Introduction
Primary gallbladder carcinoma (PGC) is the most
common and aggressive malignancy in the biliary system. The
evolution of PGC is a complex multi-step process, including the
malignant transformation of normal cells and malignant tumor cell
proliferation, invasion and metastasis. This process is not only
regulated by oncogenes and anti-oncogenes, but also closely depends
on the tumor stromal microenvironment. Due to lacking
characteristic signs or symptoms, patients diagnosed as PGC have
mostly been in advanced stage with a low resection ratio (1–3). With
the in-depth understanding of the immune system and its regulation,
immunotherapy has become the focus of tumor treatment due to its
promising advantages. Immunotherapy can clean away tumor cells in
stationary phase, cancer stem cells and a small amount of residual
tumor cells to prevent tumor metastasis and recurrence. However,
the clinical outcomes of most immunotherapeutic strategies have
been less effective than anticipated. It has been demonstrated that
the reasons behind the attenuated clinical outcomes of cancer
vaccines were mainly because of the tumor immune tolerance induced
by many immune tolerance factors, which originate from the tumor
and tumor microenvironment. Indoleamine 2,3-dioxygenase (IDO), as
one of the main factors, plays a crucial role in tumor-induced
immune tolerance via regulating T-cell function by macrophages and
a subset of dendritic cells (4).
IDO is a heme-containing monomeric oxidoreductase
that catalyzes the first and rate-limiting step in the degradation
of the essential amino acid tryptophan to N-formylkynurenine, and
it induces tryptophan starvation and accumulation of downstream
breakdown products. Because tryptophan is the essential amino acid
in the process of T-cell activation and proliferation, tryptophan
starvation resulting from IDO consumption inhibits T-cell
activation and proliferation. Therefore, T cells cannot synthesize
sufficient protein to proliferate and result in immune tolerance of
the local microenvironment (5,6).
IDO has been investigated in pulmonary,
hepatocellular, renal, endometrial and nasopharyngeal carcinomas
(7–10). The inhibition of IDO expression
could significantly suppress tumor growth and promote the
anti-tumor activities of various immunotherapeutic agents in
relevant animal models (11,12).
More importantly, previous studies of our group have reported that
IFN-γ induces the expression of IDO via the Janus kinase/signal
transducer and activator of transcription 1 (JAK/STAT1) signaling
pathway (13,14). Based on these backgrounds, we
wondered whether inhibition of the JAK/STAT1 signaling pathway
could down-regulate the expression of IDO in gallbladder carcinoma
cells and restore the tumor antigen-specific T-cell proliferation,
and eventually break tumor immune tolerance and enhance the effect
of tumor immunotherapy.
It is widely accepted that the JAK/STAT1 signaling
pathway of IDO expression induced by IFN-γ is as follows: the
interaction of IFN-γ with its cell surface receptor leads to the
activation of JAK1 and JAK2 (phosphorylation), which in turn
phosphorylate and activate STAT1. Phosphorylated STAT1 forms a
homodimer and translocates to the nucleus where it binds to and
activates IFN-γ-responsive specific promoters of IDO, including
members of the γ-activated sites (GAS) and the interferon
stimulated response elements (ISRE) family of enhancers, and
interferon regulatory factor genes-1 (IRF-1) to promote gene
transcription (8,15–17).
In the current study, we reported that suberoylanilide hydroxamic
acid (SAHA) down-regulated the expression of IDO induced by IFN-γ
in human gallbladder carcinoma (SGC-996) cells and therefore, may
provide potential therapeutic strategies in tumor
immunotherapy.
SAHA, a kind of histone deacetylases inhibitors
(HDACI), is a promising new anticancer agent due to its low
toxicity in cancer treatment and protective action against
intracellular events including IFN-γ-mediated signaling
transduction (18,19). HDACI are a group of compound
regulating the expression of different regulatory genes which are
responsible for cell growth, proliferation, apoptosis and
regulation of other mechanisms involved in tumor development and
growth (20–22).
However, the mechanism of the anti-tumor activity of
SAHA remains to be elucidated in more detail. Here, we treated
SGC-996 cells with SAHA and IFN-γ to investigate the role of
inhibition of the JAK/STAT1 signaling pathway by SAHA on the
down-regulation of the expression of IDO, in anticipation of
providing a new strategy to break tumor immune tolerance in
gallbladder carcinoma immunotherapy.
Materials and methods
Chemicals and reagents
IFN-γ and SAHA were respectively purchased from
Sigma-Aldrich (Deisenhofen, Germany) and Cayman Chemical (Ann
Arbor, MI, USA). Dual-luciferase assay kit and vector (pRL-TK) were
purchased from Promega (Madison, WI, USA). Vectors
(pGL3-Enhancer-GAS7 and pGL3-Enhancer-ISRE4) were kindly provided
by Professor Jun Du (Sun Yat-sen University, China). Monoclonal
anti-STAT1 (9H2) and anti-phospho-Stat1 (Tyr701) antibodies were
acquired from a commercial source (Cell Signaling Technology,
Beverly, MA). The secondary anti-mouse antibody conjugated to FITC,
DAPI dye and Lipofectamine 2000 reagent were purchased from
Invitrogen (Carlsbad, CA, USA). The polyclonal rabbit anti-human
IDO and monoclonal mouse anti-human IRF-1 antibody (H-8) were
products of Santa Cruz Biotechnology Inc. (Santa Cruz, CA,
USA).
