Introduction
Gliomas are the most common type of malignant brain
tumors and are resistant to many types of treatments, including
chemotherapy, radiation and other adjuvant therapies. Patients with
the most malignant histopathologic subtype, glioblastoma, present
with the worst prognosis, with a median survival length of less
than one year, despite aggressive surgery with adjuvant
radiotherapy and chemotherapy (1,2). In
addition, glioma cells are prone to acquire drug resistance
(3). Currently, there is still a
need to identify chemotherapeutic agents with cytotoxic effects
exclusively targeted against malignant glioma cells. Improved
chemotherapeutic regimens and other strategies are urgently
needed.
‘Apoptosis’, signifying genetically controlled
programmed cell death (PCD), not only plays a crucial role during
tissue development and homeostasis, but is also involved in a wide
range of pathologies (4). Since the
1960s, various morphological forms of PCD have been recognized.
Clarke classified cell death into 4 types, including type I PCD
(apoptosis) and type II PCD (autophagy) (5). Evasion of apoptosis is a hallmark of
most malignant cells and contributes to the insensitivity to
various current cancer therapies (6). Numerous studies have demonstrated that
most chemotherapeutic agents and certain naturally occurring
compounds induce cell death by activating the apoptotic pathways.
It is thought that apoptosis induction in tumor cells, with either
drugs or natural products, is an effective therapy for cancer and
immune system diseases.
Autophagy is one of the major regulatory mechanisms
in the degradation of intracellular proteins and organelles
(7,8). It is a highly conserved evolutionary
process which occurs in the cells of all eukaryotic organisms, from
yeasts to humans. During autophagy, the cytosol and entire
organelles become encased in double-membrane-bound vacuoles
(autophagosomes), and subsequently fuse with lysosomes to form
autolysosomes and are eventually degraded by lysosomal hydrolases
(7). Although originally
characterized as a survival response to nutrient deficiency,
autophagy is now recognized as being frequently induced in response
to a variety of stressors to maintain cellular homeostasis
(9–11). The importance of autophagy has been
emphasized in various biological fields, including cancer (10,12–14).
Furthermore, cancer cells undergo less autophagy than normal cells
(15,16). These findings indicate that
autophagy induction is an attractive modality of anticancer
therapy.
β-carboline alkaloids are present in several
medicinal plants, including Peganum harmala, Passiflora
incarnate, and Bansteriopsis caapi(17,18).
These plants have been used in traditional medicine to treat
asthma, jaundice, lumbago, and other human ailments (18–20).
The β-carboline alkaloids are also found in common plants (e.g.,
wheat, rice, soybeans, grapes) and plant-derived drinks (e.g.,
wine, beer, whisky, brandy) (21).
It is known that they also exist in mammalian tissues (22,23).
It has been reported that certain β-carboline alkaloids and their
related compounds exhibit cytotoxic effects on cancer cells
(24–26). We also previously reported that
harmol, a β-carboline alkaloid, induced apoptosis (27) and autophagic cell death in human
non-small cell lung cancer cells (28).
In the present study, we investigated the anticancer
effect of harmol on the U251MG human glioma cell line. Furthermore,
we examined the types of cell death which were induced by harmol
and the possible mechanisms.
Materials and methods
Chemicals
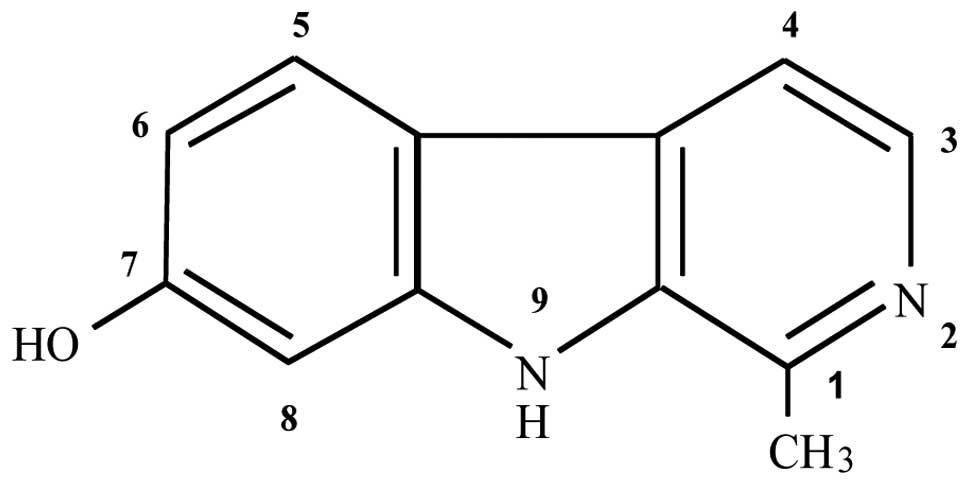
Harmol (purity, minimum 98%) (Fig. 1), dimethyl sulfoxide (DMSO),
3-methyladenine (3-MA) and chloroquine (CQ) were purchased from
Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Harmol was
dissolved in DMSO at a concentration of 200 mM, and stored at −20°C
until use. Monodansylcadaverine (MDC) was obtained from
Sigma-Aldrich (St. Louis, MO, USA). Caspase substrates
Ac-DEVD-7-amino-4-trifluoromethyl coumarin (AFC) (caspase-3),
Ac-IETD-AFC (caspase-8), and Ac-LEHD-AFC (caspase-9) were obtained
from MBL (Nagoya, Japan). A recombinant full-length human active
Akt1 protein (rAkt1) was purchased from R&D Systems, Inc.
(Minneapolis, MN, USA).
Cell lines and culture conditions
Human glioma cell line U251MG was obtained from the
Human Science Research Resources Bank (Osaka, Japan). This cell
line was cultured in Dulbecco’s modified Eagle’s medium (Wako) with
80 mg/l kanamycin sulfate (Wako) and heat-inactivated fetal bovine
serum (Biowest, Miami, FL, USA) and maintained at 37°C in an
incubator containing 95% air and 5% CO2.
Cell viability
Cell viability was assessed with the CellTiter-Blue
cell viability assay kit (Promega Corp., Madison, WI, USA). In
viable cells, resazurin, which is contained in the CellTiter-Blue
cell viability assay reagent, is metabolized to the fluorescent
product resorufin. The tumor cells (4.5×104 cells/well)
were precultured in a 48-well flat-bottom microtiter plate
overnight at 37°C in a 5% CO2 humidified chamber. Then,
various concentrations of harmol were added and the cells were
incubated for 24–48 h. After incubation, 100 μl of CellTiter-Blue
assay reagent (Promega Corp) was added to each well and the cells
were further incubated for 1.5 h. After incubation, the medium of
each well was analyzed using a Powerscan HT microplate reader (DS
Pharma Biomedical Co., Osaka, Japan) at an excitation wavelength of
560 nm and an emission wavelength of 590 nm. Cell viability was
determined based on the fluorescence intensity of non-treated
cells.
Assay for caspase activity
Caspase activity was measured using fluorogenic
peptide substrates. Both untreated control cells and cells which
had been treated with 75 μM harmol were washed with ice-cold
phosphate-buffered saline (PBS) and suspended in lysis buffer [100
mM HEPES (pH 7.5), 150 mM NaCl, 1% Nonidet P-40, 1 mM EDTA, 1 mM
dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride, 10 μM
leupeptin and 1 μM pepstatin] for 20 min on ice, followed by
centrifugation at 12000 × g for 10 min at 4°C. In total, 50–70 μg
of protein in 40 μl of buffer solution was mixed with 10 μl of 5 mM
fluorogenic report substrate, for each individual caspase:
Ac-DEVD-AFC for caspase-3, Ac-IETD-AFC for caspase-8 and
Ac-LEHD-AFC for caspase-9. The reaction mixture was added to 50 μl
of assay buffer [20 mM HEPES (pH 7.4), 0.1 M NaCl, 5 mM DTT, 0.1%
Nonidet P-40], and incubated at 37°C for 1 h. The enzymatic
product, AFC, which was released from the substrate, was excited at
400 nm to measure its emission at 505 nm. Untreated cells were used
as a control.
Visualization of MDC-labeled vacuoles in
harmol-treated A549 cells
U251MG cells were treated with 100 μM harmol for
0–12 h at 37°C, and then further treated with 30 μM MDC for 10 min.
After treatment, cells were washed 3 times with PBS and immediately
observed by fluorescence microscopy (BZ-8100, Keyence Co., Osaka,
Japan).
Electron microscopy
Cultured U251MG cells with and without 100 μM harmol
treatment were rinsed with phosphate buffer (PB) (0.1 M, pH 7.4).
Cells were then fixed with 2.5% glutaraldehyde in PB for 3 h at
4°C, rinsed with PBS for 5 min at least 3 times, and post-fixed
with 1% osmium tetroxide for 1 h at 4°C, followed by dehydration in
a graded ethanol series for 5 min at room temperature (RT). After
dehydration with absolute ethanol, cells were treated with a
mixture of absolute ethanol and Spurr resin (1:1, v/v) for 1 h at
RT. Then, the mixture was replaced with pure Spurr resin and cells
were treated for 1 h at RT, twice. The pellets were transferred to
a BEEM capsule, overlaid with new resin and subjected to resin
polymerization at 70°C for 16 h. Ultrathin sections were cut with
an Ultracut E microtome (Reichert-Jung Co., Vienna, Austria),
stained with uranyl acetate and lead citrate, and examined using a
JEM-1230 transmission electron microscope (JEOL, Tokyo, Japan).
