Introduction
Lung cancer is one of the most deadly cancers in the
world (1,2). It was estimated that more than 326,000
deaths of lung cancer in China in 2005 (3). The death rate of lung cancer in China
soared to 306.1 per 1,000,000 persons in 2008, in a sharp contrast
to 5.47 and 17.27 per 1,000,000 persons in the mid 1970s and in the
early 1990s, respectively (4).
Treatment of lung cancer remains a challenge.
Interferons (IFNs) have been used to treat various
types of cancer in clinical practice (5). Interferons, including type I, type II
and type III IFNs, are a group of secreted cytokines that possess
antiviral and antitumor capacities (5–7). The
receptors of IFNs are composed of a ligand-binding subunit and an
accessory subunit. The ligand-binding subunit is usually
type-specific, while the accessory subunit is commonly shared by
receptor complexes of various cytokines. Upon the ligation of IFNs
to their respective receptors, the receptor-associated Janus
activated kinases (JAKs) are phosphorylated and activated, which in
turn phosphorylate and activate various signal transducer and
activator of transcription (STAT) family members. STATs undergo
homo- or hetero-dimerization, translocate to nuclei and bind to the
promoter region of IFN-stimulated genes leading to downstream gene
transcription (8). Crosstalk
between the JAK-STAT pathway and other signaling pathways, such as
phosphotidylinositol 3-kinase and mitogen-activated protein kinase
pathways, modulates cellular responses to IFNs and IL-10-related
cytokines (9,10).
Type I IFNs, including IFNα and IFNβ, have intense
antiviral and antitumor activities, as well as severe and common
side effects, such as bone marrow suppression, psoriasis, thyroid
disorders, diabetes, retinal changes and psychiatric disorders,
which significantly limit their clinical applications (11,12).
Type II IFN, including only IFNγ, generally has weak antiviral and
antitumor activities (12). Type
III IFNs, including IFNλ1, IFNλ2 and IFNλ3, possess comparable
antiviral and antitumor capacities to type I IFNs, however, with
much less side effects, which makes them new hotspots in the
development of antiviral and antitumor agents (13).
Evasion of apoptosis is an important step in the
development of cancer (14).
Therapeutic activation of apoptosis in cancer cells is a potential
anticancer strategy (14). More
than 300 genes regulated by IFNs are implicated in apoptosis
(15–18). Previous studies have demonstrated
the antitumor potential of type III IFNs in mouse melanoma
(19) and fibrosarcoma (20,21),
human colorectal adenocarcinoma (22), glioblastoma (23), pancreatic neuroendocrine tumor
(24) and colorectal carcinoma
(25). There is also a report
indicating that IFNλ induces apoptosis in non-small cell lung
cancer (NSCLC) cells (26). But,
its molecular mechanisms and effects when combines with type II IFN
still remain elusive.
In this study, we selected IFNλ1 as a model to study
the antitumor effects of type III IFNs on human lung cancer. By
using an established chimeric receptor (22), we observed the apoptotic effects of
IFNλ1 on human lung cancer with a concomitant activation of STAT1.
In addition, we observed that IFNγ renders human lung cancer cells
sensitivity to IFNλ-induced apoptosis.
Materials and methods
Plasmid, cells and transfection
The plasmid FL-10R1/λR1 (10R1/λR1) was previously
described (22). Human lung cancer
cells A549 (from PriCell Research Institute, Wuhan, P.R. China)
were maintained in RPMI-1640 medium (Sigma-Aldrich) with 10%
heat-inactivated fetal bovine serum (FBS; Sigma-Aldrich). A549
cells were transfected with expression plasmid 10R1/λR1 or its
cognate vector using TransIT transfection kit (Mirus) following the
manufacturer’s instruction. G418-resistant transfectants were
selected in medium containing antibiotic G418 (400 μg/ml).
Cell cycle analysis and TUNEL assay
To determine distribution of cells through the cell
cycle, 105–106 cells (attached and floating)
were collected, rinsed with phosphate-buffered saline (PBS)
containing 5% FBS, permeabilized by incubation with 0.1% Triton
X-100 at 22°C for 5 min, washed with PBS containing 5% FBS,
precipitated by centrifugation at 2000 g, 4°C and re-suspended in
PBS containing 5% FBS, 8 mg/l PI and 40 mg/l RNase A at 22°C for 10
min in the dark. Cell distribution through the cell cycle was
analyzed by flow cytometry.
TUNEL assay to determine DNA fragmentation in
apoptotic cells was performed according to the manufacturer’s
suggested protocols (Promega). Briefly, 3–5×106 cells
were trypsinized, washed twice with cold PBS, fixed in 4%
paraformaldehyde at 4°C for 20 min, washed again with PBS and
permeabilized with 0.5 ml 0.5% saponin at 22°C for 5 min. The cells
were washed with PBS, incubated with 80 μl equilibration buffer at
22°C for 5 min, washed with PBS, re-suspended in 50 μl Nucleotide
mix and incubated in the dark at 37°C for 1 h. Cells were washed
again with PBS then analyzed by fluorescence microscopy.
To induce apoptosis, A549 cells were treated with a
combination of IFNγ (10 ng/ml) and TNFα (1 ng/ml). A549 cells
expressing 10R1/λR1 (A549/10R1/λR1) were treated with IL-10 (10
ng/ml) at 37°C for 48 h. The concentrations and durations of
cytokine treatments were indicated in other apoptosis-related
assays. Where indicated, caspase inhibitor Z-VAD-FMK (Calbiochem)
was added to the medium.
Flow cytometry and antibodies
Flow cytometry was performed as previously described
(27). FITC-conjugated mouse
anti-human activated STAT1 antibody was purchased from Cell
Signaling, USA. FITC-conjugated mouse anti-human major
histocompatibility complex class II (MHC II) antibody,
FITC-conjugated mouse anti-human caspase-3 antibody,
FITC-conjugated mouse anti-human caspase-8 antibody, APC-conjugated
mouse anti-human caspase-9 antibody and FITC-conjugated Annexin V
antibody were purchased from BD Biosciences. FITC-conjugated rabbit
anti-FLAG antibody was purchased from Sigma-Aldrich.
Phycoerythrin-conjugated mouse anti-human IFNλR1 antibody was
purchased from R&D Systems.
Proliferation and cell viability
assays
Proliferation and cell viability assays were
performed as previously described (22). Briefly, to determine cell
proliferation, equal amount of cells (105/well) were
plated in 6-well plates and treated with various concentrations of
cytokines, as indicated in the text. Floating cells were collected
and combined with adherent cells released from the wells by
trypsinization before cell counting.
To determine cell viability, an equal amount of
cells (3×104/well) were plated in all wells of 96-well
microtitre plate and treated with various concentrations of
cytokines as described in the text. Dead cells lost their
attachment. At indicated time points live adherent cells were
visualized by staining with crystal violet at 22°C for 5 min.
Statistical analysis
Statistical analyses were performed using Prism
software (GraphPad Prism). Untreated and treated groups were
compared using the Student’s t test when the data were normally
distributed. When the data were not normally distributed, the two
groups were compared using the non-parametric Mann-Whitney U test.