Cell culture
The human primary gallbladder carcinoma cell line
SGC-996 was purchased from the Academy of Life Science, Tongji
University (23). Cells were
maintained in RPMI-1640 supplemented with 10% heat-inactivated
endotoxin-free fetal bovine serum, 100 g/ml streptomycin and 100
U/ml penicillin under a humidified 5% CO2 atmosphere at
37°C in a CO2 incubator.
Western blot analysis
Western blotting was performed as previously
described (13). Briefly, cells
were lysed in lysis buffer containing 1% Nonidet P-40, 20 mM
Tris-HCl (pH 7.6), 0.15 M NaCl, 3 mM EDTA, 3 mM EGTA, 1 mM
phenylmethylsulfonyl fluoride, 20 mg/ml aprotinin, and 5 mg/ml
leupeptin. The lysates were cleared by centrifugation and denatured
by boiling in Laemmli buffer; equal amounts of protein samples were
separated on 10% sodium dodecylsulfate (SDS)-polyacrylamide gels
and electrophoretically transferred to nitrocellulose membranes.
Following blocking with 5% non-fat milk at room temperature for 2
h, membranes were incubated overnight with the primary antibody at
1:1,000 dilution at 4°C and then incubated with a horseradish
peroxidase-conjugated secondary antibody at 1:5,000 dilution for 1
h at room temperature. Specific immune complexes were detected with
the Western Blotting Plus Chemiluminescence Reagent (Life Science,
Inc., Boston, MA).
Transient transfections and reporter gene
assays
For measuring the activation of SAHA on
STAT1-dependent transcriptional activation of GAS and ISRE, cells
were plated in a 96-well plate at a density of 2.0×104
in 500 μl of growth medium per well without antibiotics.
Twenty-four hours later, cells were 80% confluent at the time of
transfection, and were transfected with 0.2 μg DNA/cm2
per plasmid [including objective plasmid and thymidine kinase (TK)]
and Lipofectamine 2000 reagent according to the manufacturer’s
instructions. Transfection efficiency was normalized by
