Introduction
Epigenetic alteration is a general phenomenon during
carcinogenesis (1). Both DNA
methylation and histone modification play critical roles in the
control of expression of oncogenes and tumor suppressor genes
(2,3). One of the most important histone
modifications is acetylation which is controlled by histone
acetyltransferases (HATs) and histone deacetylases (HDACs). These
enzymes are considered to be crucial targets for the development of
anticancer drugs. Previous studies demonstrated that HDAC
inhibitors show potent anticancer activities against different
types of human cancer (4–6).
HDAC inhibitors are classified into different groups
based on their chemical structures and their ability to inhibit
individual HDACs (7,8). The first class is short chain fatty
acids including butyrate and valproate. The second class is
hydroxamic acid derivatives such as trichostatin A, suberoylanilide
hydroxamic acid (SAHA, also known as vorinostat). The third class
is benzamide and the fourth class is cyclic tetrapeptide. Among
them, SAHA is one of the pioneer HDAC inhibitors that has entered
clinical therapy and is now approved for the treatment of cutaneous
T cell lymphoma (9,10). This drug is also undergoing clinical
trials for the treatment of solid tumors, including non-small cell
lung cancer and breast cancer. The anticancer effects of SAHA are
mediated by different mechanisms. First, SAHA directly causes
apoptosis in cancer cells (11,12).
Second, SAHA upregulates anti-proliferative genes, such as p21, to
inhibits proliferation (13,14).
Third, SAHA acts as an immune modulating drug to improve anticancer
immune surveillance (15,16). Fourth, SAHA suppresses angiogenesis
to attenuates tumor growth in vivo(17).
Lymphangiogenesis is the process by which cancer
cells promote the proliferation of lymphatic endothelial cells and
enhance the migration of these cells toward tumor part (18). Previous studies demonstrated that
induction of lymphangiogenesis is strongly associated with tumor
metastasis and poor prognosis in cancer patients (19–21).
The key step in inducing lymphangiogenesis is the production of
lymphangiogenic factors by cancer cells. Among the lymphangiogenic
factors studied, vascular endothelial growth factor-C (VEGF-C) has
received considerable attention since lymphatic endothelial cells
express high levels of its cognate receptor VEGFR3 and their
proliferation is potently stimulated by VEGF-C.
Although SAHA was able to inhibit angiogenesis in
different types of cancer, its effect on lymphangiogenesis has not
been demonstrated. In this study, we investigated the expression of
VEGF-C in breast cancer cell lines and its regulation by SAHA. In
addition, we tried to elucidate the underlying mechanism by which
SAHA modulated VEGF-C transcription.
Materials and methods
Cell culture
Human breast cancer cell lines MDA-MB-231, MCF-7,
MDA-MB-453 and the normal breast cell line M10 were purchased from
the cell bank of the National Health Research Institute (Maoli,
Taiwan). MDA-MB-231 and MDA-MB-453 cells were grown in the L15
medium (Invitrogen, Carlsbad, CA, USA) containing 10% FCS,
L-glutamate and antibiotics. MCF-7 and M10 cells were cultured in
the MEM medium (Invitrogen) containing 10% FCS and antibiotics.
BT-474 breast cancer cells were kindly provided by Dr Jin-Yuh Shew
(National Taiwan University, Taiwan) and were maintained in the
DMEM/F12 medium (Invitrogen) supplemented with 10% FCS,
L-glutamate, nonessential amino acids, sodium pyruvate and
antibiotics. All cells were cultured at 37°C in a humanized
incubator with 5% CO2.
Reagents and antibodies
SAHA was purchased from LC Laboratories (Woburn, MA,
USA). SAHA was dissolved in DMSO at a concentration of 20 mM and
stored at −70°C. It was diluted in the culture medium to different
working concentrations and used for cell treatment. Mithramycin A
was purchased from Sigma-Aldrich (St. Louis, MO, USA). Antibody
against VEGF-C was purchased from R&D Systems (Minneapolis, MN,
USA). Antibody against NF-κB (p65) and Sp3 were obtained from Santa
Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against actin
and Sp1 were purchased from Millipore (Billerica, MA, USA).
Antibody against acetyl lysine-histone 3 was purchased from Cell
Signaling Technology (Beverly, MA, USA).
Construction of VEGF-C promoter plasmids
and the luciferase assay
Genomic DNA was extracted from MCF-7 cells using an
extraction kit (Qiagen, Hilden, Germany) and the −1046/+38 region
from translational start site of VEGF-C gene (NM_005429) was
amplified by PCR using two specific primers (VEGF-C-1046-forward
5′-ATACTCGAGCTTTTACAACCCCCAGG ACA-3′ and VEGF-C-1046-reverse
5′-AAAAAGCTTAGAGA ACACGCCACAGAGAAG-3′). A 1.1-kb DNA fragment was
subcloned into the luciferase reporter gene vector pGL3 (Promega,
Madison, WI, USA) by XhoI and HindIII to yield the
luciferase reporter construct pGL3-1.1kb-VEGF-C (-1046/+38). Using
this construct as a template, two 5′-deletion constructs were
generated by the following primers: VEGF-C-439-forward
5′-ATACTCGAGCTCTCACTTCGGGGAAGG-3′ and VEGF-C-185-forward
5′-ATACTCGAGGCCCTGCAAAGTT GGGAAC-3′. MDA-MB-231 cells were seeded
into 6-well plates and transfected with 1 μg of serial VEGF-C
promoter-luciferase plasmids. After 24 h, cells were incubated
without or with SAHA (10 μM) for an additional 24 h. The luciferase
activity was detected by reporter assay system (Promega), according
to the manufacturer’s instructions, and normalized by protein
concentration in cell lysates.
Transient cell transfection
GFP-Sp1 expression vector (kindly provided by Dr
Jan-Jong Hung, National Cheng Kung University, Taiwan) and control
vector were transfected into MDA-MB-231 cells using Lipofectamine
2000 (Invitrogen). After 48 h, cells were treated with SAHA for an
additional 24 h. Conditioned medium and cell lysates were harvested
for further analysis.