All tests were two-tailed. The P-values <0.05 were considered
statistically significant.
Results
IFNλ signaling does not inhibit the
proliferation of human lung cancer cells
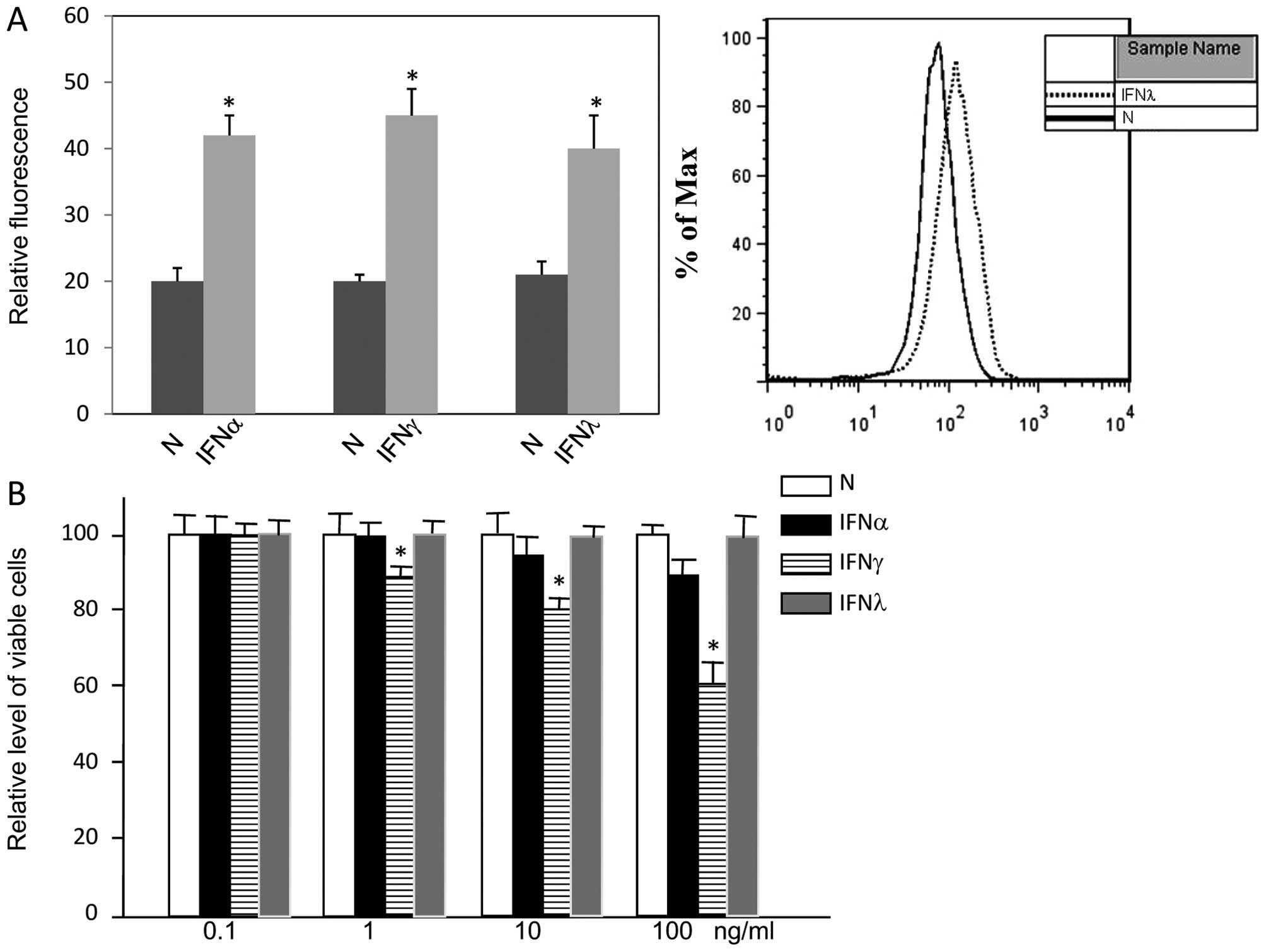
Treatment of A549 cells with either type I, type II
or type III IFNs increased the expression levels of MHC II to
equivalent levels, indicating that A549 cells responded to all
three types of IFNs (Fig. 1A). In
contrast to type II IFNs, type I and type III IFN did not inhibit
the proliferation of A549 cells, even when the concentration was up
to 100 ng/ml (Fig. 1B). In summary,
IFNλ1 did not inhibit A549 cell proliferation.
Signal induction through chimeric
receptor 10R1/λR1
The expression level of IFN receptors can determine
the responsiveness of cells to IFNs. It has been reported that the
expression level of IFNλR1, the signal competent of IFNλ receptor
complex, correlates to the ability of IFNλ to block cell
proliferation (23,28). We wondered if overexpression of
IFNλR1 could render A549 cell responsiveness to IFNλ-induced
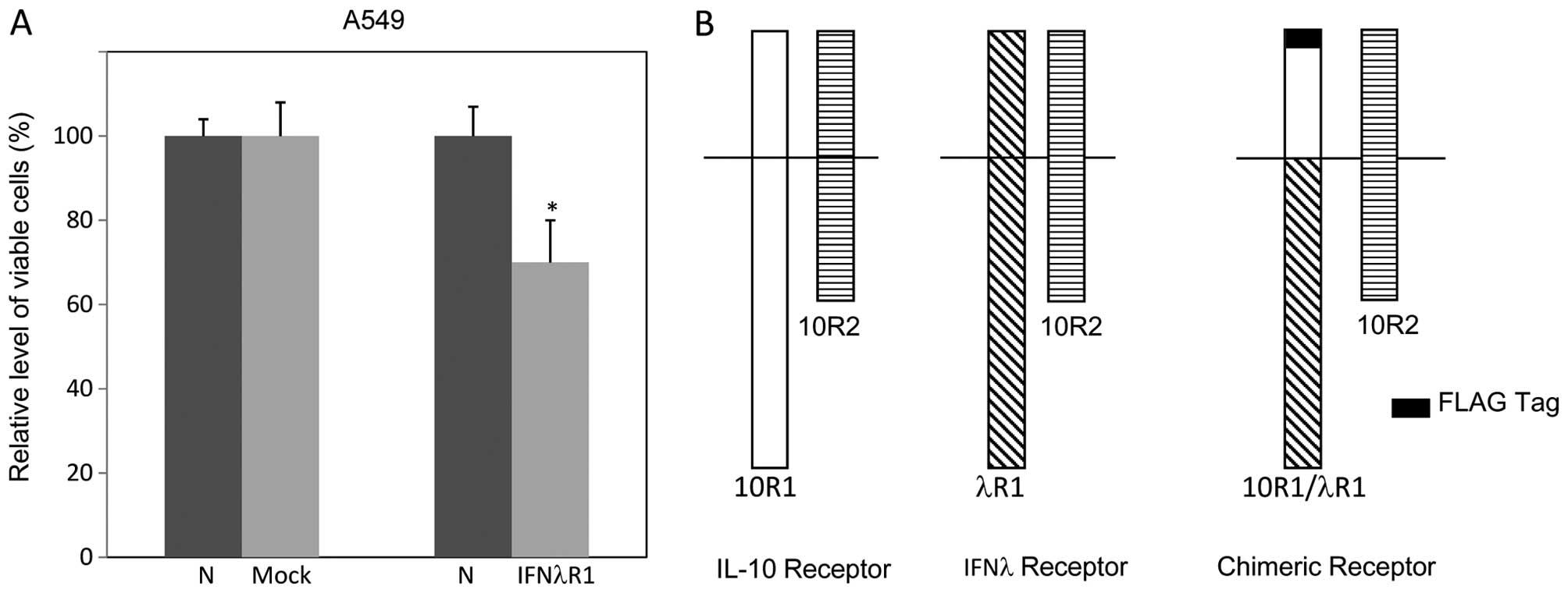
antiproliferative or apoptotic effects. However, ectopic expression
of full-length IFNλ receptor R1 (IFNλR1) in A549 cells resulted in
cell death even without the presence of IFNs. Clones expressing
detectable levels of ectopic IFNλR1 could not be obtained (Fig. 2A).