co-transfection with pRL-TK. Transcriptional activity was
determined by a luminometer, using a Dual-Luciferase Assay kit.
Results are displayed as the ratio between the activity of the
reporter plasmid and pRL-TK.
Confocal microscopy for STAT1
Cells were grown on 18×18 cm coverslips in a 6-well
plate, serum starved for 16 h, then stimulated with or without 10
μM SAHA for 2 h. Followed by being treated with or without 500 U/ml
IFN-γ for 30 min, cells were fixed in 4% paraformaldehyde and
permeabilized with 1% Triton X-100 (prepared from 30% stock
solution) for 30 min equally, then blocked with goat serum for 30
min at 37°C and incubated with anti-STAT1 antibody at 1:100
dilution for 1 h at 37°C. Coverslips were washed with PBS and
incubated with a secondary anti-mouse antibody conjugated to FITC
at 1:1000 dilution for 1 h at 37°C. After being washed 3 times with
PBS, the cells were incubated with DAPI (10 μg/ml) for 10 min to
visualize cell nuclei. The coverslips were mounted to microscopic
slides and covered with glycerol jelly, and then the slides were
analyzed by a Confocol Laser Scanning Microscope (LSM 710, Carl
Zeiss, Germany) to analyze nuclear translocation of STAT1.
Statistical analysis
All values are expressed as the means ± SEM of two
independent experiments unless otherwise specified. We analyzed
data by two-tailed unpaired Student’s t-tests between the two
groups and by one-way ANOVA followed by the Bonferroni test for
multiple comparisons. These analyses were performed using the
GraphPad Prism Software Version 5.0 (GraphPad Software Inc., La
Jolla, CA). P<0.05 was considered statistically significant.
Results
IDO expression induced by IFN-γ is dose-
and time-dependent in SGC-996 cells
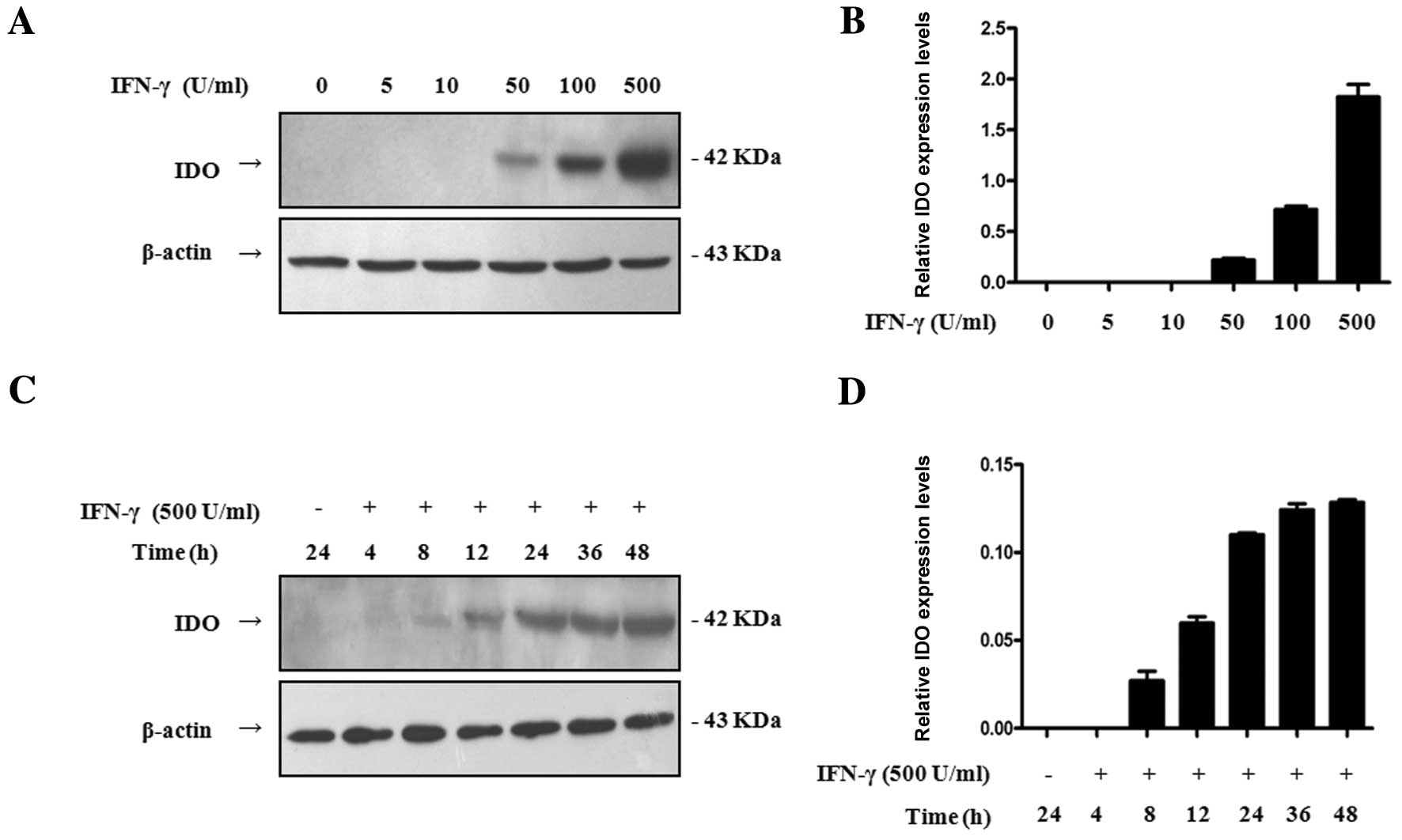
To investigate the effect of IFN-γ on the expression
of IDO in SGC-996 cells, we performed Western blot analysis to
explore the effects of varying concentrations and incubation
