Introduction
Prostate cancer is the second leading malignancy in
the Western world (1). Radiation is
the mainstay of conventional prostate cancer treatment and it is
likely to remain as such in the foreseeable future (2). Unfortunately, in our clinical
practice, radioresistant prostate cancer was observed in
approximately 20–30% of patients treated with primary radiation
therapy for clinically localized prostate cancer, leading to
cancer-related mortality in at least 27% of patients within 5 years
(3). It is known that the radiation
doses in prostate cancer treatment are generally limited to less
than 80 Gy due to intolerable toxicity at higher doses; however,
the radioresistance of prostate cancer cells has significantly
reduced the therapeutic effect of the limited amount of radiation
(4).
Previous studies have demonstrated that the DNA
damage response pathway plays a dominant role in conferring
radioresistance and its key mediator, Chk1, has been recognized as
the culprit for radioresistance development (5,6). When
activated, Chk1 phosphorylates a plethora of effector molecules
involved in cell cycle arrest, DNA repair and apoptosis, which
triggers cell cycle checkpoint and DNA repair defects, resulting in
radiation hypersensitivity (7).
Thus, restraining Chk1 activity may translate into improvement in
the overall efficacy of radiation.
Apart from Chk1, it is believed that cancer stem
cells are predisposed to radioresistance due to their preferential
activation of the DNA damage checkpoint response and increasing DNA
repair capacity (8–10). Indeed, radiation has been shown to
kill differentiated tumor cells while sparing the rare cancer stem
cells (11). Since cancer stem
cells possess the capacity of self-renewal, multilineage
differentiation and maintained proliferation, the presence of a
small fraction of cancer stem cells that have survived radiation
tend to put patient at a higher risk of tumor regrowth and
recurrence (12–14).
In our previous study, we reported that Chk1
knockdown improved the radiosensitivity of glioblastoma stem-like
cells (15). These aforementioned
data lead us to infer that restraining Chk1 activity may sensitize
prostate cancer stem cells to radiation therapy; however, evidence
supporting this hypothesis is lacking. In this study, using Chk1
knockdown, we investigated whether restraining Chk1 activity is
associated with the radiosensitization of prostate cancer stem
cells. Our results demonstrate that the isolated
CD133+CD44+ subpopulation from DU145 human
prostate cancer cells present the key biological properties of
cancer stem cells. Chk1 knockdown potentiates the cytotoxic effects
of radiation on CD133+CD44+ prostate cancer
stem cells by abrogating G2 arrest, as well as increasing
apoptosis. Furthermore, the Cdc25c-Cdc2 pathway may be associated
with Chk1 knockdown-mediated cell cycle arrest abrogation, while
the induced apoptosis may be associated with caspase-2 activation
in CD133+CD44+ prostate cancer stem cells. To
our knowledge, this study presents the first description of the
effects of Chk1 restraining activity on the radiosensitivity of
prostate cancer stem cells, and may provide a broad therapeutic
paradigm against prostate cancer.
Materials and methods
Cell culture
The DU145 human prostate cancer cell line was
obtained from ATCC and was maintained in RPMI-1640 medium
supplemented with 10% fetal bovine serum in a humidified incubator
(37°C, 5% carbon dioxide). Purified
CD133+CD44+ prostate cancer stem cells were
cultured DMEM/F12 medium supplemented with 20 ng/ml EGF, 10 ng/ml
bFGF, 5 mg/ml insulin and 0.4% BSA. For sphere formation, cells
were suspended and plated at 1,000 cells/ml per well in 24-well
low-attachment plates and were analyzed after 7–14 days. For
differentiation, 5% fetal bovine serum was added and the growth
factor was deleted from the culture medium. The
CD133+CD44+ prostate cancer stem cells were
then cultured for analysis.
shRNA preparation
As described in our previous study (15), we designed the interferential
sequence Chk1-shRNA based on the target sequences of the Chk1 gene
(GenBank GI: 166295195). The Chk1 shRNA sequences were as follows:
5′-CCG GCT GCA AAT AGT AGT TCC TGA ACT CGA GTT CAG GAA CTA CTA TTT
GCA GTT TTTG-3′ (forward) and 5′-AAT TCA AAA ACT GCA AAT AGT AGT
TCC TGA ACT CGA GTT CAG GAA CTA CTA TTT GCAG-3′ (reverse). The mock
shRNA sequences were: 5′-GAT CCC CGT TCT CCG AAC GTG TCA CGT TTC
AAG-3′ (forward) and 5′-AGA ACG TGA CAC GTT CGG AGA ATT TTT TGG
AAA-3′ (reverse). The above-mentioned shRNAs were ligated into the
pLKO.1-TRC vector and the Chk1-shRNA, and the mock-Chk1 plasmids
were then formed. The cells were transiently transfected with 2 μg
plasmids via Lipofectamine 2000 (Invitrogen) according to the
manufacturer’s instructions. The knockdown efficiency was confirmed
by RT-PCR and western blot analysis.
Isolation
DU145 cells were cultured under normal conditions.
Subsequently, 108 cells were suspended in magnetic
microbeads buffer in a final volume of 600 μl. FcR blocking reagent
(200 μl) and 200 μl CD44 MACS microbeads were then added to the
suspension sequentially. The suspension was then mixed and
incubated for 15 min at 4°C. The column was subsequenlty placed in
the magnetic flied of a MACS Separator and washed with 3 ml buffer.
For the following step, the cell suspension was loaded onto the
column and the negative cells were allowed to pass though and the
column was then rinsed with 3 ml buffer three times. The column was
placed on a collection tube and 5 ml of buffer were pipetted onto
the column and the positive fraction was flushed out. The positive
fraction was then eluted with buffer and the CD44+ cells
were obtained. The cells were sorted again with CD133 MACS
microbeads as described above.
Flow cytometry
For surface marker identification, the expression of
the CD133 and CD44 markers on DU145 cells was determined by flow
cytometry after surface staining with anti-human CD133 and CD44
antibodies, respectively. Flow cytometry was performed on a
FACSCanto II (BD Biosciences) and analyzed using BD FCSDiva
Software and FCS Express 4 software (De Novo Software, Los Angeles,
CA). For cell cycle analysis, the cells were harvested and fixed in
75% ethanol at 4°C overnight. Next day, cells were suspended in 20
μl PBS and 10 μl RNase A (5 mg/ml). After 30-min incubation, PI
(500 μg/ml) was added and the cells were incubated for 30 min in
the dark for analysis. For apoptosis analysis, the cells were
suspended in Annexin V-FITC binding buffer (195 μl) and Annexin
V-FITC (5 μl) in the dark for 10 min. The cells were centrifuged
and suspended in binding buffer (190 μl) and 10 μl PI solution on
ice in the dark for analysis.