Reverse transcription-polymerase chain
reaction (RT-PCR)
MDA-MB-231, MCF-7, BT-474 and M10 breast cells were
seeded into the 6-well plates. Following overnight incubation,
cells were treated with different concentrations of SAHA for 24 h.
Total RNA was isolated by RNeasy mini kit (Qiagen) and mRNAs were
reverse-transcribed to cDNA by MMLV reverse transcriptase (Promega)
using oligo-dT primers according to the manufacturer’s
instructions. PCR reaction was performed under the following
conditions: initialization step for 5 min at 95°C, 30 cycles of
amplification, with 40 sec at 95°C for denaturation and 40 sec at
60°C for annealing and 40 sec at 72°C for elongation, and 7 min at
72°C for further extension. The PCR primers used are shown in
Table I.
 | Table IPCR primers used in this study. |
Table I
PCR primers used in this study.
| VEGF-C | F:
5′-CAGTTACGGTCTGTGTCCAGTGTAG-3′ |
| VEGF-C | R:
5′-GGACACACATGGAGGTTTAAAGAAG-3′ |
| Sp1 | F:
5′-TATAGCAAATGCCCCAGGT-3′ |
| Sp1 | R:
5′-TTGCCATACACTTTCCCACA-3′ |
| Sp3 | F:
5′-CCTGCAGATATTAGGATCAAGG-3′ |
| Sp3 | R:
5′-GCCTCTGTAATTCATCACTTCG-3′ |
| RelA | F:
5′-TCAATGGCTACACAGGACCA-3′ |
| RelA | R:
5′-ATCTTGAGCTCGGCAGTGTT-3′ |
| Actin | F:
5′-TGTTACCAACTGGGACGACA-3′ |
| Actin | R:
5′-GGGGTGTTGAAGGTCTCAAA-3′ |
Enzyme-linked immunosorbent assay (ELISA)
for VEGF-C
MDA-MB-231 cells were incubated under various
concentrations of SAHA in L15 medium for 24 h. The conditioned
medium was collected and centrifuged at 1500 rpm for 5 min to
remove cell debris. VEGF-C concentration was measured using
Quantikines Human VEGF-C Immunoassay kit from R&D.
Western blotting
Cells were seeded into the 6-well plates. Following
overnight incubation, cells were treated with different
concentrations of SAHA for 6, 12 or 24 h. Cells were washed with
cold PBS and lysed in RIPA buffer (50 mM Tris-HCl, pH 7.4, 50 mM
NaCl, 1 mM EDTA, 0.5 M sucrose, 0.25% sodium deoxycholate, 10%
glycerol, 1% NP-40; protease inhibitors were added prior to use) on
ice for 10 min. Cell debris was removed by centrifugation and
cellular proteins were further fractionated by SDS-PAGE and
transferred onto polyvinylidene difluoride membranes (Millipore).
After incubation with 5% non-fat milk in TBST, the membranes were
probed with different primary antibodies followed by horseradish
peroxidase-conjugated anti-mouse or anti-rabbit antibodies.
Enhanced chemiluminescence (ECL) reagent (Millipore) was used to
detect the blots according to the manufacturer’s instructions.
Statistical analysis
Data were expressed as the means ± SE. Student’s
t-test was used to evaluate the differences between various
experimental groups. p<0.05 was considered to indicate a
statistically significant difference.
Results
VEGF-C expression is inhibited by SAHA in
breast cancer cell lines
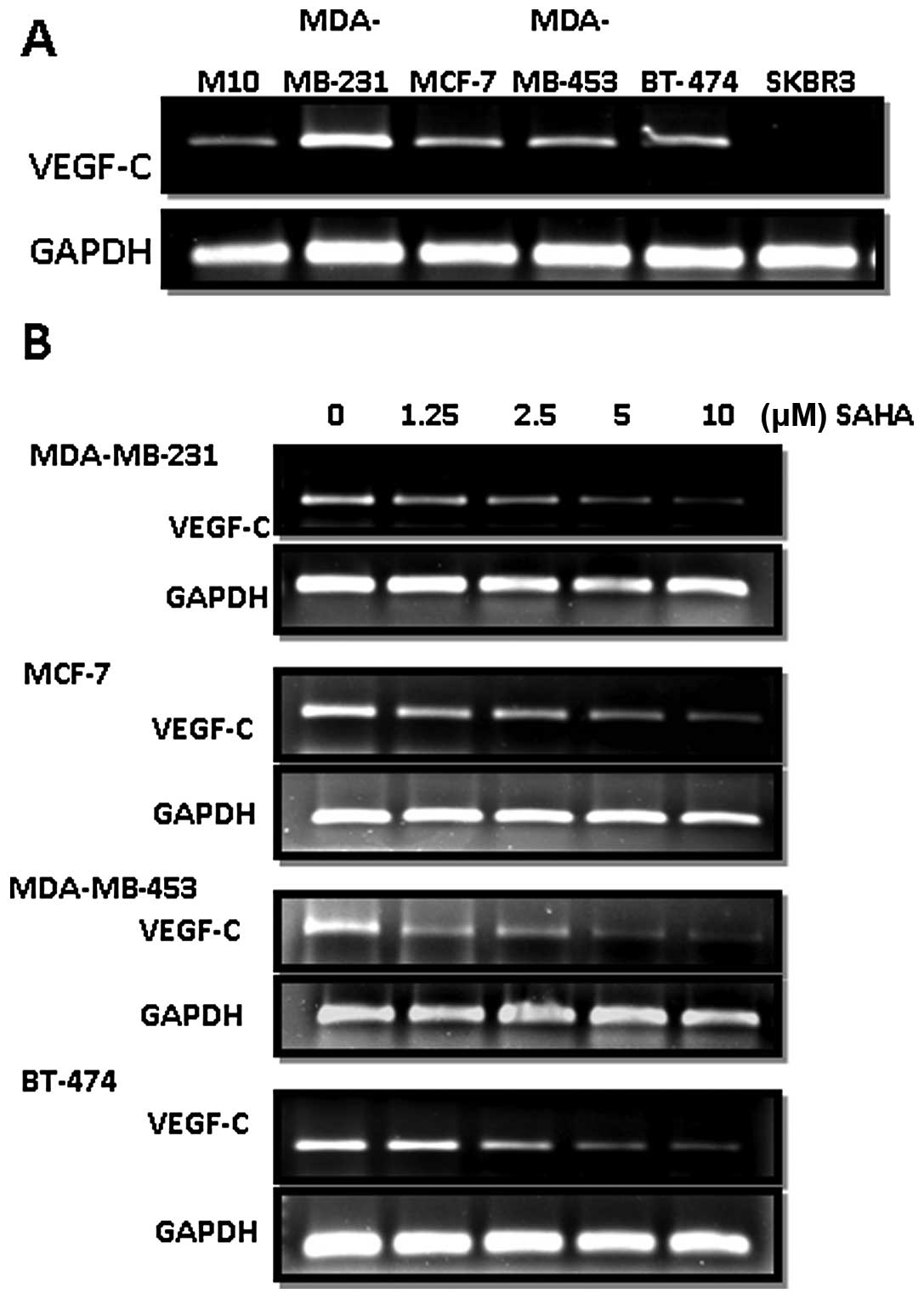
As shown in Fig. 1A,
we found that all the cell lines, except SKBR3, investigated in
this study expressed higher levels of VEGF-C than the normal breast
epithelial cell line M10. SAHA dose-dependently downregulated the
expression of VEGF-C in MDA-MB-231, MCF-7, MDA-MB-453 and BT-474
cells (Fig. 1B).
