1. Introduction
Angiogenesis is a tightly regulated process dealing
with the development of new blood vessels from a pre-existing
vascular network. In solid tumor growth, angiogenesis is critical
for tumor development, progression and metastasis (1,2).
Several types of leukemias similar to solid tumors were also
reported to have high in bone marrow microvessel density (MVD)
(3). This suggests that
angiogenesis plays an important role in the progression of
hematolymphoid malignancies. Often, angiogenesis is maintained by a
balance of endogenous antiangiogenic and proangiogenic factors.
However, the exact mechanism triggering vascular endothelial growth
factor (VEGF) expression in hematolymphoid tumors is unknown;
different mechanisms similar to those observed in solid tumors are
anticipated. In recent years, the expression level of VEGF/VEGF
receptor (VEGFR) in patients with different hematolymphoid tumors
has been detected and related to reduced survival and lower
remission rates (4). Studies have
demonstrated that VEGF/VEGFR-related pathways are the most relevant
regulators of neoangiogenesis, vasculogenesis and recruitment of
endothelial progenitor cells. Furthermore, VEGF/VEGFR interactions
may stimulate proliferation, migration and survival of
leukemia/lymphoma cells by autocrine and paracrine loops (3). According to a number of studies, acute
leukemia cells secrete significant amounts of VEGF in the serum and
malignant hematopoietic cells were found to express VEGF and its
receptors (5). The clinical outcome
of this approach has also been confirmed, and recently coordinated
efforts in research have resulted in a number of novel
antiangiogenic agents (6). This
review focuses on the current knowledge of angiogenesis and
antiangiogenic therapies in hematolymphoid malignancies. It mainly
demonstrates the role of VEGF/VEGFR in hematological malignancies
including its important role in growth, proliferation, survival, as
well as the correlation of VEGF/VEGFR with the treatment, relapse
or prognosis of leukemia. The progress of VEGF and its receptors as
attractive targets for therapies are discussed together with their
clinical application in leukemic diseases.
2. Brief review of VEGF/VEGFR
VEGF, a glycoprotein, was first purified from the
in vitro culture medium of bovine pituitary
folliculo-stellate cells by Ferrara and Henzel (7). Named after its mitogenic activity for
vascular endothelial cells, VEGF was considered a highly specific
co-mitogen for the vascular endothelial cells that promoted
vascular permeability (8). The five
subtype members in the VEGF family are VEGF-A, PlGF, VEGF-B, VEGF-C
and VEGF-D. VEGF-A is the prototype of the ligand and is commonly
known as VEGF. All these ligands promote endothelium regeneration
and increase vascular permeability via binding to three
transmembrane receptor tyrosine kinases which are VEGFR-1, -2 and
-3. VEGFR-1 ligands include VEGF-A, -B, and placental growth factor
(PlGF). VEGFR-2 (also known as KDR in human and Flk-1 in mice) has
ligands of VEGF-A, -C, -D and is predominantly expressed in
vascular endothelial cells. The activation of VEGFR-2 is necessary
and sufficient in order to mediate VEGF-dependent angiogenesis and
the induction of vascular permeability (8,9). The
binding of VEGFR-3 to the VEGF homologues VEGF-C and VEGF-D is
largely restricted to lymphatic endothelial cells and plays an
important role in the regulation of lymphangiogenesis (9,10).
Receptor tyrosine kinases are expressed in adult
endothelial cells except the brain. VEGFR-1 is also expressed in
hematopoietic, monocytes and smooth muscle cells (11,12).
VEGFR-2 is expressed mostly in vascular endothelial cells, and also
in neuronal, megakaryocytes and hematopoietic stem cells (13). Although the exact contribution of
VEGFR-1 signaling to angiogenesis is unclear, it has been shown
that VEGFR-1 directly cooperates with VEGFR-2 via
heterodimerization similar to the binding of two additional VEGF
homologues, VEGF-B and PIGF (12).
It has been discovered by Brekken et al (14) that VEGF-2 plays a significant role
in VEGF-induced angiogenesis and the binding between VEGF and the
VEGFR-2 may activate the mitogen-activated protein kinase (MAPK)
system via protein kinase C (PKC) or the Ras protein to induce the
proliferation of vascular endothelial cells.
It has been identified that VEGF is the specific
growth factor for angiogenesis, while other growth factors
including fibroblast growth factor (FGF) and platelet-derived
growth factor (PDGF) have no specificity since they act on several
different types of cells including vascular endothelial cells.
Moreover, VEGF may regulate the development of hemopoietic stem
cells and remodel both the extracellular matrix and the
regeneration of inflammatory cytokines (15,16).
Several leukemic cell strains and primary cells synthesizing and
secreting VEGF may affect and modulate the malignant biological
behavior of leukemic cells by two positive-feedback loops which are
paracrine and autocrine (17,18).
VEGF secreted by leukemic cells interacts with relevant receptors
on the endothelial cell surface and stimulates endothelial cells to
produce growth factors that act on leukemic cells resulting in an
increase in their proliferative activity and drug resistance.
Hence, antiangiogenesis therapy based on the principle of
inhibiting the physiological function of VEGF has become a novel
target for antiangiogenic therapy (19).
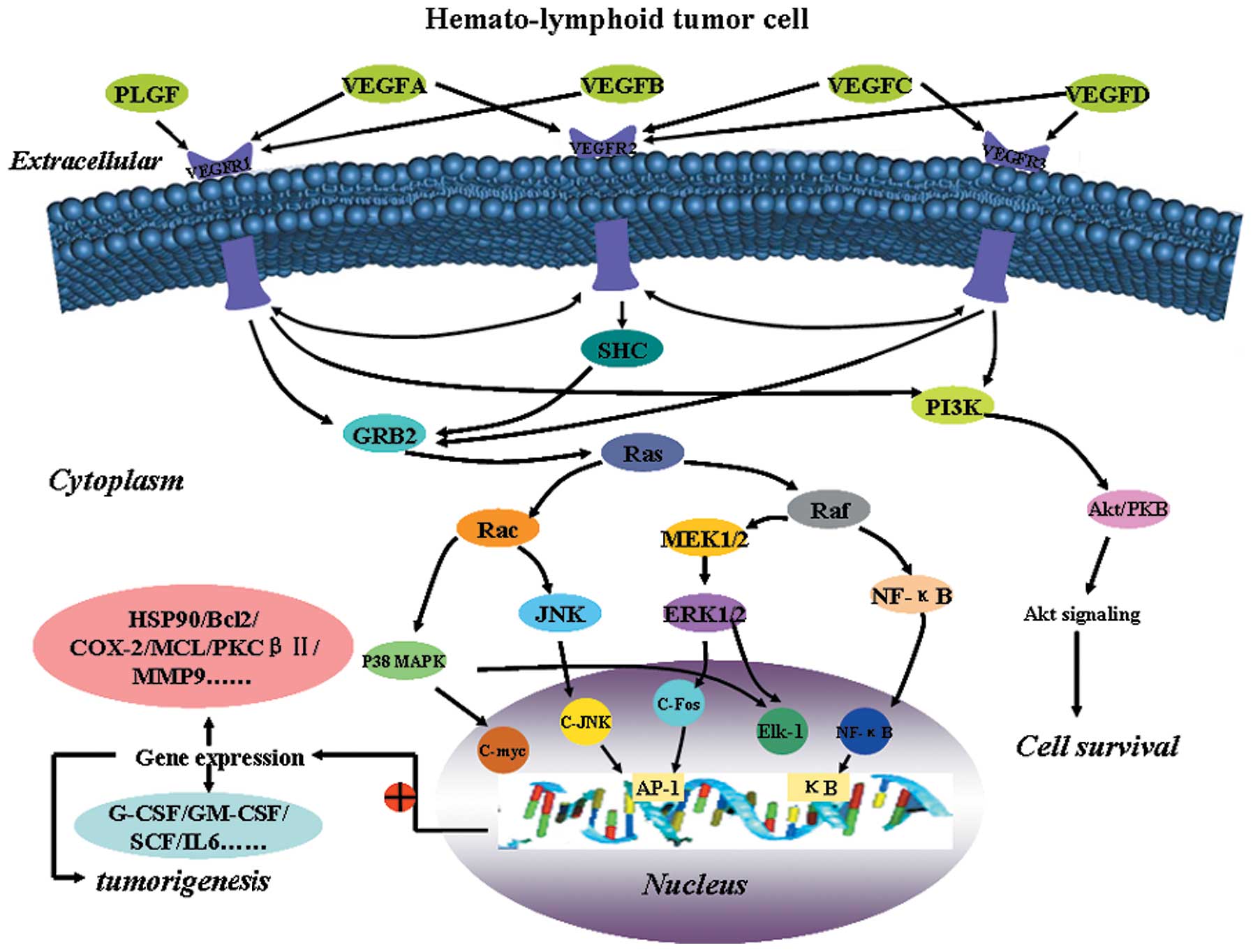
3. Signaling pathway of VEGF/VEGFR in
leukemia
Upstream regulators target VEGF/VEGFR
expression in leukemic diseases
Much research has focused on the factors regulating
VEGF expression and their functions in leukemic diseases from
different aspects. VEGF expression is regulated by several
intrinsic and extrinsic factors, with hypoxia and hypoglycemia
being the major stimuli (20).