EdU assay
The 96-well plate was centrifuged (1,000 rpm for 5
min). Subsequently, 100 μl EdU (50 μM) were added into each well
followed by mixture. After incubation for 2 h, the supernatant was
removed. The cells were washed with PBS and fixed using 4% buffered
formaldehyde for 30 min. The cells were cultured in 4% glycine for
5 min. After washing with PBS, the cells were permeabilized with
0.5% Triton X-100 for 5 min followed by washing with PBS. The cells
were then stained with Apollo staining reagent (100 μl) and
incubated for 30 min. After permeabilization for 30 min, the cells
were washed with 100 μl methanol, followed by washing with PBS. The
cells were then incubated with 100 μl Hoechst 33342 for 30 min.
After washing with PBS, the wells were viewed and photographed
using a confocal fluorescence microscope (Olympus FV500; Olympus,
Tokyo, Japan). The staining positive rate was counted as positive
cells/overall cells ×100%. For each sample, the cell number was
counted at least three times.
Western blot analysis and RT-PCR
The procedure and reagent for western blot analysis
and RT-PCR has been described in our previous study (16). The anti-Chk1 (1:1,000; Santa Cruz
Biotech, Santa Cruz, CA), anti-Rad51 (1:1,000; Santa Cruz Biotech),
anti-pH3 (1:500; Upstate Biotechnology, Lake Placid, NY),
anti-γ-H2AX (1:500; Upstate Biotechnology, Milford, MA),
anti-caspase-2 (1:1,000; Cell Signaling, Danvers, MA), anti-pCdc25C
(1:1,000; Cell Signaling), anti-pCdc2 (1:1,000; Cell Signaling) and
anti-GAPDH antibodies (1:3,000; Santa Cruz Biotech) were used. In
this study, we employed the following primers for RT-PCR: human
Chk1 forward, 5′-ATG CTC GCT GGA GAA TTG C-3′ and reverse, 5′-ATA
AGG AAA GAC CTG TGC GG-3′; and human GAPDH forward, 5′-ACG GAT TTG
GTC GTA TTGGG-3′ and reverse, 5′-TGA TTT TGG AGG GAT GTCGC-3′.
Statistical analysis
Statistical analyses were performed using
statistical software SPSS 13.0. Data are expressed as means ± SD.
The student’s t-test and variance analysis were used in this study.
P-values <0.05 were considered to indicate statistically
significant differences.
Results
Isolation and identification of prostate
cancer stem cells
Since Chk1 inhibition radiosensitizes tumor cells in
a p53-dependent manner with an obvious effect on p53 mutant tumor
cells (17–19), in this study, we employed the p53
mutant DU145 human prostate cancer cell line to isolate prostate
cancer stem cells. As CD133 and CD44 have been recognized as the
markers of prostate cancer stem cells (20–24),
we analyzed the ratio of the CD133+CD44+
subpopulation in DU145 cells using flow cytometry prior to
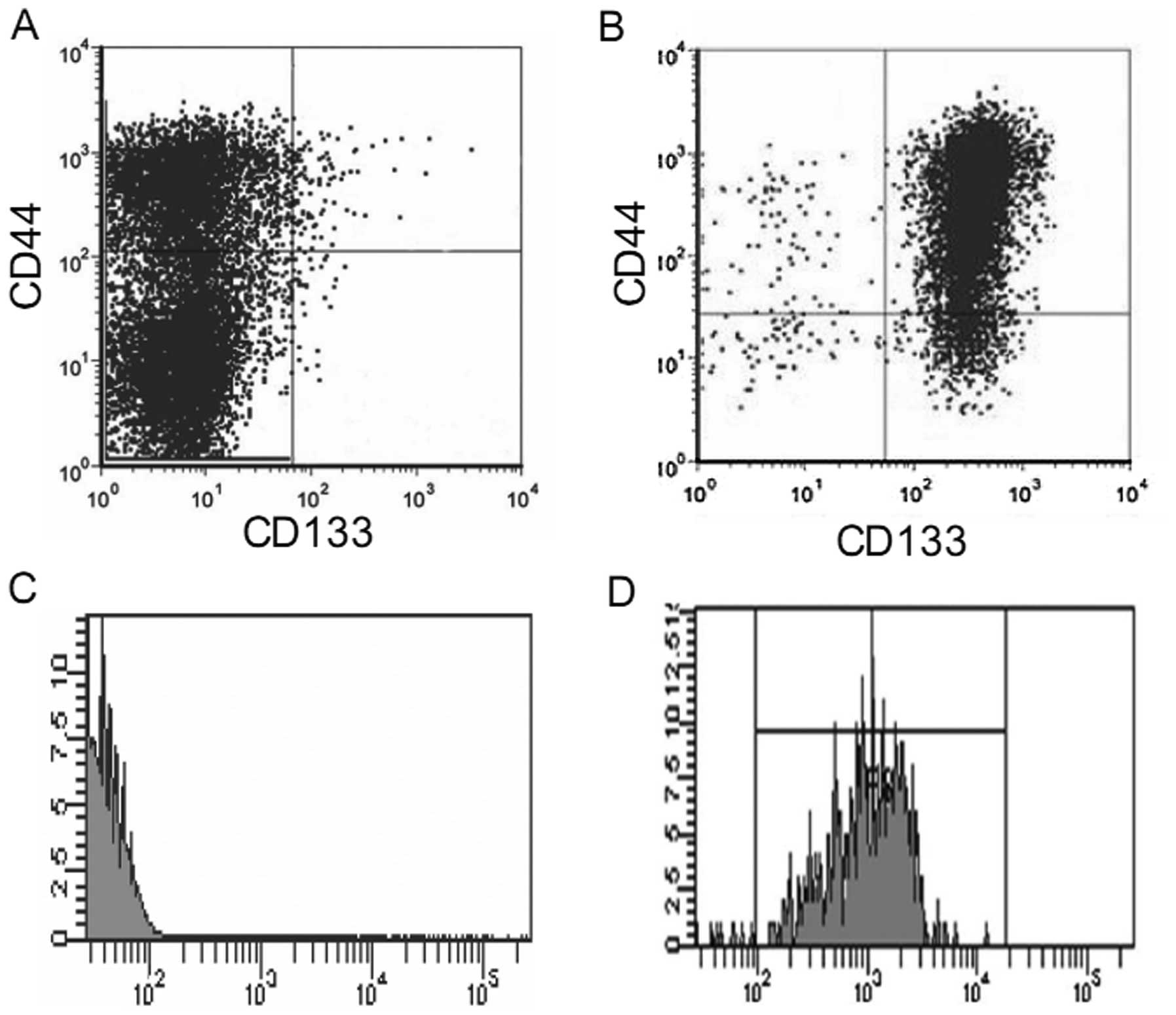
isolation. As shown in Fig. 1A, the
percentages of the CD133+CD44+,
CD44+ and CD133+ cell subpopulations in the
total DU145 cells were 0.51±0.14, 32.21±6.1 and 0.81±0.23%,
respectively, confirming the existence of a subpopulation of
prostate cancer stem cells in the DU145 cells. We then isolated
CD133+CD44+ cells from the DU145 cells using
CD44 microbeads and CD133 microbeads sequentially. As there is
significantly greater number of CD44+ than
CD133+ cells in the DU145 cell line, we first used CD44
microbeads to isolate the CD44+ cells from the total
DU145 cells. The obtained purified CD44+ cells were
resuspended and sorted again using CD133+ microbeads.
