Introduction
Lung cancer is a leading cause of cancer-related
death both in the US and worldwide. NSCLC represents 80% of lung
cancers. It is estimated that 40% of patients who present with
NSCLC are already at an advanced inoperable stage of the disease
(1). For those patients who present
with late stage disease, therapy consists of a combination of
radiation and chemotherapy. Unfortunately, many NSCLC
subpopulations exhibit intrinsic radiation resistance which leads
to local recurrence, lymphovascular invasion and distant metastatic
disease.
A fusion protein of echinoderm microtubule
associated protein like-4 (EML4) and anaplastic lymphoma kinase
(ALK) has been found in NSCLC patients (2). It is estimated that approximately 5%
of NSCLC cases harbor an EML4-ALK fusion (3,4). The
EML4-ALK fusion functions in a manner similar to EGFR mutations;
that is, EML4-ALK constitutively activates a tyrosine kinase
receptor leading to cancer dependence on overactive mitogenic
pathways (5). Transgenic mice that
express the EML4-ALK fusion protein grow numerous lung
adenocarcinomas (6). This
constitutive mechanism represents a prime target for
chemotherapy.
PF-02341066, a novel dual c-Met and ALK inhibitor,
has recently been evaluated in both preclinical and clinical
trials. Preclinical studies have shown that treatment with ALK
inhibitors can lead to drastic tumor regression in in vivo
xenograft models (7). A phase I
trial of PF-02341066 revealed impressive results with a 53%
response rate and a disease control rate of 79% (3). PF-02341066 is currently under
evaluation as a secondary agent as well as a single-drug therapy in
phase III and phase II trials, respectively.
While PF-02341066 has shown significant and
promising results as a chemotherapeutic agent, it has not been
evaluated, to date, in conjunction with radiation in NSCLC models.
In this study, we evaluated PF-02341066 as a potential
radiation-sensitizing agent in 5 different established NSCLC cell
lines (H460, A549, H3122, H2228 and H1993) with varying expression
levels of c-Met and EML4-ALK (8).
Materials and methods
Cell culture and reagents
Human NSCLC cell lines H460, A549, H3122, H1993 and
H2228 were kindly provided by Dr John D. Minna at the UT
Southwestern Medical Center, Dallas, TX. These cell lines were
maintained in RPMI-1640 with 10% FBS and 50 units/ml penicillin and
50 μg/ml streptomycin in 5% carbon dioxide at 37°C.
PF-02341066 (MW, 450.3) was obtained from Pfizer
Inc., dissolved in DMSO to give a stock solution of 10 mM and
stored at −20°C. Cells were irradiated using a 137Cs
source (Mark 1–68 irradiator, J.L. Shepherd and Associates, San
Fernando, CA) at a dose rate of 3.47 Gy/min (9).
Clonogenic survival assay
Exponentially growing cells were treated with
PF-02341066 for 2 h and then treated with increasing doses of IR
(0, 2, 4, 6 and 8 Gy). Cells were trypsinized and counted using a
particle counter (Beckman Coulter, Inc.), diluted serially to
appropriate concentrations and plated into a 60-mm dish in
triplicate. After 7 or 14 days of incubation, the colonies were
fixed and stained with 4% formaldehyde in PBS containing 0.05%
crystal violet. Colonies containing >50 cells were counted. The
surviving cell fraction was calculated as: (Mean colony
counts)/[(cells inoculated) × (plating efficiency)], in which
plating efficiency was defined as (Mean colony counts)/(cells
inoculated for unirradiated controls). The data are presented as
the mean ± SD of at least 3 independent experiments. The curve S =
e −(αD + βD2) was fitted to the experimental data using
a least square fit algorithm using the program SigmaPlot (Systat
Software, Inc.) as previously described (9). The radiation dose enhancement ratio
(DER) was calculated as the dose (Gy) for radiation alone divided
by the dose (Gy) for radiation plus drugs (normalized for drug
toxicity) resulting in a surviving cell fraction of 0.25.
Clonogenic survival assay was also performed to determine the
growth inhibitory response (50%) of these NSCLC cells using
increasing dosages of PF-02341066. Inhibitory dose concentrations
were determined using a 4 parameter variable slope regression
model.
Immunoblot assay
Cell lysates were prepared from each sample as
previously described (10). An
equal amount of total protein (20 μg) was subjected to a 10%
SDS-PAGE for immunoblot analysis and probed with primary antibodies
as indicated. β-actin was used for the loading control.
Cell cycle analysis
Cell cycle assays were performed with propidium
iodide (PI, 100 μg/ml) as previously described (10). At least 20,000 cells were counted;
the proportion of cells of different phases was gated and
calculated using the software FlowJo 8.7.1 (Tree Star, Inc.).
DNA double-strand break (DSB) repair
assay
DSB repair assay was performed by counting
phospho-γH2AX foci following treatment with IR alone, drug alone or
IR + PF-02341066, as previously described (10). Cells were plated on
poly-lysine-coated coverslips and were allowed to attach and then
treated as indicated. Cells were fixed in 4% formaldehyde/PBS for
30 min, permeabilized in 0.5% Triton X-100 in PBS for 1 h, and
blocked in 5% bovine serum albumin and 1% normal goat serum for 1 h
at room temperature. Cells were then incubated with the primary
antibody, anti-phospho-Histone γH2AX (Ser139; 1:2,000) for 1 h.
Rhodamine red-conjugated goat anti-mouse was used as a secondary
antibody. Cells were mounted in a Vectashield mounting medium
containing 4′,6-diamidino-2-phenylindole (DAPI). Phospho-γH2AX foci
were examined using a fluorescence microscope (CRG Precision
Electronics). The number of phospho-γH2AX foci was determined at
each time point (average of 50 nuclei), and the percentage of
remaining foci was plotted against time to obtain DSB repair
kinetics. Data are represented as the mean ± SEM.
