylate/salicin-related natural products and willow-derived
phenolics can influence intracellular redox balance and cancer-cell
stress responses, supporting redox modulation as a plausible
contribution to their bioactivity (23). Excessive ROS can overwhelm
antioxidant defenses, promote mitochondrial dysfunction and
precipitate regulated cell death programs (including apoptosis)
when redox homeostasis collapses (24). In this context, ROS induction may
represent an early event contributing to the anticancer activity of
D-salicin by pushing malignant cells beyond their oxidative stress
buffering capacity (22).
Consistent with this interpretation, NAC co-treatment markedly
blunted D-salicin-driven ROS accumulation across
IC25-IC75 conditions, supporting an
NAC-sensitive oxidative component in Ishikawa cells. The
differential ROS response between Ishikawa and HDF cells may
reflect inherent differences in baseline antioxidant capacity and
redox buffering between malignant and non-malignant cells. Cancer
cells, including endometrial carcinoma lines, frequently exhibit
elevated baseline ROS levels and reduced antioxidant reserves
compared with normal counterparts, rendering them more susceptible
to further oxidative insult. This concept of a redox ‘threshold
effect’, whereby cancer cells operate closer to their oxidative
tolerance limit, may explain the preferential ROS accumulation and
downstream cytotoxic response observed in Ishikawa cells under
identical D-salicin exposure conditions (22). Consistent with this interpretation,
the present study demonstrated a more pronounced ROS elevation in
Ishikawa cells compared with HDF cells under the same treatment
conditions, supporting a tumor cell-specific redox
vulnerability.
Consistent with ROS-mediated cytotoxicity, a
significant increase in caspase-3 levels in Ishikawa cells at
higher D-salicin concentrations was observed, indicating engagement
of apoptotic execution pathways (25). Activation of caspase-3 represents a
terminal step in the apoptotic cascade, leading to the proteolytic
cleavage of essential cellular substrates and execution of cell
death (26).
Salicin/salicylate-related compounds can promote apoptotic cell
death with caspase pathway involvement, often alongside oxidative
stress signaling and suppression of pro-survival pathways (27). The absence of caspase-3 activation
in HDF cells further underscores the cancer-selective pro-apoptotic
effect of D-salicin and supports the interpretation that oxidative
stress-induced apoptosis contributes to its anticancer activity
(28). However, while caspase-3
activation supports engagement of apoptotic signaling, it does not
fully distinguish apoptosis from other antiproliferative
mechanisms. Although caspase-3 was not evaluated under NAC
co-treatment in the present dataset, the NAC-sensitive ROS and
p-ERK1/2 responses are compatible with redox-dependent upstream
events that can converge on apoptotic execution pathways (29). Importantly, although modest changes
in oxidative stress markers were also observed in HDF cells, these
effects were substantially less pronounced and were not associated
with downstream cytotoxic or apoptotic responses, supporting a
relative degree of tumor selectivity. Accordingly, the selectivity
of D-salicin should be considered relative rather than absolute and
interpreted within the context of differential cellular sensitivity
to oxidative stress.
Immunofluorescence analysis revealed that D-salicin
suppressed ERK1/2 phosphorylation in Ishikawa cells in a
concentration-dependent manner, while ERK signaling remained
largely unchanged in normal fibroblasts. The RAS-RAF-MEK-ERK
(MAPK/ERK) cascade is a core signaling module that regulates
cellular proliferation, growth, and survival, and dysregulated
ERK1/2 activity is widely implicated in cancer progression and
therapeutic resistance (6,30). Accordingly, the observed decrease in
p-ERK1/2 in Ishikawa cells represents a disruption of pro-survival
signaling that plausibly contributes to the cytotoxic response
under D-salicin exposure. Notably, NAC co-treatment partly restored
p-ERK1/2 levels at the IC50 condition, providing
functional support that ERK suppression is at least partly
redox-linked under these experimental conditions (13,31).
It should be noted that the present study provided associative
rather than direct mechanistic evidence for ROS-ERK coupling; NAC
co-treatment offers functional support but does not exclude
alternative upstream mediators. Complementary approaches, including
pharmacological ERK activators or inhibitors and genetic modulation
strategies, would be required to establish direct causality and
will be addressed in future studies.
Collectively, the present data supported a model in
which D-salicin elevated intracellular ROS levels beyond a
tolerable threshold in endometrial cancer cells, leading to
attenuation of ERK1/2-mediated survival signaling and activation of
caspase-3-associated apoptotic signaling. The limited effects
observed in normal fibroblasts suggest the existence of a
therapeutic window, a desirable characteristic for further
translational exploration. The observation that NAC attenuates
D-salicin-induced ROS and partially reverses p-ERK1/2 suppression
further supports involvement of a redox-sensitive ROS-ERK axis in
the anticancer response.
From a translational perspective, the
IC50 of D-salicin determined in the present study (~48
µM) warrants consideration in the context of physiological
achievability. While in vitro IC50 values do not
directly predict effective in vivo concentrations due to
differences in bioavailability, protein binding and tissue
distribution, the concentration range employed here was broadly
comparable with those reported for other phenolic glycosides with
demonstrated anticancer activity in cell-based models. However, it
remains uncertain whether the IC50 concentration can be
safely achieved in vivo. Nevertheless, in vivo
pharmacokinetic and pharmacodynamic studies are required to
evaluate whether therapeutically relevant plasma concentrations of
D-salicin are achievable, and to assess its safety profile in more
complex biological systems.