Subsequently, using flow cytometry, we determined the purity of the
CD133+CD44+ cells in the obtained isolated
cells and found that the purity of the
CD133+CD44+ cells ranged between 85–92%
(Fig. 1B-D).
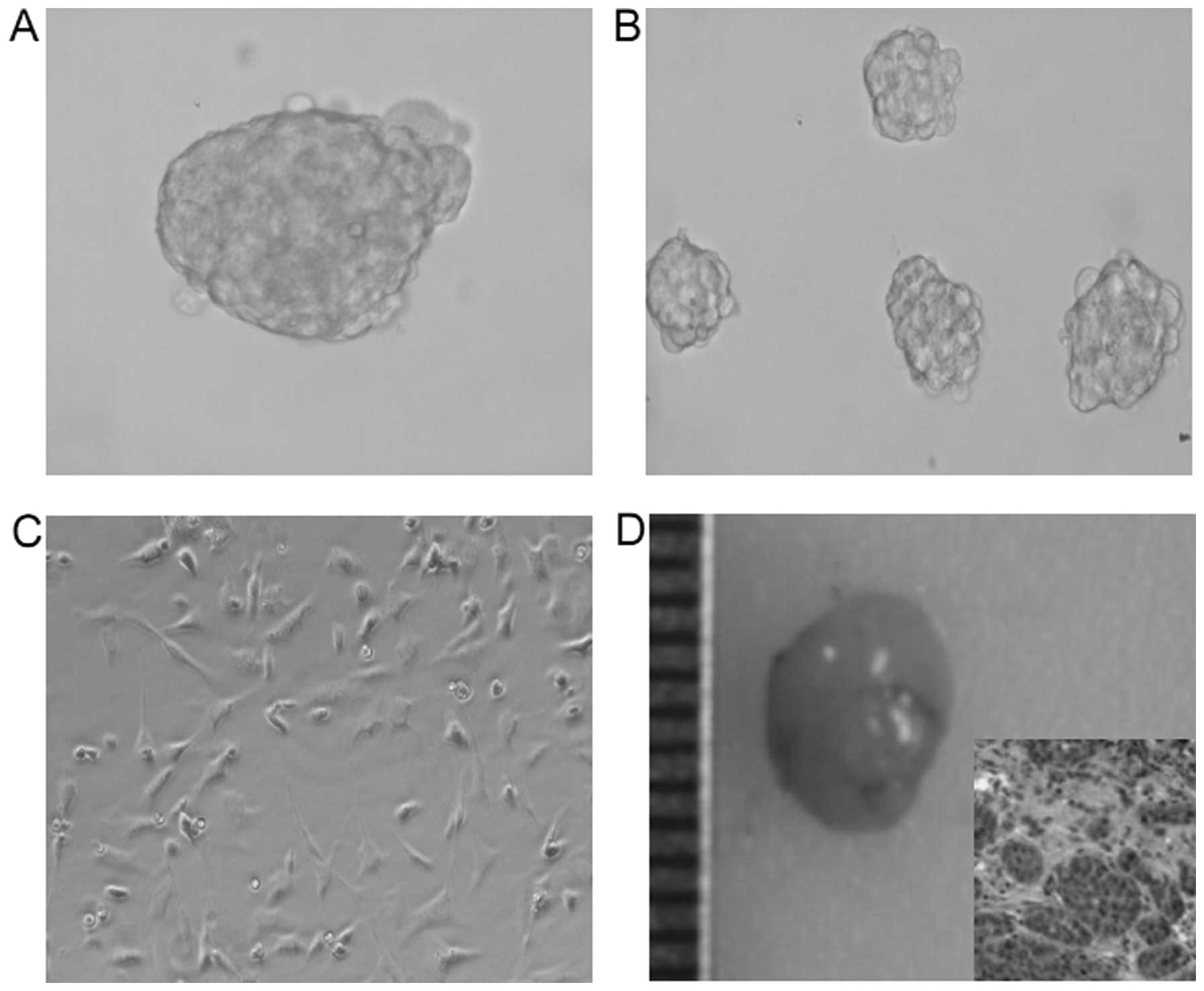
After isolation, we identified the stem cell
properties in the CD133+CD44+ isolated cells.
As expected, a small number (500 cells) of
CD133+CD44+ isolated cells generated
prostaspheres (Fig. 2A).
Furthermore, some spheroids generated new prostaspheres, indicating
the self-renewal ability of these cells (Fig. 1B). In addition, after exposure to
normal medium, many spheroid cells grew as a flat monolayer with a
morphology similar to DU145 cells after 10 days, showing the
differentiation capacity (Fig. 1C).
More importantly, a very small number of cells (6,000 cells) formed
xenograft tumors in nude mice (Fig.
1D). These observation support the evidence that
CD133+CD44+ cells isolated from DU145 cells
have cancer stem cell properties.
