Introduction
Unlike tumour suppressive apoptosis and autophagic
cell death, necrosis promotes tumour growth, progression, and
aggressiveness (1,2). The tumour promoting activity of
necrosis is thought to be mediated by a nuclear protein high
mobility group box 1 (HMGB1), which exerts pro-inflammatory and
tumour-promoting cytokine activities when released into the
extracellular spaces due to the rupture of the plasma membrane by
necrosis (1–4). In solid tumours, the cells in the
inner regions experience metabolic stress such as hypoxia and
glucose deprivation (GD) resulting from insufficient
vascularization. Although most cells adapt to this environment to
obtain more aggressive properties, those in the core region die by
necrosis, thereby forming the necrotic core (5). Because necrosis is linked to tumour
growth and development, it is an important cell death type in
tumour cell biology; however, the molecular mechanism underlying
necrotic cell death remains largely unknown.
Early growth response 1 (Egr-1) is a 3 Cys2-His2
type COOH-terminal zinc-finger transcription factor that binds to
GC-rich recognition motifs (5′-GCGT/GGGGCG-3′ or
5′-TCCT/ACCTCCTCC-3′) (6,7). Egr-1 is induced by a number of
different stimuli, such as anti-cancer drugs and growth factors and
inhibits or stimulates tumour growth depending the cellular context
and the duration of Egr-1 induction (6–13).
Egr-1 was able to directly regulate multiple tumour suppressors
including p53, TGF-β1, and PTEN to induce apoptotic cell death
(14). In addition, Egr-1 is
induced by hypoxia and plays a critical role(s) in hypoxia-induced
tumour progression, survival, and angiogenesis (15–18).
Furthermore, Egr-1 is involved in hepatocyte growth factor
(HGF)-induced cell scattering, migration, and invasion via Snail
activation (19). While transient
induction of Egr-1 is known to activate angiogenesis, sustained
Egr-1 expression induces antiangiogenesis, growth arrest, and
apoptosis (20). Thus, Egr-1 is
thought to act as a crucial regulator of tumour cell death, growth,
invasion, and angiogenesis.
In this study, we tried to identify the mechanism
underlying metabolic stress-induced necrosis. Previously, we showed
that GD induced necrosis in several tumour cell types including
A549, MDA-MB-231, and HepG2 cells and activation of protein kinase
C by treatment of phorbol 12-myristate 13-acetate (PMA) switched
GD-induced necrosis to apoptosis in A549 cells (21). By cDNA microarray analysis, we found
that Egr-1 expression was increased by GD, but not by GD+PMA. In
this study, we evaluated the possible role(s) of Egr-1 in necrosis.
We found that Egr-1 shRNA prevented necrosis, whereas Egr-1
overexpression made tumour cells more sensitive to GD, thereby
leading to necrosis. In addition, Egr-1 shRNA suppressed the growth
of multicellular tumour spheroids (MTSs), an in vitro tumour
model system. Taken together, these results demonstrate that Egr-1
plays an important role(s) in GD-induced necrosis and tumour
progression.
Materials and methods
Cell culture, chemical treatment, and
multicellular tumour spheroid (MTS) culture
A549, MDA-MB-231, HepG2, HCT116, and HeLa cells were
obtained from American Type Culture Collection, and maintained in
RPMI-1640 or DMEM supplemented with 10% (v/v) heat-inactivated
fetal bovine serum (Hyclone, Logan, UT, USA) and 1%
penicillin-streptomycin (Hyclone) in a 37°C humidified incubator
with 5% CO2. The cells were treated with GD, reactive
oxygen species [ROS, including H2O2 and
menadione (an O2- generator)], or other
chemicals as described previously (22). Multicellular tumour spheroid culture
was performed using MCF-7 cells (provided by Dr J.I. Yook,
University of Yonsei, Korea) as described previously (22) and MTSs dissociation into
subpopulations of cells from four different locations was conducted
as described by LaRue and colleagues (23).
Microarray
Microarray was performed to screen the
differentially expressed genes using Operon Human Whole 35K Oligo
chips (GenoCheck, Korea) (22). The
Affymetrix microarray data have been deposited in the Gene
Expression Omnibus (GEO) database (GEO accession no. GSE24271).
Western blotting, HMGB1 release assay,
RT-PCR, and real-time PCR
Western blotting were performed using the following
antibodies: Egr-1 (Santa Cruz, CA); α-tubulin (Biogenex, CA); HMGB1
(BD Pharmingen, CA); CuZnSOD (Santa Cruz, CA); ERK1/2 (Cell
Signaling, MA). The HMGB1 release assay was carried out as
described previously (21,22). Transcript levels were assessed with
reverse transcription-polymerase chain reaction (RT-PCR) with
primers for Egr-1 and GAPDH (Table
I). Quantitative real-time PCR was conducted in a LightCycler
(Roche Diagnostics, Mannheim, Germany) using a SYBR Green kit
(Roche Diagnostics) with primers for Egr-1 and β-actin (Table I).
 | Table ISequences used in this study for
RT-PCR, real-time PCR, and shRNA interference. |
Table I
Sequences used in this study for
RT-PCR, real-time PCR, and shRNA interference.
| | Sequence 5′→3′ | Annealing
temperature (°C) |
|---|
| RT-PCR |
| GAPDH |
| NM_002046.3 | Sense |
GTGGTCTCCTCTGACTTCAAC | |
| Antisense |
TCTCTTCCTCTTGTGCTCTTG | |
| Egr-1 |
| NM_001964.2 | Sense |
ATTCTGAGGCCTCGCAAGTA | 54 |
| Antisense |
CACTGCTTTTCCGCTCTTTC | |
| Real-time PCR |
| β-actin |
| NM_001101.3 | Sense |
ACTCTTCCAGCCTTCCTTCC | |
| Antisense |
TGTTGGCGTACAGGTCTTTG | |
| Egr-1 | Sense |
AGGACAGGAGGAGGAGATGG | 62 |
| Antisense |
GGAAGTGGGCAGAAAGGATTG | |
| shRNA
interference |
| Con shRNA | |
AATTCTCCGAACGTGTCACGT | |
| Egr-1 shRNA | |
AAGTTACTACCTCTTATCCAT | |
Hoechst 33342 (HO)/propidium iodide (PI)
staining and ROS staining
To determine the cell death mode, Hoechst 33342 (HO)
and propidium iodide (PI) double staining was performed (21,22).