periods of IFN-γ on the expression of IDO. SGC-996 cells were
treated with IFN-γ at different doses for 24 h or 500 U/ml IFN-γ
for different times. There was no IDO expression observed without
IFN-γ treatment, but IFN-γ significantly enhanced IDO expression in
a dose-dependent manner, beginning with 50 U/ml (Fig. 1A and B). In addition, IDO was
induced by exposure to 500 U/ml IFN-γ for 8 h and was enhanced by
continuous exposure to IFN-γ (Fig. 1C
and D). Thus, IDO expression in SGC-996 cells was an inducible
event, and the expression of IDO induced by IFN-γ was dose- and
time-dependent.
SAHA down-regulates the expression of IDO
induced by IFN-γ in a dose-dependent manner
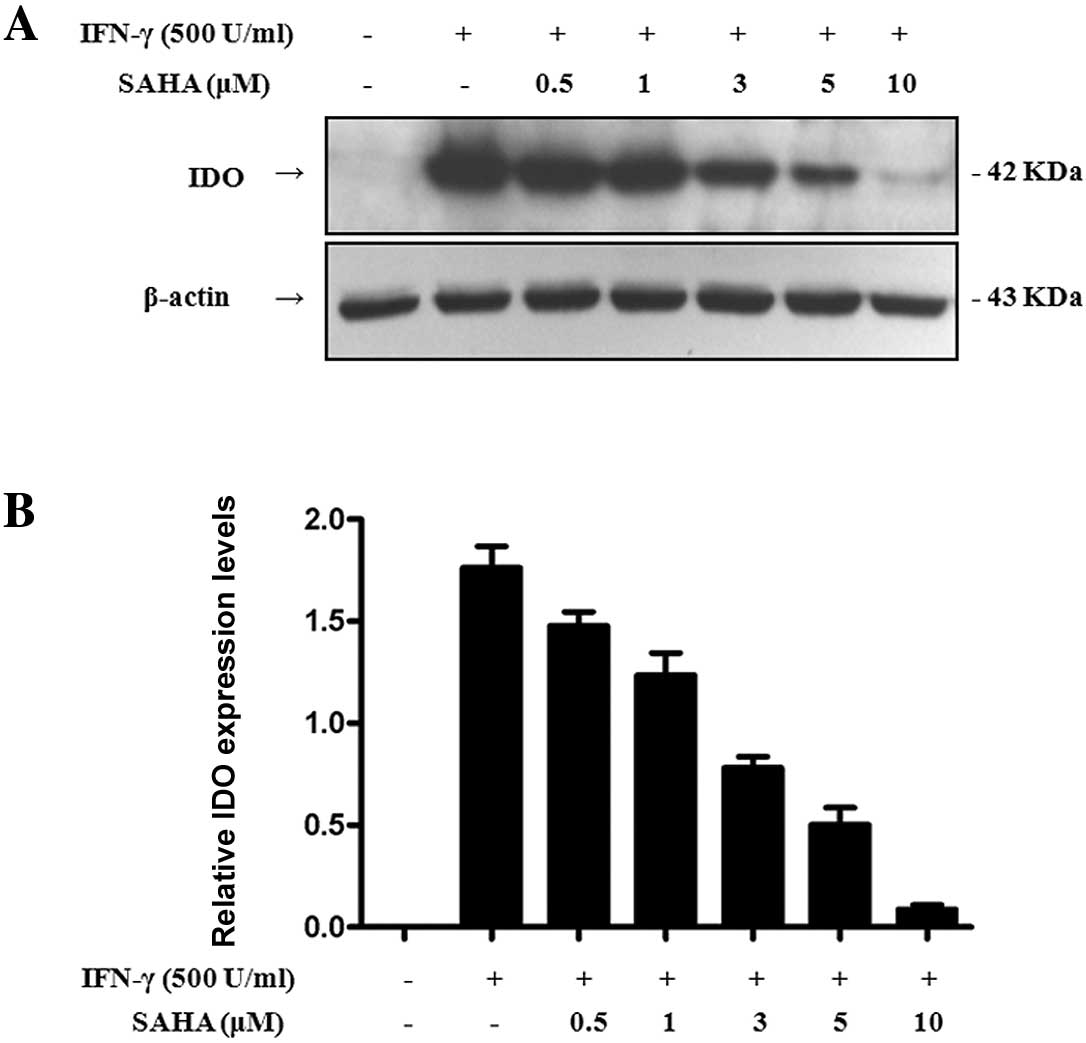
We further explored the influence of SAHA on IDO
expression. SGC-996 cells were pre-treated with various
concentrations of SAHA for 2 h, and then treated with IFN-γ for 24
h. We performed Western blot analysis to detect IDO expression in
total cell lysates. Treatment with SAHA (up to 0.5 μM) dramatically
reduced IDO expression induced by 500 U/ml IFN-γ (Fig. 2). With increasing concentrations of
SAHA, IDO expression decreased. IDO expression was almost
completely inhibited at a concentration of 10 μM. Therefore, SAHA
inhibited the expression of IDO induced by IFN-γ in a
dose-dependent manner.
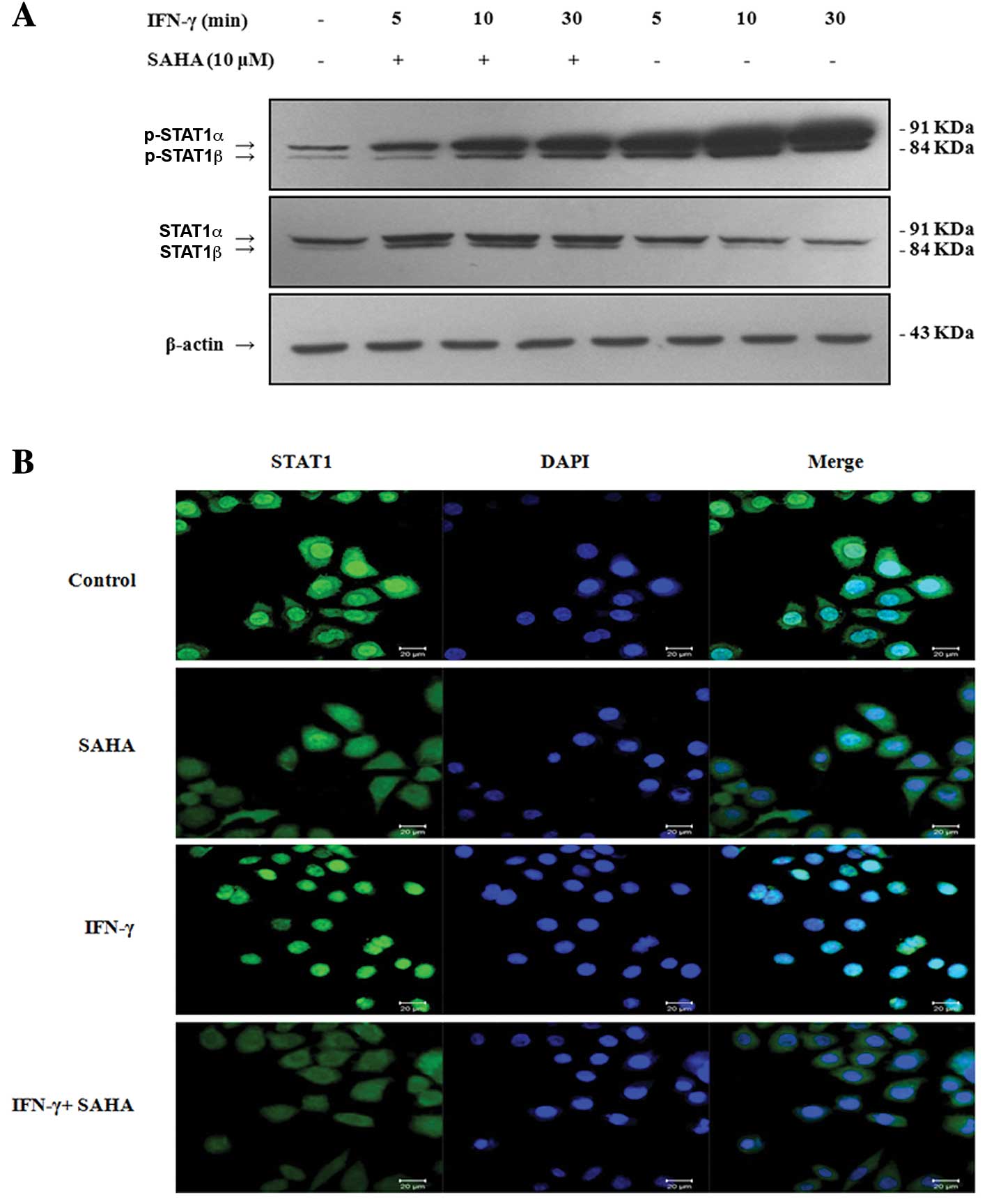
SAHA inhibits IFN-γ-induced STAT1
phosphorylation and nuclear translocation
IFN-γ is known to induce IDO expression via
activation of STAT1 (24). STAT1
has two isoforms, STAT1α (91 kDa) and STAT1β (84 kDa). Both
isoforms can be activated by IFN-γ. Phosphorylation of STAT1 at
Tyr701 induces STAT1 dimerization, nuclear translocation and DNA
binding. STAT1 phosphorylation acts as an active transcription
factor in the nucleus. Thus, we wondered whether SAHA blocked STAT1
Tyr701 phosphorylation and inhibited its nuclear translocation.