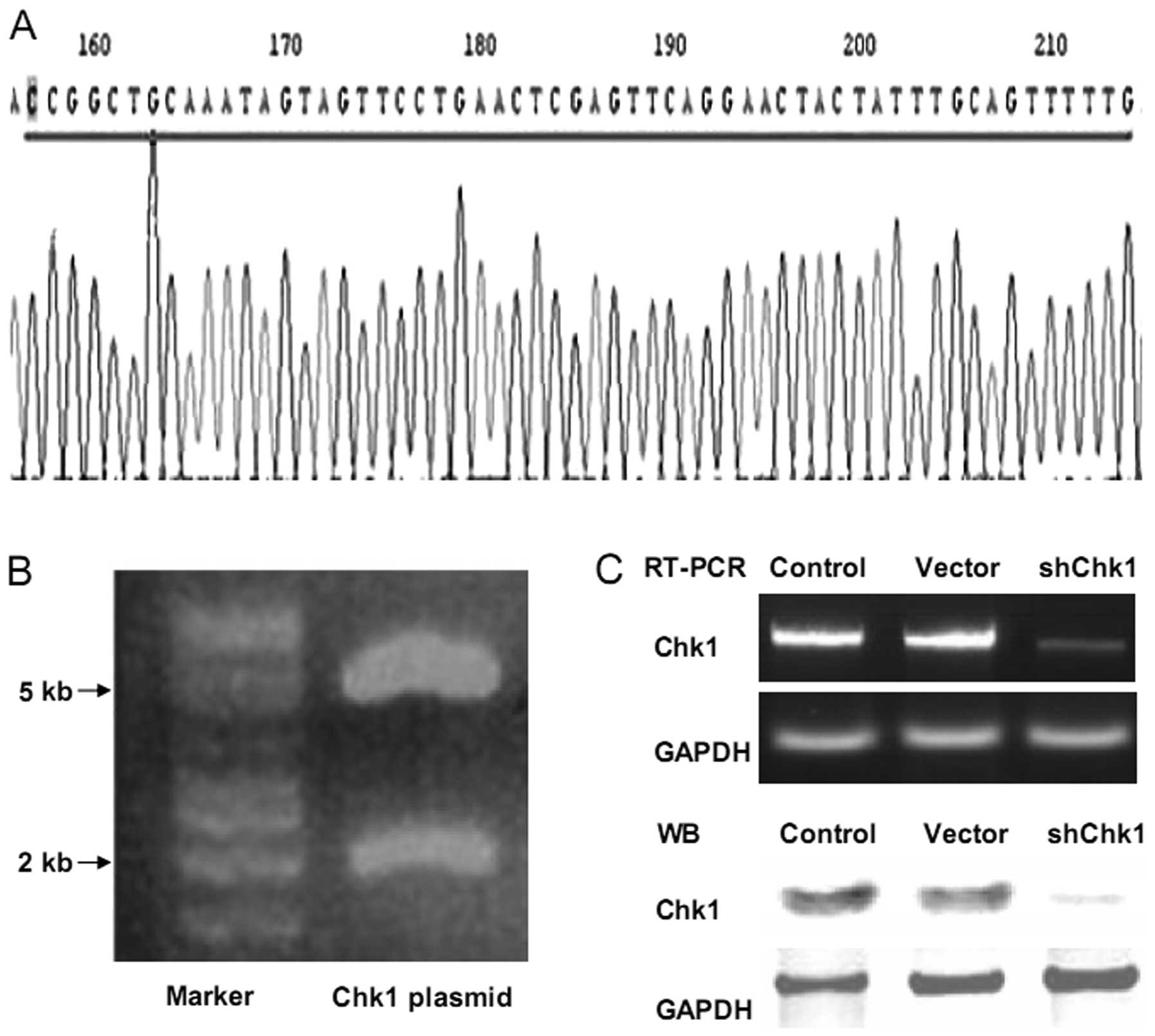
Knockdown of Chk1 in prostate cancer stem
cells using Chk1 shRNA
In our previous study, the shRNA-Chk1 plasmid was
used successfully to knockdown Chk1 in glioblastoma stem-like cells
(15). In this study, the same
plasmid was employed to knockdown Chk1 in
CD133+CD44+ cells. The electrophoresis
pattern of the shRNA-Chk1 plasmid digested for sequencing analysis,
confirmed the base sequences (Fig. 3A
and B). Using the Lipofectamine 2000 system, the cells were
transiently transfected with the pLKO.1-Chk1 and pLKO.1-mock
plasmids, and the inhibition efficiency was confirmed using RT-PCR
and western blot analysis (Fig.
3C).
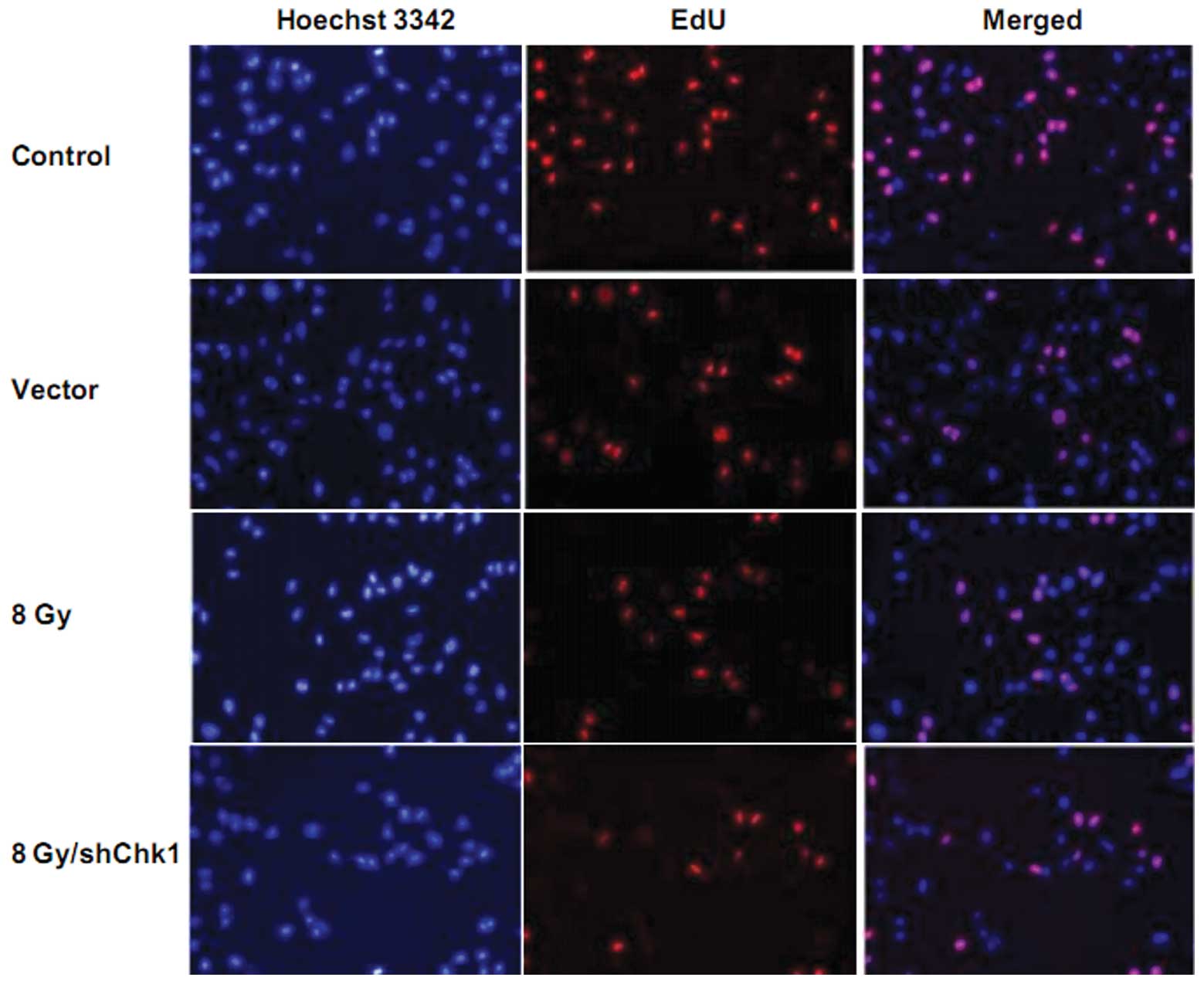
Chk1 knockdown radiosensitizes prostate
cancer stem cells
In order to investigate the effect of Chk1 knockdown
on the radiosensitivity of CD133+CD44+ cells,
EdU cell proliferation assay was performed. Chk1 knockdown cells,
vector cells and control cells were radiated at a dose of 8 Gy. As
shown in Fig. 4, 72 h after
radiation, EdU assay showed that the growth rate of the Chk1
knockdown cells was significantly lower than that of the vector and
control cells after radiation. No obvious difference was observed
between the growth rates of the vector and control cells (growth
rate: shChk1 plus radiation group vs. control, vector and radiation
groups, 9.23±2.15 vs. 50.59±4.27, 48.21±5.83 and 21.58±4.92%,
respectively; P<0.05).