Tumor growth delay (TGD)
Female athymic nude mice (nu/nu, 5–6 weeks) were
injected (1×106 cells in 100 μl) s.c. into the right
posterior flanks. Treatment was initiated when tumors reached a
diameter 2–3 mm in size. Treatment groups (5 animals each) included
untreated control (0.9% saline), PF-02341066 alone (50 mg/kg/day
for 5 days, p.o.), radiation [2 Gy/day, 5 days, X-RAD 320
(Precision X-Ray, North Branford, CT] and combined treatment of
PF-02341066 and irradiation. The drug was administered 1 h before
radiation. Tumors were measured 3 times per week using a Vernier
caliper. Results were evaluated with the formula: Volume = 0.5abc
(a, width, b, length and c, thickness). Tumor growth delay (TGD)
was calculated as the time for treated tumors to reach 1,000
mm3 minus the time for control tumors to reach the same
volume. Enhancement factor (EF) was then determined as follows: EF
= (TGDdrug + IR - GDdrug)/GDIR as
previously described (10,11). All the experiments were conducted
under the Institutional Animal Care and Use Committee of UT
Southwestern Medical Center, Dallas, TX approved guidelines for
animal welfare.
Apoptosis assay
Cells were plated in a 100-mm dish and 24 h later
were treated with IR alone, drug alone or IR + drug. Floating and
attached cells were harvested post treatment as indicated. After
centrifugation (200 × g, 5 min), the medium was removed, and the
cell pellet was carefully resuspended in 5 ml PBS. Phycoerythrin
(PE) Annexin V apoptosis detection kit I (BD Biosciences) was used
to identify apoptotic cells by flow cytometry. Cells that stained
positive for PE Annexin V and negative for 7-AAD (right bottom
quadrant) were undergoing apoptosis. The proportion of apoptotic
cells were gated and calculated by FlowJo 8.7.1 as previously
described (10).
Statistical analysis
Data are presented as the means ± SD or SEM, as
noted, of at least 3 independent experiments. Results were tested
for significance using either Mantel-Cox log-rank test,
Mann-Whitney rank sum test, or t-test as noted.
Results
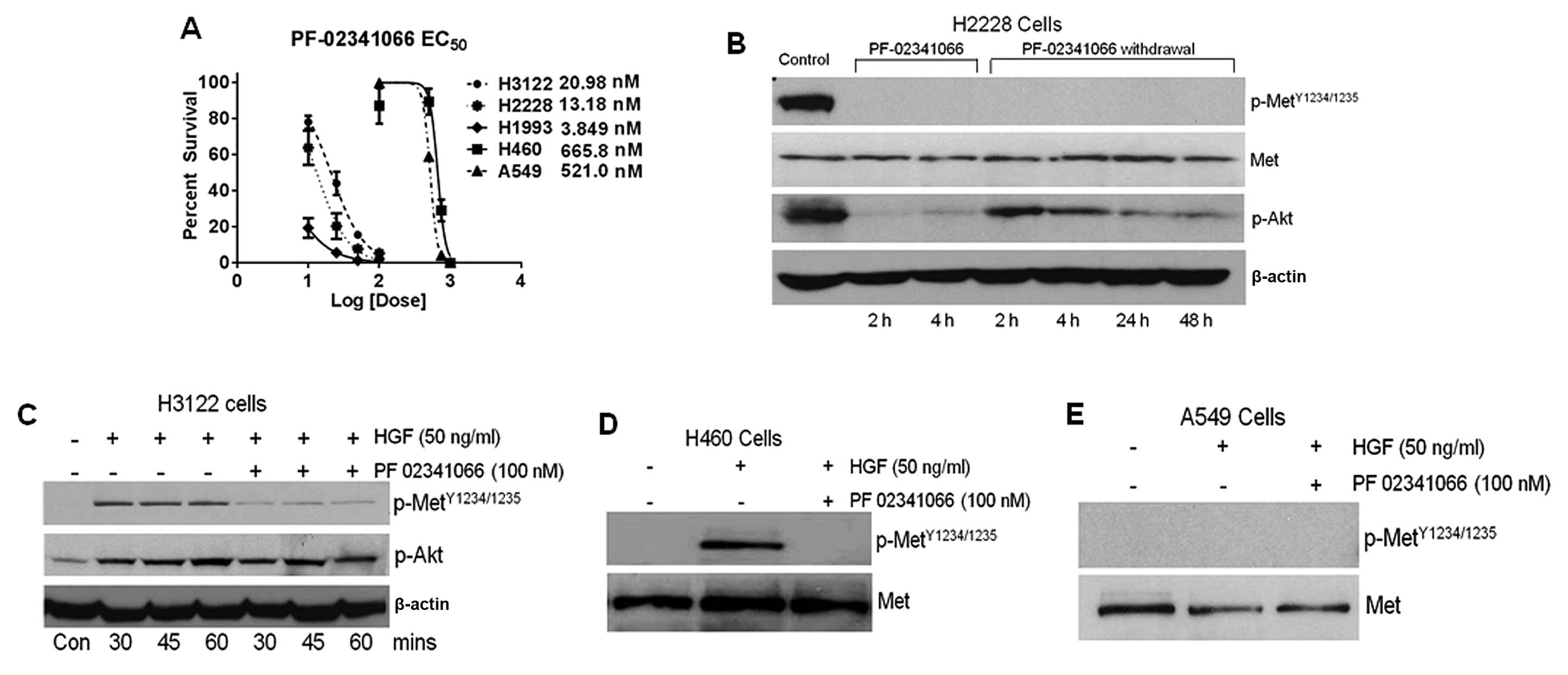
Specificity of PF-02341066 in NSCLC
The toxicity of PF-02341066 was reported at 50%
colony survival and determined in a set of several NSCLC cell
lines. Drug toxicity varied greatly among the cell lines; H2228
(13.18 nM), H3122 (20.98 nM) and H1993 (3.85 nM) were highly
sensitive to PF-02341066 whereas, H460 (666 nM) and A549 (521 nM)
were resistant (Fig. 1A).