Western blot analysis
Whole cell proteins were isolated from untreated and
harmol-treated U251MG cells. After treatment, the cells were
centrifuged at 300 × g for 5 min, and the pellet was lysed in a
buffer containing 25 mM HEPES (pH 7.4), 150 mM NaCl, 0.5% Triton
X-100, 10% glycerol, 1 mM DTT, 1 mM sodium orthovanadate, 25 mM
β-glycerophosphate, 1 mM NaF and 5 μl/ml protease inhibitor
cocktail (Wako). The protein content of each lysate was determined
using a BCA protein assay kit (Pierce Biotechnology Inc., Rockford,
IL, USA). Protein lysates were then mixed with an equal volume of
gel loading buffer [20% glycerol, 4% sodium dodecyl sulfate (SDS),
100 mM Tris, 5% β-mercaptoethanol and 0.01% bromophenol blue]
before being boiled for 5 min. After boiling, 40 μg of protein was
subjected to SDS-polyacrylamide gel electrophoresis. Proteins were
then transferred onto a polyvinylidene fluoride membrane (PVDF; GE
Healthcare UK Ltd., Buckinghamshire, UK). Blots were then blocked
for 1 h at RT in 5% non-fat dry milk diluted in Tris-buffered
saline supplemented with 0.1% Tween-20 (TBS-T) (ICN Biomedicals
Inc., Aurora, OH, USA). The following primary antibodies were
incubated overnight at RT in TBS-T as follows: rabbit
anti-poly-(ADP-ribose)-polymerase (PARP) (Cell Signaling Technology
Inc., Danvers, MA, USA; 1:1000), rabbit anti-Akt (Cell Signaling
Technology; 1:1000), rabbit anti-phospho-Akt (p-Akt) (Cell
Signaling Technology; 1:1000), rabbit anti-mammalian target of
rapamycin (mTOR) (Cell Signaling Technology; 1:1000), rabbit
anti-phospho-mTOR (p-mTOR) (Cell Signaling Technology; 1:1000),
rabbit anti-70 kDa ribosomal protein S6 kinase (P70S6K) (Cell
Signaling Technology; 1:1000), rabbit anti-phospho-P70S6K
(p-P70S6K) (Cell Signaling Technology; 1:1000), rabbit
anti-eukaryotic translation initiation factor 4E-binding protein 1
(4E-BP1) (Cell Signaling Technology; 1:1000), rabbit
anti-phospho-4E-BP1 (p-4E-BP1) (Cell Signaling Technology; 1:1000),
mouse anti-microtubule-associated protein 1 light chain 3 (LC3)
(MBL; 1:1000), and mouse anti-β-actin (Sigma-Aldrich; 1:3000).
Peroxidase-conjugated secondary antibodies were incubated for 1 h
at RT as follows: goat anti-mouse IgG (Jackson Immunoresearch
Laboratories Inc., West Grove, PA, USA; 1:5000) or goat anti-rabbit
IgG (Cell Signaling Technology; 1:2000). Blots were then washed
with TBS-T and developed using Immobilon chemiluminescent substrate
(Millipore Corp., Billerica, MA, USA).
Survivin siRNA transfection
Knockdown of survivin expression in U251MG cells was
achieved by transfection of siRNA. The control and survivin siRNAs
(Cell Signaling Technology) were diluted to a final concentration
of 20 nM in Opti-Mem I (Invitrogen Corp., Carlsbad, CA, USA), and
transfection was performed with cells at 40–50% confluency using
Lipofectamine 2000 transfection reagent (Invitrogen) according to
the manufacturer’s protocol.
Statistical analysis
Results are presented as means ± SD. Statistical
comparisons of the results were carried out using analysis of
variance. A P-value of <0.05 was considered to indicate a
statistically significant difference. Differences between the means
of control and treated cells were analyzed by Dunnett’s test.
Results
Cytotoxic effects of harmol on U251MG
cells
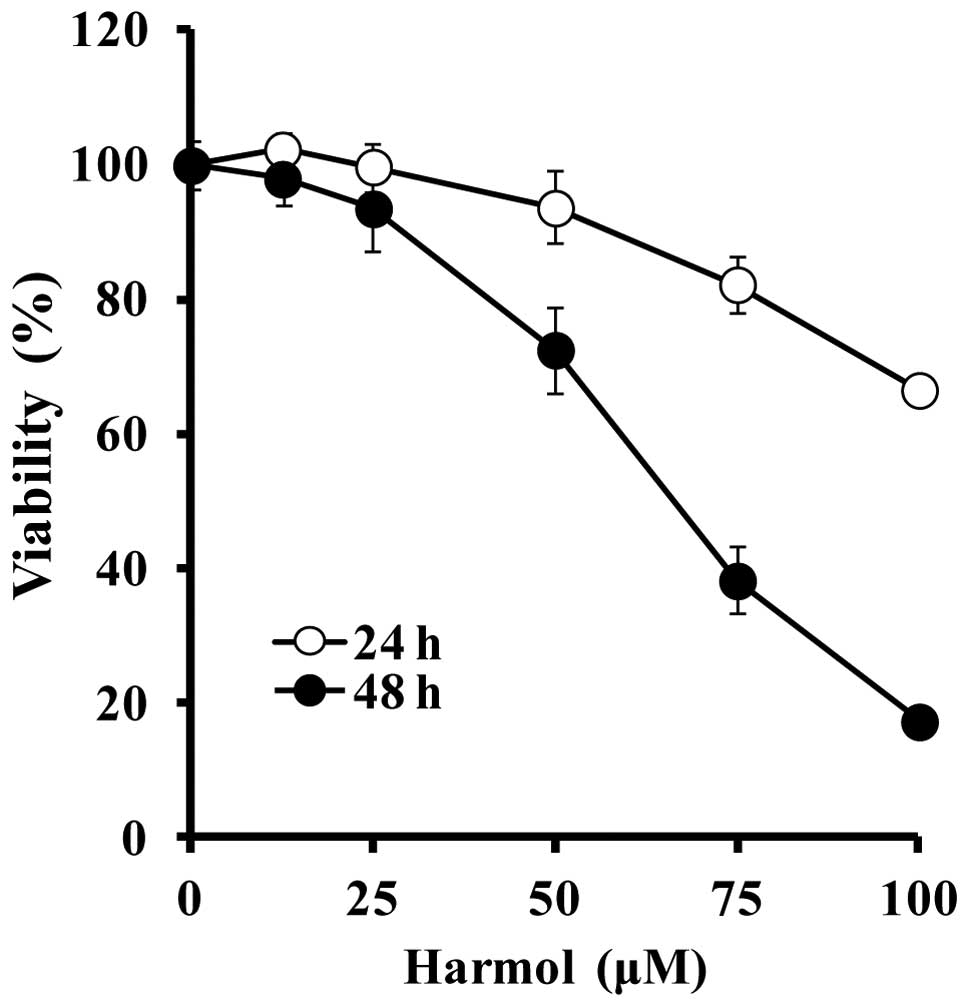
Continuous treatment of U251MG human glioma cells
with harmol for 24 and 48 h induced growth inhibition and cell
death in a time- and dose-dependent manner (Fig. 2). The cell viabilities of U251MG
cells exposed to harmol at various concentrations of 12.5, 25, 50,
75, and 100 μM in triplicate for 24 h were 102.3, 99.8, 93.7, 82.3,
and 66.7%, respectively. After treatment with harmol for 48 h, the
cell viabilities decreased to 98.0, 93.6, 72.5, 38.2, and 17.3%,
respectively (Fig. 2).
Changes in caspase activities and
poly-(ADP-ribose)-polymerase (PARP) cleavage
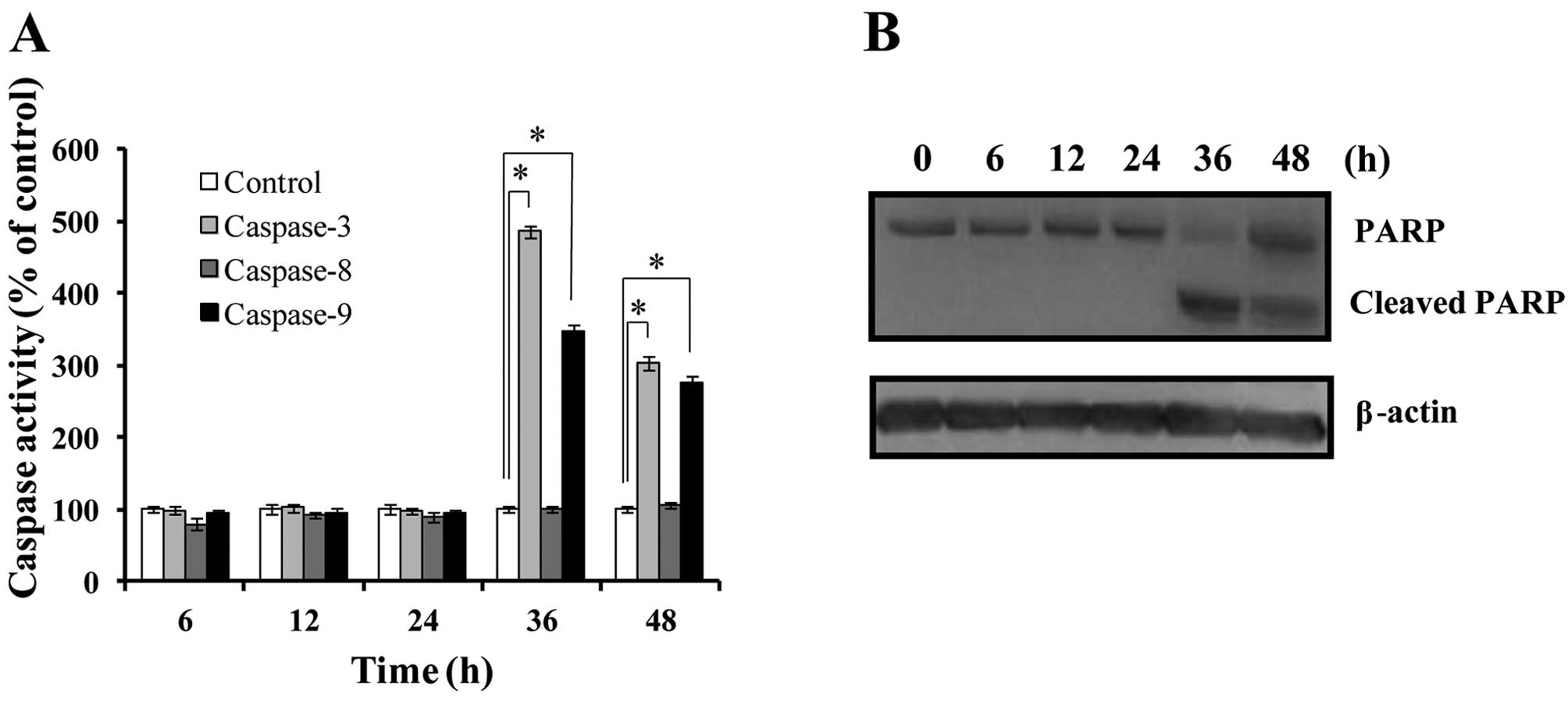
To confirm whether the cell death induced by harmol
treatment was due to apoptosis, the activities of 3 types of
caspases were examined (Fig. 3A).