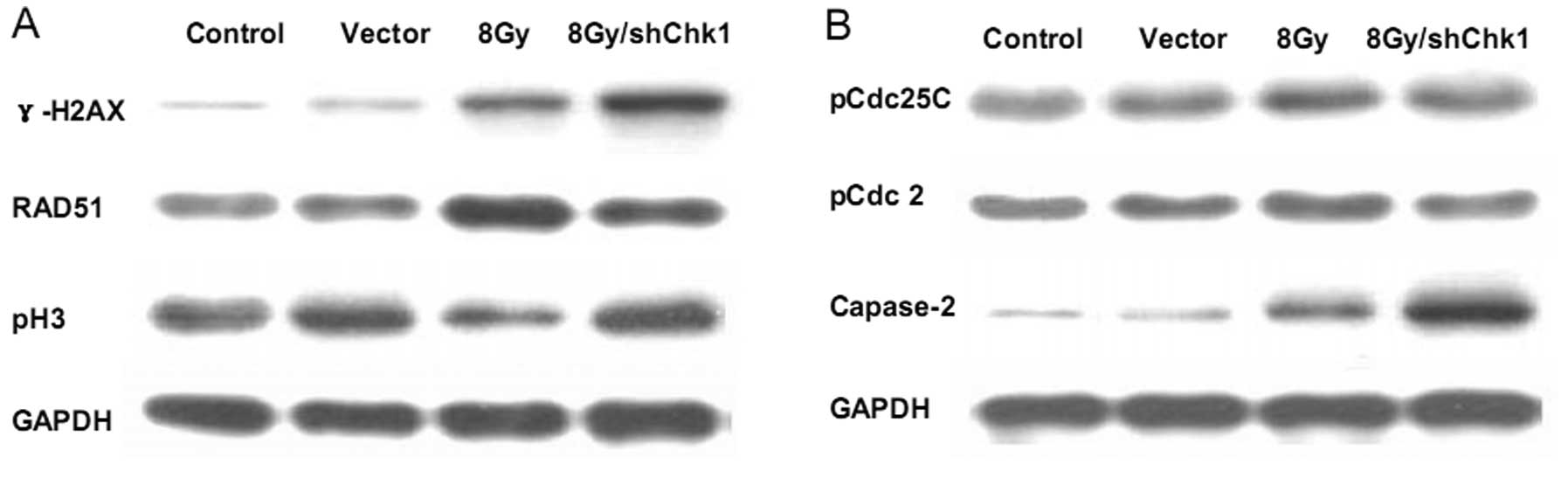
As γ-H2AX is considered an indicator of DNA damage
(25), in order to further
determine the effects of Chk1 knockdown on DNA damage in
CD133+CD44+ cells, we measured γ-H2AX protein
expression in the different groups of cells. Western blot analysis
showed that radiation resulted in a moderate increase in γ-H2AX
protein expression in the CD133+CD44+ cells,
while Chk1 knockdown significantly accelerated the
radiation-induced accumulation of γ-H2AX protein expression
(Fig. 7A). These results indicated
that Chk1 knockdown significantly exacerbated the radiation-induced
DNA damage in CD133+CD44+ cells and
sensitized the CD133+CD44+ cells to
radiation.
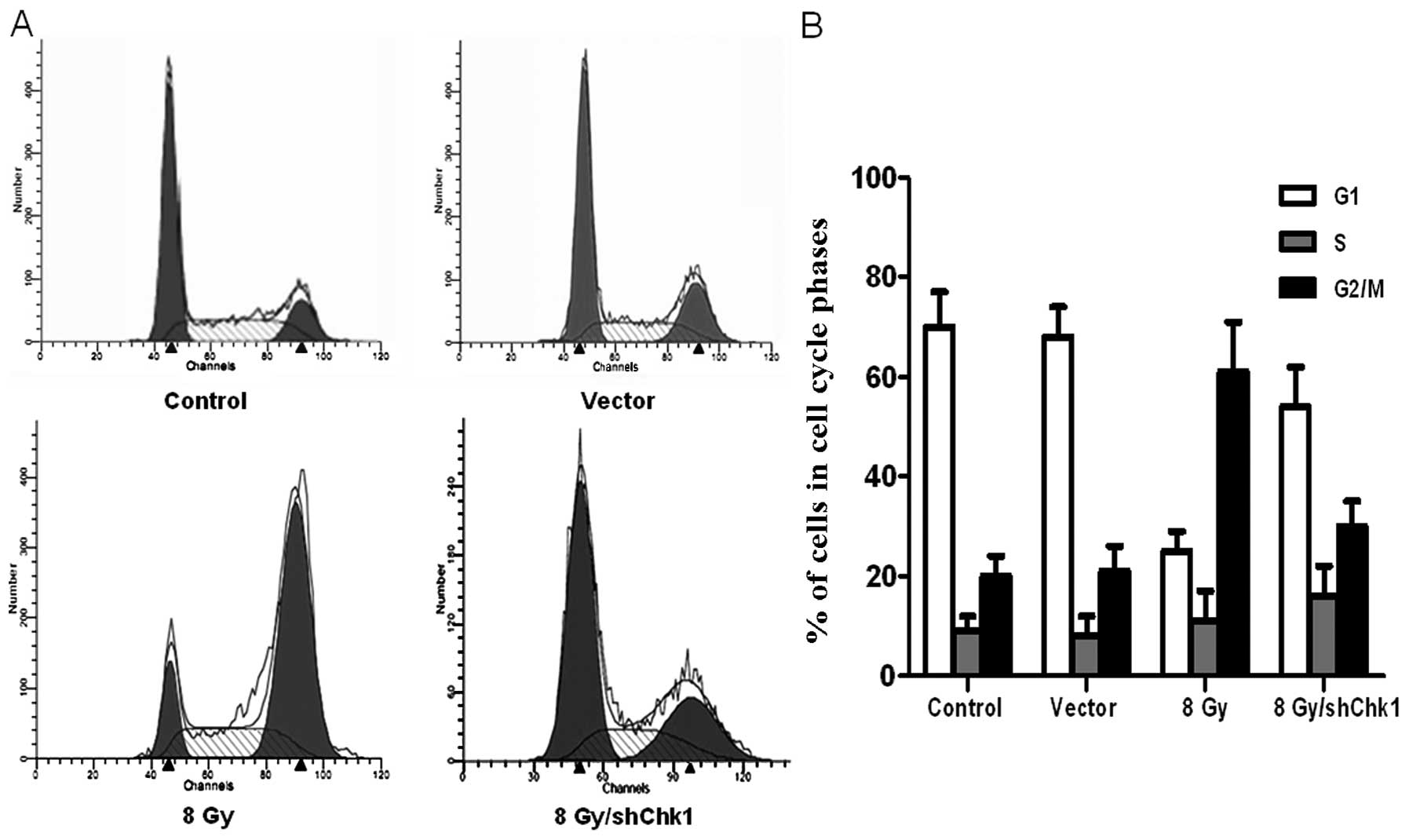
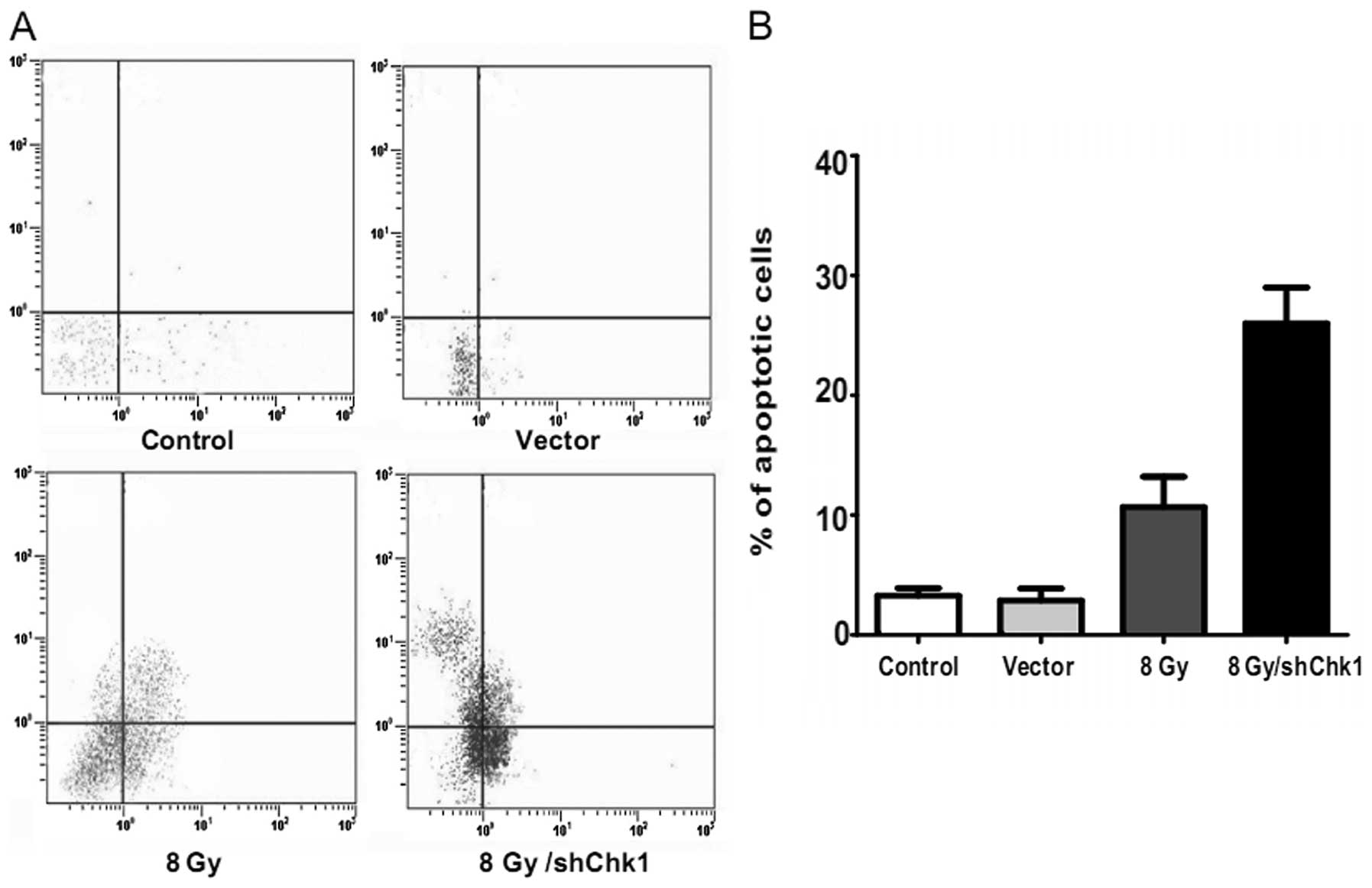
Chk1 knockdown abrogates
radiation-induced cell cycle arrest and facilitates
radiation-induced apoptosis
Chk1 knockdown, vector and control cells were
radiated at dose of 8 Gy. Another group of
CD133+CD44+ cells were cultured as the
untreated controls without radiation. After 48 h, flow cytometry
was used to examine the cell cycle distribution and cell apoptosis
in the four groups (Figs. 5 and
6). Compared to the untreated
control cells, radiation resulted in a high accumulation of
CD133+CD44+ cells in the G2/M phase, while
radiation only caused a slight G2/M phase accumulation in the Chk1
knockdown CD133+CD44+ cells (percentage of
cells in the G2/M phase: shChk1 plus radiation group vs. control,