PF-02341066 specificity was then determined by the
phosphorylation status of the c-Met receptor at
Tyr1234/1235 residues before and after treatment with
HGF using immunoblot analysis. For this study, H3122 and H2228
cells were used specifically because of their differential level of
endogenous phosphorylated c-Met as shown in Fig. 1B and C. H3122 cells demonstrated
induction of phosphorylation within 30 min after addition of HGF
(Fig. 1C). HGF-induced c-Met
phosphorylation in H3122 was completely inhibited when the drug was
added 2 h prior to the addition of HGF. Akt phosphorylation was
also increased upon addition of HGF, however, treatment with
PF-02341066 did not block Akt phosphorylation. The endogenous level
of phospho-c-Met was significantly higher in H2228 cells and there
was no further enhancement of phosphorylation after addition of HGF
(Fig. 1B). PF-02341066 completely
prevented endogenous c-Met phosphorylation in H2228 cells within 2
h. In addition, the c-Met receptor remained unphosphorylated up to
48 h after removal of the drug from the medium. However, Akt, which
is a downstream target of c-Met, displayed a high level of
phosphorylation in H2228 cells; p-Akt levels were reduced by
PF-02341066 treatment. phospho-Akt, did, however, reappear after
removal of the drug. Furthermore, a small amount of pAkt was noted
after a 4-h drug treatment. PF-02341066 also prevented c-Met
phosphorylation in H460 cells (Fig.
1D). HGF-mediated induction of c-Met phosphorylation was not
observed in A549 cells (Fig. 1E).
These results are also summarized in Table IA.
 | Table ICharacterization and cellular
apoptosis of NSCLC cells following IR and PF-02341066
treatment. |
Table I
Characterization and cellular
apoptosis of NSCLC cells following IR and PF-02341066
treatment.
| A, Characterization
of NSCLC cells |
|---|
|
|---|
| Cells | EML4-Alk fusion | LD50