In 2D culture, cells were seeded at a density of 2.5×105
cells/ml in 35-mm dishes. After 24 h, the cells were treated with
GD for the indicated times and then stained with HO (1 μg/ml) and
PI (5 μg/ml) for 15 min. In 3D culture, equal numbers of spheroids
were transferred to 1.2% agarose-coated 60-mm dishes and
trypsinized and then stained with HO/PI, The stained cells were
observed under a fluorescence microscope and apoptotic and necrotic
cells were scored. Intracellular H2O2,
O2- and mitochondrial ROS measurement were
conducted as described previously (21,22).
Egr-1 transfection and short hairpin RNA
(shRNA) interference
pcDNA3.1-Egr-1, constructed by inserting the Egr-1
open reading frame into plasmid pcDNA3.1/NEO expression vector
(Invitrogen), was provided by Dr Thomas E. Eling (Laboratory of
Molecular Carcinogenesis, National Institute of Environmental
Health Sciences, USA). The vectors pcDNA3.1 and pcDNA3.1-Egr-1 were
transfected into MCF-7 cells using jetPEI (Polyplus transfection)
according to manufacturer’s protocol. Egr-1 shRNA target sequences
were designed and verified as specific for Egr-1 by Blast search
against the human genome and real-time PCR, respectively (Table I). The vectors pSUPER-control shRNA
and pSUPER-Egr-1 shRNA were transfected using jetPEI and stable
cell lines were selected using 1–2 mg/ml G418. Several stable
clones were isolated after shRNA transfection and individually
characterized.
Statistical analysis
Data were analyzed by the Student’s t-test and
P<0.05 was considered statistically significant.
Results and Discussion
Induction of Egr-1 by metabolic stress
and ROS
We analyzed the gene expression profiling of A549
cells that were treated with GD or GD+PMA by cDNA microarrays
(21). One of GD-induced genes was
Egr-1 (Fig. 1A); Egr-1 level was
increased 3.4-fold during necrosis, whereas its level was not
significantly changed during apoptosis. The induction of Egr-1 by
GD was also observed in MDA-MB-231 and HepG2 cells that underwent
necrosis upon GD, as revealed by RT-PCR (Fig. 1B). We further examined GD induction
of Egr-1 in other cancer cells. Western blot analysis showed the
induction of Egr-1 by GD in MDA-MB-231 and HepG2 cells, but not in
MCF-7 cells that exhibit necrosis to a much lower degree than
MDA-MB-231 and HepG2 cells upon GD and in HCT116 and HeLa cells
that exhibit apoptosis upon exposure to GD (Fig. 1C). Real-time PCR confirmed the
induction of Egr-1 by GD in MDA-MB-231 and HepG2 cells (3- to
4-fold), and but not in MCF-7, HCT116, and HeLa cells (Fig. 1D). GD is known to induce necrosis by
increasing mitochondrial ROS production. Thus, we examined the
possible role(s) of ROS in GD-induced Egr-1 expression. Egr-1
induction by GD was inhibited by treatment with the antioxidant
N-acetylcysteine (NAC) in MDA-MB-231 and HepG2 cells (Fig. 1D). In addition,
H2O2 and menadione (an
O2- generator) increased Egr-1 mRNA
expression in MCF-7 cells as determined by real-time PCR (Fig. 1E), indicating the redox-sensitivity
of Egr-1 expression. H2O2 at non-toxic doses
has been shown to induce the accumulation of mRNA for Egr-1 gene in
mammalian cells (24).