SGC-996 cells were pre-incubated with or without 10 μM SAHA for 2 h
and then treated with 500 U/ml IFN-γ for 5, 10 or 30 min. We
performed Western blot analysis to detect the phosphorylation of
STAT1 with an anti-phospho-STAT1 (Tyr701) antibody as described
above. Phosphorylated STAT1 was expressed without stimulation by
IFN-γ in SGC-996 cells. Stimulation of cells with IFN-γ for 5 min
alone brought about a rapid increase in tyrosine phosphorylation of
STAT1. The expression of phosphorylated STAT1 increased with the
stimulation time of IFN-γ. However, SAHA significantly inhibited
this increase (Fig. 3A). To further
investigate whether SAHA inhibited STAT1 nuclear translocation,
SGC-996 cells were grown on coverslips, pre-incubated with or
without 10 μM SAHA for 2 h, and then treated with or without 500
U/ml IFN-γ for 30 min. STAT1 nuclear localization was assessed by
confocal microscopy with an anti-STAT1 antibody. STAT1 was
localized exclusively in the cytoplasm of untreated cells, and SAHA
did not alter the basal subcellular localization of STAT1. On the
contrary, cells treated with IFN-γ for 30 min showed a significant
nuclear translocation of STAT1. However, IFN-γ-induced nuclear
translocation of STAT1 was considerably reduced after pre-treatment
with SAHA for 2 h (Fig. 3B). In
summary, our data suggested that SAHA inhibits phosphorylation and
nuclear translocation of STAT1, and this inhibition may result in
down-regulation of IDO expression.
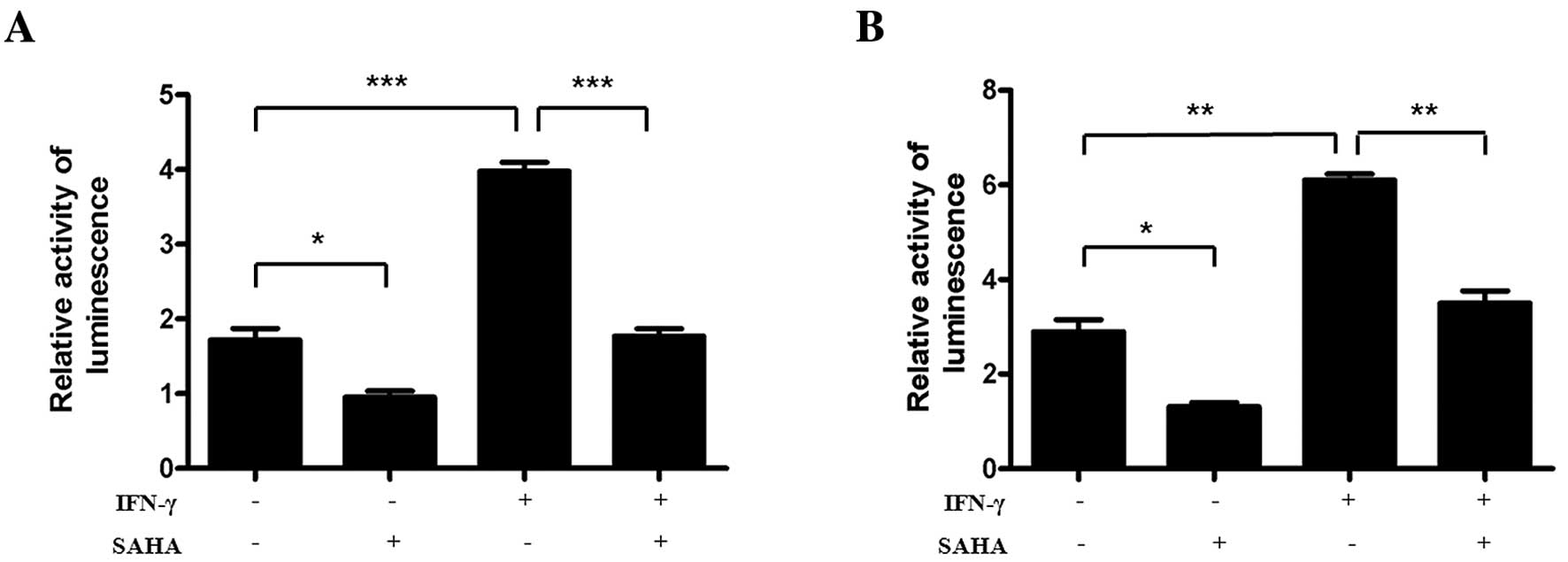
SAHA blocks IFN-γ-induced activation of
GAS and ISRE
Following the determination that SAHA inhibits the
tyrosine phosphorylation of STAT1 and its translocation to nucleus,
we examined whether the inhibitory effect of SAHA could block the
transcriptional activation of STAT1 in the nucleus. The reporter
gene plasmids pGL3-Enhancer-GAS7-luc and pGL3-Enhancer-ISRE4-luc
were transfected into SGC-996 cells. The transfection efficiency
was normalized by co-transfection with pRL-TK. The results showed
that IFN-γ significantly enhanced the activity of the
pGL3-Enhancer-GAS7-luc and the pGL3-Enhancer-ISRE4-luc via
Dual-Glo-Luciferase analysis. Furthermore, SAHA sharply inhibited
the activity of pGL3-Enhancer-GAS7-luc and pGL3-Enhancer-ISRE4-luc
(Fig. 4). These facts suggested
that SAHA down-regulated IFN-γ-induced expression of IDO via
blocking STAT1-driven transcriptional activity, mainly the activity
of GAS and ISRE.