(nM) | SF2 | DER | Endogenous
pc-Meta | HGF inducible
pc-Meta |
|---|
| H1993 | Negative | 3.90 | 0.48 | 1.00 | + | No |
| H2228 | Positive | 13.18 | 0.76 | 1.00 | ++ | No |
| H3122 | Positive | 20.98 | 0.64 | 1.10 | - | Yes |
| H460 | Positive | 665.80 | 0.54 | 0.95 | - | Yes |
| A549 | Negative | 521.00 | 0.73 | 1.08 | - | No |
|
| B, Cellular apoptosis
of NSCLC cells |
|
| Treatment | H460 | A549 | H3122 | H1993 | H2228 |
|
| Control | 0.89 | 1.84 | 1.19 | 2.09 | 2.44 |
| Radiation | 9.83 | 4.01 | 3.43 | 6.28 | 13.4 |
| PF-02341066
alone | 1.54 | 5.09 | 1.72 | 4.65 | 5.37 |
| Radiation +
PF-02341066 | 7.32 | 12.07 | 4.93 | 6.69 | 19.74 |
PF-02341066 does not affect radiation
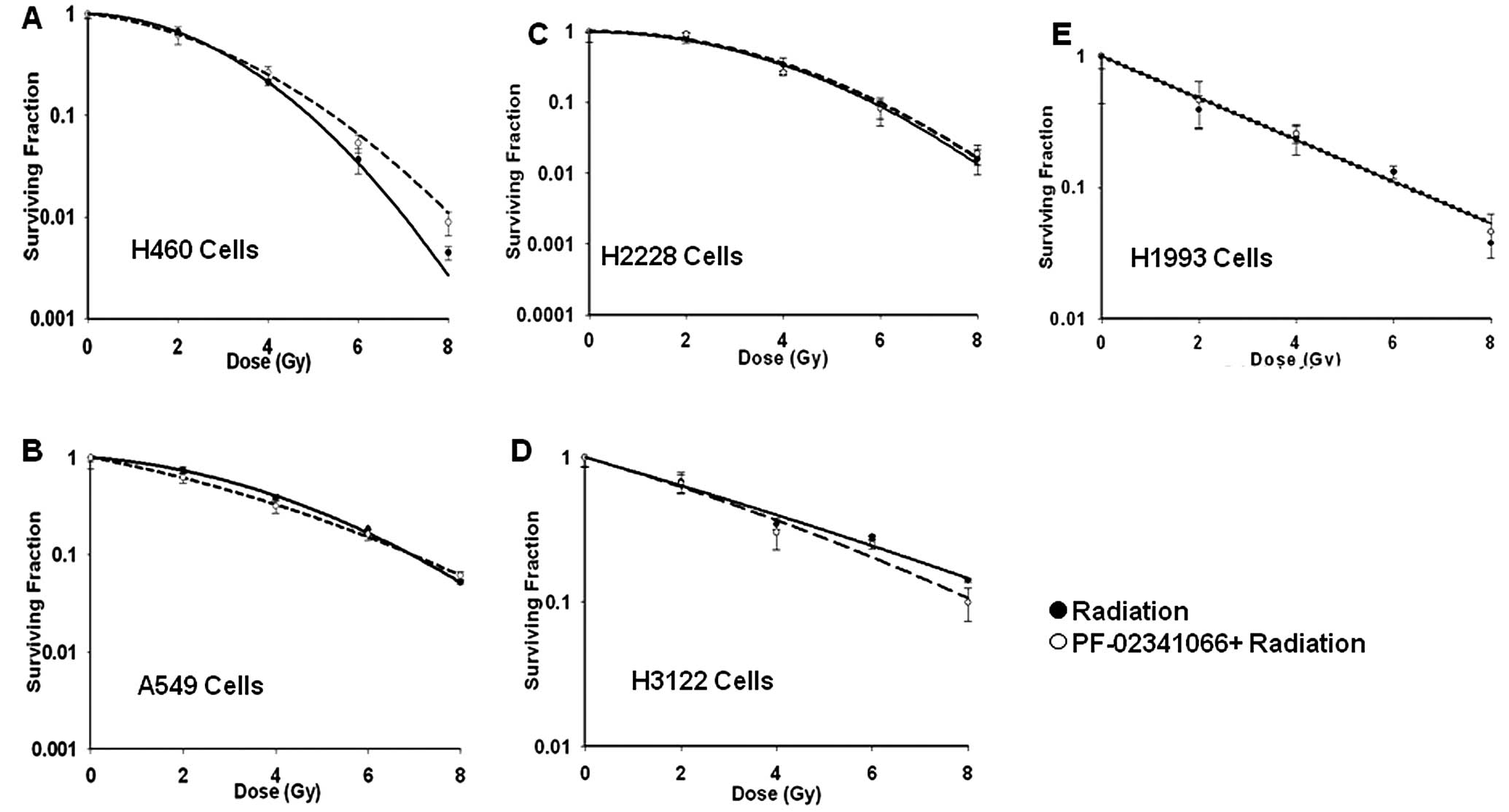
sensitivity in vitro or in vivo
The modulation of radiation sensitivity by
PF-02341066 was investigated in all 5 NSCLC cell lines (Fig. 2). Cells were treated with the drug
for 2 h before being treated with increasing doses of IR (0, 2, 4,
6 and 8 Gy). The maximally tolerated dose used in phase II trials
of PF-02341066 resulted in trough plasma concentrations of 57 nM
(12); therefore, cell lines with
EC50 below the MTD were treated at the EC50
concentration while cell lines that were resistant were treated at
100 nM; slightly less than twice the clinically relevant
concentration. Intrinsic radiation response of each cell line was
different (Fig. 2) and their
corresponding SF2 values are shown in Table I. Notably, when these cells were
treated with PF-02341066 followed by IR, very little or no change
was noted between treated and untreated SF2 values
(Table I) for any of these cell
lines (P>0.05 for all SF2 data). Further assays were
performed in which cells were either treated concurrently or
sequentially with radiation prior to PF-02341066 to modulate the
sensitivity; however, no enhancement was observed regardless of the
