Introduction
Breast cancer is a common type of cancer affecting
Taiwanese women and its incidence and mortality rates increase year
by year (1). To eradicate cancer
and its recurrence, a multifaceted therapeutic approach has been
employed and includes targeting pathways that promote or sustain
cancer cell growth and invasion (2,3). These
therapeutic strategies are successful in preventing tumor
progression with a minimal effect on normal cells (4). Several pathways are potential targets
for cancer treatments, including pathways that induce cell
apoptosis, prevent cell proliferation, modulate cellular re-dox
states and detoxify carcinogens. These therapeutic targets are
considered clinically useful (5).
New practical strategies have been used for cancer
treatment, including the inhibition of cell cycle of tumor cells
and induction of apoptosis in tumor cells. For example, through
phosphorylation of target proteins to interfere with the
interactions of cyclins with CDKs, it is possible to regulate the
cell cycle and to stop the growth of progressive tumors (6). Therefore, it provides an opportunity
for those chemicals capable of interfering with the abnormal cell
proliferation to be used as chemotherapeutic agents. Similarly,
intervention in the programmed cell death, known as apoptosis, is
another possible strategy for controlling cell growth in cancer
therapies (7). Two distinct
apoptotic pathways have been identified: the intrinsic apoptotic
pathway (also known as the p53-mitochondrial pathway) and the
extrinsic pathway (activated by ‘death receptors’ and their
corresponding ligands) (8). Unlike
the extrinsic pathway, whose apoptotic pathway begins outside the
cell through the activation of specific pro-apoptotic receptors,
the intrinsic apoptotic pathway involves the regulation of
anti-apoptotic (Bcl-2) and pro-apoptotic (Bax and Bad) proteins of
the Bcl-2 family to trigger the release of cytochrome c from
the mitochondria, subsequently culminating in caspase-9-dependent
apoptosis (9). The induction of
apoptosis in tumor cells has become one of the most widely-used
strategies of cancer therapy.
Targeting the angiogenesis of tumor cells is another
common strategy for cancer treatment (10). Several pro-angiogenic factors,
including vascular endothelial growth factor (VEGF), basic
fibroblast growth factor (bFGF), placental growth factor, and
platelet-derived endothelial growth factor, are known factors of
tumor angiogenesis (11). These
pro-angiogenic factors activate quiescent endothelial cells and
promote their migration into the tumor (11). Therefore, preventing angiogenesis
and consequent metastasis in tumors has become an attractive
strategy in tumor treatment (12).
In 2004, Miller reported that certain chemotherapeutic agents
routinely used for breast cancer treatments have anti-angiogenic
activity (12). Ma and Waxman also
conceived the potential benefits of targeting angiogenesis in
chemotherapy (11). Taken together,
an agent combined with both anti-proliferative and anti-angiogenic
activities would make an ideal drug for cancer therapy.
The carbazole alkaloids are widely distributed
compounds in nature and have certain physiological roles in plants
and microorganisms. Several structurally different carbazole
alkaloids from either natural or synthesized sources have shown
diverse pharmacological effects (13–16).
Some of these carbazoles and their derivatives exhibit cytotoxic
activities related to the inhibition of DNA-dependent enzymes such
as topoisomerase I/II and telomerase (13–16).