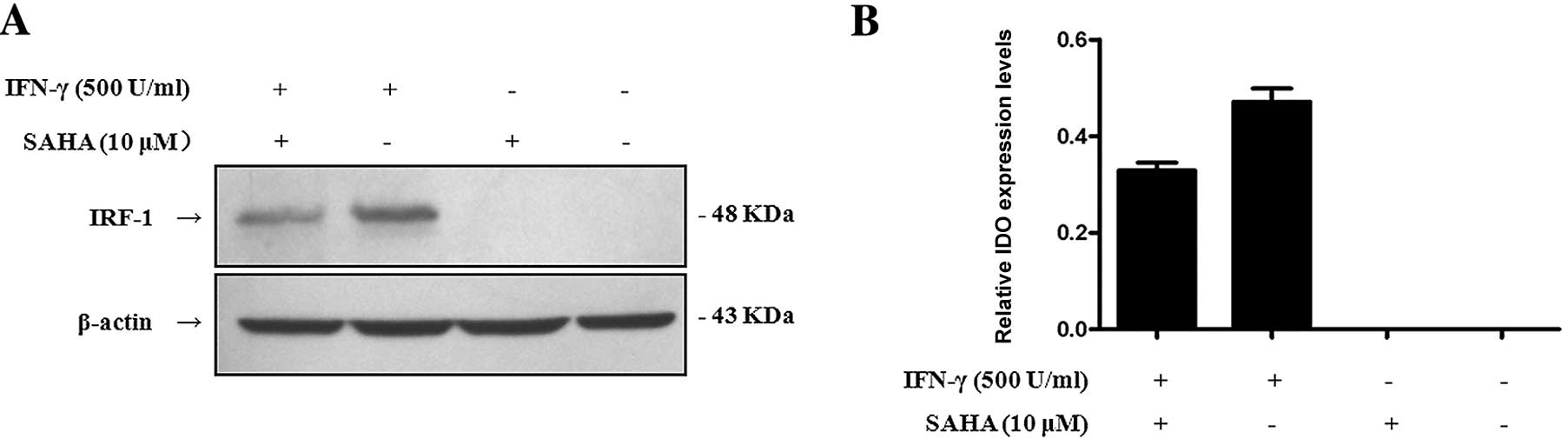
SAHA down-regulates the expression of the
IRF-1 protein induced by IFN-γ
IRF-1 is an essential factor for IFN-γ-induced
expression of IDO (8). Therefore,
we ttempted to determine whether SAHA affected the expression of
the IRF-1 protein. Cells were pre-treated with or without 10 μM
SAHA for 2 h and then treated with or without 500 U/ml IFN-γ for 8
h. We used Western blot analysis to detect IRF-1 protein in total
cell lysates with a monoclonal mouse anti-human IRF-1 antibody. The
results showed that IFN-γ dramatically enhanced IRF-1 expression,
but SAHA inhibited the expression of IRF-1 which was induced by
IFN-γ (Fig. 5). It could therefore
be concluded that SAHA down-regulated the expression of IDO via
blocking activation of IRF-1.
Discussion
IDO is an important immunogenic regulatory factor in
the process of tumor development and tumor immune tolerance
(25,26). Overexpression of IDO is closely
related to poor prognosis of patients with malignant tumors
(27). Furthermore, it is well
known that IFN-γ induces IDO expression via the JAK/STAT1 signaling
pathway. Consequently, blocking of the JAK/STAT1 signaling pathway
to break immune tolerance may present an option to improve the
clinical immunotherapeutic effect in gallbladder carcinoma
treatments. However, possible strategies to induce this inhibition
remain to be figured out.
IDO has been investigated in many tumors other than
gallbladder carcinoma as described above. Therefore, we first
detected whether IDO was expressed in SGC-996 cells. In the present
study, we demonstrated that IDO was not expressed without IFN-γ
treatment. However, after stimulation with 50 U/ml IFN-γ for 24 h
or 500 U/ml IFN-γ for 8 h, the expression of IDO could be
significantly detected. IDO expression induced by IFN-γ was dose-
and time-dependent in the gallbladder carcinoma SGC-996 cells.
Based on the fact that IDO is expressed in SGC-996
cells, we intended to find whether SAHA could inhibit IDO
expression in order to improve the immunotherapy of gallbladder
carcinoma. In this study, we discovered that SAHA down-regulated
the expression of IDO in a dose-dependant manner. Our observations
suggest that SAHA may be used in the clinic to improve the
efficiency of gallbladder carcinoma immunotherapy.
The IDO gene had a putative promoter region, and
this promoter contains a series of putative transcription factor
binding sites, including two ISRE regions, a GAS sequence near
ISRE-1 and ISRE-2, and two AP-1 binding sites (28,29).