We previously managed this obstacle in other cell
lines with a FLAG-tagged chimeric receptor 10R1/λR1 (Fig. 2B), via which treatment of IL-10
could induce intracellular IFNλ signaling (6,22).
Therefore, we generated A549 cells expressing 10R1/λR1
(A549/10R1/λR1) and its cognate vector. To characterize the
signaling mediated by 10R1/λR1 in A549 cells, Cl.1, a clonal
population expressing 10R1/λR1 at high level, and Cl.2, a clonal
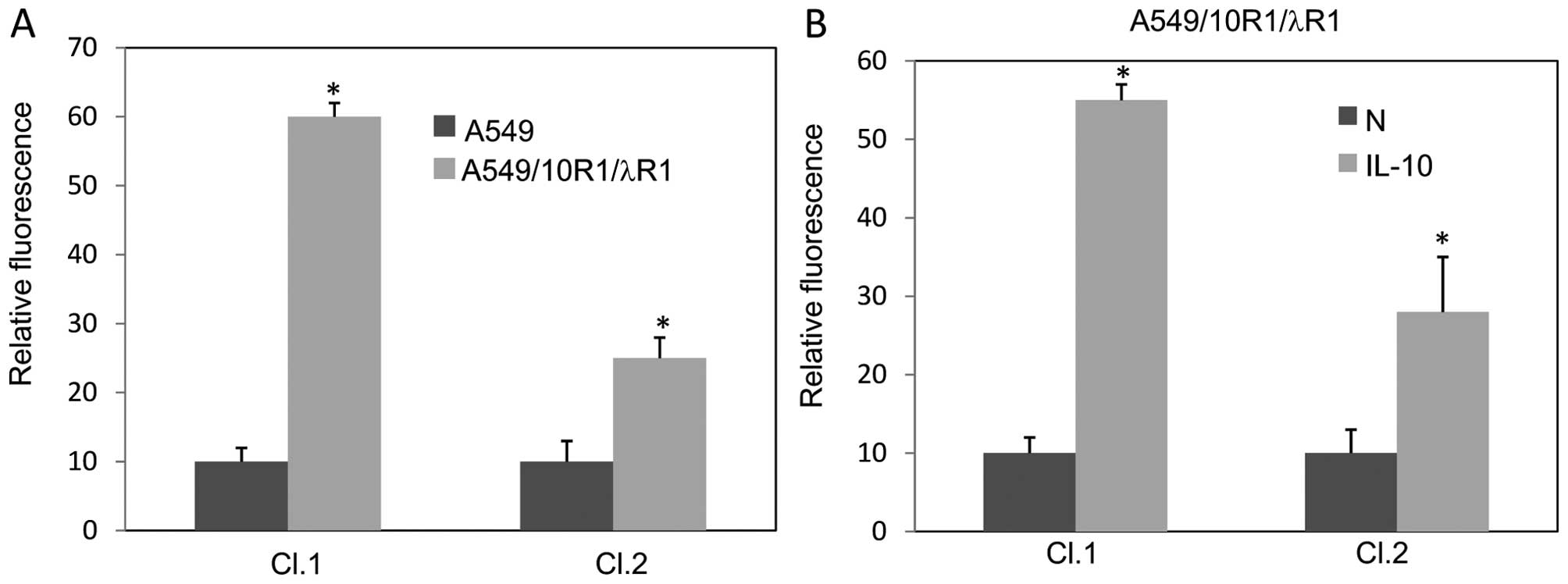
population expressing 10R1/λR1 at low level, were selected.
Expression levels of the FLAG-tagged proteins were examined by flow
cytometry (Fig. 3A). In response to
a low concentration of IL-10 when the possible apoptotic response
was minimal, i.e., at the concentration of 0.3 ng/ml for 72 h, the
cell line Cl.1 and Cl.2 showed increased MHC II expression (about
5.5- and 2.8-fold, respectively) when compared with cells treated
by mock solution (Fig. 3B).