treatment condition (data not shown).
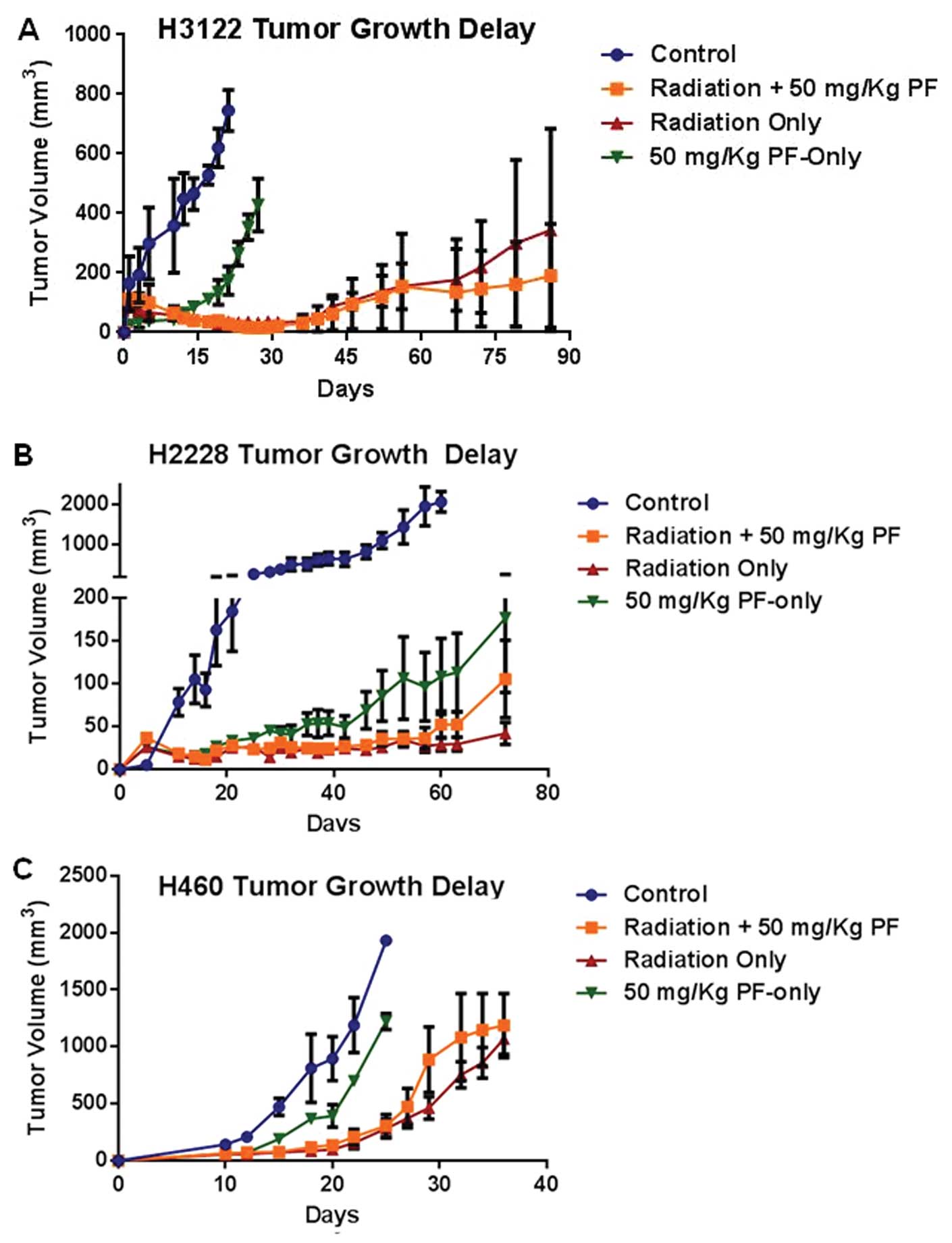
Next, the radiation sensitivity modulation by
PF-02341066 was studied in in vivo xenograft models of 3
different cell lines, H3122, H2228 and H460 (Fig. 3). H3122 tumors that were treated
with IR (2 Gy × 5) or IR (2 Gy × 5) plus PF-02341066 (50 mg/kg,
recommended dose) showed significant growth delay; however, no
difference was appreciated between the two groups (Fig. 3A). It was also noted that following
treatment with PF-02341066 alone, the time for a tumor to reach 500
mm3 was significantly different when compared with the
control (28 vs. 15 days, respectively) (P=0.01). The H2228 in
vivo experiment, following treatment with IR and IR +
PF-02341066 yielded similar results as H3122 with significant
growth arrest noted with no appreciable difference between the IR
and IR + drug groups (Fig. 3B).
However, treatment with PF-02341066 alone showed significant TGD in
the H2228 xenografts (Fig. 3B).
These in vivo results further recapitulate the in
vitro surviving fraction data which showed that no significant
radiation dose enhancement occurred when PF-02341066 was added to
the treatment. The drug resistant cell line, H460, was also tested
in the xenograft experiment. Treatment with IR alone and IR plus
PF-02341066 resulted in TGD of 30 and 27 days, respectively
(Fig. 3C). The effect of radiation
and radiation plus drug was not significantly different (P=0.58).
It is important to note that in the H460 model, the effect of
radiation treatment was significantly different than treatment with
PF-02341066 alone (P=0.008).
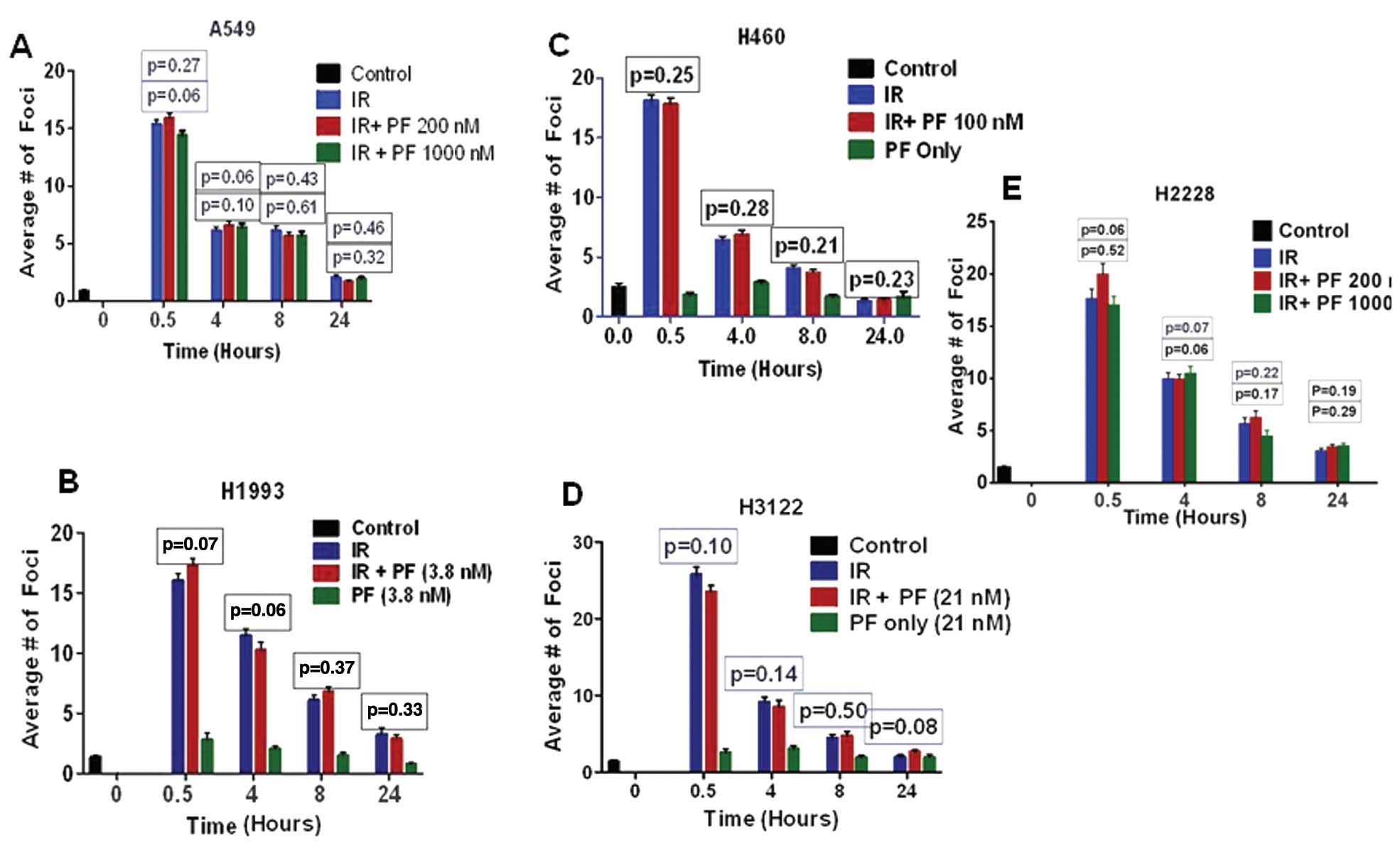
Effect of PF-02341066 on IR-induced DNA
repair kinetics
Previous reports have suggested that c-Met and Alk
kinases provide a survival advantage to cancer cells through their
effects on DNA DSB repair (13–15).