In addition, other reported cytotoxic effects of carbazoles include
anti-proliferation (17,18), anti-angiogenesis (19), anti-HIV-activities (20,21)
and anti-estrogenic activities (22). Previous studies have shown that
LCY-2-CHO, an asymmetric substituted carbazole derivative, showed
evident apoptosis effects in three leukemia cells, but had only
slight effects on adherent cells including PC3 and MCF-7 (18). Another asymmetric substituted
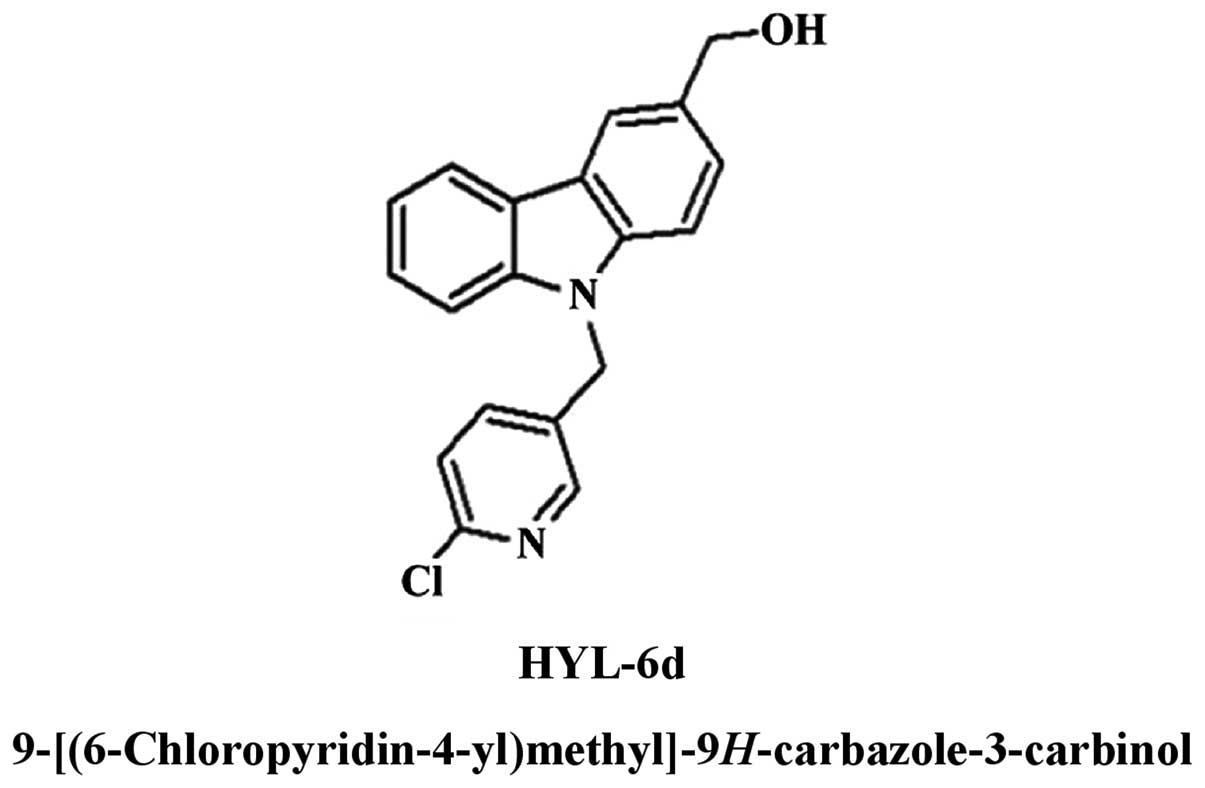
9H-carbazole derivative, 9-[(6-chloropyridin-4-yl)
methyl]-9H-carbazole-3-carbinol (HYL-6d) (Fig. 1), synthesized by the laboratory of
Dr L.J. Huang, has demonstrated a greater cytotoxic effect on six
adherent human cancer cell lines, particularly on MCF-7 cells
(Fig. 2). The aim of the present
study was to determine the underlying mechanisms of the in
vitro cytotoxic effects of HYL-6d. This study analyzed
different cell subsets of cell cycle in HYL-6d-treated human breast
cancer MCF-7 cells, and determined whether HYL-6d induces apoptosis
through the molecular parameters related to the Bcl-2 family, p53
and caspase pathways. Furthermore, we found that HYL-6d could
suppress human umbilical vein endothelial cell (HUVEC)
proliferation, migration, and tube formation in vitro.
Materials and methods
Reagents
HYL-6d (purity >95%), was synthesized and
dissolved in dimethylsulfoxide (DMSO) (Sigma, St. Louis, MO, USA).
Further dilutions were made immediately prior to each experiment
with a final DMSO concentration of <0.1% in culture media.