Reduction of VEGF-C production by
SAHA
We next investigated whether production of the
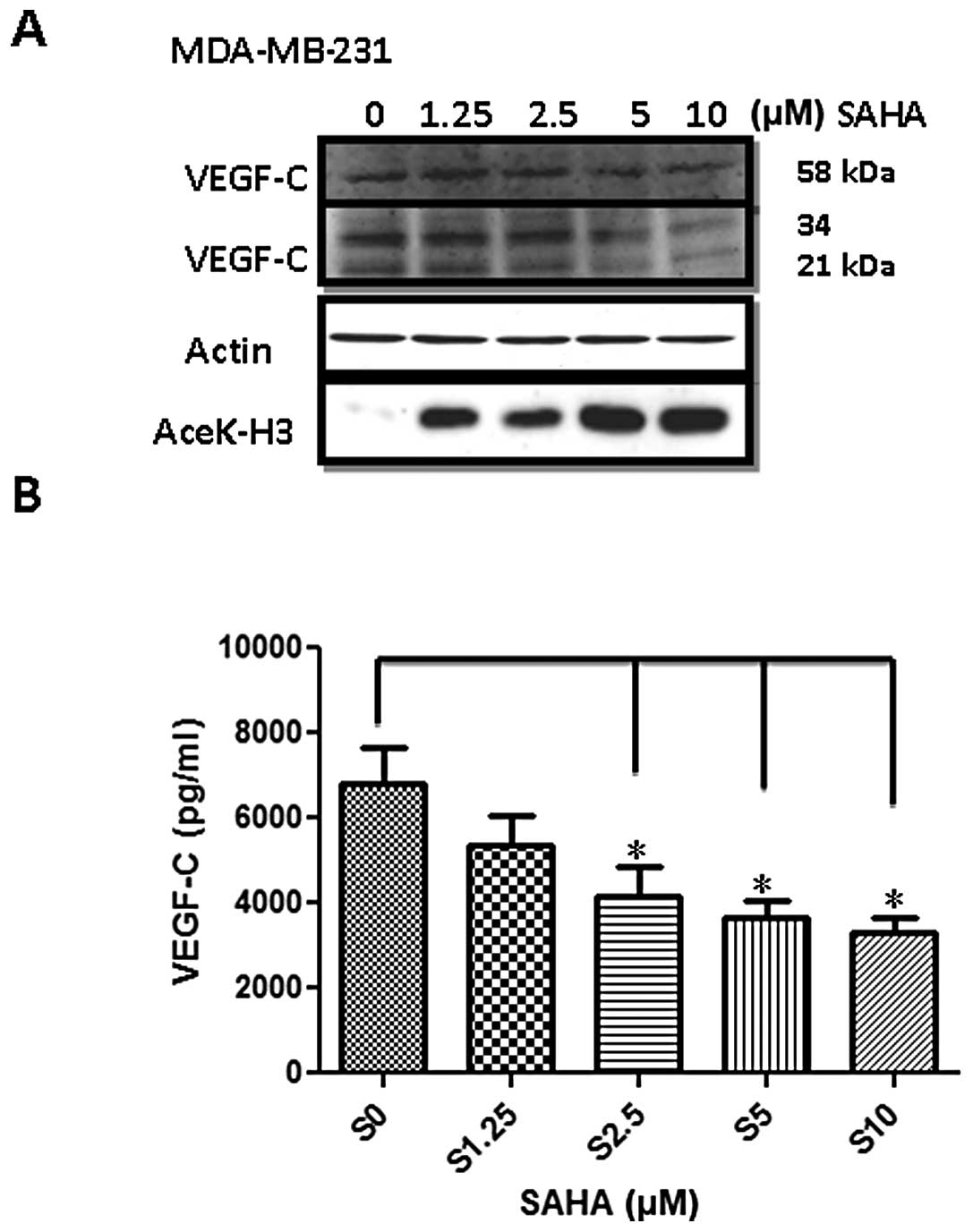
VEGF-C protein was indeed inhibited. Since MDA-MB-231 cells
expressed the highest amount of VEGF-C, we used this cell line as a
model in the subsequent experiments. Two approaches were performed.
First, cellular VEGF-C protein level was determined by western
blotting. As shown in Fig. 2A,
three isoforms of VEGF-C with a molecular weight of 58, 34 and 21
kDa were reduced by SAHA in a dose-dependent manner. We also found
that histone H3 acetylation was significantly increased indicating
SAHA at these concentrations indeed exerted potent inhibitory
effects on HDACs. In addition, VEGF-C released into the conditioned
medium detected by ELISA assay was also reduced accordingly.