Therefore, c-Met inhibition leads to changes in DNA double-strand
break (DSB) repair kinetics (Fig.
4). To measure the effect of PF-02341066 on the repair of
IR-induced DNA DSB repair, the different NSCLC cell lines were
exposed to 2 Gy of radiation and fixed in paraformaldehyde at the
times indicated for γH2AX staining (Fig. 4). c-Met/EML4-ALK inhibition by this
drug did not induce foci formation in any of the cell lines tested
(Fig. 4). Previous reports with
different c-Met inhibitors have shown radiation-sensitizing effects
in glioma models. To examine whether the previously reported effect
could be replicated with supra-physiologic doses, we treated A549,
a non-responding cell line, and H2228, a c-Met/EML4-Alk-positive
cell line, with various high doses of PF-02341066 (Fig. 4A and E). Treatment with this drug,
regardless of dose, did not modify IR-induced DNA DSB repair
kinetics in any of the cell lines tested in this study (Fig. 4).
Effect of PF-02341066 and radiation on
cell cycle arrest in NSCLC cells
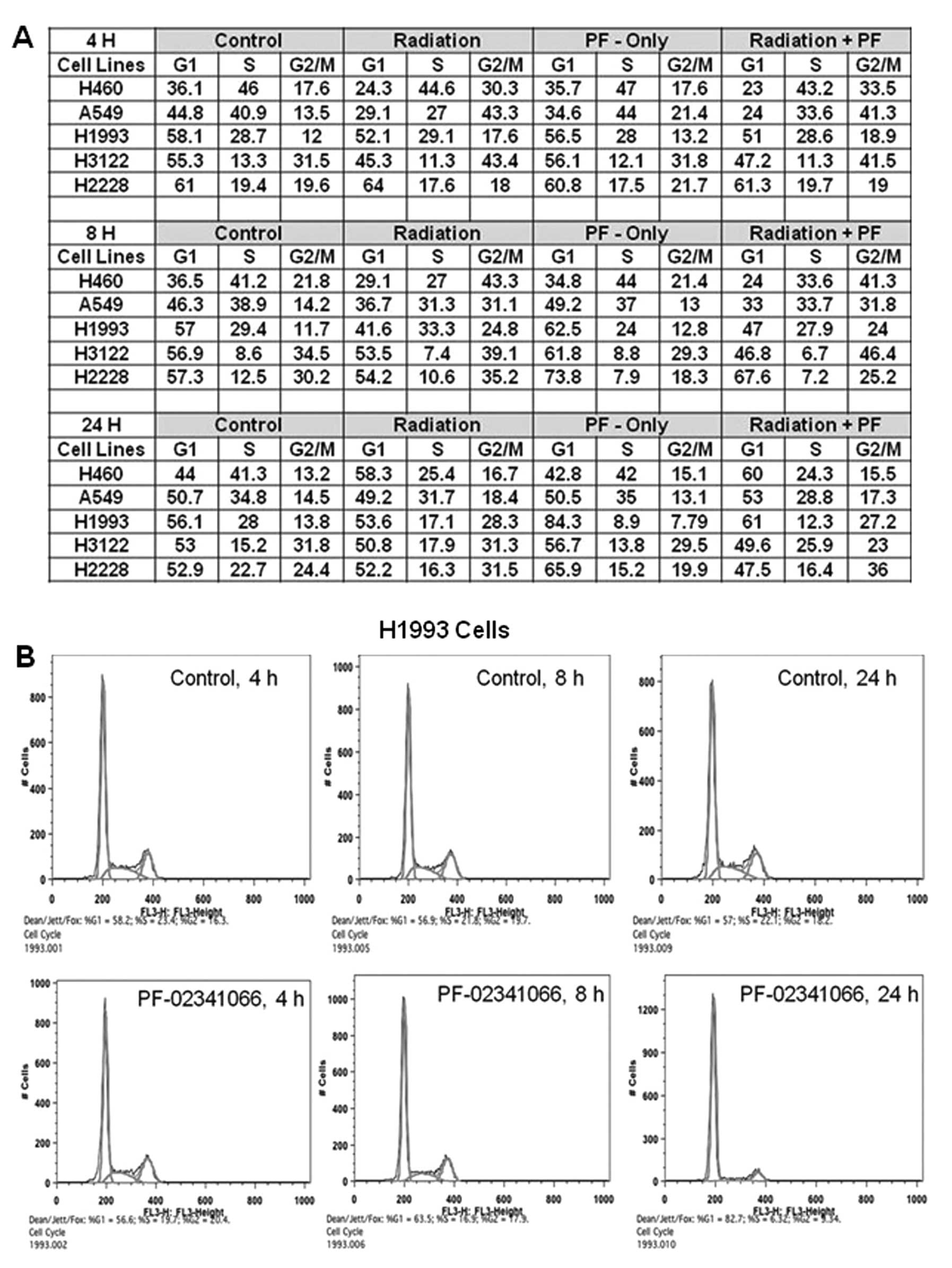
In this experiment each cell line showed increased
G2/M arrest in response to IR at 4 and 8 h (Fig. 5). The effect of the combined
treatment of PF-02341066 and IR did not significantly differ from
that of IR alone in any of the cell lines with respect to G2/M
arrest. It is interesting to note that in cell lines that expressed
endogenous phospho-c-Met, H2228 and H1993, drug treatment alone
induced G1 arrest (Fig. 5). The G1
arrest was most pronounced at 8 h for H2228 cells (57–74%) and 24 h
for H1993 cells (56–84%). In H2228 cells, it appeared that G1
arrest attenuated the G2M arrest normally observed with radiation
alone. This result was further verified in H2228 cells by
demonstrating a decreased number of cells in M-phase after
treatment with PF-02341066 using phospho-Histone3 analysis (data
not shown).