In the process of the IFN-γ-induced expression of IDO, STAT1 acts
as a critical transcriptional factor. STAT1 is phosphorylated at
the cytoplasmic tail of the IFN-γ receptor, dimerizes and
translocates to the nucleus, where it binds GAS to activate IDO and
IRF-1 gene expression directly. On the other hand, STAT1
contributes indirectly to the activation of IDO via inducing the
production of IRF-1, which binds to ISRE-1 and ISRE-2 elements in
the IDO regulatory region resulting in activation of IDO gene
expression (8,17,30–32).
According to our data, SAHA inhibited the
IFN-γ-induced phosphorylation of STAT1, which plays an important
role in the regulation of IDO gene transcription. The inhibition of
STAT1 nuclear translocation resulted in an increase of STAT1
accumulation in the cytoplasm and disruption of the regulation of
the IDO gene transcription, directly down-regulating the expression
of IDO. In addition, we analyzed the roles of GAS and ISRE, which
regulated the expression of IDO in SGC-996 cells. Cells treated
with IFN-γ significantly induced transcriptional activity of the
reporter plasmids pGL3-Enhancer-GAS7 and pGL3-Enhancer-ISRE4
(Fig. 4), suggesting the importance
of GAS and ISRE in the IFN-γ-induced expression of IDO.
Furthermore, SAHA significantly inhibited
IFN-γ-induced transcriptional activation of the reporter plasmid
pGL3-Enhancer-GAS7 and pGL3-Enhancer-ISRE4 (Fig. 4). This phenomenon could be explained
by the fact that SAHA inhibited STAT1 phosphorylation and nuclear
translocation (Fig. 3A), which was
important for STAT1 binding of GAS and ISRE to activate the IDO
expression. Inhibition of GAS activation could directly
down-regulate the expression of IDO. Unlike GAS, activation of ISRE
required both phosphorylated STAT1 and IRF-1. Thus, the fact that
SAHA inhibited the activation of ISRE could result from the
inhibition of phosphorylated STAT1 and IRF-1. The IRF-1 promoter
region contains GAS elements which are important for IRF-1
expression. In this study we found that SAHA decreased the IRF-1
protein level induced by IFN-γ (Fig.
5). This phenomenon could be explained by the fact that SAHA
inhibited STAT1 phosphorylation and activation of GAS, followed by
down-regulation of the expression of IRF-1 protein.
In conclusion, we demonstrated for the first time
that SAHA down-regulated the expression of IDO induced by IFN-γ via
interfering with the phosphorylation and nuclear translocation of
STAT1 inhibiting its binding to GAS, ISRE and IRF-1 elements in the
IDO regulatory region. These discoveries suggest that the JAK/STAT1
signaling pathway plays a key role in the expression of IDO in
gallbladder carcinoma cells. Therefore, regulating JAK/STAT1
signaling pathway may provide a new gallbladder carcinoma
immunotherapeutic strategy to break tumor immune tolerance and
contribute to the development of clinical cancer immunotherapy.
Acknowledgements
This study was funded by the National Natural
Science Foundation of China (no. 30840096, no. 30873032 and no.
81071712), the National Science and Technology Major Project of
China (no. 2009ZX09103-040), the National Basic Research Program of
China (2011CB935800) and the Bureau of Guangdong Provincial Science
and Technology (no. 2008A030201007).
Abbreviations:
|
PGC
|
primary gallbladder carcinoma
|
|
HDACI
|
histone deacetylases inhibitors
|
|
SAHA
|
suberoylanilide hydroxamic acid
|
|
IDO
|
indoleamine 2,3-dioxygenase
|
|
IFN-γ
|
interferon-γ
|
|
JAK
|
Janus kinase
|
|
STAT1
|
signal transducer and activator of
transcription 1
|
|
GAS
|
γ-activated sites
|
|
ISRE
|
interferon-stimulated response
elements
|
|
IRF-1
|
interferon regulatory factor
genes-1
|
References
|
1
|
Jiao XY, Ren JL, Chen HY, et al: Ala499Val
(C>T) and Lys939Gln (A>C) polymorphisms of the XPC gene:
their correlation with the risk of primary gallbladder
adenocarcinoma: a case-control study in China. Carcinogenesis.
32:496–501. 2011.
|
|
2
|
Jiao XY, Huang JF, Wu SL, et al: HOGG1
Ser326Cys polymorphism and susceptibility to gallbladder cancer in
a Chinese population. Int J Cancer. 121:501–505. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Reid KM, Ramos-De la Medina A and Donohue
JH: Diagnosis and surgical management of gallbladder cancer: a
review. J Gastrointest Surg. 11:671–681. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Munn DH and Mellor AL: Indoleamine
2,3-dioxygenase and tumor-induced tolerance. J Clin Invest.
117:1147–1154. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Takikawa O: Biochemical and medical
aspects of the indoleamine 2,3-dioxygenase-initiated l-tryptophan
metabolism. Biochem Biophys Res Commun. 338:12–19. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Soliman H, Mediavilla-Varela M and Antonia
S: Indoleamine 2,3-dioxygenase: is it an immune suppressor? Cancer
J. 16:354–359. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Karanikas V, Zamanakou M, Kerenidi T, et
al: Indoleamine 2,3-dioxygenase (IDO) expression in lung cancer.