Cell lines and cell cultures
Five human cancer cell lines, including breast
carcinoma (MCF-7 and MDA-MB-231), cervical carcinoma (CaSki and
HeLa) and ovarian carcinoma (SKOV3) were obtained from the
Bioresource Collection and Research Centre, Taiwan (BCRC; formerly
the Culture Collection and Research Centre). Human ovarian
carcinoma 2774 and HUVECs were provided by China Medical
University, Taichung, Taiwan. MCF-7, MDA-MB-231 and HeLa cells were
routinely cultured in Dulbecco’s modified Eagle’s medium (DMEM;
Gibco-BRL, Grand Island, NY, USA). CaSki cells were maintained in
RPMI-1640 medium (Gibco-BRL). SKOV3 and 2774 cells were maintained
in DMEM/F12 (Gibco-BRL), supplemented with 10% fetal bovine serum
(FBS; Kibbutz Beit Haemek, Israel), 5% glutamine, 100 U/ml
penicillin, and 100 μg/ml streptomycin (Gibco-BRL). HUVECs were
cultured in M199 medium (Sigma) containing 20% FBS and 15%
endothelial cell growth supplements. Cells were incubated at 37°C
in a humidified atmosphere containing 5% CO2. Medium was
changed every 2 days, and cells were passaged after treatment with
a 0.05% trypsin/0.02% EDTA solution. Experiments were conducted on
HUVECs that had gone through two to five passages.
Cell viability assay
For cell viability assays, cells were seeded at a
density of 5×104 cells/well in 12-well culture plates.
After 24 h, cells were treated with various concentrations of
HYL-2d-6d (from 1 to 100 μM in 1 μl DMSO) for the indicated times.
Cell viability was determined using the
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
(Sigma) assay and the trypan blue exclusion assay.
Cell cycle analysis
MCF-7 cells were treated with or without 30 μM
HYL-6d and harvested at different times. After washing with
phosphate-buffered saline (PBS), the cells were fixed in 70%
ethanol/PBS for 30 min on ice. Approximately 4×105 cells
were centrifuged and the cell pellets were re-suspended in PBS and
further treated with RNase (DNase free, 100 μg/ml, final
concentration in PBS) and propidium iodide (40 μg/ml, final
concentration in PBS) at room temperature for 30 min. The cells
were centrifuged and the cell pellets were re-suspended in PBS. The
cell suspension was passed through a 19-gauge needle and kept on
ice until analysis. The number of cells in different phases of the
cell cycle was analyzed using a FACSCalibur flow cytometer
(Becton-Dickinson, San Jose, CA, USA). Distributions of different
cell cycle phases were determined using CellFIT software
(Becton-Dickinson).
Apoptosis
Cell apoptosis was determined using TUNEL/DAPI
double stains and DNA fragmentation. HYL-6d-treated and untreated
cells were harvested at 36 h, after washing twice with PBS. These
cells were first fixed with 2% formaldehyde for 30 min and then
permeabilized by treating with 0.1% Triton X-100 in PBS for 10 min
at room temperature. After washing with PBS, the TUNEL assay was
performed according to the manufacturer’s instructions (Boehringer
Mannheim, Germany). Briefly, cells were incubated in TUNEL reaction
buffer in a 37˚C humidified chamber for 60 min in the dark, then
rinsed twice with PBS and incubated with DAPI (1 μg/ml) at 37°C for
30 min. Cells were visualized using a fluorescence microscope.
TUNEL positive cells were considered apoptotic.
For DNA fragmentation assay, MCF-7 cells were grown
in the presence or absence of 30 μM HYL-6d for 36 h, and treated
cells were harvested and lysed in extraction buffer (50 mM Tris pH
7.5, 10 mM EDTA, and 0.3% Triton X-100) for 30 min on ice. RNAs
were removed by treating with RNase (100 μg/ml) for 30 min at 55°C.
Cell lysates were subsequently treated with proteinase K (400
μg/ml) for 1 h at 55°C. The supernatants were then prepared by
centrifugation, and the soluble proteins were extracted by
phenol/chloroform. DNA was ethanol-precipitated and subjected to
electrophoresis on a 2% agarose gel.