Combination therapy results in additive
increases in apoptosis
It was previously shown that cells containing ALK
fusions undergo increased apoptosis following a 48-h PF-02341066
treatment (16). Therefore, we
aimed to ascertain whether the combination of radiation and
PF-02341066 has an additive or supra-additive effect on apoptosis.
In those cells considered resistant to PF-02341066, H460 and A549,
48 h of treatment produced little apoptosis: 1.3 and 5.9%,
respectively, when compared to the untreated samples, while an
additive increase in apoptosis was noted when PF-02341066 was
combined with radiation. In A549 cells, this additivity was
achieved at concentrations of drug greater than clinically
achievable. In H1993, H3122 and H2228 cells, a greater degree of
apoptosis was noted; 2.25, 5.31 and 5.9%, respectively. However,
when PF-02341066 was combined with radiation the percentage of
apoptosis noted was roughly additive (Table IB).
Discussion
c-Met activity has been associated with increased
radiation resistance (17,18). Furthermore c-Met is known to be an
upstream activator of Akt which has also been linked to radiation
resistance (15,19). In addition, recent studies have
implied that the EML4-ALK fusion protein may interact with many
similar pathways similar to c-Met (13,14).
We, therefore, investigated whether c-Met and EML4-ALK inhibition
by PF-02341066 leads to increased radiation sensitivity in NSCLC
cells.
Initially, we determined the toxicity of PF-02341066
in a panel of NSCLC cell lines (Fig.
1A). It became apparent that the cell lines could be divided
into responders (H2228, H1993 and H3122) and non-responders (A549
and H460). Not surprisingly, the c-Met-positive cell lines H2228
(which is also EML4-ALK-positive) and H1993 responded well to
PF-02341066 treatment. Among the non-responding cell lines, H460
has been classified as EML4-ALK-positive but contains a different
variant than that of the H2228 and H3122 cell lines (8). However, the classification of H460 as
an EML4-ALK carrier remains controversial with some studies failing
to find this fusion (20). It is
important to note that the EC50 concentrations of the
non-responding cell lines were well above the maximally tolerated
dose (plasma concentration of 57 nM) as determined in phase II
trials of PF-02341066 (12). In
addition, although we used a concentration of 100 nM for treatment,
we confirmed that this dose did effectively inhibit c-Met
phosphorylation (Fig. 1D).
We clearly demonstrated that, within the context of
the cell lines we studied, treatment with PF-02341066 did not
increase radiation sensitivity. For 5 cell lines used in this study
H2228, H3122, H1993, H460 and A549; there was no significant dose
enhancement in vitro. When tested in in vivo models,
there was statistically significant increase in radiation response
with treatment. The dose used in this experiment, 50 mg/kg, has
been shown to be the cut-off dose in which further dose escalation
results in marginal increases in plasma concentration (7). c-Met has been implicated in modulating
DNA repair kinetics which is thought to provide a survival
advantage to cancer cells (15).
However, we showed that c-Met inhibition through PF-02341066 does
not affect DNA repair kinetics. These data are further supported by
cell cycle analysis which failed to show an increased proportion of
cells in G2M arrest indicating that cells that are damaged are able
to repair at the same rate regardless of c-Met inhibition. It is
interesting to note that H2228 and H1993 cells showed increased G1
arrest following treatment of the drug alone; however, this
increased G1 arrest did not result in significant changes in
radiation response. The G1 arrest noted for H2228 and H1993 cells
represents the most likely mechanism for decreased
clonogenicity.
Finally, we assayed for PF-02341066 specificity
in vivo. We report that in H2228, a cell line that has high
levels of endogenous c-Met, PF-02341066 irreversibly inhibits c-Met
activity. We showed, however, that Akt phosphorylation was only
transiently affected and phosphorylation reappeared after 4 h of
treatment. In addition, once the drug was removed from the medium
there was a strong p-Akt rebound. In the cell lines that did not
express endogenous phospho-c-Met, HGF did induce c-Met
phosphorylation which was inhibited by PF-02341066. However, in
these cell lines, the level of p-Akt was largely unaffected by
treatment. The inability to keep p-Akt suppressed throughout the
treatment course may represent an escape pathway for these NSCLC
cells. Although we were inhibiting an upstream modulator of Akt, it
appeared that the cells were able to compensate through separate
and redundant pathways thereby reactivating the Akt pathway, a
pathway known to increase radiation resistance.