Treatment of 10 μM SAHA reduced VEGF-C concentration by 50%
(Fig. 2B).
SAHA represses VEGF-C promoter activity
via the −185/+38 of the promoter region
Since SAHA reduces VEGF-C mRNA levels, this drug
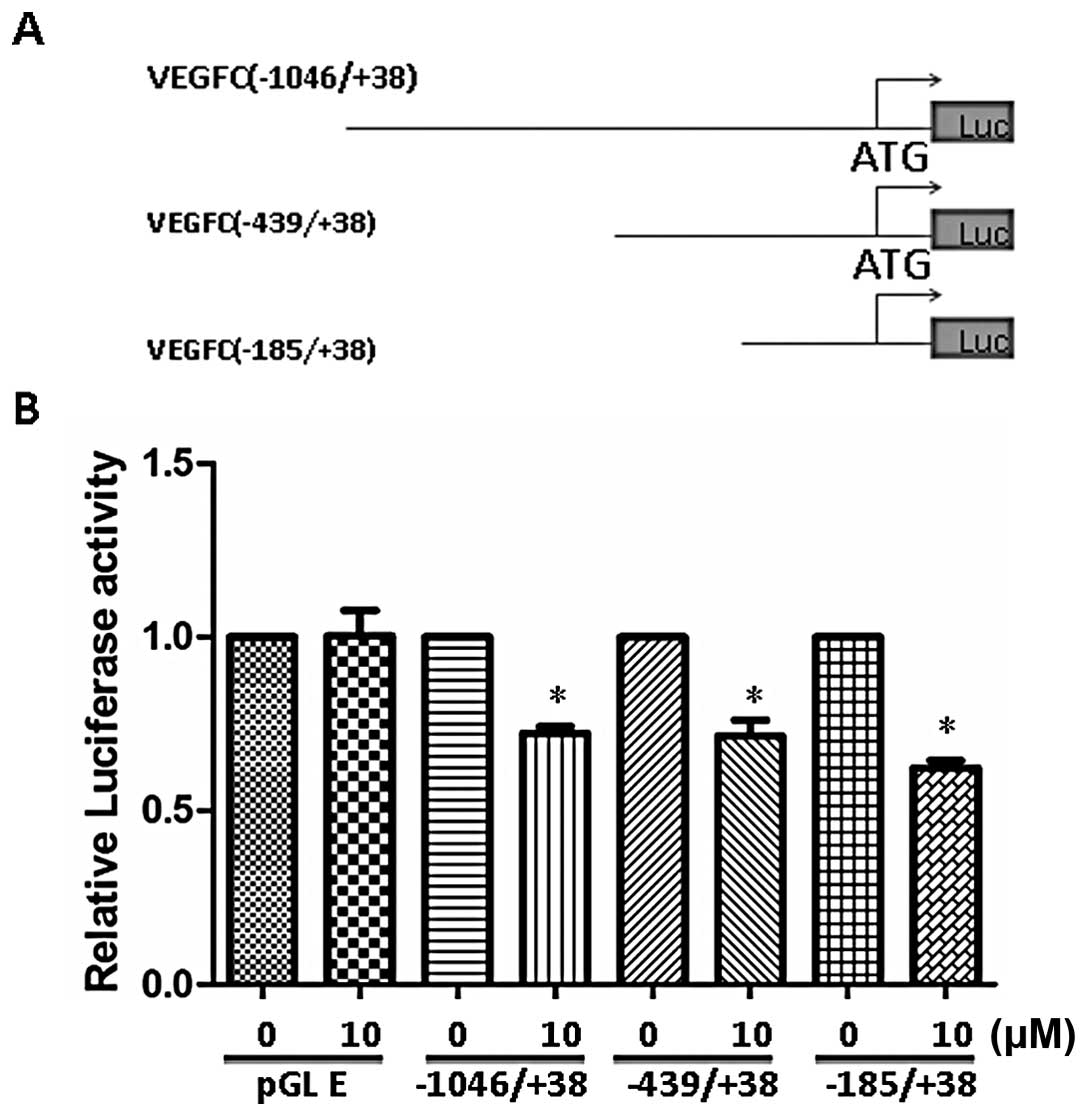
might directly inhibit VEGF-C transcription. We cloned human VEGF-C
promoter region between −1046/+38 bp region from translational
start site and generated different deletion promoter-luciferase
constructs (Fig. 3A). These
constructs were transfected into MDA-MB-231 and the effect of SAHA
was examined. Our data demonstrated that SAHA at the concentration
of 10 μM inhibited the full-length (−1046/+38) promoter by 30–40%
(Fig. 3B). Deletion of promoter to
−185 region did not affect SAHA-induced inhibition indicating the
responsive elements were located between −185/+38 region.