Harmol showed no noticeable effect on the activities of these
caspases within the 24-h treatment. However, in the 36-h treatment,
activities of caspase-3 and -9 were elevated to 486 and 346%,
respectively, while caspase-8 activity showed no significant
change. We also examined the effects of harmol on PARP cleavage.
PARP is a key participant in DNA base excision repair and in
maintaining genome integrity. PARP is well known as an endogenous
substrate for caspase-3 and an early marker of apoptosis (29). After treatment with harmol for 6–48
h, the cleaved form of PARP (89 kDa) was detected at 36 and 48 h
following treatment of the cell lysate (Fig. 3B). These data indicated that
treatment with harmol for over 36 h induced apoptosis in U251MG
cells.
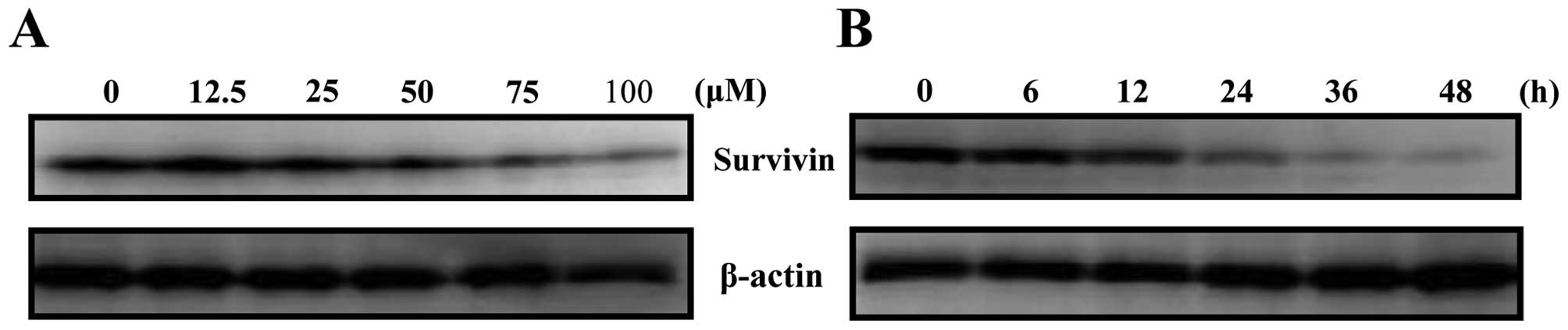
Harmol treatment suppresses survivin
protein expression
While harmol was shown to induce apoptosis in U251MG
cells, the mechanisms of inducing this phenomenon were unclear.
Afterwards, we investigated the effect of harmol treatment on the
expression of survivin protein, which is a known anti-apoptotic
protein. From the results, harmol treatment was shown to suppress
the expression of survivin protein in a dose- and time-dependent
manner (Fig. 4).
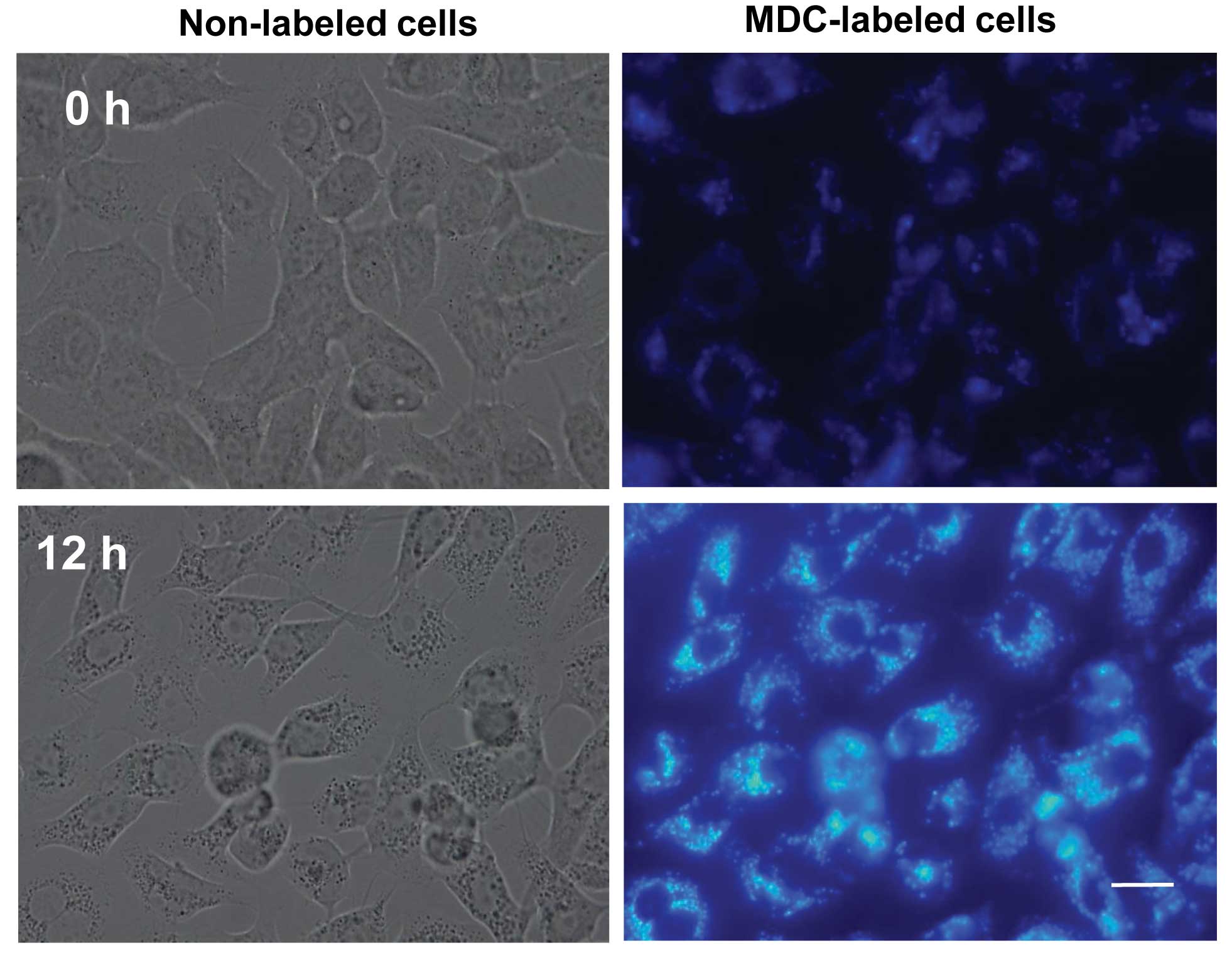
Formation of autophagic vacuoles
following treatment with harmol
Phase contrast microscopy of harmol-treated U251MG
cells showed numerous, high-density vacuoles (Fig. 5, left lower panel). Based on the
above findings, we would have expected these vacuoles to be formed
upon harmol treatment; they were not detected in untreated cell
autophagosomes. The fluorescent compound MDC is a specific marker
for autophagosomes (30), and is
commonly used to stain autophagic vesicles. We studied the
incorporation of MDC in cells where autophagy was stimulated by
harmol treatment. U251MG cells were treated with 100 μM harmol for
12 h and then analyzed by fluorescence microscopy. As shown in
Fig. 5 (right upper panel), in
control cells (0 h treatment), MDC-labeled vacuoles were scarcely
detected. On the other hand, in cells which were treated with
harmol for 12 h, numerous MDC-labeled fluorescent dots were clearly
detected (Fig. 5, right lower
panel). Although MDC-labeled fluorescent dots were observed in
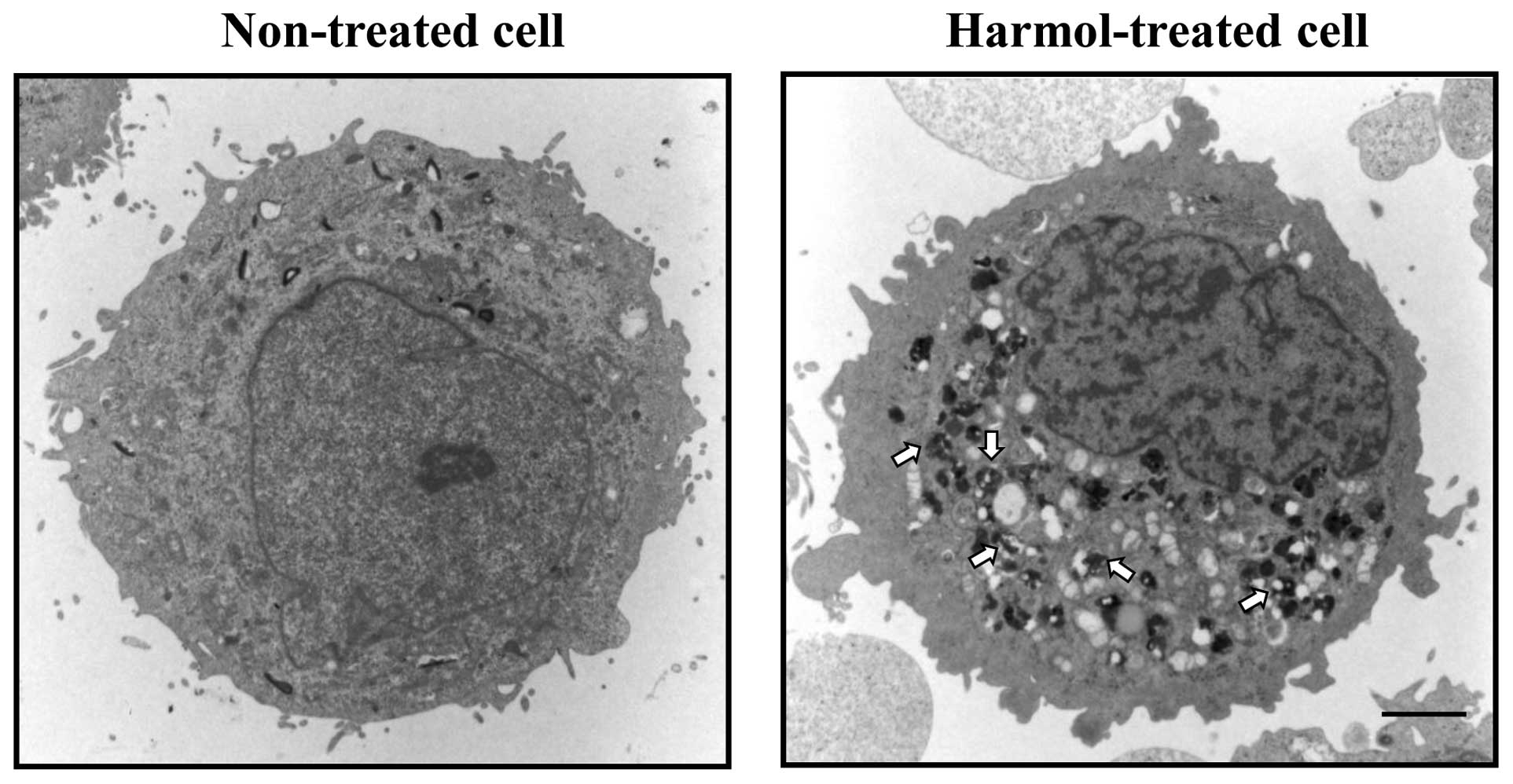
harmol-treated U251MG cells, in order to identify them more
precisely, we performed electron microscopy to obtain
ultrastructural information regarding the morphology of
harmol-induced autophagy in U251MG cells. Fig. 6 shows an electron micrograph of a
non-treated U251MG cell (left panel) and a U251MG cell treated with