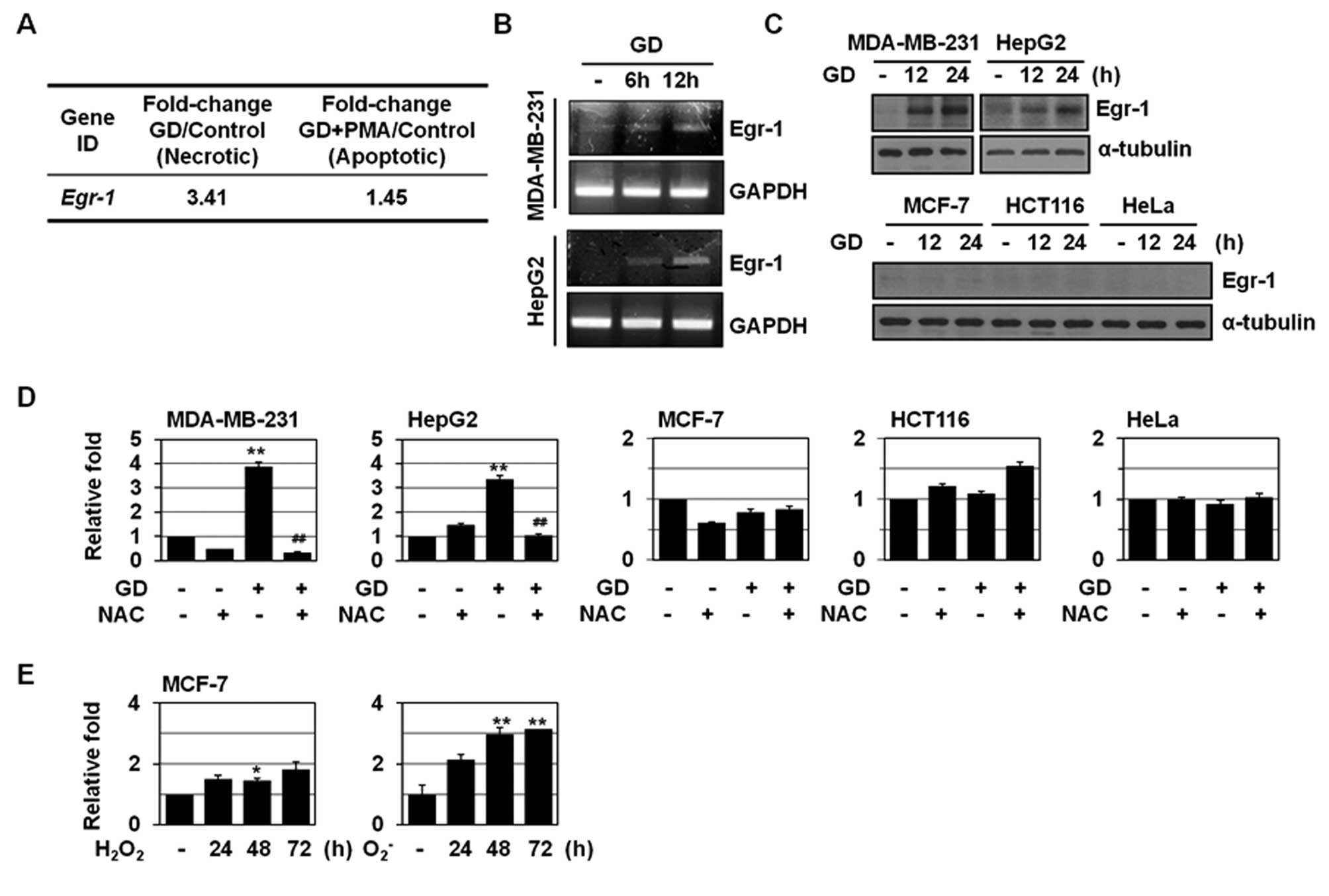 | Figure 1Induction of Egr-1 during GD-induced
necrosis. (A) A549 cells were pretreated with PMA and treated with
GD for 12 h and microarray analysis was performed. The numbers mean
fold increase in expression as compared with GD-untreated control
cells. (B) MDA-MB-231 and HepG2 cells were exposed to GD medium for
the indicated times, and then analyzed by RT-PCR for Egr-1 and
GAPDH. (C) Several cancer cells including MDA-MB-231, HepG2, MCF-7,
HCT116, and HeLa were exposed to GD for the indicated times, and
then analyzed by western blotting for Egr-1 and α-tubulin. (D)
Cancer cells were pretreated with NAC (10 mM) for 1 h, exposed to
GD medium for 12 h, and then analyzed by real-time PCR for Egr-1
and β-actin. Results are expressed as mean ± SE.
**P<0.01 versus untreated; ##P<0.01
versus GD-treated cells. (E) MCF-7 cells were treated with
H2O2 (300 μM) or menadione
(O2-, 10 μM) for the indicated times and then
analyzed by real-time PCR for Egr-1 and β-actin. Results are
expressed as mean ± SE. *P<0.05,
**P<0.01 versus untreated. |
Egr-1 shRNA prevented metabolic
stress-induced necrosis and HMGB1 release
To explore the role(s) of Egr-1 in necrosis, we
examined the effects of Egr-1 shRNA, directed to the C-terminal
region of human Egr-1 mRNA sequences (position from 1630 to 1648 in
human cDNA, Table I), on GD-induced
necrosis. HO/PI double staining method was used to identify
apoptosis as well as necrosis. While HO penetrates non-selectively
plasma membrane of both damaged and intact cells and binds to DNA,
causing a blue nuclear fluorescence, PI penetrates only cells with
damaged-membranes, causing red nuclear fluorescence. Thus, the cell
death mode could be discriminated morphologically by nuclear
fluorescence images: intact blue nuclei, condensed/fragmented blue
nuclei, condensed/fragmented pink nuclei, and intact pink nuclei
indicated viable, early apoptotic, late apoptotic (secondary
necrotic), and necrotic cells, respectively. Egr-1 shRNA appeared
to effectively knock down Egr-1 mRNA levels in MDA-MB-231 cells, as
determined by RT-PCR (Fig. 2A).
Egr-1 shRNA significantly inhibited GD-induced cell rounding (data
not shown) and increase in cell population that had intact pink
nuclei in HO/PI staining in MDA-MB-231 cells (Fig. 2B), without increasing the population
of the cells with condensed/fragmented blue nuclei and apoptotic
bodies (data not shown). Thus, knockdown of Egr-1 appeared to
inhibit GD-induced necrosis without switching to apoptotic cell
death as an alternative death mechanism. We also observed that
Egr-1 shRNA suppressed GD-induced release of HMGB1 into the
extracellular space (Fig. 2C).
Previously, we showed that in contrast to a general concept that
necrotic cell death causes the release of most cellular proteins
due to cell membrane rupture, only a restricted set of cellular
proteins such as HMGB1 and CuZnSOD were selectively released during
GD-induced necrosis (25). Egr-1
shRNA appeared to suppress GD-induced release of CuZnSOD into the
extracellular space (Fig. 2C).