Often, hypoxia-induced transcription of VEGF mRNA is mediated by
the binding of hypoxia-inducible factor-1 (HIF-1) (21); intratumoral hypoxia and HIF-1
mediation have been discovered to be a key angiogenesis triggering
event (22,23). This basic helix-loop-helix
transcription factor may not only contribute to hypoxia-induced
VEGF production, but may also play a critical role in the
oncogene-dependent expression of VEGF (24). Additional recent data suggest that
HIF-1α may also be involved in BCR/ABL-dependent expression of VEGF
in Ba/F3 cells (25,26). HIF-1α also accounts for the
molecular mechanism of autocrine regulation of VEGF in chronic
lymphocytic leukemia (CLL) B cells. Ghosh et al (24) reported that CLL B cells express
constitutive levels of HIF-1α under normoxia. The stabilized HIF-1α
may form an active complex with the transcriptional coactivator
p300 and phosphorylated-STAT3 at the VEGF promoter and recruit RNA
polymerase II to upregulate VEGF transcription. Consequently, VEGF
is secreted at higher levels in CLL B cells. The authors examined
the status of the von Hippel-Lindau gene product (pVHL) that dealt
with HIF-1α degradation and discovered it was at a notably lower
level in CLL B cells compared with that in normal B cells. This
initial evidence explained the aberrant autocrine VEGF secretion in
CLL cells. Besides BCR/ABL, microvesicles (MVs) released by
malignant cancer cells constituting an important part of the tumor
microenvironment may also activate the HIF-1α pathway with VEGF
production in B-cell CLL patients. Ghosh et al (27) demonstrated that MVs circulating in
the plasma of B-cell CLL patients exhibited a phenotypic shift from
the predominant platelet derived in early stage to leukemic B-cell
derived at advanced stage. Furthermore, the total MV level in
patients with CLL was higher compared to that of healthy patients.
Apart from being a factor in angiogenesis these results indicate
that VEGF is also an essential mediator in other clinical
phenomena.
As described, VEGF production is associated with the
constitutive activity of Janus kinase 3 (Jak3) and the c-Jun
N-terminal kinases (JNKs). Jak3 has been suggested to play a key
role in the transformation of CTCL T cells since Jak3 inhibitors
trigger apoptosis and inhibit cell growth and spontaneous cytokine
production of malignant T cells (28–30).
It is proposed that the oncogene Stat3, the primary target of
Jak3-mediated transformation, requires coactivators such as HIF-1α
to induce VEGF expression. Additionally, c-Jun phosphorylation and
its ability to bind to a VEGF promoter element relating to JNK
activity and VEGF production indicate that JNK induced VEGF
expression is caused by an increase in c-Jun/AP-1 activity.
Activation of the JNK/AP-1 signaling pathway has previously been
implicated in the induction of VEGF transcription (31). However, the JNKs have been shown to
promote VEGF expression through other mechanisms, such as
increasing VEGF mRNA stability (32). Therefore, inhibition of
VEGF-inducing pathways or neutralization of VEGF itself may imply
novel therapeutic modalities in cutaneous T-cell lymphoma (CTCL)
(33). In a previous study,
researchers discovered that lysophosphatidic acid (LPA) protected
CLL cells from apoptosis through a higher expression of LPA
receptors and autocrine production of VEGF. Kumar et al
(34) reported that an increase in
VEGF by LPA was mediated through the activation of JNK and
transcription factor NF-κB since blocking JNK or NF-κB activation
may inhibit LPA to induce VEGF expression. Furthermore, it was
demonstrated that LPA protected cells from apoptosis by blocking
the activation of both VEGFR-1 and VEGFR-2 via the VEGF receptor
kinase inhibitor. Knocking down the expression of VEGFR-1 and
inhibiting the activation of NF-κB and JNK may also block LPA to
avoid apoptosis. We hypothesize that LPA contributing to VEGF
production in B cell malignancies leads to cell survival (35).
Leukemia is an angiogenesis-dependent malignancy
(36,37) and angiogenesis is strictly dependent
on Akt/NF-κB activation. The inhibitors of the NF-κB pathway
decreasing VEGF secretion in leukemic cells and inhibiting
endothelial cell activities may cause the interruption of a
reciprocal stimulatory loop between leukemic and endothelial cells.
Different reports demonstrating the activation of NF-κB in lymphoid
and myeloid malignancies underscore the implication in malignant
transformation (38). While the
overexpression of NF-κB may lead to chemoresistance, the
appropriate inhibition of this pathway may lead to successful
therapy. Moreover, the transcription of VEGF by the classical NF-κB
target gene may be repressed, which is one aspect of the
participation in tumorigenesis of adult T-cell leukemia (ATL)
(39). NF-κB is also activated by
the phosphoinositide 3-kinase (PI3K)/Akt signaling pathway, which
is also crucial to several aspects of cell growth, survival and
apoptosis. PI3K/Akt activation has been implicated in both the
pathogenesis and the progression of a variety of neoplasms which
includes leukemias (40).
Accumulated evidence over the years has indicated that the PI3K/Akt
signal transduction pathway is a major factor for cancer resistance
in conventional therapies. Indeed, pharmacologic inhibitors of
PI3K/Akt have been found to potentiate the apoptotic action of
anti-leukemic drugs (41).
Therefore, targeting NF-κB activation as well as its upstream
regulator Akt may constitute an additional strategy to improve
conventional therapies.
Several extensively characterized transcription
factors which include activator protein-1 (AP-1), NF-κB and
stimulatory protein-1 (SP-1) modulate VEGF expression. The
activation and binding of these transcription factors in tumor
cells contribute to VEGF transcription and tumor metastasis
(42–44). Among those, AP-1 is a critical
factor in the regulation of VEGF gene expression. Pollmann et
al (45) determined that VEGF
gene expression is mostly regulated by AP-1 and c-Jun in human
promyelocytic leukemia (HL-60) cells. Poulaki et al
(46) demonstrated that the
insulin-like growth factor-1 receptor (IGF-1R) in thyroid tumor
cell membranes may promote VEGF production by enhancing the
activity of transcription factor AP-1 (46). Rutin in combination with vitamin E
has been demonstrated to synergistically inhibit oxidative damage.
Chuang et al (47) reported
that rutin in combination with vitamin E attenuated VEGF expression
in HL-60 cells by decreasing the activity of AP-1.
The BTB domain (named after the Drosophila
transcription factors Bric-a-brac, Tramtrack and Broad) of
promyelocytic leukemia zinc finger (PLZF) as a novel apoptotic and
antiangiogenic protein, may directly inhibit tube formation and
migration of endothelial cells on Matrigel in vitro. To
date, the BTB domain is reported to reduce the expression of VEGF,
p-Ak, and p-eNOS in HUVECs. Akt and eNOS play significant roles in
angiogenesis stimulated by VEGF which is known to stimulate
Akt-dependent phosphorylation of eNOS. These observations reveal
that the BTB domain has little or no effect on non-phosphorylated
Akt and eNOS. These data indicate that VEGF is essential to the BTB
domain function and they form a positive feedback loop to
facilitate leukemia-related disease (48) (Fig.
1).