harmol (right panel). Although the electron micrograph of the
harmol-treated U251MG cell showed numerous cytoplasmic
phagolysosomes that contained highly electron-dense materials
(Fig. 6, right panel), few
phagolysosomes were observed in the control cell (Fig. 6, left panel).
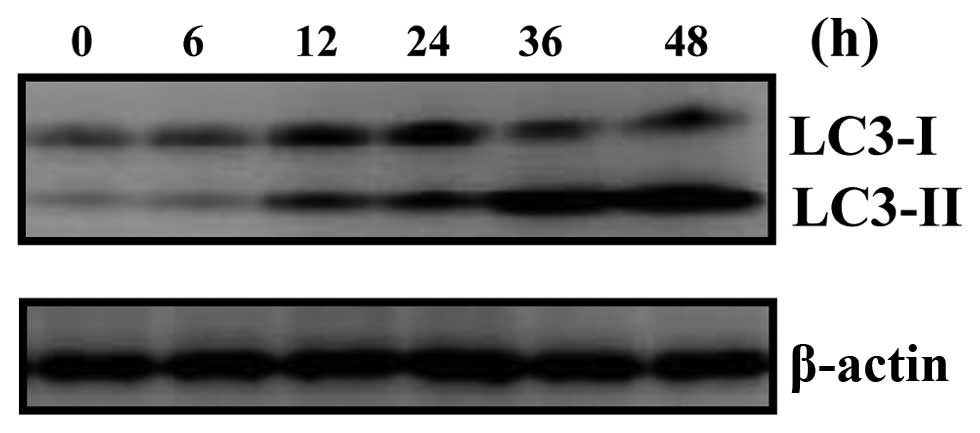
Quantitative detection of
microtubule-associated protein 1 light chain 3 (LC3)
To quantify the incidence of harmol-induced
autophagy, we examined the expression of LC3-I and LC3-II proteins
using western blot analysis, as these proteins are essential to the
formation of autophagosomes (especially LC3-II) and have been
widely used for estimating the number of autophagosomes or the
incidence of autophagy (31). The
expression of LC3-II protein in harmol-treated cells was detected
within 12 h, and increased during treatment in a time-dependent
manner (Fig. 7). Taken together,
these results also indicated that harmol induced autophagy, and
that harmol-induced autophagy preceded apoptosis in U251MG cells
(Fig. 3 vs. Figs. 5–7).
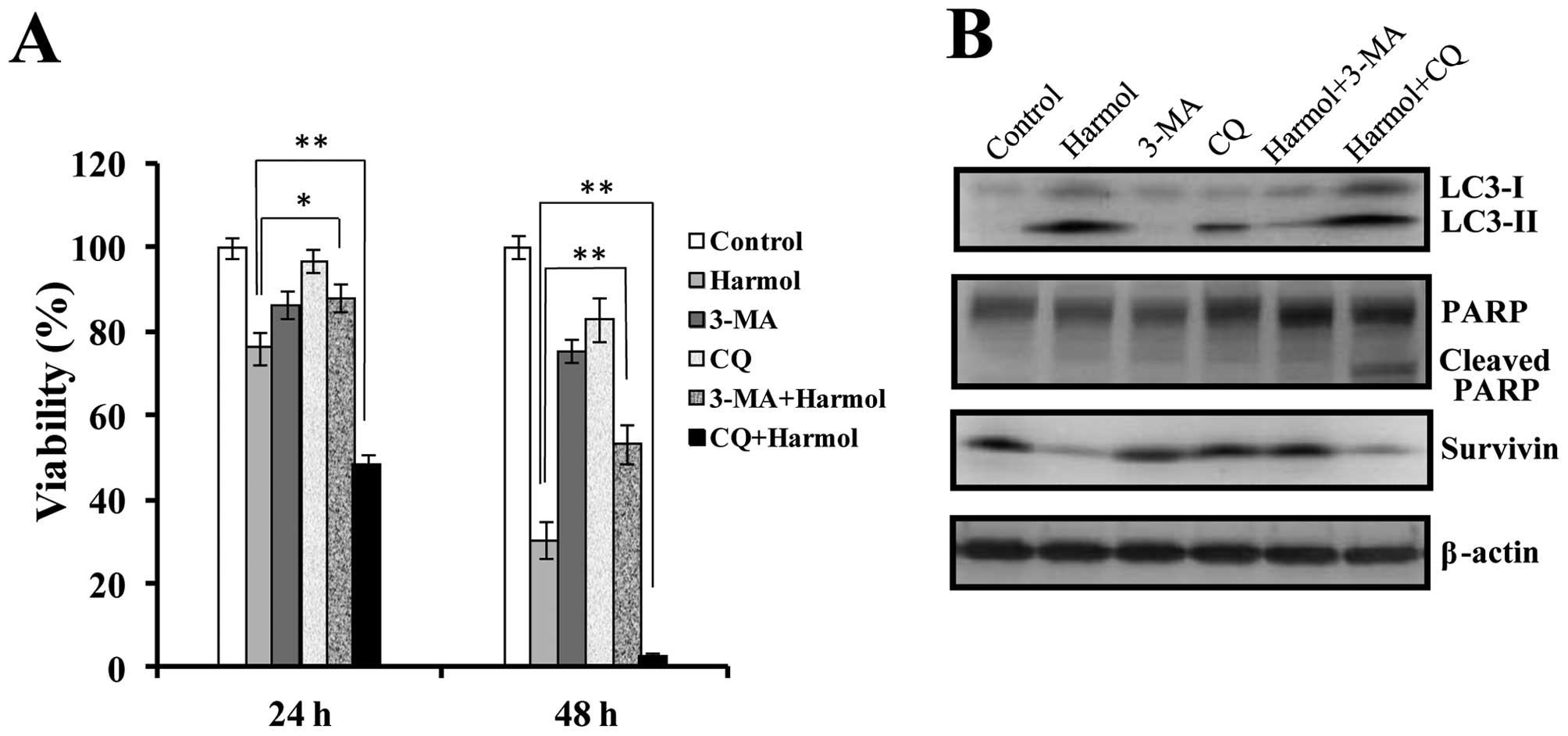
Role of autophagy in harmol-mediated cell
death
Autophagy plays an important role in the regulation
of cell survival or death (7,8). To
determine whether the induction of autophagy by harmol is related
to survival or death mechanisms, we investigated the effect of
harmol on cell survival in the presence of autophagy inhibitors in
U251MG cells. As shown in Fig. 8A,
3-MA, a specific inhibitor of the early stage autophagic process
(32), reduced the harmol-induced
cell death in U251MG cells. On the other hand, the late stage
autophagy inhibitor CQ (33)
increased the harmol-induced cell death. On western blot analysis,
although treatment with harmol alone induced the expression of
LC3-II protein (known as an autophagy marker), co-treatment with
harmol and 3-MA notably suppressed LC3-II protein expression
(Fig. 8B). In contrast,
co-treatment with harmol and CQ induced progressive accumulation of
LC3-II throughout the 24 h duration assay (Fig. 8B). Following treatment with harmol
alone for 24 h, although PARP cleavage was not induced (Figs. 3 and 8B), CQ treatment in the presence of harmol
induced PARP cleavage. Regarding the anti-apoptotic protein
survivin, although >24 h of treatment with 100 μM harmol
strongly suppressed the expression of survivin protein (Fig. 6), the combination treatment of
harmol with 3-MA did not suppress the expression of survivin
protein (Fig. 8B). However,
co-treatment with harmol and CQ did not suppress the reduction of
survivin protein expression in U251MG cells (Fig. 8B).