SAHA reduces Sp1-mediated VEGF-C
expression
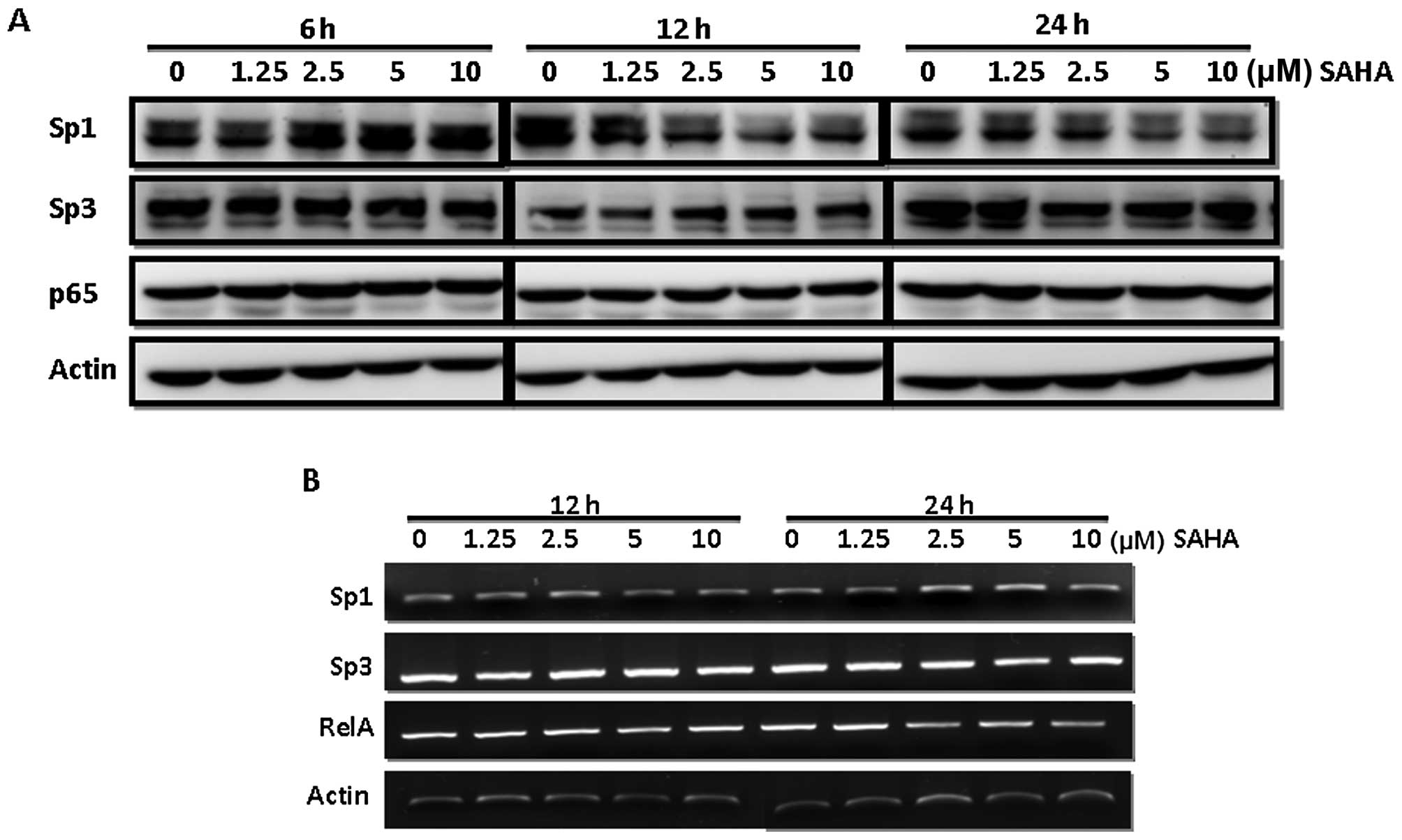
Bioinformatics search revealed several potential
transcription factor binding sites including Sp1, AP-2 and NF-κB
within this region. Notably, we found that SAHA caused reduction of
Sp1 but not Sp3 and NF-κB protein levels in MDA-MB-231 cells
(Fig. 4A). Our data demonstrated
that the Sp1 protein level was significantly decreased at 12 h
after SAHA addition. However, no significant change at the mRNA
level of Sp1, Sp3 and NF-κB was found until a 24-h treatment
suggesting SAHA inhibited Sp1 via a post-translational regulation
(Fig. 4B). To confirm the
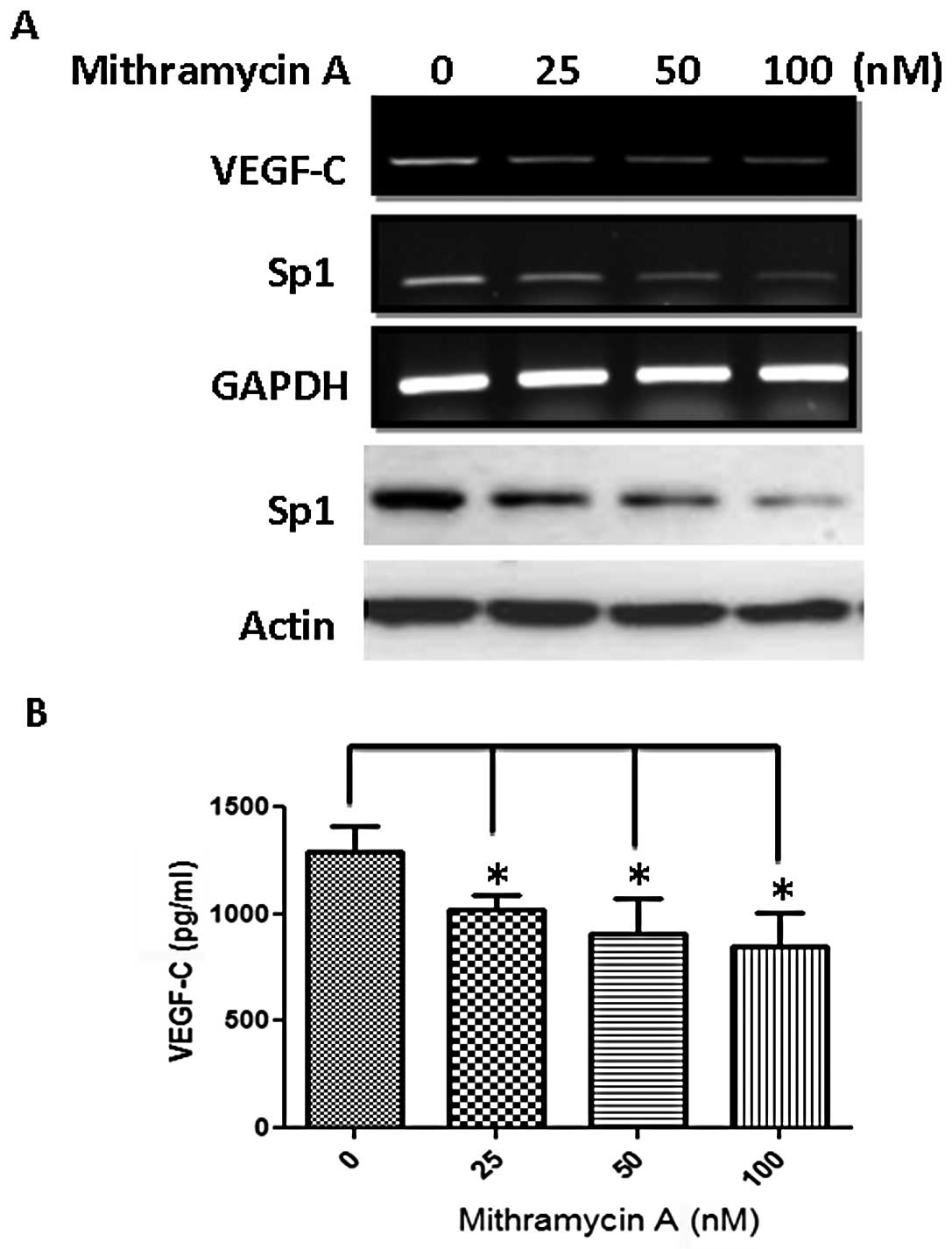
importance of Sp1, we treated cells with an Sp1 inhibitor
mithramycin A and found that this inhibitor reduced Sp1 protein and
attenuated VEGF-C mRNA expression dose-dependently (Fig. 5A). The amount of VEGF-C in the
conditioned medium was also significantly reduced (Fig. 5B).