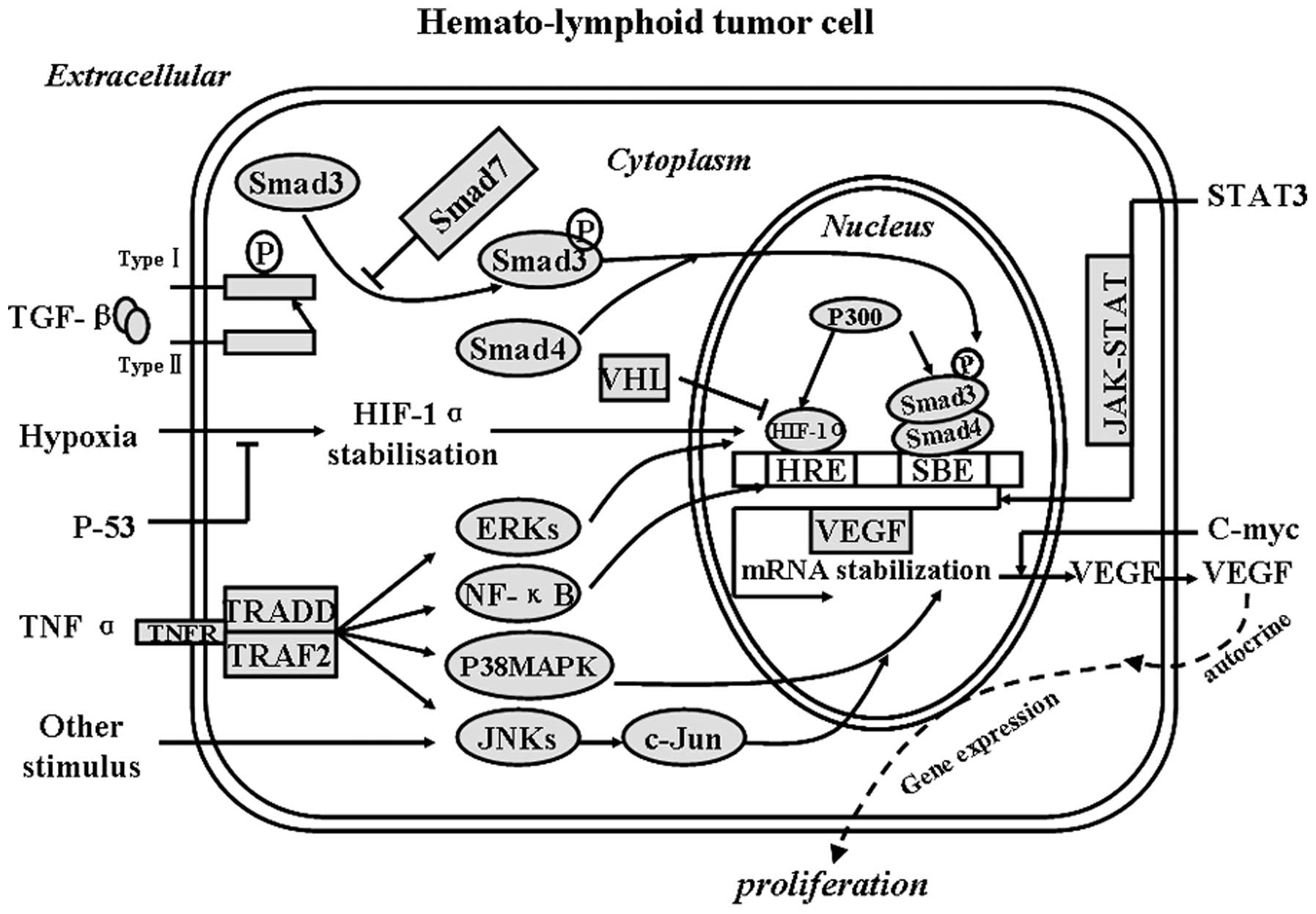 | Figure 1Upstream regulators target VEGF/VEGFR
expression in leukemias. Smads, signal transducers and
transcription factors in the TGF-β signaling pathway; TRADD,
protein which binds adaptor protein TRAF2 and then suppresses
TRAF2-mediated apoptosis; TRAF2, TNF receptor-associated factor 2,
which mediates the signal transduction from members of the TNF
receptor superfamily; VHL, von Hippel-Lindau tumor-suppressor also
known as pVHL; ERKs, extracellular signal-regulated kinases;
P38MAPK, P38 mitogen-activated protein kinases; JNKs, c-Jun
N-terminal kinases; p300, a transcriptional coactivator. TGF-β,
hypoxia and TNF-α induce the expression of VEGF and VEGFR in
hematolymphoid tumor cells through the Smad family, HIF-1α, ERKs,
NF-κB, p38-MAPK and JNKs, respectively, which, ultimately, affects
VEGF/VEGFR expression. |
BCR/ABL (an oncogene fusion protein consisting of
BCR and ABL, which is associated with the Philadelphia chromosome)
functions as a constitutive tyrosine kinase leading to
autophosphorylation (49) and
activates multiple signaling molecules including p21Ras (50), signal transducer and activator of
transcription 5 (STAT5) (51–53)
and phosphoinositide 3-kinase (PI3-kinase) (54). Using both single-marker analysis and
haplotype analysis, BCL-2 SNP was found to demonstrate consistent
association with susceptibility to chronic myeloid leukemia (CML)
(55). Recently, it was reported
that treatment with Bcr-Abl-targeting siRNAs and imatinib resulted
in an enhanced VEGF suppression in K562 cells (56). Little is known about the biochemical
mechanisms and signaling pathways contributing to BCR/ABL-induced
expression of angiogenic growth factors in CML cells. Böhm et
al (57) reported that BCR/ABL
induces VEGF production in CML cells through a pathway involving
PI3-kinase and mammalian target of rapamycin (mTOR). mTOR has
recently been implicated in leukemic cell growth, tumor-associated
angiogenesis and the expression of VEGF in acute myeloid leukemia
(AML). mTOR-targeting drugs exert anti-leukemic effects on AML
cells in vitro through multiple actions, including direct
inhibition of proliferation, induction of apoptosis and suppression
of VEGF (57).
Downstream target genes of VEGF in
leukemias
In addition to stimulating angiogenesis, other
studies have demonstrated that VEGF may directly stimulate
proliferation of several types of leukemia cells. For example, VEGF
stimulates multiple myeloma (MM) cells to migrate, proliferate and
survive on fibronectin via autocrine and paracrine loops, which
usually contributes to the binding of VEGF and VEGFR-2 (58). As to the modulation of its
downstream target gene, a great deal of progress has been made.
VEGF induces the expression of heat shock protein 90 (Hsp90), which
binds Bcl-2 and Apaf-1 to increase leukemic cell resistance to
serum deprivation-induced apoptosis (59). Moreover, VEGF may play an important
role in the growth of hematologic neoplasms via a paracrine
mechanism. When endothelial cells are exposed to recombinant human
VEGF, they may increase mRNA expression of several hematopoietic
growth factors, including G-CSF, GM-CSF, stem cell factor (SCF) and
IL-6, which act as growth factors for myeloid and lymphoid cells
(60,61). In other words, VEGF may promote
tumorigenesis by enhancing the production of hematopoietic growth
factors. Both VEGF and PKCβII were reported to have higher levels
of expression related to disease stage and tumor burden (18,62,63).
The regulation of PKCβ expression in CLL cells by VEGF was
important since the level of PKCβ expression in malignant cells in
a Tcl1 mouse model determined the development and progression of
the disease. Controlling PKCβ expression is likely to be an
important step in CLL pathogenesis. VEGF, due to its role in
stimulating PKCβ expression, is therefore central to the
pathogenesis of the disease (64).
Migration of B-cell chronic lymphocytic leukemia (B-CLL) cells
involves several molecules, including matrix metalloproteinase-9
(MMP-9) and VEGF. Downregulation of MMP-9 by VEGF significantly
inhibited the migration of B-CLL cells through human umbilical vein
endothelial cells. STAT1 was found to be responsible for MMP-9
downregulation since STAT1 gene silencing restored MMP-9 production
and B-CLL cell migration in the presence of VEGF. The VEGF/VEGFR-2
axis is upstream of STAT1 tyrosine phosphorylation thus the
inhibition of B-CLL cell migration is ultimately due to the effect
of VEGF (65).
VEGF-C has been recognized as a tumor
lymphangiogenic factor based on the effects of activated VEGF-R3 on
lymphatic endothelial cells. VEGF-C enhances c-Jun binding to the
cyclic adenosine 3′,5′-monophosphate-response element of the
cyclooxygenase-2 (COX-2) promoter and induces COX-2 expression,
which may catalyze one of the rate-limiting steps in prostanoid
biosynthesis (66) and enhance the
survival and proliferation of malignant cells, while negatively
influencing anti-tumor immunity. In addition, the VEGF-R3/JNK/AP-1
pathway also participates in the induction of COX-2 by VEGF-C in
leukemic cells. VEGF-R2 activation may induce the upregulation of
COX-2 via p38 MAPK and JNK signaling pathways in human vascular
endothelial cells (67). In line
with this study, VEGF-A may also induce the upregulation of COX-2
in leukemic cells. By acting in an autocrine/paracrine manner,
VEGF-C may contribute to tumor angiogenesis through the induction
of COX-2/prostanoids in subsets of leukemia. A previous study
demonstrated that there is a significantly higher induction of
VEGF-C by COX-2 in another context (68). Hence, the cross talk between VEGF-C
and COX-2 may initiate a positive feedback loop, resulting in
enhanced expression of COX-2 and increased synthesis of
prostanoids, which lead to a further increase in VEGF-C
activity.
The expression of the antiapoptotic myeloid cell
leukemia-1 (MCL-1) gene, leading to the enhanced survival of tumor
cells, is a novel prognostic factor in B-CLL (69,70).
VEGF and interleukin-6 (IL-6) are able to upregulate MCL-1 via
autocrine signaling loops. VEGF may be a positive autocrine
regulator of MCL-1 in B-CLL. In addition, specific downregulation
of MCL-1 gene expression was discovered to promote apoptosis and
death of primary B-CLL cells, suggesting the possibility of the
inhibition of VEGF, and its pathway may prove useful in the
treatment of B-CLL patients (71)
(Fig. 2).