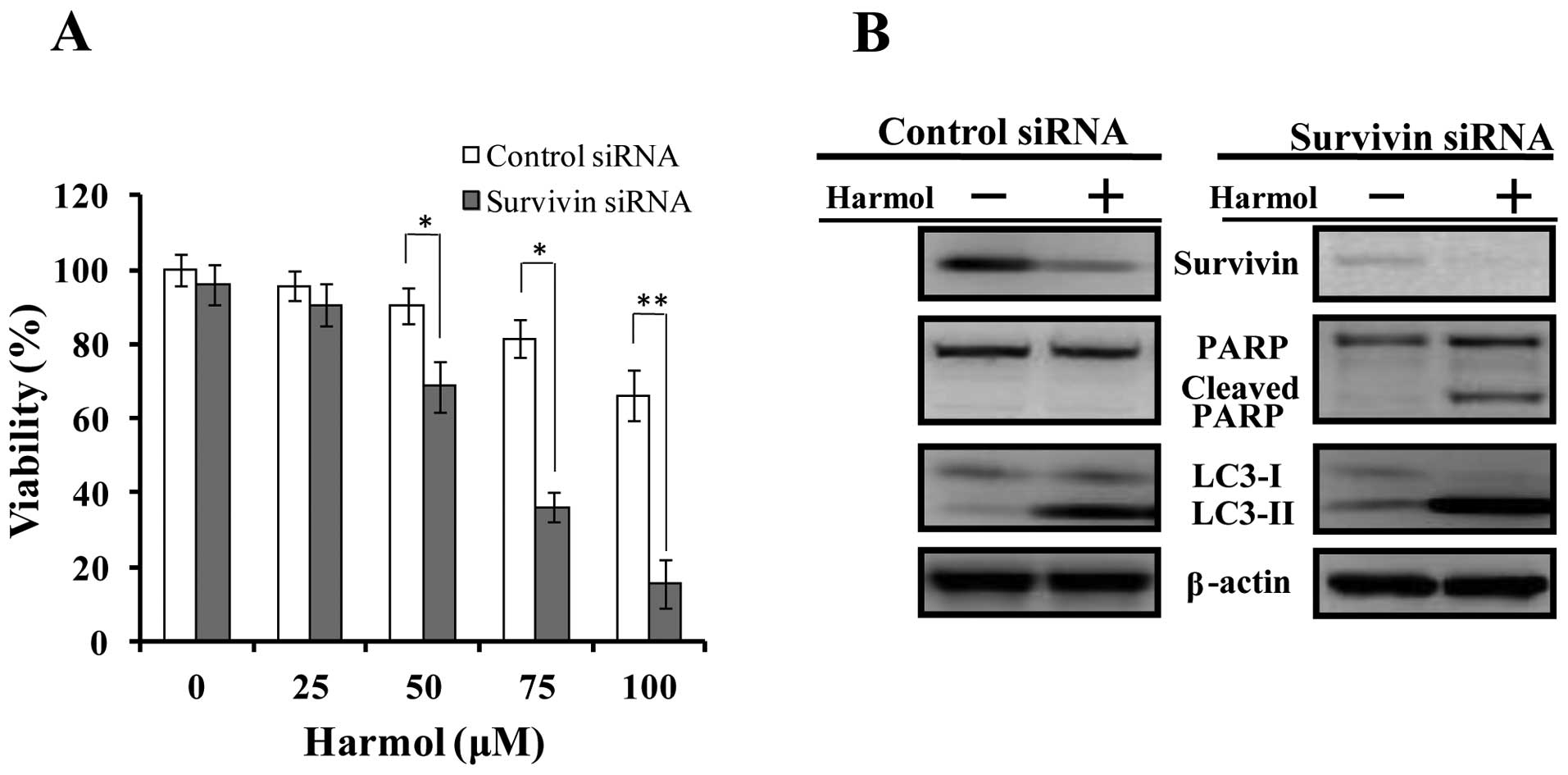
Genetic suppression of survivin protein
expression increases harmol-induced apoptosis
To demonstrate whether harmol-induced apoptosis is
related to the suppression of survivin protein expression, we
performed siRNA-mediated knockdown of survivin in U251MG cells. As
a result, although in control-siRNA transfected U251MG cells, PARP
cleavage was not detected within the 24 h treatment of harmol, in
cells where the expression of survivin protein was supressed by
transfection with survivin siRNA, PARP cleavage was detected within
24 h following treatment with harmol (Fig. 9B). In addition, siRNA depletion of
survivin protein induced autophagy, which was confirmed by LC3-II
protein expression (Fig. 9B, the
non-treated control of the survivin-siRNA transfected cell sample
was compared with the non-treated control of the control-siRNA
transfected cell sample). Furthermore, in the cytotoxicity
experiment, the suppression of survivin expression by siRNA
increased the harmol-induced cytotoxicity (Fig. 9A).
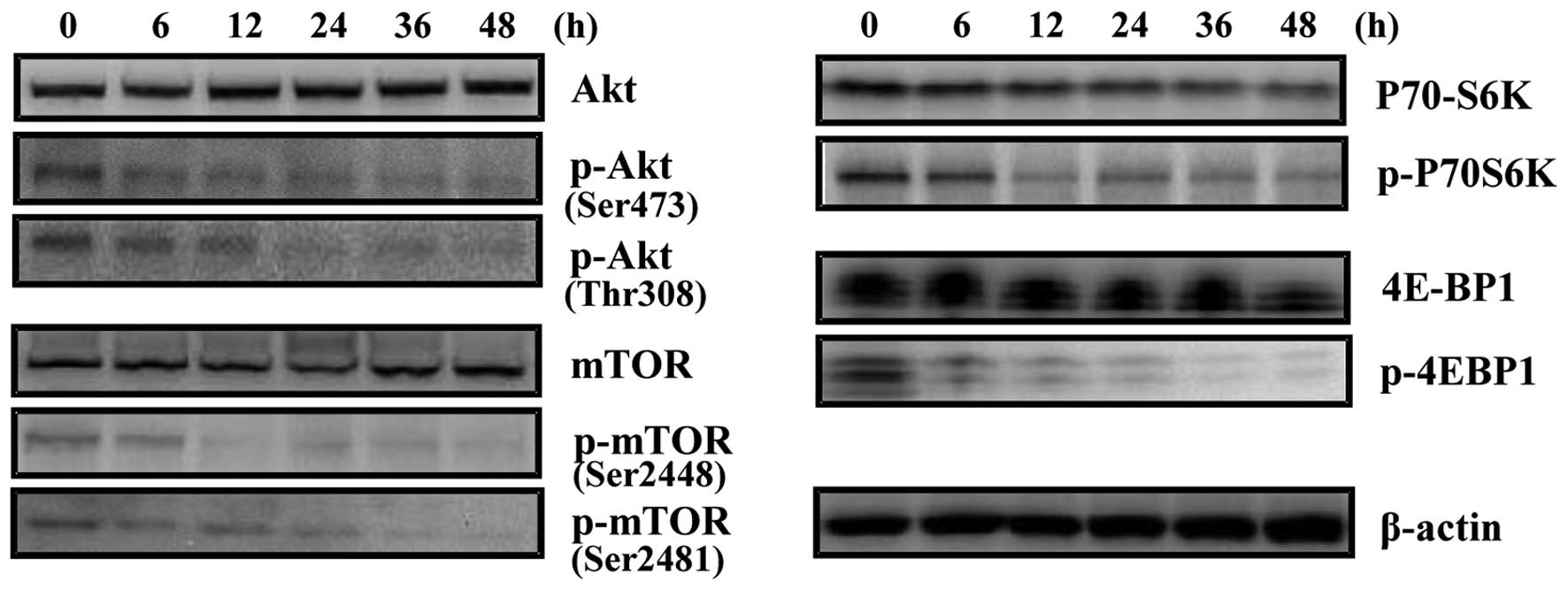
Akt/mTOR signaling pathway is involved in
harmol-induced autophagy in U251MG cells
Our next objective was to identify and characterize
the molecular pathways involved in harmol-induced autophagy. The
classical pathway that regulates autophagy involves the
serine/threonine kinase, mTOR (34). The Akt/mTOR/P70S6K, 4E-BP1 pathway
is the main regulatory pathway that negatively regulates autophagy.