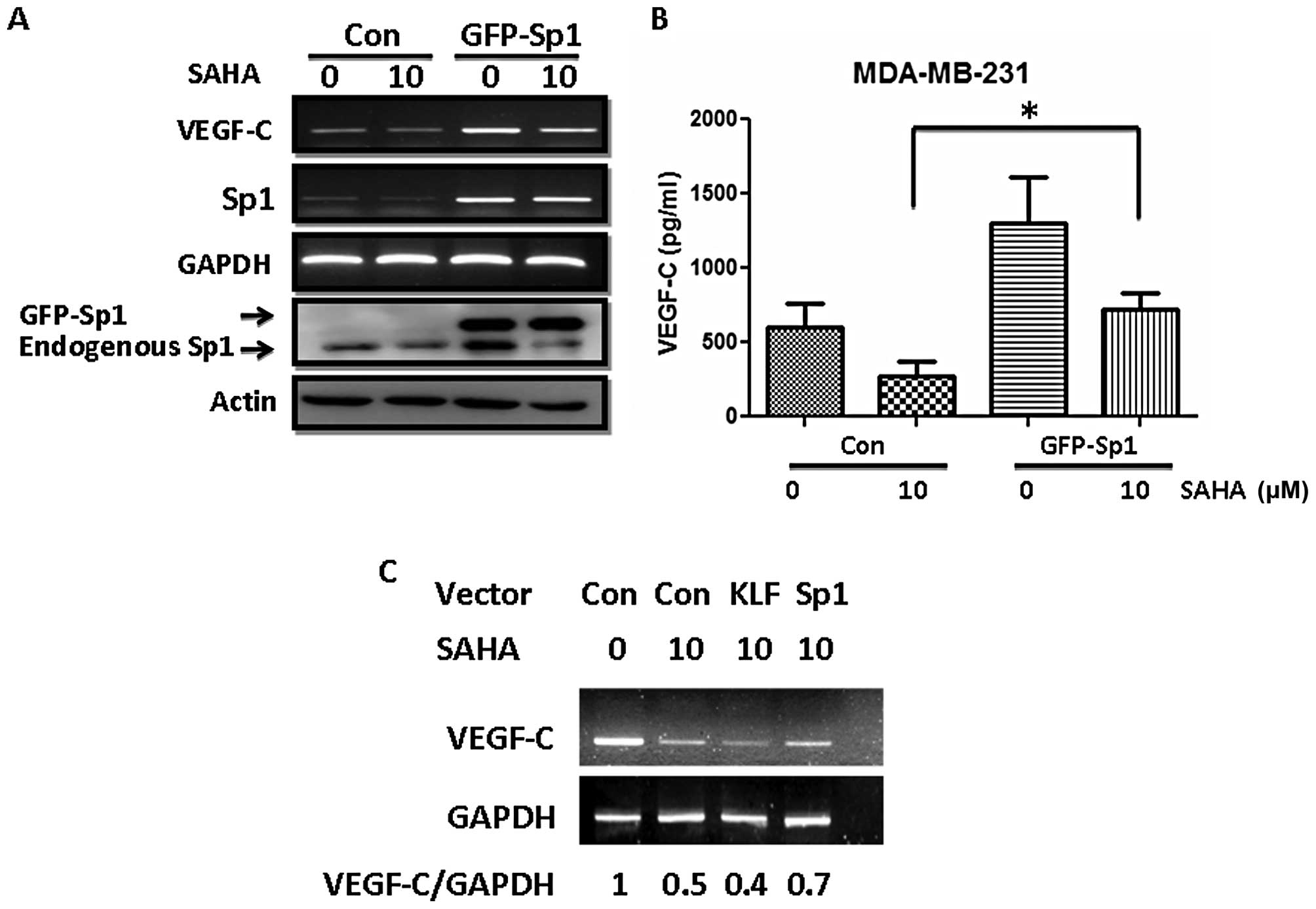
Enforced expression of Sp1 partially
reverses the inhibition of VEGF-C by SAHA
The aforementioned data suggest that SAHA inhibited
VEGF-C expression partly via Sp1. Consistent with this hypothesis,
enforced expression of Sp1 in MDA-MB-231 cells partly reversed the
inhibition of VEGF-C by SAHA (Fig.
6A). The VEGF-C released into the medium was also restored by
Sp1 overexpression (Fig. 6B, lanes
2 and 4). Ectopic expression of Sp1 alone increased VEGF-C mRNA
expression and the amount of VEGF-C in the conditioned medium.
However, this upregulation was still attenuated by SAHA as this
drug caused significant reduction of endogenous Sp1 protein as
shown in Fig. 6A. The effect of Sp1
is specific since expression of KLF10, another member of the
Kruppel-like/Sp1 gene family, could not antagonize the inhibition
of VEGF-C by SAHA (Fig. 6C).
Collectively, our results suggest that SAHA induced downregulation
of Sp1 protein via a post-translational mechanism which led to
reduction of Sp1-driven VEGF-C expression in breast cancer
cells.