4. In vivo studies of VEGF/VEGFR in
patients with leukemia
VEGF/VEGFR in acute lymphocytic leukemia
(ALL)
To the best of our knoweldge, Perez-Atayde et
al (72) first demonstrated
that leukemia progression was correlated to increased bone marrow
vascularization. It was demonstrated that ALL patients had an
increased blood vessel content compared to their normal
counterparts. There were elevated levels of proangiogenic growth
factors including basic fibroblast growth factor (bFGF) and VEGF in
urine and peripheral blood samples from ALL patients. The factors
were correlated with bone marrow angiogenesis (72,73).
The existence of an ‘angiogenesis switch’ first proposed for solid
tumors is suggested to apply to hematolymphoid malignancies
(3). The ‘angiogenesis switch’ in
leukemias is indicated to increase bone marrow microvessel density
(MVD), the expression of HIF-1, multiple proangiogenic factors
(VEGF, bFGF and angiopoietin-2) and soluble VEGFR, but a decrease
in the expression of endogenous angiogenesis inhibitors (74,75).
In a recent study, MVD was found to be higher in T-ALL compared to
B-ALL patients (76). In the B-ALL
group, cases with t(12;21) were characterized by a low MVD, while
patients with hyperdiploid leukemia displayed a high MVD (3). The correlation between MVD and white
blood cell count (WBC) may be a determination of high-risk for
B-ALL patients. In addition, patients with a high marrow reticulin
fiber density and high MVD exhibit an unfavorable outcome,
indicating the possible cellular origin of certain proangiogenic
factors (76). The following
clinical studies support the capacity of leukemia cells to produce
proangiogenic growth factors including VEGF and bFGF in
vitro (77).
To further detect whether tumor progression is in
concert with the induction of tumor angiogenesis in leukemia,
angiogenesis was characterized by immunohistochemical staining of
factor VIII (FVIII) in bone marrow biopsies and was quantified by
MVD assessment in patients with newly diagnosed ALL or after the
completion of remission induction chemotherapy. The results
suggested that the leukemia was angiogenesis-dependent. This
increased the possibility for antiangiogenic drugs to treat
leukemia (78). The high plasma
VEGF concentration was linked to leukemic cell invasion in adult
T-cell leukemia (ATL) and the major source was from ATL cells
themselves (79). Noteworthy, all
cell lines were observed to express mRNA and protein of VEGFR-1. In
clinical specimens, similar results were also detected in all
(100%) of 11 and 8 (73%) of 11 ATL patients, respectively. VEGF was
effectively bound only to VEGFR-1-expressing cells. Further study
demonstrated that the key role of VEGF in ATL is to assist in cell
invasion, not proliferation. ATL cells upregulate their own
chemotaxis to facilitate the invasion into various organs through
the combination of VEGF and VEGFR-1 (80). Besides that, the angiogenic effect
of VEGF seems to be dependent on nitric oxide (NO). The
associations among functional polymorphisms in VEGF (−2578C>A,
−1154G>A and −634G>C) and NOS3 (−786T>C, intron 4 b>a
and Glu298Asp) were examined with the prognosis of childhood ALL.
The results indicate that polymorphisms of VEGF and NOS3 genes are
highly associated with the risk of relapse, therefore it may be a
prognostic sign in childhood ALL (81).
VEGF/VEGFR in acute myeloid leukemia
In recent studies, the expression of VEGF/VEGFR in
AML patients has been detected. The increased levels of plasma VEGF
have been correlated with reduced survival and lower remission
rates (4), and the level of
plasma/serum VEGF was found to be related to the number of
circulating blasts (82). In
addition to the modulation of bone marrow angiogenesis by VEGF from
leukemia cells, it was confirmed that the expression of
endothelial-specific tyrosine kinase receptors, such as VEGFR-1, -2
and -3, are also observed in leukemic cells (83,84).
De Bont et al (85) reported
that new vessel formation was the result of angiogenesis and
vasculogenesis. The degree of neovascularization in the bone marrow
was correlated with VEGF expression in the leukemic cells. The bone
vessel count (MVC) was higher in 23 cases of AML patients compared
to that in the normal controls, and the cell proliferation, bone
marrow angiogenesis and expression of VEGF were correlated to each
other in patients with AML (85).
To further assess cellular VEGF levels and their prognostic
significance in newly diagnosed AML, a radioimmunoassay (RIA) was
performed to quantify VEGF levels in stored samples from 99
patients diagnosed with AML. Although the patients with an
increased VEGF level had a shorter survival, there was no evidence
to demonstrate the significant relationship between VEGF level and
WBC or blast count in AML. In contrast, the results suggest that
the cellular VEGF level is an independent predictor of outcome in
AML (86). Interestingly, the
co-expression of CD147 and VEGF may indicate a poor prognosis in
AML and may be a highly sensitive marker for predicting the
clinical outcome of patients (87).
As to the induction of angiogenesis by VEGF in AML,
Fielder et al (88)
investigated the expression of VEGF and its receptors in fresh
leukemic blasts and addressed the possible loops for the
stimulation of AML blasts. The autocrine VEGF worked through
VEGFR-2, by activating eNOS to produce NO through PI3-K/Akt kinase,
and maintained clonogenic cell growth in the OCI/AML-2 cell line
(89). Also, Fielder et al
(90) reported that VEGFR-3
represented a cloned member of class III receptor tyrosine kinase
with VEGFR-1 and VEGFR-2. The ligand of VEGFR-3 has been identified
as VEGF-C that shares sequence homology with VEGF and PlGF. VEGF-C
expression was discovered in leukemic samples of 4 out of 7
VEGFR-3-positive and 4 out of 6 VEGFR-3-negative patients (90). MVD was increased in the bone marrow
of patients with AML, supporting the hypothesis that angiogenesis
plays an important role in AML (61). High VEGF-C mRNA expression in AML
blasts is related to drug resistance in vitro and in
vivo. The prognostic significance and associated gene
expression profiles of VEGF-C with long-term outcome remain to be
defined. However, several effects of VEGF-C on the treatment and
gene expression profiles were investigated by using microarray
analysis in 525 adult and 100 pediatric AML patients. The results
showed that increased VEGF-C predicted adverse long-term prognosis,
which provided additional information to well-known factors
(91).
VEGF/VEGFR in chronic myeloid leukemia or
chronic lymphocytic leukemia
Aguayo et al (36) evaluated the blood vessels in 145
bone marrow biopsies and the levels of VEGF, bFGF, TNF-α, TGF-α and
HGF in 417 plasma samples. Except for CLL, vascularity was
significantly higher in all leukemias compared with the control
bone marrows. The highest number of blood vessels and the largest
vascular area were discovered in CML. At the same time the highest
levels of VEGF in plasma were detected in CML, while the highest
levels of bFGF were in CLL. The level of HGF was highest in chronic
myelomonocytic leukemia (CMML). Molica et al (92) evaluated the levels of VEGF in serum
in B-CLL, and the results indicated that increased serum levels of
VEGF may be considered in predicting the risk of disease
progression. Ferrajoli et al (93) discovered VEGFR-2 was a high-affinity
VEGF receptor that plays a role in de novo blood vessel
formation and hematopoietic cell development. Cellular VEGFR-2
levels may serve as a prognostic factor in CLL. All evidence
supports that VEGF is a critical microenvironmental factor, and
VEGF inhibition may be a promising new therapeutic approach in CLL.
For example, vatalanib and pazopanib seem to be effective and safe
candidates. More evaluation will be required to confirm these
results.
5. Antiangiogenic therapy in leukemias
Antiangiogenic therapy in animal models
with leukemias
According to the relationship between VEGF/VEGFR and
leukemias, antiangiogenic therapy was used to treat leukemias in
animal models. Dias et al (94) reported, using an in vivo
model of human leukemia, that blocking angiogenesis induced by the
interaction of leukemia-derived VEGF with murine VEGFR-2 delays
leukemic growth. Although it was not sufficient for its eradication
from inoculated mice, these results demonstrated that targeting
VEGF-induced angiogenesis may be at least partially effective in
delaying the progression of leukemia. Interestingly, long-term
remission was achieved only when mice were treated with
neutralizing monoclonal antibody (mAb) against murine and human
VEGFR-2, blocking the paracrine and autocrine VEGF-VEGFR-2
signaling pathways. On the other hand, mAbs against murine or human
VEGFR-1 had no effect towards improving the survival of the
leukemic mice, suggesting that the VEGF/VEGFR-2 pathway is more
important for the proliferation and engraftment of acute leukemias
in vivo. However, it is also possible that several leukemias
may depend on VEGFR-1 signaling (95). In order to further examine the role
of VEGF in AML progression in vivo, a number of researchers
established two mouse models. In a murine chloroma model, the
delivery of VEGF using microencapsulation technology resulted in
enhanced tumor growth and vascularization, whereas treatment with a
VEGF antagonist soluble NRP-1 (sNRP-1) inhibited tumor angiogenesis
and growth. In a systemic leukemia model, the survival of mice
injected with adenovirus (Ad) encoding for Fc-sNRP-1 was
significantly prolonged as compared with mice injected with
Ad-LacZ. Further analysis showed a reduction in circulating
leukemic cells and infiltration of liver and spleen as well as bone
marrow neovascularization and cellularity (96). Liu et al (97) demonstrated that adenoviral gene
therapy with antiangiogenic fragments of thrombospondin-1 inhibited
leukemic xenograft growth in mice, which addressed the possibility
that reduced production of angiogenesis inhibitors by leukemic
cells may trigger the onset of the neovascularization process, by
shifting the local (bone marrow) angiogenesis balance.