However, several pathways appear to regulate autophagy in mammalian
cells. Thus, we investigated whether the autophagy induced by
harmol is mediated by an mTOR-dependent or -independent pathway
using western blot analysis (Fig.
10). Harmol treatment caused a significant time-dependent
decrease in the phosphorylation of Akt protein in U251MG cells.
Equally, harmol treatment diminished the levels of the
phosphorylated (activated) form of mTOR, a downstream target of Akt
which may inhibit cell growth and induce autophagy. Furthermore,
examination of the phosphorylation of 2 downstream effectors of
mTOR signaling, P70S6K and 4E-BP1, showed that both p-P70S6K and
p-4E-BP1 decreased in a time-dependent manner, revealing a potent
inhibitory effect of harmol treatment on the Akt/mTOR signaling
pathway.
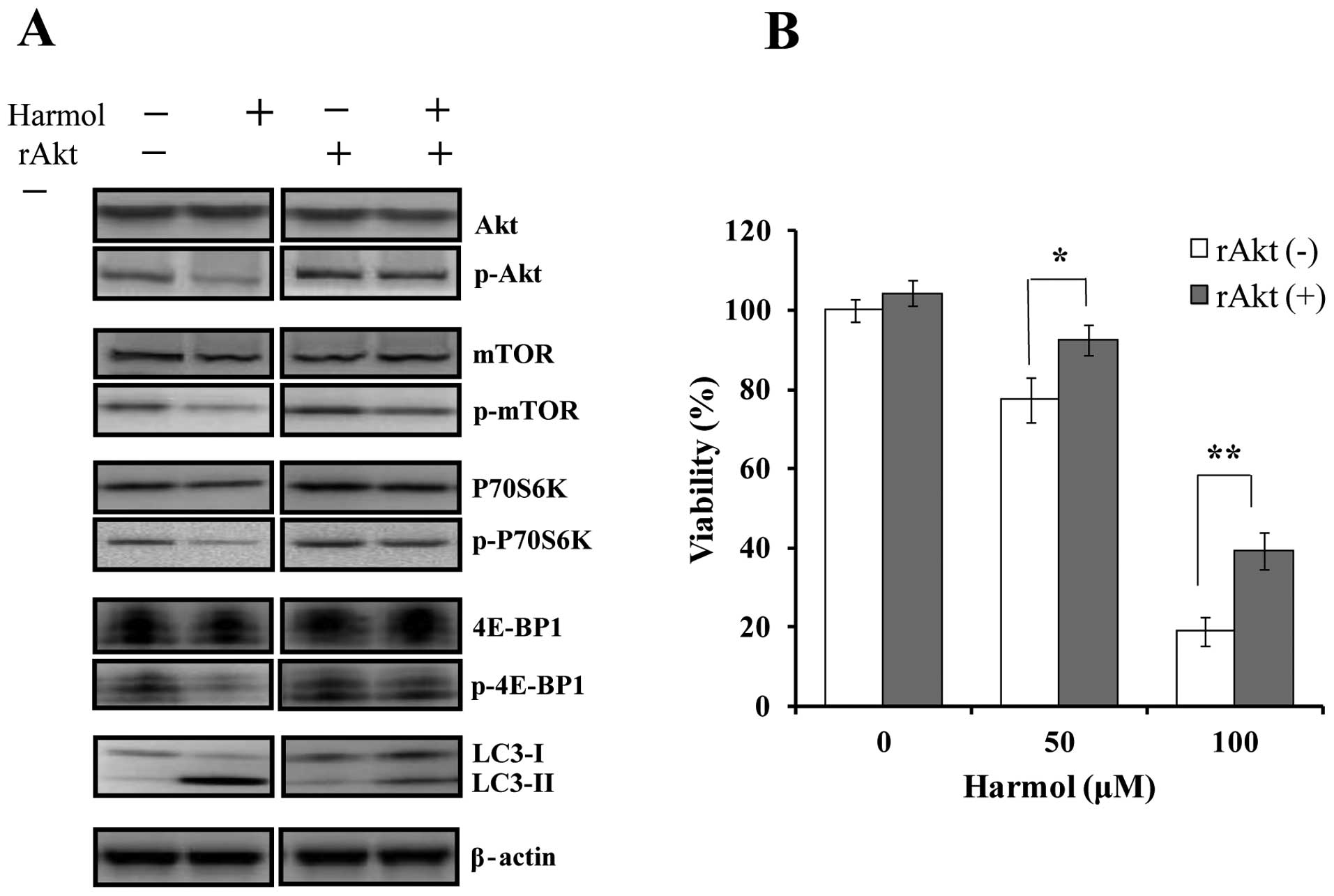
Activation of the Akt pathway inhibits
harmol-induced autophagy and cytotoxicity in U251MG cells
It has been reported that the Akt/mTOR pathway
mediates autophagy induced by various anticancer therapies and
natural products (35–37). Thus, we used rAkt1 to activate the
Akt pathway as described previously (38). In Akt pathway-activated U251MG cells
treated with rAkt1, we examined the effect on harmol-induced
autophagy and cell death. U251MG cells were treated with 100 μM
harmol, 500 ng/ml rAkt1, or both, for 12 h for western blotting.
The addition of rAkt1 inhibited the harmol-induced decrease in
phosphorylated Akt, mTOR and P70S6K in U251MG cells (Fig. 11A). Furthermore, the addition of
rAkt1 significantly decreased harmol-induced autophagy, confirmed
by LC3-II protein expression (Fig.
11A). In the cytotoxicity experiment, the addition of rAkt1
significantly suppressed harmol-induced cytotoxicity in U251MG
cells (Fig. 11B).
Discussion
Previous studies have demonstrated that harmol
induced apoptosis in human lung H596 cells through the
caspase-8-dependent pathway, but independently of Fas/Fas ligand
interaction (27). Furthermore,
harmol induced autophagic cell death in human lung A549 cells
(28). In the present study, we
found that harmol induced autophagy and apoptotic cell death in
U251MG cells.
Defects in the regulation of apoptosis are common
phenomena in many types of cancer and are also the critical steps
in tumorigenesis and resistance to therapy (39). There are two key signaling
mechanisms through which cells may be triggered to undergo
apoptosis. In the intrinsic pathway, diverse stimuli, such as
reactive oxygen species, DNA-damaging reagents, and Ca2+
mobilizing agents provoke cell stress or damage, typically by
activating one or more members of the BH3-only protein family,
leading to the release of cytochrome c. Cytochrome c
then binds to apoptosis-activating factor 1 (Apaf-1) and
procaspase-9, resulting in the activation of caspase-9 by
proteolytic cleavage. The extrinsic pathway starts with death
receptor ligation or Fas/Fas ligand interaction, followed by
oligomerization of the receptor, use of Fas-associated death domain
(FADD) protein, and activation of caspase-8 (40). Caspase-8 and -9 have been generally
defined as initiator caspases, and can in turn activate caspase-3,
the executor of apoptosis (41,42).
As shown in Fig. 3, over 36 h of
treatment with harmol increased the activity of both caspase-9 and
-3, and induced PARP cleavage. However, no noticeable change in the
activity of caspase-8 was observed. From these results, it is
assumed that harmol-induced apoptosis was associated with an
intrinsic pathway instead of an extrinsic pathway, such as Fas/Fas
ligand interaction or the TNF receptor pathway. Although the
reasons for the differences in the responses to harmol treatment in
U251MG, H596 and A549 cells are unclear, these phenomena in each of
these cells might be based on the characteristics of each cell line
and the involved stimuli.
Survivin is the smallest member of the inhibitor of
apoptosis protein (IAP) family that is selectively overexpressed in
most common types of human cancers, but not in normal adult
tissues, and has been implicated in the control of cell division,
inhibition of apoptosis, and tumor cell resistance to certain
anticancer agents and ionizing radiation (43–48).
Therefore, these findings make survivin an attractive anticancer
drug target. In U251MG cells, we found that harmol suppressed the
expression of survivin protein and induced apoptosis associated
with activation of caspases and cleavage of PARP, consistent with
the role of survivin in antagonizing caspases, thereby blocking
apoptosis. Although treatment with harmol alone induced cell death
following a 36-h treatment (Fig.
3), the knockdown of survivin expression by specific siRNA
sensitized U251MG cells to harmol-induced cell death. Cleaved PARP,
a hallmark of apoptosis, was detected within 24 h (Fig. 9B). From these results, the
possibility remains that harmol-induced apoptosis is due to the
downregulation of survivin protein expression.
Of note, although harmol treatment induced not only
apoptosis, but also autophagy (Figs.
5–7), the suppression of
survivin protein expression by siRNA also induced autophagy
(Fig. 9B, comparing the LC3-II band
of the non-treated control sample of control siRNA with the LC3-II
band of the non-treated control sample of survivin siRNA).
Furthermore, survivin siRNA treatment increased harmol-induced
autophagy, as evidenced by conversion of the autophagosomal marker
LC3 protein from the cytosolic form (LC3-I) to the membrane-bound,
phosphatidylethanolamine-conjugated form (LC3-II), associated with
a size shift detected by western blotting (Fig. 9B). In brief, it appears that at
first, harmol induces autophagy (induced within 12 h by treatment
with harmol) (Fig. 7) and
subsequently reduces the expression of survivin protein. Next, the
reduction of survivin protein expression increases autophagy, and
finally, apoptosis is induced. Autophagy plays a critical role in
the regulation of cell survival or death (12). Therefore, to investigate whether the
induction of autophagy by harmol has a cytoprotective function or
leads to cell death (i.e., autophagic cell death), we treated
U251MG cells with harmol and measured the effects on cell survival
or apoptosis in the presence or absence of pharmacological
autophagy inhibitors. Although 3-MA treatment significantly reduced
harmol-induced cell death, CQ treatment highly enhanced
harmol-induced cell death (Fig. 8).