vector and radiation groups, 32.13±4.54 vs. 20.85±3.27, 21.64±4.91
and 62.43±8.12%, respectively; P<0.05). In addition, the Chk1
knockdown CD133+CD44+ cells displayed a
significantly higher apoptotic rate than the other groups which
received radiation (apoptotic percentage: shChk1 plus radiation
group vs. control, vector and radiation groups, 26.47±4.31 vs.
3.35±0.47, 3.21±0.82 and 12.28±3.63%, respectively; P<0.05).
Since histone H3 becomes phosphorylated during
mitosis, the phosphorylation of histone H3 (pH3) has traditionally
been recognized as a marker of cells in mitosis (26). Therefore, the effect of Chk1
knockdown on pH3 expression in radiated
CD133+CD44+ cells was determined (Fig. 7A). We found that radiation decreased
pH3 expression in CD133+CD44+ cells, while
Chk1 knockdown reversed the downregulation of pH3 in response to
radiation, which indicates that Chk1 knockdown may abrogate the
radiation-induced G2/M checkpoint and force the cells into
premature cell cycle progression as evidenced by the renewal of
mitotic progression. As RAD51 is a protein marker associated with
DNA damage repair (27,28), we determined RAD51 protein
expression and found decreased RAD51 protein accumulation in the
radiated Chk1 knockdown CD133+CD44+ cells
(Fig. 7A). These results suggest
that Chk1 knockdown enhances the efficacy of radiation therapy in
CD133+CD44+ cells by modifying the cell
cycle, DNA damage repair ability and apoptosis activity.