Characteristics of apoptosis induced by
IFNλ signaling
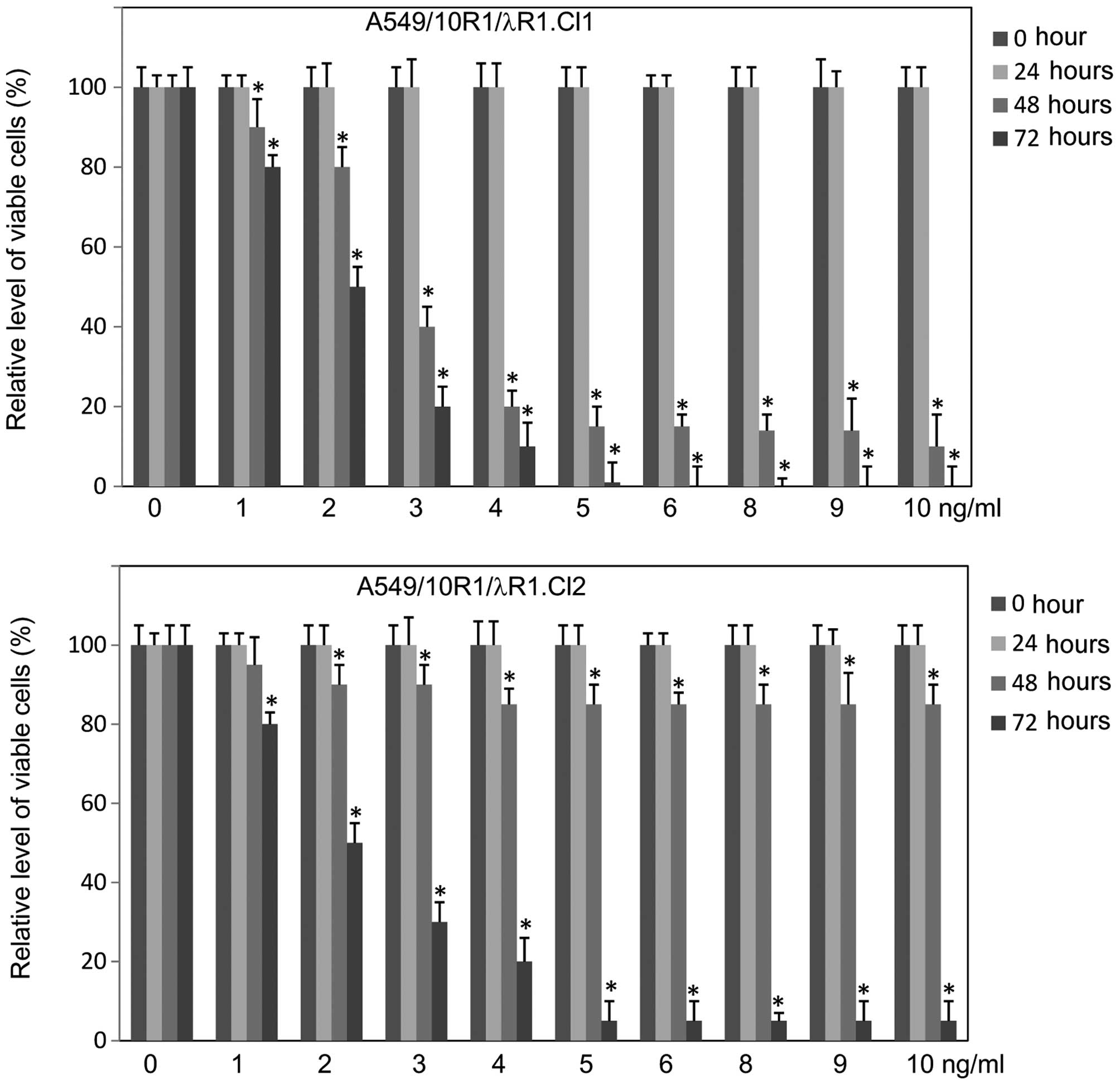
To determine the antiproliferative or apoptotic
effects of IFNλ signaling in A549 cells, we treated the cell line
Cl.1 and Cl.2 with IL-10 at various concentrations and determined
cell viability at various time points. IL-10 induced a strong
antiproliferative response and cell death in a dosage- and
time-dependent manner. The cell death was more intense in Cl.1
cells than in Cl.2 cells (Fig.
4).
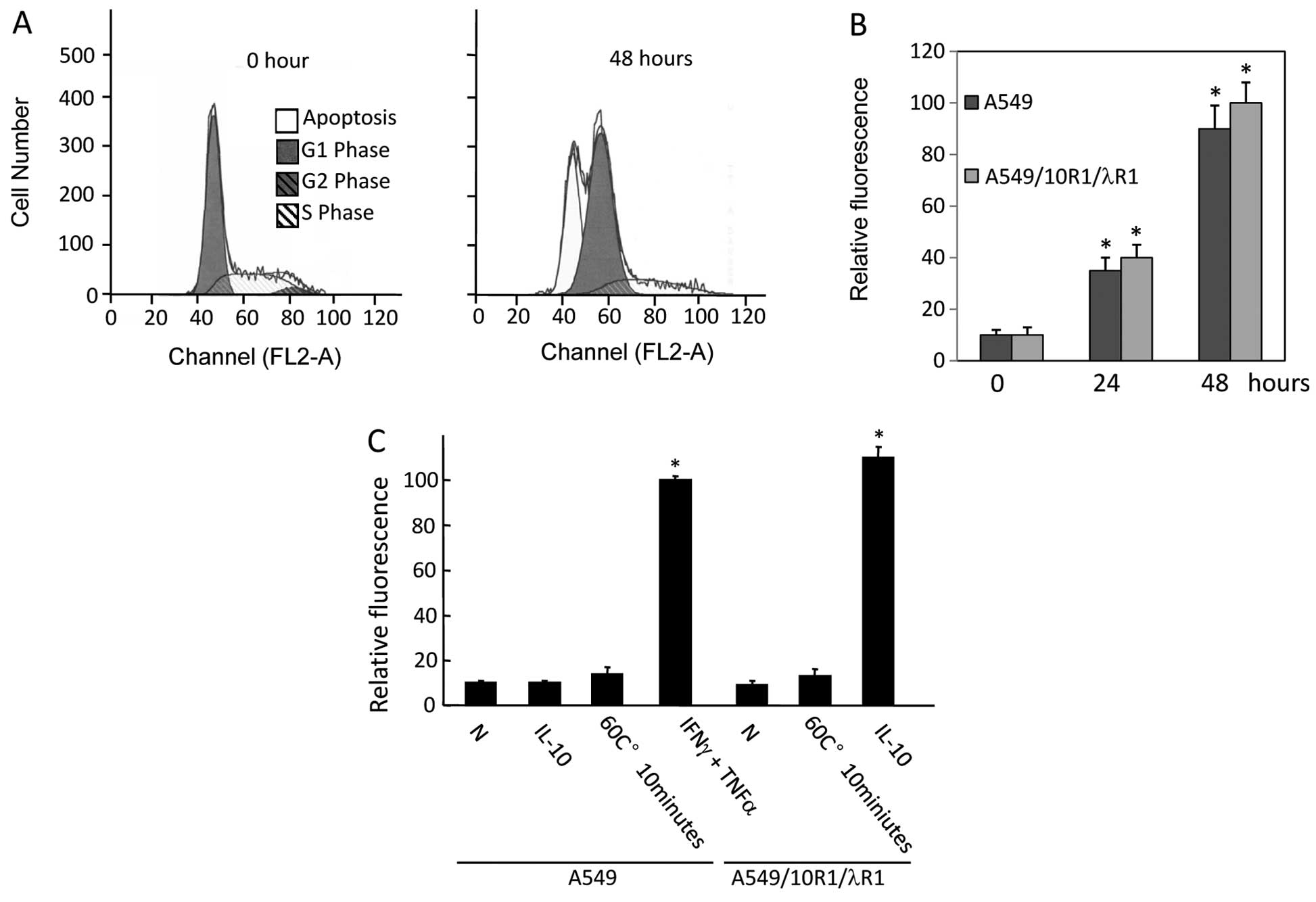
IFNλ signaling may affect cell progression through
the cell cycle (22). We treated
A549 cells expressing 10R1/λR1 with IL-10 (10 ng/ml). Cell cycle
analyses by PI staining were performed at 0 and 48 h. In response
to IL-10 stimulation (10 ng/ml), a majority of A549 cells
expressing 10R1/λR1 was in G0/G1 phase or dead at 48 h. Cells in G2
phase disappeared completely (Fig.
5A).
Upon apoptosis, cells may undergo numerous
physiological changes, including the redistribution of
phosphatidylserine (PS) to the external cell surface, activation of
caspases and DNA fragmentation (5).