Even though PF-02341066 is an effective therapy able
to suppress tumor growth in NSCLC tumors that are either EML4-ALK-
or c-Met-positive, it did not affect tumor cell radiation
resistance in any appreciable manner. We showed that PF-02341066
did not affect radiation sensitivity, DNA repair kinetics, the cell
cycle distribution or increased apoptosis in a supra-additive
manner. PF-02341066 may be able to escape the radiation-sensitizing
effects of c-Met inhibition by reactivating the Akt pathway further
downstream or through a separate and redundant pathway.
Acknowledgements
This study was supported by funding from the Flight
Attendant Medical Research Institute (D.S.), W81XWH-11-1-0270
(D.S.), Pfizer Inc. (D.S.), Clinical and Translational Science
award grant #5TL1 RR024984 (V.T.) and the Clinical Research
Fellowship from the Doris Duke Charitable Foundation.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Soda M, Choi YL, Enomoto M, et al:
Identification of the transforming EML4-ALK fusion gene in
non-small cell lung cancer. Nature. 448:561–566. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kwak EL, Bang YJ, Camidge DR, et al:
Anaplastic lymphoma kinase inhibition in non-small-cell lung
cancer. N Engl J Med. 363:1693–1703. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rodig SJ, Mino-Kenudson M, Dacic S, et al:
Unique clinicopathologic features characterize ALK-rearranged lung
adenocarcinoma in the western population. Clin Cancer Res.
15:5216–5223. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Choi YL, Takeuchi K, Soda M, et al:
Identification of novel isoforms of the EML4-ALK transforming gene
in non-small cell lung cancer. Cancer Res. 68:4971–4976. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Soda M, Takada S, Takeuchi K, et al: A
mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci
USA. 105:19893–19897. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yamazaki S, Vicini P, Shen Z, et al:
Pharmacokinetic/pharmacodynamic modeling of crizotinib for
anaplastic lymphoma kinase inhibition and antitumor efficacy in
human tumor xenograft mouse models. J Pharmacol Exp Ther.
340:549–557. 2011. View Article : Google Scholar
|
|
8
|
Lin E, Li L, Guan Y, et al: Exon array
profiling detects EML4-ALK fusion in breast, colorectal, and
non-small cell lung cancers. Mol Cancer Res. 7:1466–1476. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kodym E, Kodym R, Reis AE, Habib AA, Story
MD and Saha D: The small-molecule CDK inhibitor, SNS-032, enhances
cellular radiosensitivity in quiescent and hypoxic non-small cell
lung cancer cells. Lung Cancer. 66:37–47. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kong Z, Raghavan P, Xie D, et al:
Epothilone B confers radiation dose enhancement in DAB2IP gene
knock-down radioresistant prostate cancer cells. Int J Radiat Oncol
Biol Phys. 78:1210–1218. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim JC, Saha D, Cao Q and Choy H:
Enhancement of radiation effects by combined docetaxel and
flavopiridol treatment in lung cancer cells. Radiother Oncol.
71:213–221. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ou SH: Crizotinib: a novel and
first-in-class multitargeted tyrosine kinase inhibitor for the
treatment of anaplastic lymphoma kinase rearranged non-small cell
lung cancer and beyond. Drug Des Devel Ther. 5:471–485.
2011.PubMed/NCBI
|
|
13
|
Bai RY, Ouyang T, Miething C, Morris SW,
Peschel C and Duyster J: Nucleophosmin-anaplastic lymphoma kinase
associated with anaplastic large-cell lymphoma activates the
phosphatidylinositol 3-kinase/Akt antiapoptotic signaling pathway.
Blood. 96:4319–4327. 2000.PubMed/NCBI
|
|
14
|
Chen Z, Sasaki T, Tan X, et al: Inhibition
of ALK, PI3K/MEK, and HSP90 in murine lung adenocarcinoma induced
by EML4-ALK fusion oncogene. Cancer Res. 70:9827–9836. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fan S, Ma YX, Gao M, et al: The
multisubstrate adapter Gab1 regulates hepatocyte growth factor
(scatter factor)-c-Met signaling for cell survival and DNA repair.
Mol Cell Biol. 21:4968–4984. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Christensen JG, Zou HY, Arango ME, et al:
Cytoreductive antitumor activity of PF-2341066, a novel inhibitor
of anaplastic lymphoma kinase and c-Met, in experimental models of
anaplastic large-cell lymphoma. Mol Cancer Ther. 6:3314–3322. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
De Bacco F, Luraghi P, Medico E, et al:
Induction of MET by ionizing radiation and its role in
radioresistance and invasive growth of cancer. J Natl Cancer Inst.
103:645–661. 2011.PubMed/NCBI
|
|
18
|
Aebersold DM, Kollar A, Beer KT, Laissue
J, Greiner RH and Djonov V: Involvement of the hepatocyte growth
factor/scatter factor receptor c-met and of Bcl-xL in the
resistance of oropharyngeal cancer to ionizing radiation. Int J
Cancer. 96:41–54. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brognard J, Clark AS, Ni Y and Dennis PA:
Akt/protein kinase B is constitutively active in non-small cell
lung cancer cells and promotes cellular survival and resistance to
chemotherapy and radiation. Cancer Res. 61:3986–3997.
2001.PubMed/NCBI
|
|
20
|
McDermott U, Iafrate AJ, Gray NS, et al:
Genomic alterations of anaplastic lymphoma kinase may sensitize
tumors to anaplastic lymphoma kinase inhibitors. Cancer Res.
68:3389–3395. 2008. View Article : Google Scholar : PubMed/NCBI
|