These results indicate that Egr-1 may be involved in metabolic
stress-induced necrosis.
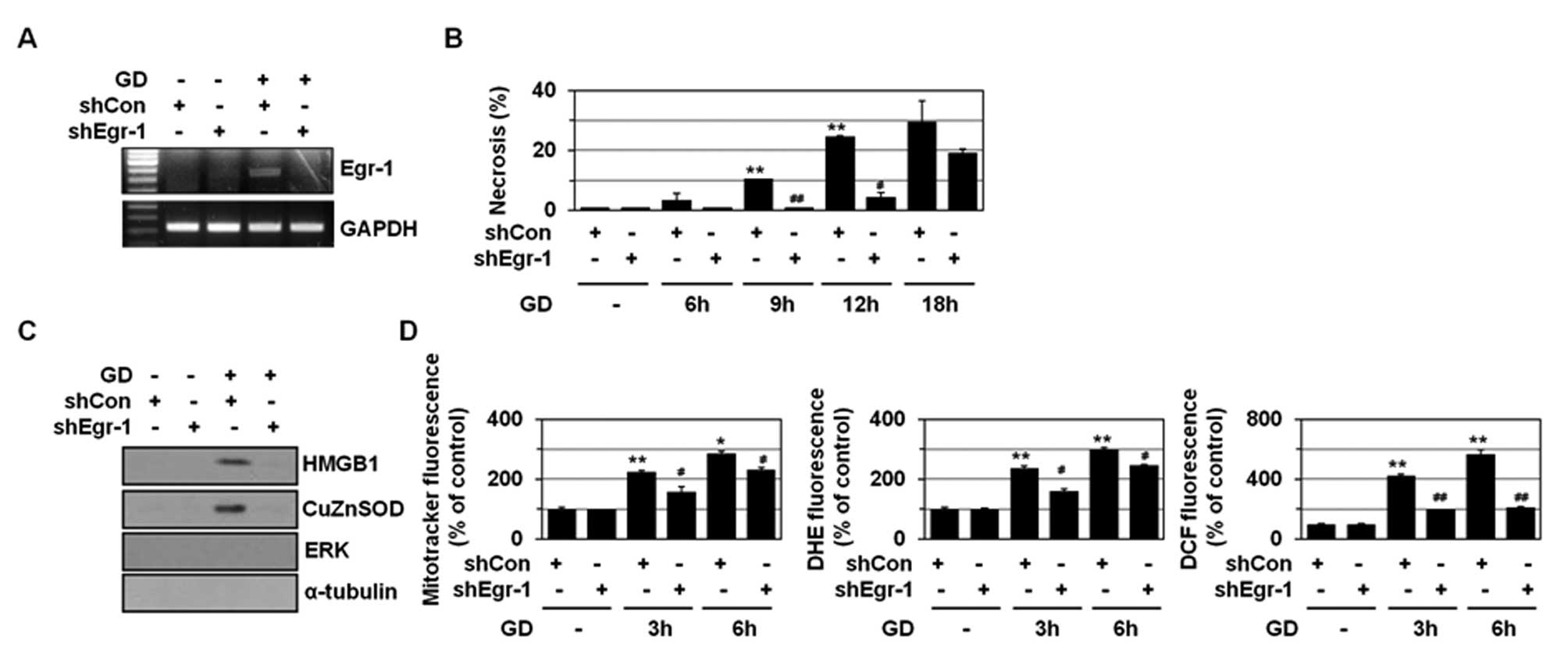 | Figure 2Egr-1 plays a role(s) in GD-induced
necrosis. (A) MDA-MB-231 cells were stably transfected with control
or Egr-1 shRNA and exposed to GD for 12 h and then analyzed by
RT-PCR using primers for Egr-1. (B) MDA-MB-231 cells stably
transfected with control or Egr-1 shRNA were exposed to GD for the
indicated times, and stained with HO/PI, and observed by
fluorescence microscopy, and apoptotic and necrotic cells were
scored. Results are expressed as mean ± SE from 500 to 800 cells
per treatment group and from three independent experiments.
**P<0.01 versus control; #P<0.05,
##P<0.01 versus control shRNA. (C) MDA-MB-231 cells
stably transfected with control or Egr-1 shRNA were exposed to GD
for 12 h and the media were analyzed by western blotting with
antibodies against HMGB1, CuZnSOD, ERK, and α-tubulin. (D)
MDA-MB-231 cells stably transfected with control or Egr-1 shRNA
were exposed to GD for 3 or 6 h, and mitochondrial ROS and
O2- and intracellular
H2O2 production was measured using the
MitoTracker Red CM-H2XRos, DHE, and DCFH-DA,
respectively, under a fluorescent microscope (X200, Carl Zeiss).
Results are expressed as mean ± SE. *P<0.05,
**P<0.01 versus untreated; #P<0.05,
##P<0.01 versus control shRNA. |
Mitochondrial O2- is induced
upon GD and mediates GD-induced necrosis and cytotoxicity (26–28).
As shown in Fig. 2D, GD
significantly enhanced the production of mitochondrial ROS,
O2-, and intracellular
H2O2, as revealed by staining with three
different fluorogenic probes including MitoTracker Red
CM-H2XRos, HE, and DCFH-DA, respectively. Egr-1
interference blocked GD-induced production of mitochondrial ROS,
O2-, and intracellular
H2O2 (Fig.
2D), indicating that Egr-1 may control necrosis through
regulating GD-induced mitochondrial ROS production.