Cancer Biol Ther. 6:1258–1262. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pine R: Convergence of TNFalpha and
IFNgamma signalling pathways through synergistic induction of
IRF-1/ISGF-2 is mediated by a composite GAS/kappaB promoter
element. Nucleic Acids Res. 25:4346–4354. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yoshida N, Ino K, Ishida Y, et al:
Overexpression of indoleamine 2,3-dioxygenase in human endometrial
carcinoma cells induces rapid tumor growth in a mouse xenograft
model. Clin Cancer Res. 14:7251–7259. 2008. View Article : Google Scholar
|
|
10
|
Liu P, Xie BL, Cai SH, et al: Expression
of indoleamine 2,3-di-oxygenase in nasopharyngeal carcinoma impairs
the cytolytic function of peripheral blood lymphocytes. BMC Cancer.
9:4162009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Miyazaki T, Moritake K, Yamada K, et al:
Indoleamine 2,3-dioxygenase as a new target for malignant glioma
therapy. Laboratory investigation. J Neurosurg. 111:230–237. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Muller AJ, DuHadaway JB, Donover PS,
Sutanto-Ward E and Prendergast GC: Inhibition of indoleamine
2,3-dioxygenase, an immunoregulatory target of the cancer
suppression gene Bin1, potentiates cancer chemotherapy. Nat Med.
11:312–319. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zeng J, Cai S, Yi Y, et al: Prevention of
spontaneous tumor development in a ret transgenic mouse model by
ret peptide vaccination with indoleamine 2,3-dioxygenase inhibitor
1-methyl tryptophan. Cancer Res. 69:3963–3970. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang KS, Li GC, He YW, et al: Curcumin
inhibiting the expression of indoleamine 2,3-dioxygenase induced by
IFN-γ in cancer cells. Zhong Yao Cai. 31:1207–1211. 2008.(In
Chinese).
|
|
15
|
Platanias LC: Mechanisms of type-I- and
type-II-interferon-mediated signalling. Nat Rev. 5:375–386.
2005.PubMed/NCBI
|
|
16
|
Klampfer L, Huang J, Sasazuki T, Shirasawa
S and Augenlicht L: Inhibition of interferon gamma signaling by the
short chain fatty acid butyrate. Mol Cancer Res. 1:855–862.
2003.PubMed/NCBI
|
|
17
|
Chon SY, Hassanain HH, Pine R and Gupta
SL: Involvement of two regulatory elements in
interferon-gamma-regulated expression of human indoleamine
2,3-dioxygenase gene. J Interferon Cytokine Res. 15:517–526. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yamaguchi H, Chen CT, Chou CK, et al:
Adenovirus 5 E1A enhances histone deacetylase inhibitors-induced
apoptosis through Egr-1-mediated Bim upregulation. Oncogene.
29:5619–5629. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun PC, Tzao C, Chen BH, Liu CW, Yu CP and
Jin JS: Suberoylanilide hydroxamic acid induces apoptosis and
sub-G1 arrest of 320 HSR colon cancer cells. J Biomed Sci.
17:762010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu WS, Parmigiani RB and Marks PA: Histone
deacetylase inhibitors: molecular mechanisms of action. Oncogene.
26:5541–5552. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vigushin DM and Coombes RC: Histone
deacetylase inhibitors in cancer treatment. Anticancer Drugs.
13:1–13. 2002. View Article : Google Scholar
|
|
22
|
Frew AJ, Johnstone RW and Bolden JE:
Enhancing the apoptotic and therapeutic effects of HDAC inhibitors.
Cancer Lett. 280:125–133. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zong H, Yin B, Chen J, Ma B, Cai D and He
X: Over-expression of c-FLIP confers the resistance to
TRAIL-induced apoptosis on gallbladder carcinoma. Tohoku J Exp Med.
217:203–208. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Qing Y and Stark GR: Alternative
activation of STAT1 and STAT3 in response to interferon-gamma. J
Biol Chem. 279:41679–41685. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kobayashi N, Kubota K, Kato S, et al:
FOXP3+ regulatory T cells and tumoral indoleamine
2,3-dioxygenase expression predicts the carcinogenesis of
intraductal papillary mucinous neoplasms of the pancreas.
Pancreatology. 10:631–640. 2010.
|
|
26
|
Munn DH: Indoleamine 2,3-dioxygenase,
tumor induced tolerance and counter regulation. Curr Opin Immunol.
18:220–225. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Urakawa H, Nishida Y, Nakashima H,
Shimoyama Y, Nakamura S and Ishiguro N: Prognostic value of
indoleamine 2,3-dioxygenase expression in high grade osteosarcoma.
Clin Exp Metastasis. 26:1005–1012. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dai W and Gupta SL: Regulation of
indoleamine 2,3-dioxygenase gene expression in human fibroblasts by
interferon-gamma. Upstream control region discriminates between
interferon-gamma and interferon-alpha. J Biol Chem.
265:19871–19877. 1990.
|
|
29
|
Kadoya A, Tone S, Maeda H, Minatogawa Y
and Kido R: Gene structure of human indoleamine 2,3-dioxygenase.
Biochem Biophys Res Commun. 189:530–536. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Konan KV and Taylor MW: Importance of the
two interferon-stimulated response element (ISRE) sequences in the
regulation of the human indoleamine 2,3-dioxygenase gene. J Biol
Chem. 271:19140–19145. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Boehm U, Klamp T, Groot M and Howard JC:
Cellular responses to interferon-gamma. Annu Rev Immunol.
15:749–795. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jeong YI, Kim SW, Jung ID, et al: Curcumin
suppresses the induction of indoleamine 2,3-dioxygenase by blocking
the Janus-activated kinase-protein kinase Cdelta-STAT1 signaling
pathway in interferon-gamma-stimulated murine dendritic cells. J
Biol Chem. 284:3700–3708. 2009. View Article : Google Scholar
|