Chk1 knockdown not only reduces the
radiation-induced phosphorylation of Cdc25C and Cdc2 but also
increases the cleavage of caspase-2
pCdc25C and pCdc2 have been reported to be involved
in Chk1-mediated cell cycle arrest (29–31),
while caspase-2 has been associated with Chk1-mediated apoptosis
(32). Therefore, in order to
further explore the mechanistic bases of Chk1 knockdown in
CD133+CD44+ cells, we determined the
expression of these proteins in our study using western blot
analysis (Fig. 7B). After radiation
exprosure, the CD133+CD44+ cells demonstrated
increased Cdc25C and Cdc2 phosphorylation, while the Chk1 knockdown
reduced the radiation-induced phosphorylation of Cdc25C and Cdc2.
Furthermore, radiation increased the cleavage of caspase-2 in the
CD133+CD44+ cells, while Chk1 knockdown
significantly enhanced the radiation-induced caspase-2 cleavage
accumulation. These data suggest the involvement of the Cdc25C-Cdc2
pathway in the mechanism of Chk1-mediated cell cycle arrest and the
association between Chk1-induced apoptosis and caspase-2
activation.
Discussion
Prostate cancer, one of the most common forms of
neoplasia, is the second leading cause of cancer related mortality
in the Western world (1,4). Although radiation therapy has long
been adopted as standard therapy for localized prostate cancer, the
long-term effects are relatively poor due to the radioresistance of
prostate cancer cells (3).
Following radiation exposure, the survived and repopulating
prostate cancer cells modulate many molecular pathways (mainly the
Chk1 pathway) to overcome the radiation cytotoxic effects, leading
to the development of radioresistance (33). The activation of the Chk1 pathway is
crucial for the proper coordination of checkpoint and DNA repair
processes, which allow for tumor cell survival following radiation
(27,33,34).
Based on these data, inhibiting Chk1 activity is
believed to sensitize tumor cells to radiation and a considerable
amount of evidence has confirmed that it is indeed the case in many
tumor cell lines (18,35,36).
Cancer stem cells are more radioresistant than bulk cancer cells
due to their preferential activation of the DNA damage checkpoint
response and increasing DNA repair capacity (11,12).
Moreover, cancer stem cells can generate tumors in very small
numbers (13). These properties
significantly contribute to tumor regrowth and radioresistance.
Thus, we hypothesized that Chk1 knockdown may enhance the radiation
sensitivity of cancer stem cells, thereby providing a more
efficient way for prostate cancer eradication.
The identification of prostate cancer stem cells has
shown that prostate cancer stem cells express a number of cell
surface markers, including CD44, CD133, intergrins, breast cancer
resistance protein (BCRP) and Sca-1 (20,24).
The CD44+ prostate cancer cell population is enriched in
tumorigenic progenitor cells (23,24).
CD133+ prostate cancer cells are more proliferative,
clonogenic and tumorigenic than bulk prostate cancer cells
(22). Since DU145 cells with
stem-like properties have been reported to be enriched with CD133,
CD44 and integrin α2β1 (24), we
attempted to isolate prostate cancer cells from DU145 human
prostate cancer cells. However, in our study, following isolation
of the CD133+CD44+ cells using magnetic
microbeads, the integrin α2β1 isolation was not achieved, as the
triple CD133−, CD44− and integrin-α2β1 positive DU145 cells always
eventually died. In our preresearch, we also attempted, but failed
to isolate prostate cancer cells from primary cell cultures of
human prostate tumor tissues. These failures may be attributed to
our limited cancer stem cell isolation and culture technology, as
well as the imperfect framework for accessing tumor tissue samples.
Thus, we only obtained the CD133+CD44+
subpopulation from the DU145 cells in our study. Apart from
isolation, we also identified the cancer stem cell properties of
the CD133+CD44+ cells obtained in our study.
The isolated CD133+CD44+ cells were able to
generate prostaspheres, differentiated into cells with a morphology
similar to unsorted DU145 cells and, most importantly, formed
tumors on transplantation with a small number of cells. Since
cancer stem cells have the capacity for self-renewal, multilineage
differentiation and maintaining proliferation, our data indicate
that CD133+CD44+ cells derived from DU145
cells have cancer stem cell properties and are responsible for the
development of prostate cancer.
In this study, we transiently transfected
CD133+CD44+ cells using Chk1 shRNA in order
to explore the potential radiosensitizing effect of Chk1 knockdown
on prostate cancer stem cells. We found that Chk1 knockdown
decreased RAD51 expression, abrogated the G2/M checkpoint and
increased γ-H2AX expression and apoptosis in
CD133+CD44+ cells following radiation. These
data indicate that Chk1 knockdown potentiates the cytotoxic effects
of radiation by abrogating the G2/M checkpoint, inhibiting DNA
damage repair and promoting premature mitosis, which in turn
results in increased apoptosis. Moreover, the abrogation of
radiation-induced G2/M arrest and the promotion of
radiation-induced apoptosis by Chk1 knockdown was associated with
the inactivation of phosphorylated Cdc25C and Cdc2, as well as the
activation of caspase-2. These results are consistent with those
from previous studies supporting the key role of Chk1 inhibition in
radiosensitizing a variety of tumor cell lines (5,17).
Our study had certain inherent limitations. Our data
only provided in vitro evidence of the radiosensitizing
effect of Chk1 knockdown on the isolated
CD133+CD44+ prostate cancer stem cells,
lacking direct evidence in vivo, as the in vivo study
is currenlty ongoing on in our laboratory. Nevertheless, the
present study suggests for the first time, that Chk1 knockdown
radiosensitizes prostate cancer stem cells. The specific molecular
mechanism behind the radiosensitizing effects of Chk1 knockdown on
prostate cancer stem cells may be linked to the abrogation of the
G2/M checkpoint, the inhibition of DNA damage repair and the
promotion of apoptosis. Since the safety of tumor-directed gene
therapy has been supported by various clinical trials and cancer
stem cells have a potent tumor-initiating capacity, as well as
intrinsic radioresistance (9,37–40),
enhancing the radiation sensitivity of cancer stem cells via Chk1
knockdown may prove to be a novel therapeutic approach to improve
the poor prognosis of radioresistant prostate cancer patients.
Further research is warranted to define the optimal clinical
settings where such a therapeutic strategy can be applied.
References
|
1
|
Neppl-Huber C, Zappa M, Coebergh JW, et
al: Changes in incidence, survival and mortality of prostate cancer
in Europe and the United States in the PSA era: additional
diagnoses and avoided deaths. Ann Oncol. 23:1325–1334. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Heidenreich A, Bellmunt J, Bolla M, et al:
EAU guidelines on prostate cancer. Part 1: screening, diagnosis,
and treatment of clinically localised disease. Eur Urol. 59:61–71.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bonkhoff H: Factors implicated in
radiation therapy failure and radiosensitization of prostate
cancer. Prostate Cancer. 2012:5932412012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Beckendorf V, Guerif S, Le PE, et al: 70
Gy versus 80 Gy in localized prostate cancer: 5-year results of
GETUG 06 randomized trial. Int J Radiat Oncol Biol Phys.