Discussion
Pathological studies have demonstrated that
expression of VEGF-C and its receptor VEGFR3 is associated with
angiogenesis, lymphangiogenesis and lymph node metastasis in breast
cancer (22,23). By using an orthotopic
transplantation model, Skobe et al clearly showed that
induction of lymphangiogenesis by VEGF-C promotes breast cancer
metastasis (24). Therefore,
targeting VEGF-C/VEGFR3 signaling axis is important for breast
cancer therapy. Since VEGFR3 is a tyrosine kinase, tyrosine kinase
inhibitors are potential candidates to inhibit VEGFR3 activity.
Sorafenib was initially identified as a potent inhibitor of c-RAF.
However, it also inhibits VEGFR2 and VEGFR3 at concentrations of
approximately 6–10 nM (25). This
drug is now used in the clinic for the treatment of solid tumors.
Other kinase inhibitors including sunitinib, AMG706, axitinib, and
AZD2171 have also shown inhibitory activity against VEGFR3 and are
now under different phases of clinical trials (26). Another strategy to suppress VEGFR3
activity is by blocking antibodies. A recombinant bispecific
antibody has been developed to neutralize the biological activities
of VEGFR2 and VEGFR3 (27). Roberts
et al also demonstrated that inhibition of VEGFR3 activation
with the antagonistic antibody potently suppresses lymph node and
distant metastasis in an orthotopic spontaneous breast cancer model
(28). An antibody that inhibits
homodimerization of VEGFR3 and its heterodimerization with VEGFR2
has recently been shown to suppress both angiogenesis and
lymphangiogenesis in vivo(29).
The targeting of VEGF-C has not received significant
attention in the past decade. By using antibody phage-display,
Rinderknecht et al identified a VEGF-C-blocking antibody
which could effectively inhibit the interaction between VEGF-C and
VEGFR3 and suppress its downstream signaling (30). The underlying mechanism mediating
the upregulation of VEGF-C in cancer cells is largely unclear. A
pioneer study of the genomic organization of human and mouse VEGF-C
genes revealed that the upstream sequences contain conserved
putative binding sites for Sp1, AP-2, and NF-κB transcription
factors but not TATA box (31). In
the present study, we demonstrated for the first time that a
clinically used histone deacetylase inhibitor SAHA was able to
inhibit Sp1-mediated expression of VEGF-C in human breast cancer
cells and reduced VEGF-C concentration in the conditioned medium.
Results of this study suggest that multiple transcription factors
may be simultaneously involved in the regulation of VEGF-C. In
addition, we demonstrated that the Sp1 specific inhibitor
mithramycin A also attenuated VEGF-C expression suggesting a
critical role of Sp1 in VEGF-C transcription. Our results provide a
new strategy to suppress the expression of a lymphangiogenic factor
in cancer cells by using HDAC inhibitors. Reduction of the
production of VEGF-C will attenuate lyphangiogenesis and lymphatic
metastasis in vivo. Since SAHA has already been approved for
cancer treatment in the clinic, it will be of benefit to test
whether this drug, in addition to its cytotoxic effect, also shows
anti-lymphangiogenic activity in animals and patients.
Sp1 is a ubiquitous transcription factor expressed
in mammalian cells and was initially recognized as a constitutive
activator of housekeeping genes. However, recent studies
demonstrated that Sp1 is involved in the control of tissue-specific
and inducible genes and these target genes play critical roles in
proliferation, differentiation, apoptosis and oncogenesis (32). In addition, increased Sp1 protein
was found in various types of human cancer suggesting a possible
oncogenic role (33,34). Our finding that SAHA causes
downregulation of Sp1 protein is noteworthy. Although the
underlying mechanism remains unclear, two potential mechanisms may
be involved. First, a previous study identified lysine-703 as a
major acetylation site of Sp1. Inhibition of HDAC activity by SAHA
may directly affect the acetylation status of Sp1 and subsequently
change protein stability (35).
Second, heat shock protein 90 (hsp90) is an important regulator of
Sp1 stability (36). SAHA has been
demonstrated to inhibit the HDAC6-hsp90 chaperone complex to induce
the degradation of client proteins such as mutant p53 (37). Therefore, it seems possible that
SAHA may inhibit hsp90 and then affect Sp1 degradation.
In the present study, we demonstrated that SAHA is a
potent inhibitor of VEGF-C expression in breast cancer cells and we
showed that the inhibition is mediated by repression of Sp1. Our
results also suggest that SAHA may exert anti-lymphangiogenic
activity in cancer treatment.
Acknowledgements
This study was supported by grant CA-101-PP-39 from
the National Health Research Institutes and DOH 101-TD-C-111-002
and DOH 101-TD-C-111-004 from the Department of Health, Taiwan,
R.O.C.