Externalization of PS and DNA fragmentation assays are often
performed to indicate induced apoptosis in cells. Externalization
of PS can be detected by Annexin V. DNA fragmentation can be
detected by TUNEL assay, in which the ends of DNA fragments can be
fluorescently labeled by terminal deoxynucleotidyl transferase.
PS externalization was observed in A549/10R1/λR1
cells in response to IL-10 treatment (10 ng/ml) as early as 24 h
and further increased at 48 h (grey bar in Fig. 5B). A similar response was observed
in the A549 cells expressing vector treated by a combination of
IFNγ (10 ng/ml) and TNFα (1 ng/ml), an established treatment to
induce apoptosis in A549 (black bar in Fig. 5B) (29).
DNA fragmentation was examined in A549/10R1/λR1
cells in response to IL-10 treatment (10 ng/ml) at 72 h (Fig. 5C). In contrast to A549 cells
expressing vector treated by IL-10 (second column in Fig. 5C), we observed substantial DNA
fragmentation in A549/10R1/λR1 cells treated by IL-10, indicating
the DNA fragmentation resulted from chimeric receptor-mediated IFNλ
signaling. The A549 cells expressing 10R1/λR1 or vector were heated
at 60°C for 10 min as a non-apoptotic cell death control and no DNA
fragmentation was observed (Fig.
5C).
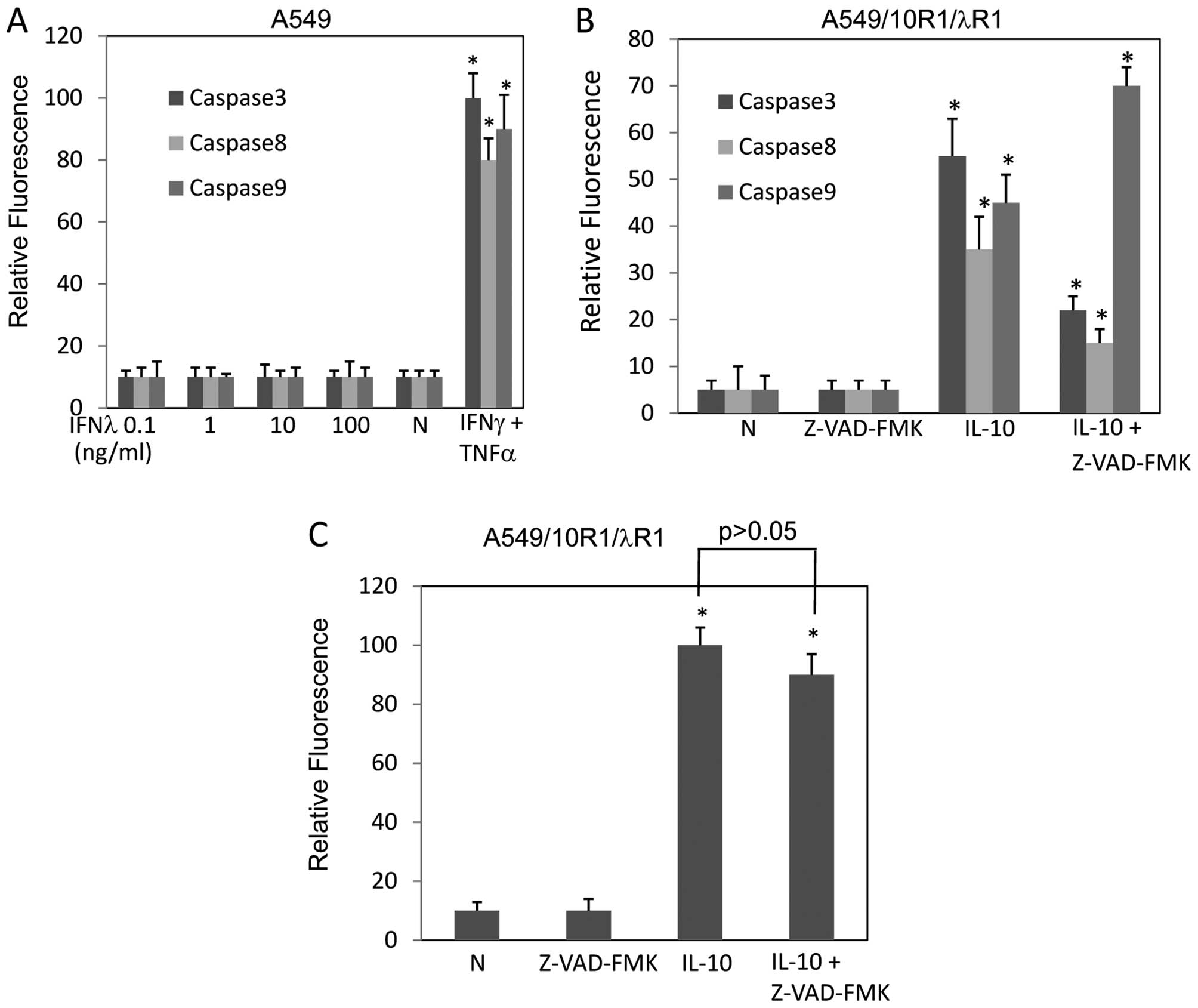
Activation of caspase-3, caspase-8 and caspase-9
were not detectable in A549 cells expressing vector in response to
IFNλ treatment up to 100 ng/ml (Fig.
6A). In contrast, activation of caspase-3, caspase-8 and
caspase-9 (about 12.2-, 6.3- and 8.1-fold, respectively) were
observed in A549/10R1/λR1 cells in response to IL-10 stimulation
(10 ng/ml), suggesting that the caspases were activated by IFNλ
signaling (Fig. 6B). In addition,
pancaspase inhibitor Z-VAD-FMK inhibited the activation of
caspase-3 and caspase-8, but surprisingly promoted caspase-9
activation. This might be a result of compensatory activation,
since caspase-3, a substrate of caspase-9, was inhibited by
Z-VAD-FMK (Fig. 6B). Because
caspase-3 activation is indispensable for caspase-8- and
caspase-9-mediated apoptosis, Z-VAD-FMK was capable of inhibiting
caspase cascade. However, TUNEL assay indicated that Z-VAD-FMK did
not prevent these cells from apoptosis induced by IFNλ signaling,
indicating that caspase-3 may not be critical for the apoptosis
induced by IFNλ signaling (Fig.
6C). As in Fig. 6B, Z-VAD-FMK
did not completely inhibit caspase-3 activation. We increased
Z-VAD-FMK concentration to 100 μM, but still failed to see a full
inhibition (data not shown). Results from a previous study have
shown by ELISA that Z-VAD-FMK (100 μM) inhibited caspase-3
completely in A549 cells (30). So,
Z-VAD-FMK should be able to block caspase-3 activation. Therefore,
apoptosis induced by IFNλ signaling should be
caspase-independent.
STAT1 is activated by IFNλ signaling
STATs, especially STAT1, are important components in
IFN signaling. To investigate the molecular mechanism of apoptosis
induced by IFNλ signaling, we examined the STAT1 activation in
parental A549 cells treated by IFNλ1 and A549/10R1/λR1 cells
treated by IL-10. STAT1 activation was much stronger in
A549/10R1/λR1 cells treated by IL-10 than that in parental A549
cells treated by IFNλ1 (Fig. 7A).