Western blot analysis
HYL-6d-treated cells were also used for western blot
analysis. Following incubation, 5×104 cells were lysed
in a 100 μl modified protein lysis buffer (50 mM Tris-HCl pH 7.4,
150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 5% 2-mercaptoethanol, 1% Triton
X-100, 0.25% sodium-deoxycholate, 5 μg/ml leupeptin, 5 μg/ml
aprotinin, 10 μg/ml soybean trypsin inhibitor, and 0.2 mM
phenylmethylsulfonylfluoride). Protein concentrations were measured
using the Bradford method (Bio-Rad Laboratories, Hercules, CA,
USA). Equal amounts of sample lysates were separated by SDS-PAGE on
an 8–12% polyacrylamide gel, and the gel was transferred to a PVDF
membrane (NEN Life Science Products Inc., Boston, MA, USA). The
proteins were detected using mouse and rabbit specific primary
antibodies (BD Biosciences Pharmingen, San Diego, CA, USA; Santa
Cruz Biotechnology, Santa Cruz, CA, USA) followed by RDye 800
anti-mouse Molecular Probes (Rockland Immunochemicals Inc.,
Gilbertsville, PA, USA), and Alexa Fluor® 700
anti-rabbit (Molecular Probes, Eugene, OR, USA) as fluorescent
secondary antibodies. The resulting membrane was imaged using the
Odyssey Infrared Imaging System (LI-COR Biosciences, St. Lincoln,
NE, USA) and analyzed with their software program as specified in
the Odyssey software manual. An equal loading in the lanes was
evaluated by probing with an anti-β-actin antibody (Santa Cruz
Biotechnology).
HUVEC proliferation assay
DNA synthesis in proliferating cells was determined
by measuring BrdU incorporation with the commercial Cell
Proliferation ELISA system (Chemicon). Confluent HUVECs were
trypsinized, suspended in M199 medium supplemented with 20% FBS,
and seeded at 2×104 cells/well in 96-well plates. After
24 h, cells were washed twice with PBS and starved with 2% FBS-M199
medium for 24 h. These cells were then incubated with or without
the indicated reagents and growth factors (such as VEGF and bFGF at
10 ng/ml). After 24 h, cells were analyzed using the BrdU Cell
Proliferation kit and quantified with an ELISA reader at
OD450/540. Culture medium without any additions was used
as a control for non-specific binding.
Migration assay
To measure the ability of HUVECs to migrate, cells
were seeded into a Boyden chamber with 8 μm pore polycarbonate
filters (Costar, Cambridge, MA, USA). Briefly, HUVECs were
pretreated with various concentrations of HYL-6d. After 24 h,
HUVECs were detached by trypsin and resuspended in a serum-free
medium. VEGF or bFGF was diluted to 10 ng/ml in 100 μl M199/2% FBS
and added to the lower well chamber as a chemoattractant, and
HUVECs were seeded on the upper chamber at a density of
1×105 cells/well in 150 μl of serum-free medium. The
chamber was incubated for 4 h at 37°C. Following incubation, HUVECs
in the upper surface of the membrane were carefully removed with a
cotton swab and cells that migrated to the lower surface of the
membrane were fixed with methanol and stained with 5% Giemsa
solution. Migratory cells on the lower surface of the membrane
filter were counted in six randomly chosen high-power fields
(magnification, ×400). Cell migration was calculated as the
difference between the number of migrated cells in the
HYL-6d-treated samples and the number of migrated cells in the
control samples. Each experiment was carried out in triplicate.
Matrigel capillary tube formation
assay
A 24-well culture plate was coated with 250 μl of
Matrigel (Becton-Dickinson, Bedford, MA, USA), and incubated for 1
h to allow solidification. HUVECs (2×105 cells/ml) were
treated with or without HYL-6d (3–30 μM) and VEGF or bFGF (10
ng/ml). After 24 h, trypsin-harvested HUVECs were suspended in 500
μl M199/2% FBS. The suspension was placed onto the surface of the
Matrigel and incubated for 24 h. The cell morphology was evaluated
using a phase-contrast microscope, and cells were photographed. The
lengths and areas of tube-structured cells were then quantified
using a MetaFluor Imaging System (Meta Imaging Series; Molecular
Devices Corp., Silicon Valley, CA, USA).
Statistical analysis
All data presented in this study (as the means ± SD
of nine replicates) are from three separate experiments. Figures
are representative of at least three independent experiments with
similar patterns. The Student’s t-test was used for statistical
analysis between the HYL-6d-treated group and the control
group.