In addition, although harmol treatment decreased the expression of
survivin, 3-MA treatment in the presence of harmol suppressed the
reduction of survivin expression (Fig.
8B). On the other hand, CQ treatment in the presence of harmol
did not suppress the reduction of survivin expression (Fig. 8B). The 3-MA molecule, a specific
inhibitor of the early-stage autophagic process, is known as a PI3K
inhibitor. Class III phosphatidylinositol-3-kinase (class III PI3K)
is closely associated with autophagosome membrane formation
(10). Therefore, treatment with
3-MA inhibits the formation of autophagic vacuoles. On the other
hand, the late-stage autophagy inhibitor CQ blocks the fusion of
autophagosomes and lysosomes, thereby suppressing the degradation
of the contents within the autophagosome. Considering this
information, the possibility remains that when harmol-induced
autophagosome formation is inhibited, suppression of survivin
protein expression is not induced. In contrast, if an autophagosome
is formed, suppression of survivin protein expression occurs, and
subsequently apoptosis is induced. In brief, it is assumed that
harmol-induced autophagy plays pro-apoptotic roles. However, the
detailed mechanisms of these phenomena are unclear. Additional
experiments are required to clarify these mechanisms.
Autophagy is regulated by multiple signaling
pathways as diverse as class III PI3K (10), protein kinase mTOR (7,49,50),
mitogen activated protein kinase (MAPK) (51,52),
AMP-activated protein kinase (AMPK) (53,54),
Beclin1 (55) and bcl-2 (56). In particular, Akt/mTOR signaling is
the major pathway and plays a variety of physiologic roles,
including cell growth, cell cycle regulation, migration and
survival (57–59). Furthermore, this pathway is also
frequently associated with oncogenesis (60). It is known that the Akt/mTOR pathway
negatively regulates autophagy (10). Several studies have indicated that
inhibition of the Akt/mTOR pathway is associated with the
triggering of autophagy in cancer cells (60–63).
In U251MG cells, harmol treatment inhibited the phosphorylation of
Akt (both Ser473 and Thr308), and moreover, exposure of cells to
harmol also inactivated mTOR and reduced phosphorylation of its
downstream targets p70S6K and 4E-BP1 (Fig. 10). In contrast, the activation of
the Akt pathway by rAkt1 treatment increased the phosphorylation of
mTOR and its downstream targets p70S6K and 4E-BP1 (Fig. 11A). Treatment with rAkt1 also
improved the viability of harmol-treated U251MG cells (Fig. 11B). These results are consistent
with many studies indicating that inhibition of the Akt/mTOR
pathway is associated with induction of autophagy in cancer cells
(35,36,50,61).
β-carbolines which occur naturally as harmala
alkaloids in Peganum harmala Linné, are also widely found in
other medicinal plants (17,18),
and are present endogenously in mammalian tissues (22,23).
Harmala alkaloids have been used in certain hallucinogenic
preparations by some South American and African tribes (62). Furthermore, plants containing
harmala alkaloids have long been used in traditional medicine to
treat asthma, jaundice, lumbago, and other ailments (18–20).
Moreover certain β-carboline alkaloids have a wide spectrum of
neuropharmacological and psychopharmacological actions on the
central nervous system, such as tremorogenesis (63,64),
hypothermia (65), hallucinogenesis
(66,67), monoamine oxidase inhibition
(68,69), and convulsive or anticonvulsive
actions (70). The
β-carboline-induced central nervous effects mainly occur with
β-carbolines which have a methoxyl group at the C-7 position in
their structures (71). Therefore,
β-carbolines which have a methoxyl group at the C-7 position are
not suitable for chemotherapeutic agents. On the other hand, it is
reported that harmol, which has a hydroxyl group at the C-7
position (Fig. 1), showed only
slight central depression and convulsive effects (72). Furthermore, harmol has either slight
or no tremor-inducing action (73,74).
With regard to its metabolic pathway, harmol is
conjugated with sulfate and excreted in the urine and bile; it has
a rapid rate of urinary excretion in humans after intravenous
administration (75). Therefore,
harmol is considered to have low toxicity in humans and
animals.
In conclusion, we demonstrated that harmol induces
autophagy and apoptosis in U251MG human glioma cells. As these
effects of harmol were investigated only in glioma cells, harmol’s
effects on other types of cancer should also be examined.
Furthermore, although additional investigation is needed to
precisely clarify the autophagy and apoptosis induction pathway, it
is thought to be a candidate pathway that can be targeted by a
chemotherapeutic agent in cancer treatment.
Acknowledgements
We are indebted to Dr Clifford A. Kolba and
Associate Professor Edward F. Barroga of the Department of
International Medical Communications of Tokyo Medical University
for their editorial review of the English manuscript.
References
|
1
|
DeAngelis LM: Brain tumors. N Engl J Med.
344:114–123. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Phillips HS, Kharbanda S, Chen R, Forrest
WF, Soriano RH, Wu TD, et al: Molecular subclasses of high-grade
glioma predict prognosis, delineate a pattern of disease
progression, and resemble stages in neurogenesis. Cancer Cell.
9:157–173. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kanzawa T, Kondo Y, Ito H, Kondo S and
Germano I: Induction of autophagic cell death in malignant glioma
cells by arsenic trioxide. Cancer Res. 63:2103–2108.
2003.PubMed/NCBI
|
|
4
|
Fadeel B, Orrenius S and Zhivotovsky B:
Apoptosis in human disease: a new skin for the old ceremony?
Biochem Biophys Res Commun. 266:699–717. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Clarke PG: Developmental cell death:
morphological diversity and multiple mechanisms. Anat Embryol.
181:195–213. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brown JM and Attardi LD: The role of
apoptosis in cancer development and treatment response. Nat Rev
Cancer. 5:231–237. 2005.PubMed/NCBI
|
|
7
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Levine B and Klionsky DJ: Development by
self-digestion: molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: a double-edged sword. Science. 306:990–995.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gozuacik D and Kimchi A: Autophagy as a
cell death and tumor suppressor mechanism. Oncogene. 23:2891–2906.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Debnath J, Baehrecke EH and Kroemer G:
Does autophagy contribute to cell death? Autophagy. 1:66–74. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ogier-Denis E and Codogno P: Autophagy: a
barrier or an adaptive response to cancer. Biochim Biophys Acta.
1603:113–128. 2003.PubMed/NCBI
|
|
14
|
Corcelle E, Nebout M, Bekri S, et al:
Disruption of autophagy at the maturation step by the carcinogen
lindane is associated with the sustained mitogen-activated protein
kinase/extracellular signal-regulated kinase activity. Cancer Res.
66:6861–6870. 2006. View Article : Google Scholar
|
|
15
|
Otsuka H and Moscowitz M: Differences in
the rates of protein degradation in untransformed and transformed
cell lines. Exp Cell Res. 112:127–135. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kirkegaad K, Taylor MP and Jacson WT:
Cellular autophagy: surrender, avoidance and subversion by
microorganisms. Nat Rev Microbiol. 2:301–314. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
List PH and Hörhammer L: Hager’s Hand book
for Pharmaceutical Practice. Springer-Verlag; Berlin: 1970
|
|
18
|
Naranjo C: Psychotropic properties of the
harmala alkaloids. Ethnopharmacological Search for Psychoactive
Drugs. Efron DH, Holmestedt B and Kline NS: U.S. Public Health
Service; New York: pp. 385–392. 1967
|
|
19
|
Nadikarni KM: Indian Material Medica. 1.
Popular Pakistan Limited; Bombay: pp. 927–929. 1976
|
|
20
|
Dymock W, Warden CJ and Hooper D:
Pharmacopia Indica. 1. Harmad National Foundation of Pakistan; pp.
252–253. 1976
|
|
21
|
Khan SI, Abourashed EA, Khan IA and Walker
LA: Transport of human alkaloids across Caco-2 cell monolayers.
Chem Pharm Bull. 52:394–397. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Airaksinen MM and Kari I: β-carbolines,
psychoactive compounds in the mammalian body. Part I: occurrence,
origin and metabolism. Med Biol. 59:21–34. 1981.
|
|
23
|
Beck O and Faull KF: Concentrations of the
enantiomers of 5-hydroxymethylptoline in mammalian urine:
Implications for in vivo biosynthesis. Biochem Pharmacol.
65:97–106. 1986.PubMed/NCBI
|
|
24
|
Sobhani AM, Ebrahimi SA and Mahmoudian MJ:
An in vitro evaluation of human DNA topoisomerase I inhibition by
Peganum harmala L. seed extract and its beta-carboline
alkaloids. Pharm Pharm Sci. 5:19–23. 2002.PubMed/NCBI
|
|
25
|
Lamchouri F, Setaff A, Cherrah Y, et al:
Antitumor principles from Peganum harmala seeds. Therapie.
54:753–758. 1999.PubMed/NCBI
|
|
26
|
Kuo PC, Shi LS, Damu AG, et al: Cytotoxic
and antimalarial beta-carboline alkaloids from the roots of
Eurycoma longifolia. J Nat Prod. 66:1324–1327. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Abe A and Yamada H: Harmol induces
apoptosis by caspase-8 activation independently of Fas/Fas ligand
interaction in human lung carcinoma H596 cells. Anticancer Drugs.
20:373–381. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Abe A, Yamada H, Moriya S and Miyazawa K:
The β-carboline alkaloid harmol induces cell death via autophagy
but not apoptosis in human non-small cell lung cancer A549 cells.