In contrast to the positive roles in leukemia
disease, emerging evidence from genetically modified animal models
suggests that elevated levels of VEGF, or a proangiogenic
phenotype, may impede, rather than promote, early tumor development
and progression (98). Researchers
reported a tumor inhibitory role for VEGF by demonstrating that a
2-fold overexpression of systemic levels of VEGF in mice
heterozygous for a VEGF ‘hypermorphic’ allele decelerated
tumorigenesis in a retroviral-induced, spontaneous murine leukemia
model (99). Alterations in the
innate immune function, specifically enhanced natural killer cell
activity, and increased hematopoietic progenitor cell survival were
identified as acquired phenotypes that strongly correlated with and
were likely responsible for this leukemic inhibition (99). Accordingly, Ebos et al
(100) recently showed that
blockade of the VEGF pathway, before tumor induction, led to a more
aggressive metastasis and shortened survival. Therefore, further
experimental and clinical studies are required to clarify the
controversy surrounding the dichotomous roles of VEGF in tumor
angiogenesis.
Antiangiogenic therapy in patients with
leukemias
The first antiangiogenic agent to be approved was
bevacizumab, a humanized anti-VEGF monoclonal antibody.
Administration of bevacizumab, in combination with cytotoxic
chemotherapy, confers benefits to patients with several types of
solid cancers (101–103). Additionally, two small-molecule
inhibitors targeting VEGFR and other kinases, sorafenib and
sunitinib, have been approved for treating renal cells- and
hepatocellular carcinoma (104,105). These are currently under
investigation for patients with relapsed and refractory acute
leukemia in combination with standard chemotherapy (106). The major classes of antiangiogenic
therapy include direct anti-VEGF acting molecules (anti-VEGF
antibodies, VEGF-antisense nucleotides), immunomodulatory drugs
(IMIDs) with antiangiogenic properties, receptor tyrosine kinase
inhibitors that target VEGFR signaling as well as receptors of
other (proangiogenic) factors, the anti-endothelial approach of
metronomic therapy, and other new compounds targeting signaling
downstream to proangiogenic growth factors, such as mTOR
inhibitors, histone deacetylase (HDAC) inhibitors and proteasome
inhibitors. Moreover, angiogenesis appears to be targeted even by
conventional chemotherapy in different leukemias (61) (Table
I). For example, bevacizumab is a humanized murine anti-human
VEGF monoclonal IgG1 antibody that blocks the binding of human VEGF
to its receptors VEGFR-1 and -2 (107). It was administered after
chemotherapy to 48 adults with refractory or relapsed AML. The
overall response was 23 of 48 (48%), with complete response (CR) in
16 (33%). MVD decreased in the bone marrow after bevacizumab
administration. Currently, bevacizumab is being evaluated as a
treatment option for newly diagnosed AML patients in combination
with cytarabine and idarubicin in a phase II study. Thalidomide is
administered as an antineoplastic agent after the demonstration of
its antiangiogenic activity (108). The newer IMIDs lenalidomide, a
synthetic compound derived by modifying the chemical structure of
thalidomide, was observed to have 2–3 times more potent
antiangiogenic activity than thalidomide in various in vivo
assays (109) and the
antiangiogenic activity has been demonstrated to be independent of
their immunomodulatory effects (110). In a phase II study by Thomas et
al (111), thalidomide was
analyzed in 16 patients with relapsed or refractory AML; one
patient (6%) achieved CR lasting for 36 months. There was no
correlation between the reduction in angiogenesis marker levels and
responses. In a phase I/II trial by Steins et al (112) in 20 AML patients, a partial
response was observed in four patients. In parallel, MVD
significantly decreased in five patients during treatment with
thalidomide. In a study by Barr et al (113), thalidomide was examined in
combination with fludarabine, carboplatin, and topotecan in 42
patients with poor AML prognosis and 10 of 42 (24%) patients
achieved a CR. Small tyrosine kinase inhibitors that target VEGFR
are a further important class of antiangiogenic drugs. For example,
vatalanib is an oral angiogenesis inhibitor that offers a novel
approach to inhibiting tumor growth (114) by interfering with the ATP binding
sites of VEGFR. Ongoing studies are now focusing on evaluating the
efficacy of vatalanib in combination with imatinib in a phase I/II
trial for patients with AML, PMF and blast phase of CML, or in
combination with cytosine arabinoside and daunorubicin in patients
with AML (115). Cediranib
(AZD2171, Recentin) is a potent inhibitor of both VEGFR-1 and -2
(116). In a phase I study with
cediranib in 35 AML patients, six patients experienced an objective
response. There was a correlation between cediranib exposure and
plasma VEGF levels (117). A
combination therapy of thalidomide and 5-azacytidine, a
hypomethylating drug, was assessed in 40 patients with AML
(118). Hematological improvement
was observed in 15 of 36 patients (42%), stable disease was
observed in 5 of 36 patients (14%), 10 of 36 patients (28%) had
disease progression and 6 had CR.
 | Table ISummary of clinical trials and
approved antiangiogenic therapies in patients with leukemia. |
Table I
Summary of clinical trials and
approved antiangiogenic therapies in patients with leukemia.
| Anti-VEGF
strategies | RTK inhibitors |
Immunomodulators |
|---|
|
|
|
|
|---|
| Patients | Target | Bevacizumab | Target | Vatalanib | Cediranib | Thalidomide | Lenalidomide |
|---|
| AML | VEGF-A | + | VEGFR1-3 | + | + | + | + |
| CML | VEGF-A | + | VEGFR1-3 | + | / | / | / |
| CLL | VEGF-A | + | / | + | + | + | + |
6. Conclusion
This investigation demonstrates that angiogenesis
has important biological and prognostic implications in
hematolymphoid malignancies. The autocrine regulators of
angiogenesis are essential to the development of diseases.
VEGF/VEGFR as key regulators of neoangiogenesis and vasculogenesis
have been widely studied. Although the mechanism of the high
expression of VEGF in leukemia cells has not yet been identified,
we are certain that VEGF/VEGFR interactions may stimulate
proliferation, migration and survival of leukemia/lymphoma cells by
autocrine and paracrine loops. In clinical studies, the elevated
level of VEGF may contribute to the adverse outcome by promoting
leukemic cell growth, survival, migration and reduce the
sensitivity of cells to therapeutic agent-induced apoptosis.
Therefore, the interference of VEGF/VEGFR-related pathway being an
ideal candidate in treating leukemia may induce antiangiogenesis
and inhibit growth of leukemia cells. All clinical trials causing
VEGFR-2 gene polymorphism relates to cytogenetic response
(treatment failure following imatinib therapy for CML) and the VEGF
genotype relates to the progression of advanced disease. Apart from
anti-VEGF molecules, other antiangiogenic therapies include IMIDs,
receptor tyrosine kinase inhibitors, anti-endothelial approach to
metronomic therapy, mTOR inhibitors, HDAC inhibitors and proteasome
inhibitors. A better understanding of the role of VEGF in leukemia
and additional trials combining antiangiogenic therapies will
provide a greater insight to the mechanisms required for
treatment.
Acknowledgements
This study was supported by the Key Foundation of
Shandong Science Project (2007GG2002023), The Shandong 1020 Project
(2008-2), The Natural Science Foundation of Shandong (Y2008C165)
and the Natural Science Foundation of China (30810444).
References
|
1
|
Kerbel RS: Tumor angiogenesis. N Engl J
Med. 358:2039–2049. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ellis LM and Hicklin DJ: VEGF-targeted
therapy: mechanisms of anti-tumour activity. Nat Rev Cancer.
8:579–591. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Medinger M, Fischer N and Tzankov A:
Vascular endothelial growth factor-related pathways in
hemato-lymphoid malignancies. J Oncol. 2010:7297252010. View Article : Google Scholar
|
|
4
|
Aguayo A, Kantarjian HM, Estey EH, et al:
Plasma vascular endothelial growth factor levels have prognostic
significance in patients with acute myeloid leukemia but not in
patients with myelodysplastic syndromes. Cancer. 95:1923–1930.
2002. View Article : Google Scholar
|
|
5
|
Zhu Z, Hattori K, Zhang H, et al:
Inhibition of human leukemia in an animal model with human
antibodies directed against vascular endothelial growth factor
receptor 2. Correlation between antibody affinity and biological
activity. Leukemia. 17:604–611. 2003. View Article : Google Scholar
|
|
6
|
Benouchan M and Colombo BM:
Anti-angiogenic strategies for cancer therapy. Int J Oncol.