80:1056–1063. 2011.PubMed/NCBI
|
|
5
|
Merry C, Fu K, Wang J, Yeh IJ and Zhang Y:
Targeting the checkpoint kinase Chk1 in cancer therapy. Cell Cycle.
9:279–283. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stracker TH, Usui T and Petrini JH: Taking
the time to make important decisions: the checkpoint effector
kinases Chk1 and Chk2 and the DNA damage response. DNA Repair.
8:1047–1054. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jackson SP and Bartek J: The DNA-damage
response in human biology and disease. Nature. 461:1071–1078. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Diehn M and Clarke MF: Cancer stem cells
and radiotherapy: new insights into tumor radioresistance. J Natl
Cancer Inst. 98:1755–1757. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Baumann M, Krause M and Hill R: Exploring
the role of cancer stem cells in radioresistance. Nat Rev Cancer.
8:545–554. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hittelman WN, Liao Y, Wang L and Milas L:
Are cancer stem cells radioresistant. Future Oncol. 6:1563–1576.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Visvader JE and Lindeman GJ: Cancer stem
cells in solid tumours: accumulating evidence and unresolved
questions. Nat Rev Cancer. 8:755–768. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Eyler CE and Rich JN: Survival of the
fittest: cancer stem cells in therapeutic resistance and
angiogenesis. J Clin Oncol. 26:2839–2845. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang G, Wang Z, Sarkar FH and Wei W:
Targeting prostate cancer stem cells for cancer therapy. Discov
Med. 13:135–142. 2012.PubMed/NCBI
|
|
14
|
Maitland NJ and Collins AT: Prostate
cancer stem cells: a new target for therapy. J Clin Oncol.
26:2862–2870. 2008. View Article : Google Scholar
|
|
15
|
Wu J, Lai G, Wan F, et al: Knockdown of
checkpoint kinase 1 is associated with the increased
radiosensitivity of glioblastoma stem-like cells. Tohoku J Exp Med.
226:267–274. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xiao Z, Liu Q, Zhao B, Wu J and Lei T:
Hypoxia induces hemorrhagic transformation in pituitary adenomas
via the HIF-1α signaling pathway. Oncol Rep. 26:1457–1464.
2011.PubMed/NCBI
|
|
17
|
Ma CX, Janetka JW and Piwnica-Worms H:
Death by releasing the breaks: CHK1 inhibitors as cancer
therapeutics. Trends Mol Med. 17:88–96. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Carrassa L and Damia G: Unleashing Chk1 in
cancer therapy. Cell Cycle. 10:2121–2128. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen Y and Sanchez Y: Chk1 in the DNA
damage response: conserved roles from yeasts to mammals. DNA
Repair. 3:1025–1032. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lang SH, Frame FM and Collins AT: Prostate
cancer stem cells. J Pathol. 217:299–306. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Collins AT, Berry PA, Hyde C, Stower MJ
and Maitland NJ: Prospective identification of tumorigenic prostate
cancer stem cells. Cancer Res. 65:10946–10951. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li C, Heidt DG, Dalerba P, et al:
Identification of pancreatic cancer stem cells. Cancer Res.
67:1030–1037. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tang DG, Patrawala L, Calhoun T, et al:
Prostate cancer stem/progenitor cells: identification,
characterization, and implications. Mol Carcinog. 46:1–14. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wei C, Guomin W, Yujun L and Ruizhe Q:
Cancer stem-like cells in human prostate carcinoma cells DU145: the
seeds of the cell line. Cancer Biol Ther. 6:763–768. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Syljuasen RG, Sorensen CS, Hansen LT, et
al: Inhibition of human Chk1 causes increased initiation of DNA
replication, phosphorylation of ATR targets, and DNA breakage. Mol
Cell Biol. 25:3553–3562. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Binder A and Bohm L: Influence of
irradiation and pentoxifylline on histone H3 phosphorylation in
human tumour cell lines. Cell Prolif. 35:37–47. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sorensen CS, Hansen LT, Dziegielewski J,
et al: The cell-cycle checkpoint kinase Chk1 is required for
mammalian homologous recombination repair. Nat Cell Biol.
7:195–201. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bahassi EM, Ovesen JL, Riesenberg AL,
Bernstein WZ, Hasty PE and Stambrook PJ: The checkpoint kinases
Chk1 and Chk2 regulate the functional associations between hBRCA2
and Rad51 in response to DNA damage. Oncogene. 27:3977–3985. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xiao Z, Chen Z, Gunasekera AH, et al: Chk1
mediates S and G2 arrests through Cdc25A degradation in response to
DNA-damaging agents. J Biol Chem. 278:21767–21773. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sanchez Y, Wong C, Thoma RS, et al:
Conservation of the Chk1 checkpoint pathway in mammals: linkage of
DNA damage to Cdk regulation through Cdc25. Science. 277:1497–1501.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sorensen CS, Melixetian M, Klein DK and
Helin K: NEK11: linking CHK1 and CDC25A in DNA damage checkpoint
signaling. Cell Cycle. 9:450–455. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sidi S, Sanda T, Kennedy RD, et al: Chk1
suppresses a caspase-2 apoptotic response to DNA damage that
bypasses p53, Bcl-2, and caspase-3. Cell. 133:864–877. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dumont F, Altmeyer A and Bischoff P:
Radiosensitising agents for the radiotherapy of cancer: novel
molecularly targeted approaches. Expert Opin Ther Pat. 19:775–799.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang H, Yoon SJ, Jin J, et al: Inhibition
of checkpoint kinase 1 sensitizes lung cancer brain metastases to
radiotherapy. Biochem Biophys Res Commun. 406:53–58. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Meuth M: Chk1 suppressed cell death. Cell
Div. 5:212010. View Article : Google Scholar
|
|
36
|
Smith J, Tho LM, Xu N and Gillespie DA:
The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and
cancer. Adv Cancer Res. 108:73–112. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gao Q, Zhou J, Huang X, et al: Selective
targeting of checkpoint kinase 1 in tumor cells with a novel potent
oncolytic adenovirus. Mol Ther. 13:928–937. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Freytag SO, Stricker H, Pegg J, et al:
Phase I study of replication-competent adenovirus-mediated
double-suicide gene therapy in combination with conventional-dose
three-dimensional conformal radiation therapy for the treatment of
newly diagnosed, intermediate- to high-risk prostate cancer. Cancer
Res. 63:7497–7506. 2003.
|
|
39
|
Geoerger B, van Beusechem VW, Opolon P, et
al: Expression of p53, or targeting towards EGFR, enhances the
oncolytic potency of conditionally replicative adenovirus against
neuroblastoma. J Gene Med. 7:584–594. 2005. View Article : Google Scholar
|
|
40
|
Tsuruta Y, Pereboeva L, Breidenbach M, et
al: A fiber-modified mesothelin promoter-based conditionally
replicating adenovirus for treatment of ovarian cancer. Clin Cancer
Res. 14:3582–3588. 2008. View Article : Google Scholar
|