Biol Pharm Bull. 34:1264–1272. 2011.
|
|
29
|
Oliver FJ, de la Rubia G, Rolli V,
Ruiz-Ruiz MC, de Murcia G and Murcia JM: Importance of
poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson
from an uncleavable mutant. J Biol Chem. 273:33533–33539. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Biederbick A, Kern HF and Elsässer HP:
Monodansylcadaverine (MDC) is a specific in vivo marker for
autophagic vacuoles. Eur J Cell Biol. 66:3–14. 1995.PubMed/NCBI
|
|
31
|
Kabeya Y, Mizusima N, Ueno T, et al: LC3,
a mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Seglen PO and Gordon PB: 3-Methyladenine:
specific inhibitor of autophagic/lysosomal protein degradation in
isolated rat hepatocytes. Proc Natl Acad Sci USA. 79:1889–1892.
1982. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kawai A, Uchiyama H, Takano S, Nakamura S
and Ohkuma S: Autophagosome-lysosome fusion depends on the pH in
acidic compartments in CHO cells. Autophagy. 3:154–157. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rubinsztein DC, Gestwicki JE, Murphy LO
and Klionsky DJ: Potential therapeutic applications of autophagy.
Nat Rev Drug Discov. 6:304–312. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fu L, Kim AY, Wang X, et al: Perifosine
inhibits mammalian target of rapamycin signaling through
facilitating degradation of major components in the mTOR axis and
induces autophagy. Cancer Res. 69:8967–8976. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Viola G, Bortolozzi R, Hamel E, et al:
MG-2477, a new tubulin inhibitor, induces autophagy through
inhibition of the Akt/mTOR pathway and delayed apoptosis in A549
cells. Biochem Pharmacol. 8:16–26. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hung JY, Hsu YL, Li CT, et al: 6-Shogaol,
an active constituent of dietary ginger, induces autophagy by
inhibiting the AKT/mTOR pathway in human non-small cell lung cancer
A549 cells. J Agric Food Chem. 57:9809–9816. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Takeuchi H, Kanzawa T, Kondo Y and Kondo
S: Inhibition of platelet-derived growth factor signaling induces
autophagy in malignant glioma cells. Br J Cancer. 90:1069–1075.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar
|
|
40
|
Gross A, Yin XM, Wang K, et al: Caspase
cleaved BID targets mitochondria and is required for cytochrome c
release, while BCL-XL prevents this release but not tumor necrosis
factor-R1/Fas death. J Biol Chem. 274:1156–1163. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zha J, Weiler S, Oh KJ, Wei MC and
Korsmeyer SJ: Posttranslational N-myristoylation of BID as a
molecular switch for targeting mitochondria and apoptosis. Science.
290:1761–1765. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shao RG, Cao CX, Nieves-Neira W,
Dimanche-Boitrel MT, Solary E and Pommier Y: Activation of the Fas
pathway independently of Fas ligand during apoptosis induced by
camptothecin in p53 mutant human colon carcinoma cells. Oncogene.
20:1852–1859. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Deveraux Q and Reed JC: IAP family
proteins: suppressors of apoptosis. Genes Dev. 13:239–252. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li F, Ambrosini G, Chu EY, et al: Control
of apoptosis and mitotic spindle checkpoint by survivin. Nature.
396:580–584. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
45
|
Monzo M, Rosell R, Felip E, et al: A novel
anti-apoptosis gene: re-expression of surviving messenger RNA as a
prognosis marker in non-small-cell lung cancers. J Clin Oncol.
17:2100–2104. 1999.PubMed/NCBI
|
|
46
|
Adida C, Haioun C, Gaulard P, et al:
Prognostic significance of surviving expression in diffuse large
B-cell lymphomas. Blood. 96:1921–1925. 2000.PubMed/NCBI
|
|
47
|
Kato J, Kuwabara Y, Mitani M, et al:
Expression of surviving in esophageal cancer: correlation with the
prognosis and response to chemotherapy. Int J Cancer. 95:92–95.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Altieli DC: Survivin, cancer networks and
pathway-directed drug discovery. Nat Rev Cancer. 8:61–70. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Levine B and Yuan J: Autophagy in cell
death: an innocent convict? J Clin Invest. 115:2679–2688. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Guertin DA and Sabatini DM: Defining the
role of mTOR in cancer. Cancer Cell. 12:9–22. 2007. View Article : Google Scholar
|
|
51
|
Petiot A, Pattingre S, Arico S, Meley D
and Codongo P: Diversity of signaling controls of macroautophagy in
mammalian cells. Cell Struct Funct. 27:431–441. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Pattingre S, Bauvy C and Codongo P: Amino
acids interfere with the ERK1/2-dependent control of macroautophagy
by controlling the activation of Raf-1 in human colon cancer HT-29
cells. J Biol Chem. 278:16667–16674. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Meley D, Bauvy C, Houben-Weerts JH, et al:
AMP-activated protein kinase and the regulation of autophagic
proteolysis. J Biol Chem. 281:34870–34879. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Herrero-Martín G, Høyer-Hansen M,
García-García C, et al: TAK1 activates AMPK-dependent
cytoprotective autophagy in TRAIL-treated epithelial cells. EMBO J.
28:677–685. 2009.PubMed/NCBI
|
|
55
|
Djavaheri-Mergny M, Maiuri MC and Kroemer
G: Cross talk between apoptosis and autophagy by caspase-mediated
cleavage of Beclin 1. Oncogene. 29:1717–1719. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhou F, Yang Y and Xing D: Bcl-2 and
Bcl-xL play important roles in the crosstalk between autophagy and
apoptosis. FEBS J. 278:403–413. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lin CC, Lee CW, Chu TH, et al:
Transactivation of Src, PDGF receptor, and Akt is involved in
IL-1beta-induced ICAM-1 expression in A549 cells. J Cell Physiol.
211:771–780. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Russo P, Catassi A, Cesario A and Servent
D: Development of novel therapeutic strategies for lung cancer:
targeting the cholinergic system. Curr Med Chem. 13:3493–3512.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Akca H, Tani M, Hishida T, Matsumoto S and
Yokota J: Activation of the AKT and STAT3 pathways and prolonged
survival by a mutant EGFR in human lung cancer cells. Lung Cancer.
54:25–33. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kishimoto H, Ohteki T, Yajima N, et al:
The Pten/PI3K pathway governs the homeostasis of Valpha14iNKT
cells. Blood. 109:3316–3324. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Takeuchi H, Kondo Y, Fujiwara K, et al:
Synergistic augmentation of rapamycin-induced autophagy in
malignant glioma cells by phosphatidylinositol 3-kinase/protein
kinase B inhibitors. Cancer Res. 65:3336–3346. 2005.PubMed/NCBI
|
|
62
|
Rommelspacher H, Nanz C, Borbe HO, Fehske
KJ, Müller WE and Wollert U: 1-Methyl-beta-carboline (harmane), a
potent endogenous inhibitor of benzodiazepine receptor binding.
Naunyn Schmiedebergs Arch Pharmacol. 314:97–100. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ho BT: Pharmacological and biochemical
studies with beta-carboline analogs. Curr Dev Psychopharmacol.
4:151–177. 1977.PubMed/NCBI
|
|
64
|
Poirier LJ, Sourkes TL, Bouvier G, Boucher
R and Carabin S: Striatal amines, experimental tremor and the
effect of harmaline in the monkey. Brain. 89:37–52. 1966.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Bruinvels J and Sourkes TL: Influence of
drugs on the temperature-lowering effect of harmaline. Eur J
Pharmacol. 4:31–39. 1969. View Article : Google Scholar
|
|
66
|
Zetler G, Back G and Iven H:
Pharmacokinetics in the rat of the hallucinogenic alkaloids harmine
and harmaline. Naunyn Shemiedebergs Arch Pharmacol. 285:273–292.
1974. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Grella B, Dukat M, Young R, et al:
Investigation of hallucinogenic and related beta-carbolines. Drug
Alcohol Depend. 50:99–107. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Udenfriend S, Wiycop B, Redfield BG and
Weissbach H: Studies with reversible inhibitors of monoamine
oxidase: harmaline and related compounds. Biochem Pharmacol.
1:160–165. 1958. View Article : Google Scholar
|
|
69
|
McIsaac WM and Estevez V: Structure-action
relationship of beta-carbolines as monoamine oxidase inhibitors.
Biochem Pharmacol. 15:1625–1627. 1966. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Loew GH, Nienow J, Lawson JA, Toll L and
Uyeno ET: Theoretical structure-activity studies of beta-carboline
analogs. Requirements for benzodiazepine receptor affinity and
antagonist activity. Mol Pharmacol. 28:17–31. 1985.
|
|
71
|
Kim H, Sablin SO and Ramsay RR: Inhibition
of monoamine oxidase A by β-carboline derivatives. Arch Biochem
Biophys. 337:137–142. 1997.
|
|
72
|
Gunn JA: Relations between chemical
constitution, pharmacological actions, and therapeutic uses in the
harmine group of alkaloids. Arch Int Pharmacodyn. 50:379–396.
1935.
|
|
73
|
Zetler G, Singbart G and Schlosser L:
Cerebral pharmacokinetics of tremor-producing harmala and iboga
alkaloids. Pharmacology. 7:237–248. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Yu AM, Idle JR, Krausz KW, Küpfer A and
Gonzalez FJ: Contribution of individual cytochrome P450 isozymes to
the O-demethylation of the psychotropic beta-carboline alkaloids
harmaline and harmine. J Pharmacol Exp Ther. 305:315–322. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Slotkin TA, DiStefano V and Au WY: Blood
levels and urinary excretion of harmine and its metabolites in man
and rats. J Pharmacol Exp Ther. 173:26–30. 1970.PubMed/NCBI
|