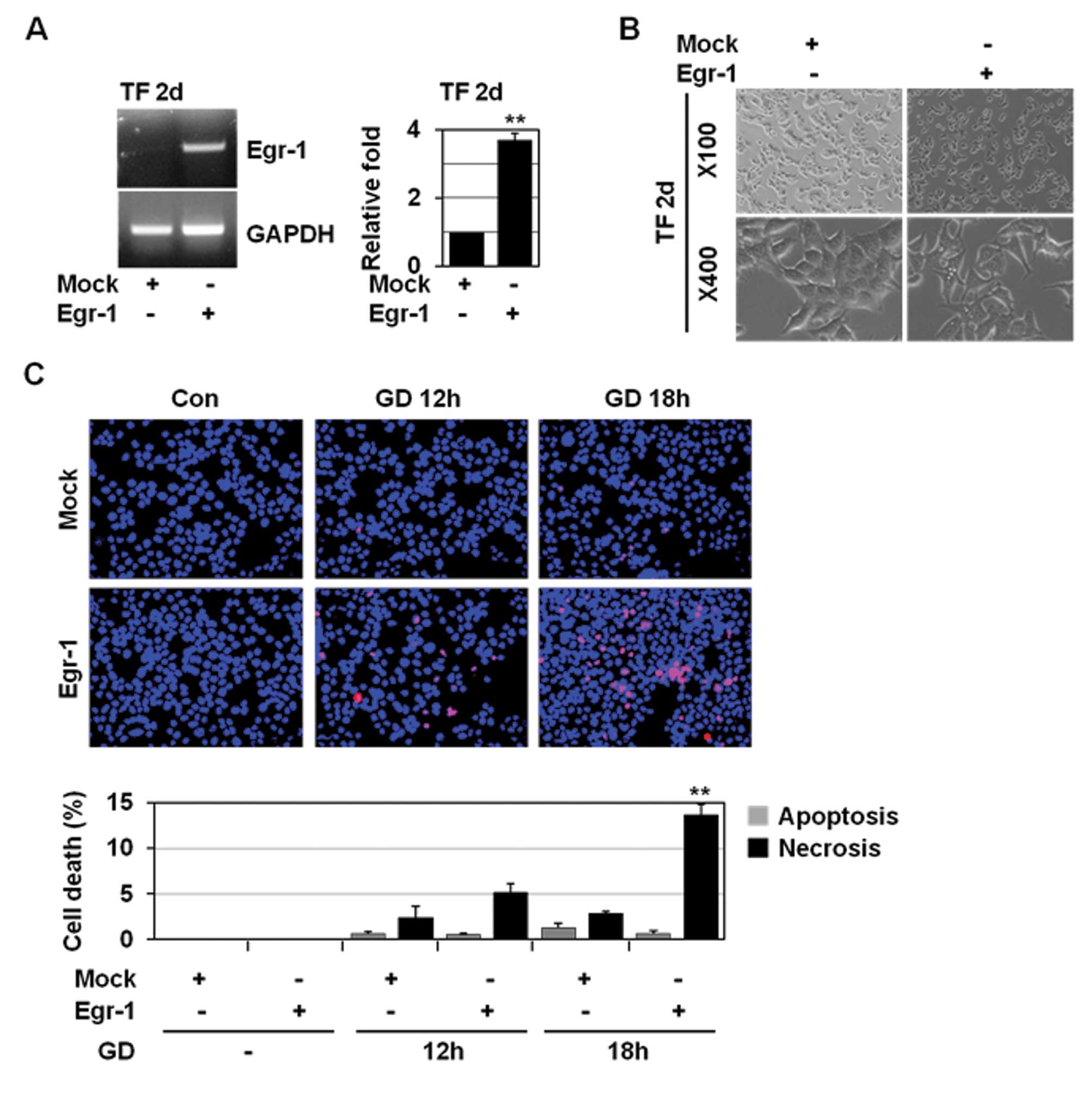
Egr-1 overexpression facilitates
GD-induced necrosis
To further examine the role of Egr-1 in necrosis, we
overexpressed Egr-1 in MCF-7 cells. Egr-1 is involved in
HGF-induced cell scattering, migration, and invasion via Snail
activation (19). Egr-1
overexpression in MCF-7 cells caused the morphological changes
including loss of intercellular adhesion and formation of a
spindle-like cell shape and pseudopodia, which represent the
morphology typical of mesenchymal cells (Fig. 3A and B). However, it did not induce
necrosis (data not shown). These results demonstrate that Egr-1 is
necessary but not sufficient to trigger necrosis. Necrosis is
closely linked to excess ROS production, mitochondrial dysfunction,
and decreased ATP production (29,30).
Thus, Egr-1 may trigger necrosis if tumour cells are under
metabolic stress. Therefore, we examined whether Egr-1
overexpression could facilitate GD-inducible necrosis in MCF-7
cells that exhibit a much lower degree (approximately 2–3%) of
necrosis upon GD. In MCF-7 cells, Egr-1 overexpression slightly,
but to a statistically significant extent (12–13%), increased
necrosis upon GD (Fig. 3C),
indicating the necrosis-facilitating activity of Egr-1.