Next we examined the duration of STAT1 activation in A549 cells
treated by IFNλ1 and A549/10R1/λR1 cells treated by IL-10. A549
cells were treated by 100 ng/ml of IFNλ1, while A549/10R1/λR1 cells
were induced with 0.5 ng/ml of IL-10, the concentration at which
IL-10 activated STAT1 in A549/10R1/λR1 cells to a similar level of
that induced by 100 ng/ml of IFNλ in A549 cells (Fig. 7A) and did not kill the cells.
Apparently, IFNλ signaling induced by IL-10 in A549/10R1/λR1 cells
was maintained much longer than that by IFNλ1 in A549 cells
(Fig. 7B). These results implied a
key role of STAT1 in apoptosis induced by IFNλ. The prolonged and
intensified signaling may determine whether IFNλ can induce
apoptosis.
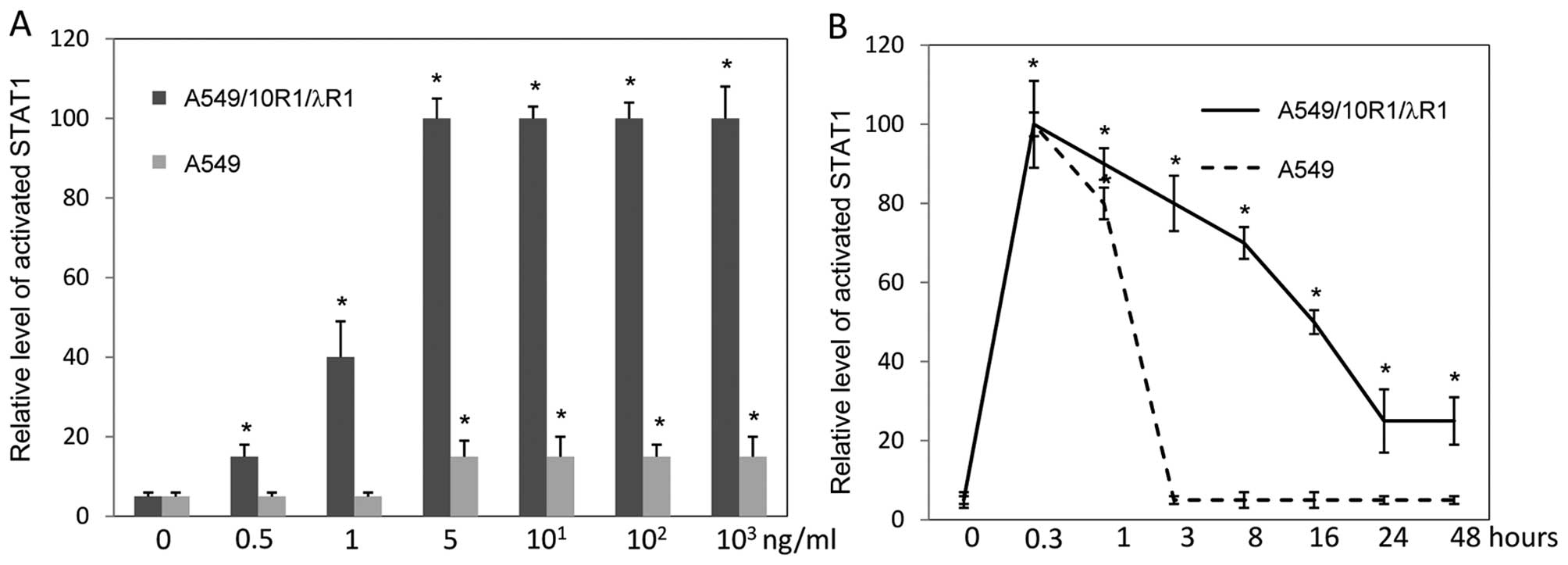 | Figure 7The effects of IFNλ signaling
intensity and duration on STAT1 activation. (A) A549 cells
expressing 10R1/λR1 (black bars) and A549 cells (grey bars) were
treated by IL-10 and IFNλ1, respectively, at various concentrations
(0, 0.5, 1, 5, 101, 102 and 103
ng/ml). Levels of activated STAT1 were examined by flow cytometry
at 15 min. (B) A549 cells expressing 10R1/λR1 and A549 cells were
stimulated by IL-10 (0.5 ng/ml) and IFNλ (100 ng/ml), respectively.
Levels of activated STAT1 were determined at time points of 0, 0.3,
1, 3, 8, 16, 24 and 48 h by flow cytometry. *P<0.05
if compared with time point 0. Data represent at least three
independent experiments. |
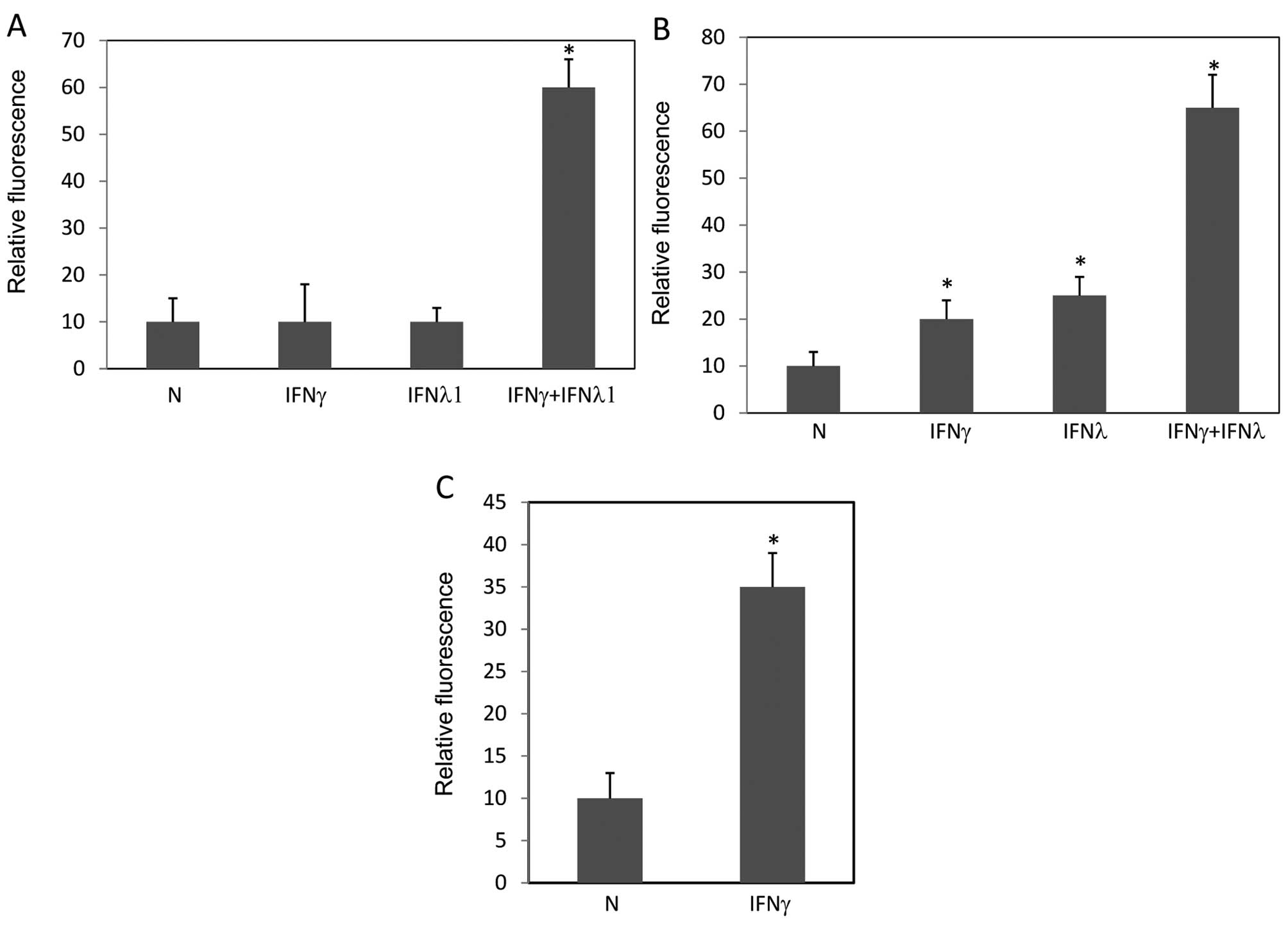
IFNγ sensitizes A549 cells to
IFNλ-induced apoptosis
It is known that IFNγ potentiates the activity of
IFNα by inducing important signaling components, such as STAT1,
STAT2 and IRF9 (31–33). These proteins are also involved in
IFNλ signaling (6). Thus, we
examined whether IFNγ could make A549 cells more sensitive to
IFNλ-induced apoptosis. First, we observed pronounced apoptosis
induced by a combination of IFNγ and IFNλ1 instead of sole
treatment of IFNγ or IFNλ1 in TUNEL assay (Fig. 8A). Next we examined the levels of
STAT1 activation by these treatments. IFNγ at low concentration
(0.1 ng/ml) sensitized STAT1 activation in response to IFNλ
(Fig. 8B). Next, since the
expression level of IFNλR1 could be a limiting factor for IFNλ
signaling to induce apoptosis, we examined expression levels of
IFNλR1 in A549 cells treated by IFNγ. IFNγ significantly increased
the expression level of IFNλR1 (Fig.
8C). In summary, these results demonstrated that IFNλ was able
to induce apoptosis in A549 cells when the cells were sensitized by
other stimuli, such as IFNγ.
Discussion
IFNs are essential members in the family of
antiviral and anticancer drugs, which are widely used in modern
clinical practice. Although type I interferons, including IFNα,
IFNβ and other members, are well studied and widely used, their
side effects are obvious. It is urgent to find alternatives with
similar functions but fewer side effects to satisfy current
clinical needs. IFNλs were discovered less than 10 years ago. Their
antiviral function is similar to that of type I interferons. In
addition, the distribution of receptors of IFNλs is much more
restricted than that of type I interferons, implying a less severe
side effect profile (23).
Therefore, IFNλs become one of the current hot points in
research.
Apoptosis is an important approach in tumor therapy
and antiviral therapy. Tumors have a variety of mechanisms to evade
apoptosis. IFNs inhibit tumor growth and clear viruses by inducing