Several limitations of the present study should be
acknowledged. First, the findings are based on in vitro
analyses using a single endometrial cancer cell line; nevertheless,
Ishikawa cells represent a well-established and widely used model
of endometrial adenocarcinoma, supporting the biological relevance
of the present findings within this system; therefore, validation
in additional endometrial cancer models with distinct molecular
backgrounds, such as HEC-1A or KLE cell lines representing
different histologic and molecular subtypes, would strengthen
generalizability. Second, although the present data indicated a
strong association between ROS accumulation and ERK1/2 inhibition,
complementary pharmacological and genetic approaches will be
valuable to establish direct causal relationships. While NAC
co-treatment provides functional support for redox involvement,
additional strategies, such as alternative antioxidants/thiol
modulators, ROS-generating controls, time-course experiments and
direct manipulation of ERK signaling, would strengthen causal
inference. Although NAC co-treatment was used to functionally
assess redox involvement, direct measurement of GSH replenishment
following NAC exposure was not performed and represents a
limitation of the current study; future studies incorporating such
analyses would further strengthen the interpretation of
redox-related effects. Assessing apoptotic readouts (such as
caspase-3 activity/cleavage or downstream substrates) under NAC
co-treatment would further clarify whether redox modulation
directly rescues apoptotic execution in this model. Moreover,
caspase-3 ELISA reflects total protein levels rather than the
cleaved/activated form; therefore, complementary approaches such as
Annexin V-propidium iodide flow cytometry or PARP cleavage analysis
would provide more definitive confirmation of apoptotic cell death.
Such complementary approaches would also more precisely delineate
the contribution of apoptosis vs. cell cycle arrest to the observed
reduction in cell viability, particularly under NAC co-treatment
conditions where ROS neutralization may engage distinct
antiproliferative mechanisms.
Additionally, ERK1/2 phosphorylation was assessed by
immunofluorescence, which provides spatial resolution but is
semi-quantitative in nature; western blot analysis would offer
complementary biochemical validation of the p-ERK1/2 findings
reported here.
Finally, in vivo studies are required to
evaluate the pharmacological feasibility, bioavailability, and
safety profile of D-salicin, as well as to determine whether the
observed redox-ERK modulation translates into antitumor efficacy in
more complex biological systems.
In conclusion, this study provided evidence that
D-salicin exerts selective anticancer effects in endometrial cancer
cells through induction of oxidative stress, suppression of ERK1/2
signaling and activation of caspase-3-associated apoptotic
signaling. The NAC-sensitive attenuation of ROS accumulation and
partial restoration of p-ERK1/2 further implicate a redox-linked
contribution to ERK pathway modulation. These findings position
D-salicin as a promising redox-modulating agent targeting the
ROS-ERK axis in endometrial cancer.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study are included
in the figures and/or tables of this article.
Authors' contributions
NSA and ACG conceived and designed the study. NSA
supervised and coordinated the research. NSA and ACG conducted the
experiments, performed the data analysis and interpreted the
findings. NSA and ACG drafted the manuscript and critically revised
it for important intellectual content. NSA and ACG confirm the
authenticity of all the raw data. Both authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Authors' information
Neziha Senem Arı ORCID ID 0000-0003-2926-6892. Ayşe
Çakır Gündoğdu ORCID ID 0000-0002-2466-9417.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI
|
|
2
|
Bray F, Laversanne M, Sung H, Ferlay J,
Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 74:229–263.
2024.PubMed/NCBI
|
|
3
|
Concin N, Matias-Guiu X, Vergote I, Cibula
D, Mirza MR, Marnitz S, Ledermann J, Bosse T, Chargari C, Fagotti
A, et al: ESGO/ESTRO/ESP guidelines for the management of patients
with endometrial carcinoma. Radiother Oncol. 154:327–353. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Panieri E and Santoro MM: ROS homeostasis
and metabolism: A dangerous liaison in cancer cells. Cell Death
Dis. 7:e22532016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
An X, Yu W, Liu J, Tang D, Yang L and Chen
X: Oxidative cell death in cancer: Mechanisms and therapeutic
opportunities. Cell Death Dis. 15:5562024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Martin-Vega A and Cobb MH: Navigating the
ERK1/2 MAPK cascade. Biomolecules. 13:15552023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Crosbie EJ, Kitson SJ, McAlpine JN,
Mukhopadhyay A, Powell ME and Singh N: Endometrial cancer. Lancet.
399:1412–1428. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shi A, Liu L, Li S and Qi B: Natural
products targeting the MAPK-signaling pathway in cancer: Overview.
J Cancer Res Clin Oncol. 150:62024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Parvin N, Aslam M, Joo SW and Mandal TK:
Nano-phytomedicine: Harnessing plant-derived phytochemicals in
nanocarriers for targeted human health applications. Molecules.