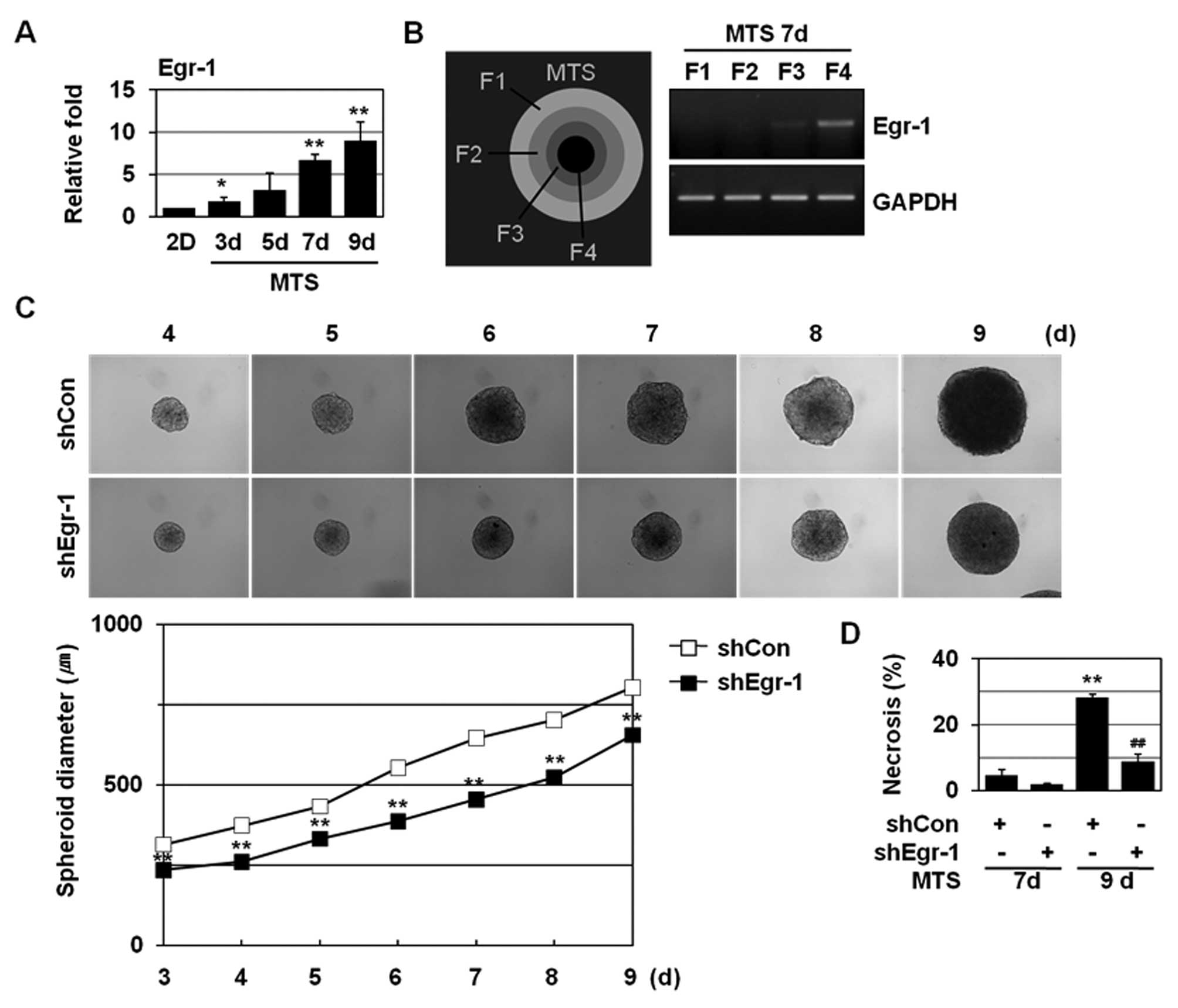
Egr-1 shRNA prevents metabolic
stress-induced necrosis in MTSs and suppresses MTS growth
We examined the effects of Egr-1 shRNA on necrosis
using MTSs. MTSs closely resemble poorly vascularized solid
tumours, and thus are used for an in vitro model of solid
tumours. MCF-7 cells formed tightly packed spheroids of a
homogeneous size and as the MCF-7 MTSs grow, a proliferation
gradient is observed, with proliferating cells at the periphery,
cell cycle arrested cells in the inner regions, and necrotizing
cells in the core regions (31,32).
Continued MTS growth leads to the formation of necrotic core due to
microenvironmental stresses including deprivation of oxygen and
nutrients. H&E and HO/PI double staining revealed the necrotic
core formation at 8–9 days of MTS culture (22,33).
Although monolayer-cultured MCF-7 cells exhibit limited levels of
necrosis upon GD, they showed prominent necrotic cell death in the
core region when cultured as MTSs. An increased expression of Egr-1
was detected with extended MTS culture (Fig. 4A); Egr-1 induction was observed at 7
and 9 day MTSs. To determine the expression of Egr-1 in MTSs, the
spheroids were selectively dissociated to yield cells from four
discrete regions within the spheroid. Enhanced Egr-1 expression was
detected in the innermost F4 fraction (Fig. 4B), indicating that Egr-1 expression
is closely related to microenvironmental stresses, such as hypoxia
and GD. As shown in Fig. 4C, Egr-1
shRNA prevented MTS growth. We further found that Egr-1 shRNA
caused a prominent reduction in the population of cells that had
pink nuclei with HO/PI staining at 9 days in MCF-7 MTS culture
(Fig. 4D). These findings
demonstrate that Egr-1 is involved in GD-induced necrosis and
tumour progression.
Biological relevance of this study
An immediate early gene Egr-1 is implicated in
tumour cell biology. While Egr-1 is known to induce apoptosis in
tumour cells, it also promotes tumour progression, survival, and
angiogenesis (6–13). In this study, we showed that Egr-1
is induced upon GD and is implicated in GD-induced necrosis. Egr-1
appeared to be induced by GD-triggered mitochondrial ROS production
and to exert a positive effect on mitochondrial ROS production, in
a forward feeding ROS-producing fashion. ROS induced under
stressful conditions are known to move from a mitochondrion to
neighboring mitochondria to enhance ROS production in a process
known as ROS-induced ROS release (RIRR) (34,35).