apoptosis (34–36). Our study explored the mechanisms of
type III interferons to induce apoptosis in lung cancer cells.
The expression levels of cytokine receptors are
critical for cytokines to activate intracellular signals. In many
cases they decide the amplitude of downstream biological effects
(23). Therefore, when we realized
that cells overexpressing IFNλ receptor were not viable, we
utilized an established chimeric receptor 10R1/λR1 to study the
apoptotic effect of IFNλs in human lung cancer cells.
We did not observe any inhibitory effect of IFNλ on
the growth of A549 cells (Fig. 1).
This was consistent with previous observations from mouse BW5147 T
cells and other cell lines (23–25,28).
Expression of the chimeric receptor 10R1/λR1 rendered cells
responsive to apoptosis induced by IFNλ signaling, as indicated by
cell cycle analysis by PI staining, TUNEL assay and Annexin V
staining (Fig. 5). This is
consistent with a previous report in colorectal carcinoma cell line
(22). Caspases are key effectors
in apoptosis, activation of which is critical for canonical
apoptosis (34). We observed that,
in contrast to parental A549 cells treated by IFNλ, A549 cells
expressing 10R1/λR1 treated by IL-10 showed pronounced activation
of caspase-3, caspase-8 and caspase-9. Although Z-VAD-FMK inhibited
the activation of caspase-8 and caspase-3, it did not prevent cell
apoptosis (Fig. 6), suggesting that
IFNλ-induced apoptosis may be caspase-independent. Interestingly,
caspase-9 activation was promoted by Z-VAD-FMK. This probably
resulted from a compensation of the suppression of its downstream
caspase-3. This is consistent with our previous report in
colorectal carcinoma cells (22).
We also demonstrated that both the intensity and duration of STAT1
activation were increased, when IFNλ signal was intensified
(Fig. 7). These results
demonstrated that IFNλ signal was able to trigger apoptosis in
human lung cancer cells, which probably was mediated via STAT1.
More detailed mechanism is under investigation.
Many experiments in this study utilized the chimeric
receptor 10R1/λR1, rather than the full length IFNλ receptor,
IFNλR1. This was because clonal populations expressing the ectopic
IFNλ receptor were not available. A549 cells expressing high levels
of IFNλ receptor may not be viable in physiological conditions. By
using the artificial chimeric receptor 10R1/λR1, we found the
intrinsic capability of IFNλ signaling on inducing apoptosis. But
there should be physiological or pathological conditions that this
apoptosis-mediating capability could be activated. Therefore, at
the end of this study, we tested the treatment combination of IFNλ
and IFNγ on A549 cells, and discovered that IFNγ upregulated
expression of IFNλR1, which facilitated IFNλ-mediated apoptosis
(Fig. 8). IFNγ-induced upregulation
of IFNλR1 did not kill the cells, possibly because the level of
IFNλR1 is not as high as in the clonal populations expressing the
ectopic IFNλ receptor. Further experiments are required to prove
this hypothesis. These results indicated the potential of IFNλR1 to
induce apoptosis in special conditions, such as a pathological
condition where IFNγ is upregulated. We know that the regulatory
effects of IFNγ are very broad. So, in our next step, we will try
to find other physiological or pathological stimulations that
upregulate IFNλ receptor specifically to further confirm our
findings.