27:563–571. 2005.PubMed/NCBI
|
|
7
|
Ferrara N and Henzel WJ: Pituitary
follicular cell secrete a novel heparin-binding growth factor
specific for vascular endothelial cell. Biochem Biophys Res Commun.
161:8511989. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ferrara N, Gerber HP and LeCouter J: The
biology of VEGF and its receptors. Nat Med. 9:669–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cao Y: Positive and negative modulation of
angiogenesis by VEGFR1 ligands. Sci Signal. 2:re12009.PubMed/NCBI
|
|
10
|
Zhang L, Zhou F, Han W, et al: VEGFR-3
ligand-binding and kinase activity are required for
lymphangiogenesis but not for angiogenesis. Cell Res. 20:1319–1331.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu Y, Hooper AT, Zhong Z, et al: The
vascular endothelial growth factor receptor (VEGFR-1) supports
growth and survival of human breast carcinoma. Int J Cancer.
119:1519–1529. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Autiero M, Luttun A, Tjwa M and Carmeliet
P: Placental growth factor and its receptor, vascular endothelial
growth factor receptor-1: novel targets for stimulation of ischemic
tissue revascularization and inhibition of angiogenic and
inflammatory disorders. J Thromb Haemost. 1:1356–1370. 2003.
View Article : Google Scholar
|
|
13
|
Holmes K, Roberts OL, Thomas AM and Cross
MJ: Vascular endothelial growth factor receptor-2: structure,
function, intracellular signalling and therapeutic inhibition. Cell
Signal. 19:2003–2012. 2007. View Article : Google Scholar
|
|
14
|
Brekken RA, Overholser JP, Stastny VA,
Waltenberger J, Minna JD and Thorpe PE: Selective inhibition of
vascular endothelial growth factor (VEGF) receptor 2 (KDR/Flk-1)
activity by a monoclonal anti-VEGF antibody blocks tumor growth in
mice. Cancer Res. 60:5117–5124. 2000.PubMed/NCBI
|
|
15
|
Katoh O, Tauchi H, Kawaishi K, Kimura A
and Satow Y: Expression of the vascular endothelial growth factor
(VEGF) receptor gene, KDR, in hematopoietic cells and inhibitory
effect of VEGF on apoptotic cell death caused by ionizing
radiation. Cancer Res. 55:5687–5692. 1995.PubMed/NCBI
|
|
16
|
Cursiefen C, Chen L, Borges LP, et al:
VEGF-A stimulates lymphangiogenesis and hemangiogenesis in
inflammatory neovascularization via macrophage recruitment. J Clin
Invest. 113:1040–1050. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Adams J, Carder PJ, Downey S, et al:
Vascular endothelial growth factor (VEGF) in breast cancer:
comparison of plasma, serum and tissue VEGF and microvessel density
and effects of tamoxifen. Cancer Res. 60:2898–2905. 2000.PubMed/NCBI
|
|
18
|
Kay NE, Bone ND, Tschumper RC, et al:
B-CLL cells are capable of synthesis and secretion of both pro- and
anti-angiogenic molecules. Leukemia. 16:911–919. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rafii S, Lyden D, Benezra R, Hattori K and
Heissig B: Vascular and haematopoietic stem cells: novel targets
for anti-angiogenesis therapy? Nat Rev Cancer. 2:826–835. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shweiki D, Itin A, Soffer D and Keshet E:
Vascular endothelial growth factor induced by hypoxia may mediate
hypoxia-initiated angiogenesis. Nature. 359:843–845. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dor Y, Porat R and Keshet E: Vascular
endothelial growth factor and vascular adjustments to perturbations
in oxygen homeostasis. Am J Physiol Cell Physiol. 280:C1367–C1374.
2001.PubMed/NCBI
|
|
22
|
Kaelin WG Jr: The von Hippel-Lindau tumor
suppressor protein and clear cell renal carcinoma. Clin Cancer Res.
13:680s–682s. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cong XL, Li B, Yang RC, Feng SZ, Chen SJ
and Han ZC: Enhanced growth suppression of Philadephia1 leukemia
cells by targeting bcr3/abl2 and VEGF through antisense strategy.
Leukemia. 19:1517–1524. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ghosh AK, Shanafelt TD, Cimmino A, et al:
Aberrant regulation of pVHL levels by microRNA promotes the
HIF/VEGF axis in CLL B cells. Blood. 113:5568–5574. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Withey JM, Marley SB, Kaeda J, Harvey AJ,
Crompton MR and Gordon MY: Targeting primary human leukemia cells
with RNA interference: Bcr-Abl targeting inhibits myeloid
progenitor self-renewal in chronic myeloid leukaemia cells. Br J
Haematol. 129:377–380. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ghosh AK, Secreto CR, Knox TR, Ding W,
Mukhopadhyay D and Kay NE: Circulating microvesicles in B-cell
chronic lymphocytic leukemia can stimulate marrow stromal cells:
implications for disease progression. Blood. 115:1755–1764. 2010.
View Article : Google Scholar
|
|
28
|
Aggarwal BB, Sethi G, Ahn KS, et al:
Targeting signal-transducer-and-activator-of-transcription-3 for
prevention and therapy of cancer:modern target but ancient
solution. Ann NY Acad Sci. 1091:151–169. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Eriksen KW, Kaltoft K, Mikkelsen G, et al:
Constitutive STAT3-activation in Sezary syndrome: tyrphostin AG490
inhibits STAT3-activation, interleukin-2 receptor expression and
growth of leukemic Sezary cells. Leukemia. 15:787–793. 2001.
View Article : Google Scholar
|
|
30
|
Nielsen M, Nissen MH, Gerwien J, et al:
Spontaneous interleukin-5 production in cutaneous T-cell lymphoma
lines is mediated by constitutively activated Stat3. Blood.
99:973–977. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Minet E, Michel G, Mottet D, et al: c-JUN
gene induction and AP-1 activity is regulated by a JNK-dependent
pathway in hypoxic HepG2 cells. Exp Cell Res. 265:114–124. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pages G, Berra E, Milanini J, Levy AP and
Pouyssegur J: Stress-activated protein kinases (JNK and p38/HOG)
are essential for vascular endothelial growth factor mRNA
stability. J Biol Chem. 275:26484–26491. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Krejsgaard T, Vetter-Kauczok CS, Woetmann
A, et al: Jak3- and JNK-dependent vascular endothelial growth
factor expression in cutaneous T-cell lymphoma. Leukemia.
20:1759–1766. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kumar SA, Hu X, Brown M, et al:
Lysophosphatidic acid receptor expression in chronic lymphocytic
leukemia leads to cell survival mediated though vascular
endothelial growth factor expression. Leuk Lymphoma. 50:2038–2048.
2009. View Article : Google Scholar
|
|
35
|
Hu X, Mendoza FJ, Sun J, Banerji V,
Johnston JB and Gibson SB: Lysophosphatidic acid (LPA) induces the
expression of VEGF leading to protection against apoptosis in
B-cell derived malignancies. Cell Signal. 20:1198–1208. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Aguayo A, Kantarjian H, Manshouri T, et
al: Angiogenesis in acute and chronic leukemias and myelodysplastic
syndromes. Blood. 96:2240–2245. 2000.PubMed/NCBI
|
|
37
|
Bellamy WT, Richter L, Sirjani D, et al:
Vascular endothelial cell growth factor is an autocrine promoter of
abnormal localized immature myeloid precursors and leukemia
progenitor formation in myelodysplastic syndromes. Blood.
97:1427–1434. 2001. View Article : Google Scholar
|
|
38
|
Braun T, Carvalho G, Fabre C, Grosjean J,
Fenaux P and Kroemer G: Targeting NF-kappaB in hematologic
malignancies. Cell Death Differ. 13:748–758. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhao T, Yasunaga J, Satou Y, et al: Human
T-cell leukemia virus type 1 bZIP factor selectively suppresses the
classical pathway of NF-kappaB. Blood. 113:2755–2764. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sujobert P, Bardet V, Cornillet-Lefebvre
P, et al: Essential role for the p110delta isoform in
phosphoinositide 3-kinase activation and cell proliferation in
acute myeloid leukemia. Blood. 106:1063–1066. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ramos AM, Fernandez C, Amran D, Sancho P,
de Blas E and Aller P: Pharmacologic inhibitors of PI3K/Akt
potentiate the apoptotic action of the antileukemic drug arsenic
trioxide via glutathione depletion and increased peroxide
accumulation in myeloid leukemia cells. Blood. 105:4013–4020. 2005.
View Article : Google Scholar
|
|
42
|
Shi Q, Le X, Abbruzzese JL, et al:
Constitutive Sp1 activity is essential for differential
constitutive expression of vascular endothelial growth factor in
human pancreatic adenocarcinoma. Cancer Res. 61:4143–4154.