Thus, Egr-1 may be implicated in the mechanisms for RIRR, which is
responsible for GD-induced necrosis. How does Egr-1 affect
mitochondrial ROS production upon GD? Mitochondrial dysfunction has
been shown to be linked to increased ROS production and necrosis
induction. For instance, tumour cells with dysregulated
mitochondria undergo necrosis in response to a glycolysis inhibitor
2-deoxyglucose or alkylating DNA damage that causes rapid ATP
depletion (36). Thus, Egr-1 may
regulate genes linked to mitochondrial dysfunction. Previously, we
showed that Snail (22) and Dlx-2
(37) are also implicated in
GD-induced necrosis; thus, Snail, Dlx-2 and Egr-1 may cooperate to
induce necrosis. We further showed that Snail suppressed
mitochondrial respiration and cytochrome C oxidase (COX) activity
by inhibiting the expression of 3 COX subunits, including COXVIc,
COXVIIa, and COXVIIc (38). Because
Egr-1 is able to induce Snail (data not shown), Snail may be
responsible for Egr-1-triggered necrosis. The mechanism whereby
Egr-1 and Snail enhance mitochondrial depression, mitochondrial ROS
production, and necrosis is under investigation.
Acknowledgements
This study was supported by the National Research
Foundation of Korea (NRF) grant funded by the Korea government
(MEST) (2009-0072912). We thank Dr Thomas E. Eling (Laboratory of
Molecular Carcinogenesis, National Institute of Environmental
Health Sciences, USA) and Dr J.I. Yook (University of Yonsei,
Korea) for providing pcDNA3.1-Egr-1 and MCF-7 cells,
respectively.
References
|
1
|
Vakkila J and Lotze MT: Inflammation and
necrosis promote tumour growth. Nat Rev Immunol. 4:641–648. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jin S and White E: Role of autophagy in
cancer: management of metabolic stress. Autophagy. 3:28–31. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lotze MT and Tracey KJ: High-mobility
group box 1 protein (HMGB1): nuclear weapon in the immune arsenal.
Nat Rev Immunol. 5:331–342. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schlueter C, Weber H, Meyer B, Rogalla P,
Roser K, Hauke S and Bullerdiek J: Angiogenetic signaling through
hypoxia: HMGB1: an angiogenetic switch molecule. Am J Pathol.
166:1259–1263. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gatenby RA and Gillies RJ: Why do cancers
have high aerobic glycolysis? Nat Rev Cancer. 4:891–899. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Adamson ED and Mercola D: Egr1
transcription factor: multiple roles in prostate tumor cell growth
and survival. Tumour Biol. 23:93–102. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Thiel G and Cibelli G: Regulation of life
and death by the zinc finger transcription factor Egr-1. J Cell
Physiol. 193:287–292. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xie B, Wang C, Zheng Z, Song B, Ma C,
Thiel G and Li M: Egr-1 transactivates Bim gene expression to
promote neuronal apoptosis. J Neurosci. 31:5032–5044. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yamaguchi H, Chen CT, Chou CK, Pal A,
Bornmann W, Hortobagyi GN and Hung MC: Adenovirus 5 E1A enhances
histone deacetylase inhibitors-induced apoptosis through
Egr-1-mediated Bim upregulation. Oncogene. 29:5619–5629. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mahalingam D, Natoni A, Keane M, Samali A
and Szegezdi E: Early growth response-1 is a regulator of
DR5-induced apoptosis in colon cancer cells. Br J Cancer.
102:754–764. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wagner M, Schmelz K, Dorken B and Tamm I:
Transcriptional regulation of human survivin by early growth
response (Egr)-1 transcription factor. Int J Cancer. 122:1278–1287.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zagurovskaya M, Shareef MM, Das A, Reeves
A, Gupta S, Sudol M, Bedford MT, Prichard J, Mohiuddin M and Ahmed
MM: EGR-1 forms a complex with YAP-1 and upregulates Bax expresion
in irradiated prostate carcinoma cells. Oncogene. 28:1121–1131.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ahmed MM: Regulation of radiation-induced
apoptosis by early growth response-1 gene in solid tumors. Curr
Cancer Drug Targets. 4:43–52. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Baron V, Adamson ED, Calogero A, Ragona G
and Mercola D: The transcription factor Egr1 is a direct regulator
of multiple tumor suppressors including TGFbeta1, PTEN, p53, and
fibronectin. Cancer Gene Ther. 13:115–124. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rong Y, Hu F, Huang R, Mackman N, Horowitz
JM, Jensen RL, Durden DL, Van Meir EG and Brat DJ: Early growth
response gene-1 regulates hypoxia-induced expression of tissue
factor in glioblastoma multiforme through hypoxia-inducible
factor-1-independent mechanisms. Cancer Res. 66:7067–7074. 2006.
View Article : Google Scholar
|
|
16
|
Liao H, Hyman MC, Lawrence DA and Pinsky
DJ: Molecular regulation of the PAI-1 gene by hypoxia:
contributions of Egr-1, HIF-1alpha, and C/EBPalpha. FASEB J.
21:935–949. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang P, Tchou-Wong KM and Costa M: Egr-1
mediates hypoxia-inducible transcription of the NDRG1 gene through
an overlapping Egr-1/Sp1 binding site in the promoter. Cancer Res.