30:31772025. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kong X, Zhang R, Shen Y, Bai Y, Dong K, Li
D and Wang Y: Preparation and inhibitory effect of salicin dimethyl
ether in laryngeal cancer cells through the apoptosis activation.
Acta Biochim Pol. 69:753–759. 2022.PubMed/NCBI
|
|
11
|
Kong CS, Kim KH, Choi JS, Kim JE, Park C
and Jeong JW: Salicin, an extract from white willow bark, inhibits
angiogenesis by blocking the ROS-ERK pathways. Phytother Res.
28:1246–1251. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Samuni Y, Goldstein S, Dean OM and Berk M:
The chemistry and biological activities of N-acetylcysteine.
Biochim Biophys Acta. 1830:4117–4129. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ezerina D, Takano Y, Hanaoka K, Urano Y
and Dick TP: N-Acetyl cysteine functions as a Fast-acting
antioxidant by triggering intracellular H2S and sulfane sulfur
production. Cell Chem Biol. 25:447–459.e4. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tenorio MCDS, Graciliano NG, Moura FA,
Oliveira ACM and Goulart MOF: N-acetylcysteine (NAC): Impacts on
human health. Antioxidants (Basel). 10:9672021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schieber M and Chandel NS: ROS function in
redox signaling and oxidative stress. Curr Biol. 24:R453–R462.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Son Y, Cheong YK, Kim NH, Chung HT, Kang
DG and Pae HO: Mitogen-activated protein kinases and reactive
oxygen species: How can ROS activate MAPK pathways? J Signal
Transduct. 2011:7926392011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Blagosklonny MV: Selective protection of
normal cells from chemotherapy, while killing drug-resistant cancer
cells. Oncotarget. 14:193–206. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jiang H, Zuo J, Li B, Chen R, Luo K, Xiang
X, Lu S, Huang C, Liu L, Tang J and Gao F: Drug-induced oxidative
stress in cancer treatments: Angel or devil? Redox Biol.
63:1027542023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gordhandas S, Zammarrelli WA, Rios-Doria
EV, Green AK and Makker V: Current evidence-based systemic therapy
for advanced and recurrent endometrial cancer. J Natl Compr Canc
Netw. 21:217–226. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mahdi H, Chelariu-Raicu A and Slomovitz
BM: Immunotherapy in endometrial cancer. Int J Gynecol Cancer.
33:351–357. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hayes JD, Dinkova-Kostova AT and Tew KD:
Oxidative stress in cancer. Cancer Cell. 38:167–197. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brandl N, Seitz R, Sendtner N, Muller M
and Gulow K: Living on the edge: ROS homeostasis in cancer cells
and its potential as a therapeutic target. Antioxidants (Basel).
14:10022025. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Aboul-Soud MAM, Ashour AE, Challis JK,
Ahmed AF, Kumar A, Nassrallah A, Alahmari TA, Saquib Q, Siddiqui
MA, Al-Sheikh Y, et al: Biochemical and molecular investigation of
in vitro antioxidant and anticancer activity spectrum of crude
extracts of willow leaves Salix safsaf. Plants (Basel).
9:12952020.PubMed/NCBI
|
|
24
|
Nakamura H and Takada K: Reactive oxygen
species in cancer: Current findings and future directions. Cancer
Sci. 112:3945–3952. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dho SH, Cho M, Woo W, Jeong S and Kim LK:
Caspases as master regulators of programmed cell death: Apoptosis,
pyroptosis and beyond. Exp Mol Med. 57:1121–1132. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Galluzzi L, Vitale I, Aaronson SA, Abrams
JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews
DW, et al: Molecular mechanisms of cell death: Recommendations of
the nomenclature committee on cell death 2018. Cell Death Differ.
25:486–541. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ausina P, Branco JR, Demaria TM, Esteves
AM, Leandro JGB, Ochioni AC, Mendonca APM, Palhano FL, Oliveira MF,
Abou-Kheir W, et al: Acetylsalicylic acid and salicylic acid
present anticancer properties against melanoma by promoting nitric
oxide-dependent endoplasmic reticulum stress and apoptosis. Sci
Rep. 10:196172020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Perillo B, Di Donato M, Pezone A, Di Zazzo
E, Giovannelli P, Galasso G, Castoria G and Migliaccio A: ROS in
cancer therapy: The bright side of the moon. Exp Mol Med.
52:192–203. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu J, Liu Q, Han J, Feng J, Guo T, Li Z,
Min F, Jin R and Peng X: N-acetylcysteine inhibits patulin-induced
apoptosis by affecting ROS-mediated oxidative damage pathway.
Toxins (Basel). 13:5952021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bahar ME, Kim HJ and Kim DR: Targeting the
RAS/RAF/MAPK pathway for cancer therapy: From mechanism to clinical
studies. Signal Transduct Target Ther. 8:4552023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Aldini G, Altomare A, Baron G, Vistoli G,
Carini M, Borsani L and Sergio F: N-acetylcysteine as an
antioxidant and disulphide breaking agent: The reasons why. Free
Radic Res. 52:751–762. 2018. View Article : Google Scholar : PubMed/NCBI
|