2001.
|
|
43
|
Hsu TC, Young MR, Cmarik J and Colburn NH:
Activator protein 1 (AP-1) and nuclear factor kappaB (NF-kappa
B)-dependent transcriptional events in carcinogenesis. Free Radic
Biol Med. 28:1338–1348. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Angel P and Karin M: The role of Jun, Fos
and the AP-1 complex in cell proliferation and transformation.
Biochim Biophys Acta. 1072:129–157. 1991.PubMed/NCBI
|
|
45
|
Pollmann C, Huang X, Mall J, Bech-Otschir
D, Naumann M and Dubiel W: The constitutive photomorphogenesis 9
signalosome directs vascular endothelial growth factor production
in tumor cells. Cancer Res. 61:8416–8421. 2001.PubMed/NCBI
|
|
46
|
Poulaki V, Mitsiades CS, McMullan C, et
al: Regulation of vascular endothelial growth factor expression by
insulin-like growth factor I in thyroid carcinomas. J Clin
Endocrinol Metab. 88:5392–5398. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chuang CH, Huang CS and Hu ML: Vitamin E
and rutin synergistically inhibit expression of vascular
endothelial growth factor through down-regulation of binding
activity of activator protein-1 in human promyelocytic leukemia
(HL-60) cells. Chem Biol Interact. 183:434–441. 2010. View Article : Google Scholar
|
|
48
|
Rho SB, Choi K, Park K and Lee JH:
Inhibition of angiogenesis by the BTB domain of promyelocytic
leukemia zinc finger protein. Cancer Lett. 294:49–56. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Konopka JB, Watanabe SM and Witte ON: An
alteration of the human c-abl protein in K562 leukemia cells
unmasks associated tyrosine kinase activity. Cell. 37:1035–1042.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Pendergast AM, Quilliam LA, Cripe LD, et
al: BCR-ABL-induced oncogenesis is mediated by direct interaction
with the SH2 domain of the GRB-2 adaptor protein. Cell. 75:175–185.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Carlesso N, Frank DA and Griffin JD:
Tyrosyl phosphorylation and DNA binding activity of signal
transducers and activators of transcription (STAT) proteins in
hematopoietic cell lines transformed by Bcr/Abl. J Exp Med.
183:811–820. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sillaber C, Gesbert F, Frank DA, Sattler M
and Griffin JD: STAT5 activation contributes to growth and
viability in Bcr/Abl-transformed cells. Blood. 95:2118–2125.
2000.PubMed/NCBI
|
|
53
|
Nieborowska-Skorska M, Wasik MA, Slupianek
A, Salomoni P, Kitamura T, Calabretta B and Skorski T: Signal
transducer and activator of transcription (STAT)5 activation by
BCR/ABL is dependent on intact Src homology (SH)3 and SH2 domains
of BCR/ABL and is required for leukemogenesis. J Exp Med.
189:1229–1242. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Skorski T, Bellacosa A,
Nieborowska-Skorska M, Majewski M, Martinez R, Choi JK, et al:
Transformation of hematopoietic cells by BCR/ABL requires
activation of a PI-3k/Akt-dependent pathway. EMBO J. 16:6151–6161.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hwan D, Kim, Xu W, et al: Lipton genetic
variants in the candidate genes of the apoptosis pathway and
susceptibility to chronic myeloid leukemia. Blood. 113:2517–2525.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Li L, Zhang R, Fang ZY, Chen JN and Zhu
ZL: Suppression of vascular endothelial growth factor (VEGF)
expression by targeting the Bcr-Abl oncogene and protein tyrosine
kinase activity in Bcr-Abl-positive leukaemia cells. J Int Med Res.
37:426–437. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Böhm A, Aichberger KJ, Mayerhofer M, et
al: Targeting of mTOR is associated with decreased growth and
decreased VEGF expression in acute myeloid leukaemia cells. Eur J
Clin Invest. 39:395–405. 2009.PubMed/NCBI
|
|
58
|
Venables JP: Unbalanced alternative
splicing and its significance in cancer. Bioessays. 28:378–386.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Podar K, Tai YT and Davies FE: Vascular
endothelial growth factor triggers signaling cascades mediating
multiple myeloma cell growth and migration. Blood. 98:428–435.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Hussong JW, Rodgers GM and Shami PJ:
Evidence of increased angiogenesis in patients with acute myeloid
leukemia. Blood. 95:390–313. 2000.PubMed/NCBI
|
|
61
|
Padró T, Ruiz S, Bieker R, Bürger H,
Steins M, Kienast J, et al: Increased angiogenesis in the bone
marrow of patients with acute myeloid leukemia. Blood.
95:2637–2644. 2000.
|
|
62
|
Abrams ST, Lakum T, Lin K, et al: B-cell
receptor signaling in chronic lymphocytic leukemia cells is
regulated by overexpressed active protein kinase CβII. Blood.
109:1193–1201. 2007.PubMed/NCBI
|
|
63
|
Gora-Tybor J, Blonski JZ and Robak T:
Circulating vascular endothelial growth factor (VEGF) and its
soluble receptors in patients with chronic lymphocytic leukemia.
Eur Cytokine Netw. 16:41–46. 2005.PubMed/NCBI
|
|
64
|
Abrams ST, Brown BR, Zuzel M and Slupsky
JR: Vascular endothelial growth factor stimulates protein kinase
CβII expression in chronic lymphocytic leukaemia cells. Blood.
115:4447–4454. 2010.
|
|
65
|
Ugarte-Berzal E, Redondo-Muñoz J, Eroles
P, et al: VEGF/VEGFR2 interaction down-regulates matrix
metalloproteinase-9 via STAT1 activation and inhibits B chronic
lymphocytic leukemia cell migration. Blood. 115:846–849. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Hata AN and Breyer RM: Pharmacology and
signaling of prostaglandin receptors: multiple roles in
inflammation and immune modulation. Pharmacol Ther. 103:147–166.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wu G, Luo J, Rana JS, Laham R, Sellke FW
and Li J: Involvement of COX-2 in VEGF-induced angiogenesis via P38
and JNK pathways in vascular endothelial cells. Cardiovasc.
69:512–519. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Su JL, Shih JY, Yen ML, et al:
Cyclooxygenase-2 induces EP1- and HER-2/Neu-dependent vascular
endothelial growth factor-C up-regulation: a novel mechanism of
lymphangiogenesis in lung adenocarcinoma. Cancer Res. 64:554–564.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Véronèse L, Tournilhac O, Verrelle P, et
al: Low MCL-1 mRNA expression correlates with prolonged survival in
B-cell chronic lymphocytic leukemia. Leukemia. 22:1291–1293.
2008.PubMed/NCBI
|
|
70
|
Pepper C, Lin TT, Pratt G, et al: Mcl-1
expression has in vitro and in vivo significance in chronic
lymphocytic leukemia and is associated with other poor prognostic
markers. Blood. 112:3807–3817. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Véronèse L, Tournilhac O, Verrelle P, et
al: Strong correlation between VEGF and MCL-1 mRNA expression
levels in B-cell chronic lymphocytic leukemia. Leuk Res.
33:1623–1626. 2009.PubMed/NCBI
|
|
72
|
Perez-Atayde AR, Sallan SE, Tedrow U,
Connors S, Allred E and Folkman J: Spectrum of tumor angiogenesis
in the bone marrow of children with acute lymphoblastic leukemia.
Am J Pathol. 150:815–821. 1997.PubMed/NCBI
|
|
73
|
Yetgin S, Yenicesu I, Etin MC and Tuncer
M: Clinical importance of serum vascular endothelial and basic
fibroblast growth factors in children with acute lymphoblastic
leukemia. Leuk Lymphoma. 42:83–88. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Dong X, Han ZC and Yang R: Angiogenesis
and antiangiogenic therapy in hematologic malignancies. Crit Rev
Oncol Hematol. 62:105–118. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Frater JL, Kay NE, Goolsby CL, Crawford
SE, Dewald GW and Peterson LC: Dysregulated angiogenesis in
B-chronic lymphocytic leukemia: morphologic, immunohistochemical,
and flow cytometric evidence. Diagn Pathol. 3:162008. View Article : Google Scholar
|
|
76
|
Norén-Nyström U, Heyman M, Frisk P, et al:
Vascular density in childhood acute lymphoblastic leukaemia
correlates to biological factors and outcome. Br J Haematol.
146:521–530. 2009.PubMed/NCBI
|
|
77
|
Chen H, Treweeke AT, West DC, Till KJ,
Cawley JC, Zuzel M and Toh CH: In vitro and in vivo production of
vascular endothelial growth factor by chronic lymphocytic leukemia
cells. Blood. 96:3181–3187. 2000.PubMed/NCBI
|
|
78
|
Folkman J: Fundamental concepts of the
angiogenic process. Curr Mol Med. 3:643–651. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Verstovsek S, Lunin S, Kantarjian H, et
al: Clinical relevance of VEGF receptors 1 and 2 in patients with
chronic myelogenous leukemia. Leuk Res. 27:661–669. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Hayashibara T, Yamada Y, Miyanishi T, et
al: Vascular endothelial growth factor and cellular chemotaxis: a
possible autocrine pathway in adult T-cell leukemia cell invasion.