67:9125–9133. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nishi H, Nishi KH and Johnson AC: Early
growth response-1 gene mediates up-regulation of epidermal growth
factor receptor expression during hypoxia. Cancer Res. 62:827–834.
2002.PubMed/NCBI
|
|
19
|
Grotegut S, von Schweinitz D, Christofori
G and Lehembre F: Hepatocyte growth factor induces cell scattering
through MAPK/Egr-1-mediated upregulation of Snail. EMBO J.
25:3534–3545. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lucerna M, Pomyje J, Mechtcheriakova D,
Kadl A, Gruber F, Bilban M, Sobanov Y, Schabbauer G, Breuss J,
Wagner O, Bischoff M, Clauss M, Binder BR and Hofer E: Sustained
expression of early growth response protein-1 blocks angiogenesis
and tumor growth. Cancer Res. 66:6708–6713. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim CH, Han SI, Lee SY, Youk HS, Moon JY,
Duong HQ, Park MJ, Joo YM, Park HG, Kim YJ, Yoo MA, Lim SC and Kang
HS: Protein kinase C-ERK1/2 signal pathway switches glucose
depletion-induced necrosis to apoptosis by regulating superoxide
dismutases and suppressing reactive oxygen species production in
A549 lung cancer cells. J Cell Physiol. 211:371–385. 2007.
View Article : Google Scholar
|
|
22
|
Kim CH, Jeon HM, Lee SY, Ju MK, Moon JY,
Park HG, Yoo MA, Choi BT, Yook JI, Lim SC, Han SI and Kang HS:
Implication of snail in metabolic stress-induced necrosis. PLoS
One. 6:e180002011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
LaRue KE, Khalil M and Freyer JP:
Microenvironmental regulation of proliferation in multicellular
spheroids is mediated through differential expression of
cyclin-dependent kinase inhibitors. Cancer Res. 64:1621–1631. 2004.
View Article : Google Scholar
|
|
24
|
Nose K and Ohba M: Functional activation
of the egr-1 (early growth response-1) gene by hydrogen peroxide.
Biochem J. 316:381–383. 1996.PubMed/NCBI
|
|
25
|
Han SI, Duong HQ, Choi JE, Lee TB, Kim CH,
Lee SY, Jeon HM, Shin SH, Lim SC and Kang HS: Hyperthermia switches
glucose depletion-induced necrosis to apoptosis in A549 lung
adenocarcinoma cells. Int J Oncol. 32:851–860. 2008.PubMed/NCBI
|
|
26
|
Ahmad IM, Aykin-Burns N, Sim JE, Walsh SA,
Higashikubo R, Buettner GR, Venkataraman S, Mackey MA, Flanagan SW,
Oberley LW and Spitz DR: Mitochondrial O2*-
and H2O2 mediate glucose deprivation-induced
stress in human cancer cells. J Biol Chem. 280:4254–4263.
2005.PubMed/NCBI
|
|
27
|
Spitz DR, Sim JE, Ridnour LA, Galoforo SS
and Lee YJ: Glucose deprivation-induced oxidative stress in human
tumor cells. A fundamental defect in metabolism? Ann NY Acad Sci.
899:349–362. 2000. View Article : Google Scholar
|
|
28
|
Aykin-Burns N, Ahmad IM, Zhu Y, Oberley LW
and Spitz DR: Increased levels of superoxide and
H2O2 mediate the differential susceptibility
of cancer cells versus normal cells to glucose deprivation. Biochem
J. 418:29–37. 2009.
|
|
29
|
Zong WX and Thompson CB: Necrotic death as
a cell fate. Genes Dev. 20:1–15. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Golstein P and Kroemer G: Cell death by
necrosis: towards a molecular definition. Trends Biochem Sci.
32:37–43. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Horning JL, Sahoo SK, Vijayaraghavalu S,
Dimitrijevic S, Vasir JK, Jain TK, Panda AK and Labhasetwar V: 3-D
tumor model for in vitro evaluation of anticancer drugs. Mol Pharm.
5:849–862. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ivascu A and Kubbies M: Diversity of
cell-mediated adhesions in breast cancer spheroids. Int J Oncol.
31:1403–1413. 2007.PubMed/NCBI
|
|
33
|
Jeong EK, Lee SY, Jeon HM, Ju MK, Kim CH
and Kang HS: Role of extracellular signal-regulated kinase (ERK)1/2
in multicellular resistance to docetaxel in MCF-7 cells. Int J
Oncol. 37:655–661. 2010.PubMed/NCBI
|
|
34
|
Zorov DB, Filburn CR, Klotz LO, Zweier JL
and Sollott SJ: Reactive oxygen species (ROS)-induced ROS release:
a new phenomenon accompanying induction of the mitochondrial
permeability transition in cardiac myocytes. J Exp Med.
192:1001–1014. 2000. View Article : Google Scholar
|
|
35
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial ROS-induced ROS release: an update and review.
Biochim Biophys Acta. 1757:509–517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zong WX, Ditsworth D, Bauer DE, Wang ZQ
and Thompson CB: Alkylating DNA damage stimulates a regulated form
of necrotic cell death. Genes Dev. 18:1272–1282. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee SY, Jeon HM, Kim CH, Ju MK, Bae HS,
Park HG, Lim SC, Han SI and Kang HS: Homeobox gene Dlx-2 is
implicated in metabolic stress-induced necrosis. Mol Cancer.
10:1132011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee SY, Jeon HM, Ju MK, Kim CH, Yoon G,
Han SI, Park HG and Kang HS: Wnt/Snail signaling regulates
cytochrome C oxidase and glucose metabolism. Cancer Res.
72:3607–3617. 2012. View Article : Google Scholar : PubMed/NCBI
|