References
|
1
|
Sarkies P and Sale JE: Cellular epigenetic
stability and cancer. Trends Genet. 28:118–127. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Esteller M and Herman JG: Cancer as an
epigenetic disease: DNA methylation and chromatin alterations in
human tumours. J Pathol. 196:1–7. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kouzarides T: Chromatin modifications and
their function. Cell. 128:693–705. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huang L and Pardee AB: Suberoylanilide
hydroxamic acid as a potential therapeutic agent for human breast
cancer treatment. Mol Med. 6:849–866. 2000.PubMed/NCBI
|
|
5
|
Kim MS, Kwon HJ, Lee YM, et al: Histone
deacetylases induce angiogenesis by negative regulation of tumor
suppressor genes. Nat Med. 7:437–443. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Butler LM, Agus DB, Scher HI, et al:
Suberoylanilide hydroxamic acid, an inhibitor of histone
deacetylase, suppresses the growth of prostate cancer cells in
vitro and in vivo. Cancer Res. 60:5165–5170. 2000.PubMed/NCBI
|
|
7
|
Marks P, Rifkind RA, Richon VM, Breslow R,
Miller T and Kelly WK: Histone deacetylases and cancer: causes and
therapies. Nat Rev Cancer. 1:194–202. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Minucci S and Pelicci PG: Histone
deacetylase inhibitors and the promise of epigenetic (and more)
treatments for cancer. Nat Rev Cancer. 6:38–51. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Marks PA and Breslow R: Dimethyl sulfoxide
to vorinostat: Development of this histone deacetylase inhibitor as
an anticancer drug. Nat Biotechnol. 25:84–90. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Duvic M and Vu J: Vorinostat: a new oral
histone deacetylase inhibitor approved for cutaneous T-cell
lymphoma. Expert Opin Investig Drugs. 16:1111–1120. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Peart MJ, Tainton KM, Ruefli AA, et al:
Novel mechanisms of apoptosis induced by histone deacetylase
inhibitors. Cancer Res. 63:4460–4471. 2003.PubMed/NCBI
|
|
12
|
Shao Y, Gao Z, Marks PA and Jiang X:
Apoptotic and autophagic cell death induced by histone deacetylase
inhibitors. Proc Natl Acad Sci USA. 101:18030–18035. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gui CY, Ngo L, Xu WS, Richon VM and Marks
PA: Histone deacetylase (HDAC) inhibitor activation of
p21WAF1 involves changes in promoter-associated
proteins, including HDAC1. Proc Natl Acad Sci USA. 101:1241–1246.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mitsiades CS, Mitsiades NS, McMullan CJ,
et al: Transcriptional signature of histone deacetylase inhibition
in multiple myeloma: Biological and clinical implications. Proc
Natl Acad Sci USA. 101:540–545. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Khan AN and Tomasi TB: Histone deacetylase
regulation of immune gene expression in tumor cells. Immunol Res.
40:164–178. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ogbomo H, Michaelis M, Kreuter J, Doerr HW
and Cinatl J Jr: Histone deacetylase inhibitors suppress natural
killer cell cytolytic activity. FEBS Lett. 581:1317–1322. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Deroanne CF, Bonjean K, Servotte S, et al:
Histone deacetylases inhibitors as anti-angiogenic agents altering
vascular endothelial growth factor signaling. Oncogene. 21:427–436.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Alitalo K, Tammela T and Petrova TV:
Lymphangiogenesis in development and human disease. Nature.
438:946–953. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mandriota SJ, Jussila L, Jeltsch M, et al:
Vascular endothelial growth factor-C- mediated lymphangiogenesis
promotes tumour metastasis. EMBO J. 20:672–682. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Maula SM, Luukkaa M, Grenman R, Jackson D,
Jalkanen S and Ristamaki R: Intratumoral lymphatics are essential
for the metastatic spread and prognosis in squamous cell carcinomas
of the head and neck region. Cancer Res. 63:1920–1926. 2003.
|
|
21
|
Dadras SS, Paul T, Bertoncini J, et al:
Tumor lymphangiogenesis: a novel prognostic indaicator for
cutaneous melanoma metastasis and survival. Am J Pathol.
162:1951–1960. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Valtola R, Salven P, Heikkila P, et al:
VEGFR-3 and its ligand VEGF-C are associated with angiogenesis in
breast cancer. Am J Pathol. 154:1381–1390. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang CA, Jedlicka P, Patrick AN, et al:
Six1 induces lymphangiogenesis and metastasis via upregulation of
VEGF-C in mouse models of breast cancer. J Clin Invest.
122:1895–1906. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Skobe M, Hawighorst T, Jackson DG, et al:
Induction of tumor lymphangiogenesis by VEGF-C promotes breast
cancer metastasis. Nat Med. 7:192–198. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Flaherty KT: Sorafenib in renal cell
carcinoma. Clin Cancer Res. 13:S747–S752. 2007. View Article : Google Scholar
|
|
26
|
Hanrahan EO and Heymach JV: Vascular
endothelial growth factor receptor tyrosine kinase inhibitors
Vandetanib (ZD6474) and AZD2171 in lung cancer. Clin Cancer Res.
13:S4617–S4622. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jimenez X, Lu D, Brennan L, et al: A
recombinant, fully human, bispecific antibody neutralizes the
biological activities mediated by both vascular endothelial growth
factor receptors 2 and 3. Mol Cancer Ther. 4:427–434. 2005.
|
|
28
|
Roberts N, Kloos B, Cassella M, et al:
Inhibition of VEGFR-3 activation with the antagonistic antibody
more potently suppresses lymph node and distant metastases than
inactivation of VEGFR-2. Cancer Res. 66:2650–2657. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
McDonald DM: New antibody to stop tumor
angiogenesis and lymphatic spread by blocking receptor partnering.
Cancer Cell. 18:541–543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rinderknecht M, Villa A, Ballmer-Hofer K,
Neri D and Detmar M: Phage-derived fully human monoclonal antibody
fragments to human vascular endothelial growth factor-C block its
interaction with VEGF receptor-2 and 3. PLoS One. 5:e119412010.
View Article : Google Scholar
|
|
31
|
Chilov D, Kukk E, Taira S, et al: Genomic
organization of human and mouse genes for vascular endothelial
growth factor C. J Biol Chem. 272:25176–25183. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li L and Davie JR: The role of Sp1 and Sp3
in normal and cancer cell biology. Ann Anat. 192:275–283. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang L, Wei D, Huang S, et al:
Transcription factor Sp1 expression is a significant predictor of
survival in human gastric cancer. Clin Cancer Res. 9:6371–6380.
2003.PubMed/NCBI
|
|
34
|
Safe S and Abdelrahim M: Sp transcription
factor family and its role in cancer. Eur J Cancer. 41:2438–2448.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hung JJ, Wang YT and Chang WC: Sp1
deacetylation induced by phorbol ester recruits p300 to activate
12(S)-lipoxygenase gene transcription. Mol Cell Biol. 26:1770–1785.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang SA, Chuang JY, Yeh SH, et al: Heat
shock protein 90 is important for Sp1 stability during mitosis. J
Mol Biol. 387:1106–1119. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li D, Marchenko ND and Moll UM: SAHA shows
preferential cytotoxicity in mutant p53 cancer cells by
destabilizing mutant p53 through inhibition of the HDAC6- Hsp90
chaperone axis. Cell Death Differ. 19:1268–1276. 2012.PubMed/NCBI
|