Clin Cancer Res. 7:2719–2726. 2001.PubMed/NCBI
|
|
81
|
Demacq C, Vasconcellos VB, Izidoro-Toledo
TC, et al: Vascular endothelial growth factor (VEGF) and
endothelial nitric oxide synthase (NOS3) polymorphisms are
associated with high relapse risk in childhood acute lymphoblastic
leukemia (ALL). Clin Chim Acta. 411:1335–1405. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Fragoso R, Elias AP and Dias S: Autocrine
VEGF loops, signaling pathways, and acute leukemia regulation. Leuk
Lymphoma. 48:481–488. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Bellamy WT: Expression of vascular
endothelial growth factor and its receptors in multiple myeloma and
other hematopoietic malignancies. Semin Oncol. 28:551–559. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Bairey O, Boycov O, Kaganovsky E, Zimra Y,
Shaklai M and Rabizadeh E: All three receptors for vascular
endothelial growth factor (VEGF) are expressed on B-chronic
lymphocytic leukemia (CLL) cells. Leuk Res. 28:243–248. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
de Bont ES, Neefjes VM, Rosati S, Vellenga
E and Kamps WA: New vessel formation and aberrant VEGF/VEGFR
signaling in acute leukemia: does it matter? Leuk Lymphoma.
43:1901–1909. 2002.PubMed/NCBI
|
|
86
|
Aguayo A, Estey E, Kantarjian H, et al:
Cellular vascular endothelial growth factor is a predictor of
outcome in patients with acute myeloid leukemia. Blood.
94:3717–3721. 1999.PubMed/NCBI
|
|
87
|
Fu J, Fu J, Chen X, Zhang Y, Gu H and Bai
Y: CD147 and VEGF co-expression predicts prognosis in patients with
acute myeloid leukemia. Jpn J Clin Oncol. 40:1046–1052. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Fiedler W, Graeven U, Ergün S, et al:
Vascular endothelial growth factor, a possible paracrine growth
factor in human acute myeloid leukemia. Blood. 89:1870–1875.
1997.PubMed/NCBI
|
|
89
|
Koistinen P, Siitonen T, Mäntymaa P, et
al: Regulation of the acute myeloid leukemia cell line OCI/AML-2 by
endothelial nitric oxide synthase under the control of a vascular
endothelial growth factor signaling system. Leukemia. 15:1433–1441.
2001. View Article : Google Scholar
|
|
90
|
Fielder W, Graeven U, Ergün S, et al:
Expression of FLT4 and its ligand VEGF-C in acute myeloid leukemia.
Leukemia. 11:1234–1237. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
de Jonge HJ, Valk PJ, Veeger NJ, et al:
High VEGFC expression is associated with unique gene expression
profiles and predicts adverse prognosis in pediatric and adult
acute myeloid leukemia. Blood. 116:1747–1754. 2010.PubMed/NCBI
|
|
92
|
Molica S, Vitelli G, Levato D, Gandolfo GM
and Liso V: Increased serum levels of vascular endothelial growth
factor predict risk of progression in early B-cell chronic
lymphocytic leukaemia. Br J Haematol. 107:605–610. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Ferrajoli A, Manshouri T, Estrov Z, et al:
High levels of vascular endothelial growth factor receptor-2
correlate with shortened survival in chronic lymphocytic leukemia.
Clin Cancer Res. 7:795–799. 2001.PubMed/NCBI
|
|
94
|
Dias S, Hattori K, Zhu Z, et al: Autocrine
stimulation of VEGFR-2 activates human leukemic cell growth and
migration. J Clin Invest. 106:511–521. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Dias S, Hattori K, Heissig B, et al:
Inhibition of both paracrine and autocrine VEGF/VEGFR-2 signaling
pathways is essential to induce long-term remission of
xenotransplanted human leukemias. Proc Natl Acad Sci USA.
98:10857–10862. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Schuch G, Machluf M, Bartsch G Jr, et al:
In vivo administration of vascular endothelial growth factor (VEGF)
and its antagonist, soluble neuropilin-1, predicts a role of VEGF
in the progression of acute myeloid leukemia in vivo. Blood.
100:4622–4628. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Liu P, Wang Y, Li YH, et al:
Adenovirus-mediated gene therapy with an antiangiogenic fragment of
thrombospondin-1 inhibits human leukemia xenograft growth in nude
mice. Leuk Res. 27:701–708. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Vecchiarelli-Federico LM, Cervi D, Haeri
M, Li Y, Nagy A and Ben-David Y: Vascular endothelial growth factor
- a positive and negative regulator of tumor growth. Cancer Res.
70:863–867. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Cervi D, Shaked Y, Haeri M, et al:
Enhanced natural-killer cell and erythropoietic activities in
VEGF-A-overexpressing mice delay F-MuLV-induced erythroleukemia.
Blood. 109:2139–2146. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason
GA, Christensen JG and Kerbel RS: Accelerated metastasis after
short-term treatment with a potent inhibitor of tumor angiogenesis.
Cancer Cell. 15:232–239. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Hurwitz H, Fehrenbacher L, Novotny W, et
al: Bevacizumab plus irinotecan, fluorouracil and leucovorin for
metastatic colorectal cancer. N Engl J Med. 350:2335–2342. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Sandler A, Gray R, Perry MC, et al:
Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell
lung cancer. N Engl J Med. 355:2542–2550. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Miller K, Wang M, Gralow J, et al:
Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic
breast cancer. N Engl J Med. 357:2666–2676. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Escudier B, Eisen T, Stadler WM, et al:
Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J
Med. 356:125–134. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Motzer RJ, Hutson TE, Tomczak P, et al:
Sunitinib versus interferon alfa in metastatic renal-cell
carcinoma. N Engl J Med. 356:115–124. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Karp JE, Gojo I, Pili R, et al: Targeting
vascular endothelial growth factor for relapsed and refractory
adult acute myelogenous leukemias: therapy with sequential
1-β-d-arabinofuranosylcytosine, mitoxantrone, and bevacizumab. Clin
Cancer Res. 10:3577–3585. 2004.PubMed/NCBI
|
|
107
|
Presta LG, Chen H, O’Connor SJ, et al:
Humanization of an anti-vascular endothelial growth factor
monoclonal antibody for the therapy of solid tumors and other
disorders. Cancer Res. 57:4593–4599. 1997.PubMed/NCBI
|
|
108
|
D’Amato RJ, Loughnan MS, Flynn E and
Folkman J: Thalidomide is an inhibitor of angiogenesis. Proc Natl
Acad Sci USA. 91:4082–4085. 1994.
|
|
109
|
Teo SK: Properties of thalidomide and its
analogues: implications for anticancer therapy. AAPS J. 7:E14–E19.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Dredge K, Marriott JB, Macdonald CD, et
al: Novel thalidomide analogues display anti-angiogenic activity
independently of immunomodulatory effects. Br J Cancer.
87:1166–1172. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Thomas DA, Estey E, Giles FJ, et al:
Single agent thalidomide in patients with relapsed or refractory
acute myeloid leukaemia. Br J Haematol. 123:436–441. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Steins MB, Padró T, Bieker R, et al:
Efficacy and safety of thalidomide in patients with acute myeloid
leukemia. Blood. 99:834–839. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Barr P, Fu P, Lazarus H, et al:
Antiangiogenic activity of thalidomide in combination with
fludarabine, carboplatin and topotecan for high-risk acute
myelogenous leukemia. Leuk Lymphoma. 48:1940–1949. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Drevs J, Müller-Driver R, Wittig C, et al:
PTK787/ZK 222584, a specific vascular endothelial growth
factor-receptor tyrosine kinase inhibitor, affects the anatomy of
the tumor vascular bed and the functional vascular properties as
detected by dynamic enhanced magnetic resonance imaging. Cancer
Res. 62:4015–4022. 2002.
|
|
115
|
Roboz GJ, Giles FJ, List AF, et al: Phase
1 study of PTK787/ZK 222584, a small molecule tyrosine kinase
receptor inhibitor, for the treatment of acute myeloid leukemia and
myelodysplastic syndrome. Leukemia. 20:952–957. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Wedge SR, Kendrew J, Hennequin LF, et al:
AZD2171: a highly potent, orally bioavailable, vascular endothelial
growth factor receptor-2 tyrosine kinase inhibitor for the
treatment of cancer. Cancer Res. 65:4389–4400. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Fiedler W, Mesters R, Heuser M, et al: An
open-label, phase I study of cediranib (RECENTIN) in patients with
acute myeloid leukemia. Leuk Res. 34:196–202. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Raza A, Mehdi M, Mumtaz M, Ali F, Lascher
S and Galili N: Combination of 5-azacytidine and thalidomide for
the treatment of myelodysplastic syndromes and acute myeloid
leukemia. Cancer. 113:1596–1604. 2008. View Article : Google Scholar : PubMed/NCBI
|