In summary, we report that the apoptotic potential
of IFNλs on human lung cancer cells in concomitant with STAT1
activation and increased expression of IFNλ receptor R1. This
apoptosis could be enhanced by IFNγ. Our results provided a
theoretical basis for IFNλs to treat lung cancers.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant# 30872226), the Project of
Beijing Municipal Science and Technology Commission (Grant#
D09050703590901), the Beijing Key Laboratory (grant# BZ0089) and
the Funding Project for Academic Human Resources Development in
Institutions of Higher Learning under the Jurisdiction of Beijing
Municipality (grant# PHR201007112).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: the impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. CA Cancer J
Clin. 61:212–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen W, Zhang S and Zou X: Estimation and
projection of lung cancer incidence and mortality in China.
Zhongguo Fei Ai Za Zhi. 13:488–493. 2010.(In Chinese).
|
|
4
|
Chinese Health Statistical Digest.
Ministry of Health; 2008, http://www.moh.gov.cn/publicfiles/business/htmlfiles/mohbgt/s8274/200805/35671.htm.
|
|
5
|
Clemens MJ: Interferons and apoptosis. J
Interferon Cytokine Res. 23:277–292. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kotenko SV, Gallagher G, Baurin VV, et al:
IFN-lambdas mediate antiviral protection through a distinct class
II cytokine receptor complex. Nat Immunol. 4:69–77. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sheppard P, Kindsvogel W, Xu W, et al:
IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat
Immunol. 4:63–68. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schindler C and Plumlee C: Inteferons pen
the JAK-STAT pathway. Semin Cell Dev Biol. 19:311–318. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Renauld JC: Class II cytokine receptors
and their ligands: key antiviral and inflammatory modulators. Nat
Rev Immunol. 3:667–676. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Platanias LC: Mechanisms of type-I- and
type-II-interferon-mediated signalling. Nat Rev Immunol. 5:375–386.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dunn GP, Bruce AT, Sheehan KC, et al: A
critical function for type I interferons in cancer immunoediting.
Nat Immunol. 6:722–729. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Negro F: Adverse effects of drugs in the
treatment of viral hepatitis. Best Pract Res Clin Gastroenterol.
24:183–192. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kelly C, Klenerman P and Barnes E:
Interferon lambdas: the next cytokine storm. Gut. 60:1284–1293.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar
|
|
15
|
Borden EC: Review: Milstein Award lecture:
interferons and cancer: where from here? J Interferon Cytokine Res.
25:511–527. 2005. View Article : Google Scholar
|
|
16
|
Doyle SE, Schreckhise H, Khuu-Duong K, et
al: Interleukin-29 uses a type 1 interferon-like program to promote
antiviral responses in human hepatocytes. Hepatology. 44:896–906.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Marcello T, Grakoui A, Barba-Spaeth G, et
al: Interferons alpha and lambda inhibit hepatitis C virus
replication with distinct signal transduction and gene regulation
kinetics. Gastroenterology. 131:1887–1898. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kalvakolanu DV: The GRIMs: a new interface
between cell death regulation and interferon/retinoid induced
growth suppression. Cytokine Growth Factor Rev. 15:169–194. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lasfar A, Lewis-Antes A, Smirnov SV, et
al: Characterization of the mouse IFN-lambda ligand-receptor
system: IFN-lambdas exhibit antitumor activity against B16
melanoma. Cancer Res. 66:4468–4477. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Numasaki M, Tagawa M, Iwata F, et al:
IL-28 elicits antitumor responses against murine fibrosarcoma. J
Immunol. 178:5086–5098. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sato A, Ohtsuki M, Hata M, Kobayashi E and
Murakami T: Antitumor activity of IFN-lambda in murine tumor
models. J Immunol. 176:7686–7694. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li W, Lewis-Antes A, Huang J, Balan M and
Kotenko SV: Regulation of apoptosis by type III interferons. Cell
Prolif. 41:960–979. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Meager A, Visvalingam K, Dilger P, Bryan D
and Wadhwa M: Biological activity of interleukins-28 and -29:
comparison with type I interferons. Cytokine. 31:109–118. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zitzmann K, Brand S, Baehs S, et al: Novel
interferon-lambdas induce antiproliferative effects in
neuroendocrine tumor cells. Biochem Biophys Res Commun.
344:1334–1341. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Brand S, Beigel F, Olszak T, et al: IL-28A
and IL-29 mediate antiproliferative and antiviral signals in
intestinal epithelial cells and murine CMV infection increases
colonic IL-28A expression. Am J Physiol Gastrointest Liver Physiol.
289:G960–G968. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fujie H, Tanaka T, Tagawa M, et al:
Antitumor activity of type III interferon alone or in combination
with type I interferon against human non-small cell lung cancer.
Cancer Sci. 102:1977–1990. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li W, Henderson LJ, Major EO and Al-Harthi
L: IFN-gamma mediates enhancement of HIV replication in astrocytes
by inducing an antagonist of the beta-catenin pathway (DKK1) in a
STAT 3-dependent manner. J Immunol. 186:6771–6778. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dumoutier L, Tounsi A, Michiels T,
Sommereyns C, Kotenko SV and Renauld JC: Role of the interleukin
(IL)-28 receptor tyrosine residues for antiviral and
antiproliferative activity of IL-29/interferon-lambda 1:
similarities with type I interferon signaling. J Biol Chem.
279:32269–32274. 2004. View Article : Google Scholar
|
|
29
|
Kim KB, Choi YH, Kim IK, et al:
Potentiation of Fas- and TRAIL-mediated apoptosis by IFN-gamma in
A549 lung epithelial cells: enhancement of caspase-8 expression
through IFN-response element. Cytokine. 20:283–288. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shankaranarayanan P and Nigam S: IL-4
induces apoptosis in A549 lung adenocarcinoma cells: evidence for
the pivotal role of 15-hydroxyeicosatetraenoic acid binding to
activated peroxisome proliferator-activated receptor gamma
transcription factor. J Immunol. 170:887–894. 2003. View Article : Google Scholar
|
|
31
|
Improta T, Pine R and Pfeffer LM:
Interferon-gamma potentiates the antiviral activity and the
expression of interferon-stimulated genes induced by
interferon-alpha in U937 cells. J Interferon Res. 12:87–94. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Levy DE, Lew DJ, Decker T, Kessler DS and
Darnell JE Jr: Synergistic interaction between interferon-alpha and
interferon-gamma through induced synthesis of one subunit of the
transcription factor ISGF3. EMBO J. 9:1105–1111. 1990.
|
|
33
|
Lehtonen A, Matikainen S and Julkunen I:
Interferons up-regulate STAT1, STAT2, and IRF family transcription
factor gene expression in human peripheral blood mononuclear cells
and macrophages. J Immunol. 159:794–803. 1997.PubMed/NCBI
|
|
34
|
Elmore S: Apoptosis: a review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schattenberg JM, Schuchmann M and Galle
PR: Cell death and hepatocarcinogenesis: dysregulation of apoptosis
signaling pathways. J Gastroenterol Hepatol. 26(Suppl 1): 213–219.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mihic LL, Bulat V, Situm M, Krolo I and
Seserko A: The role of apoptosis in the pathogenesis of malignant
melanoma. Coll Antropol. 34(Suppl 2): 303–306. 2010.
|