Results
Cytotoxicity of HYL-6d in various human
cancer cell lines
To assess the cytotoxicity of carbazole derivatives,
six human cancer cell lines from various organs were used,
including breast carcinoma (MCF-7 and MDA-MB-231), cervical
carcinoma (CaSki and HeLa) and ovarian carcinoma (SKOV3 and 2774).
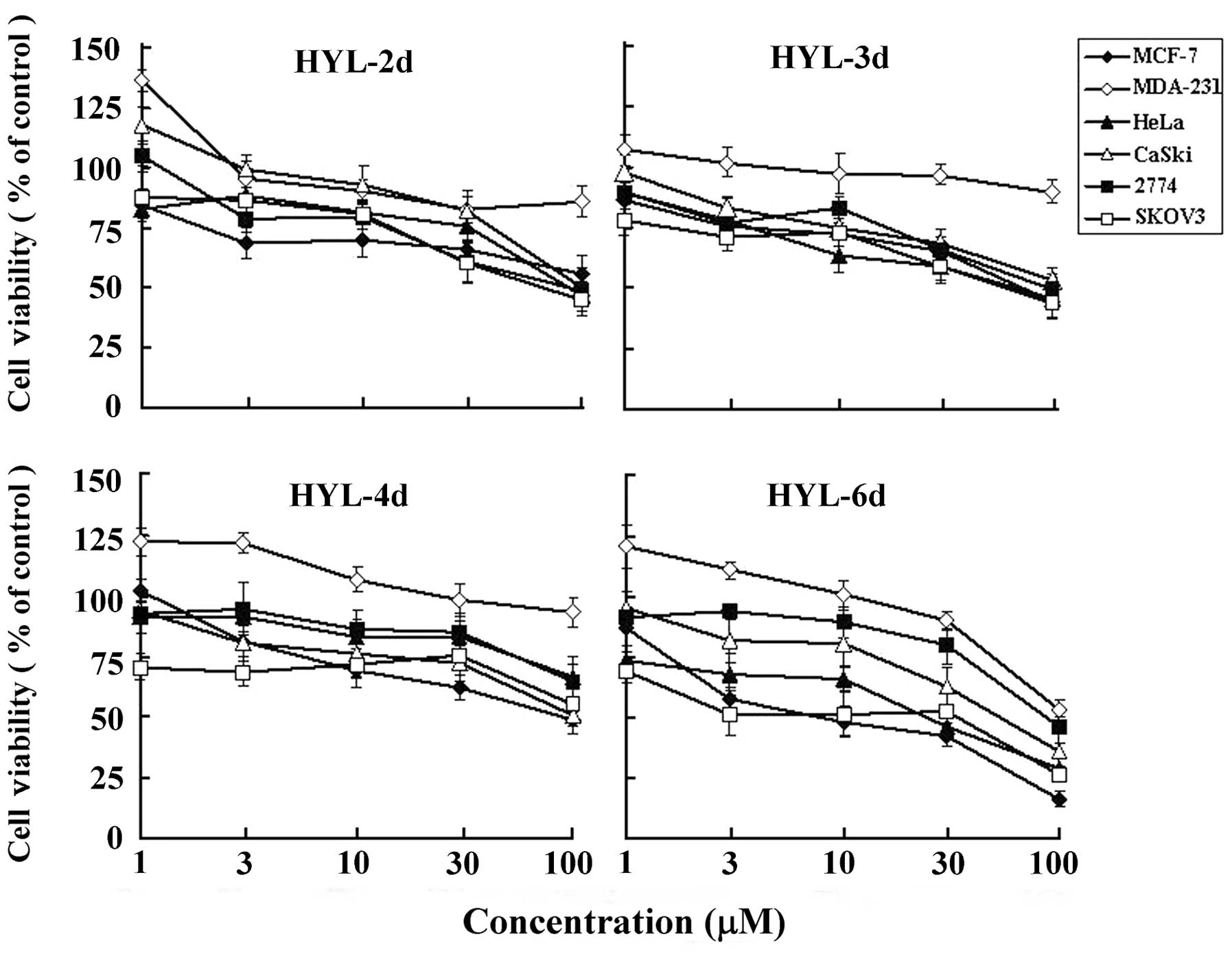
These cell lines were treated with HYL-2d, 3d, 4d and 6d at various
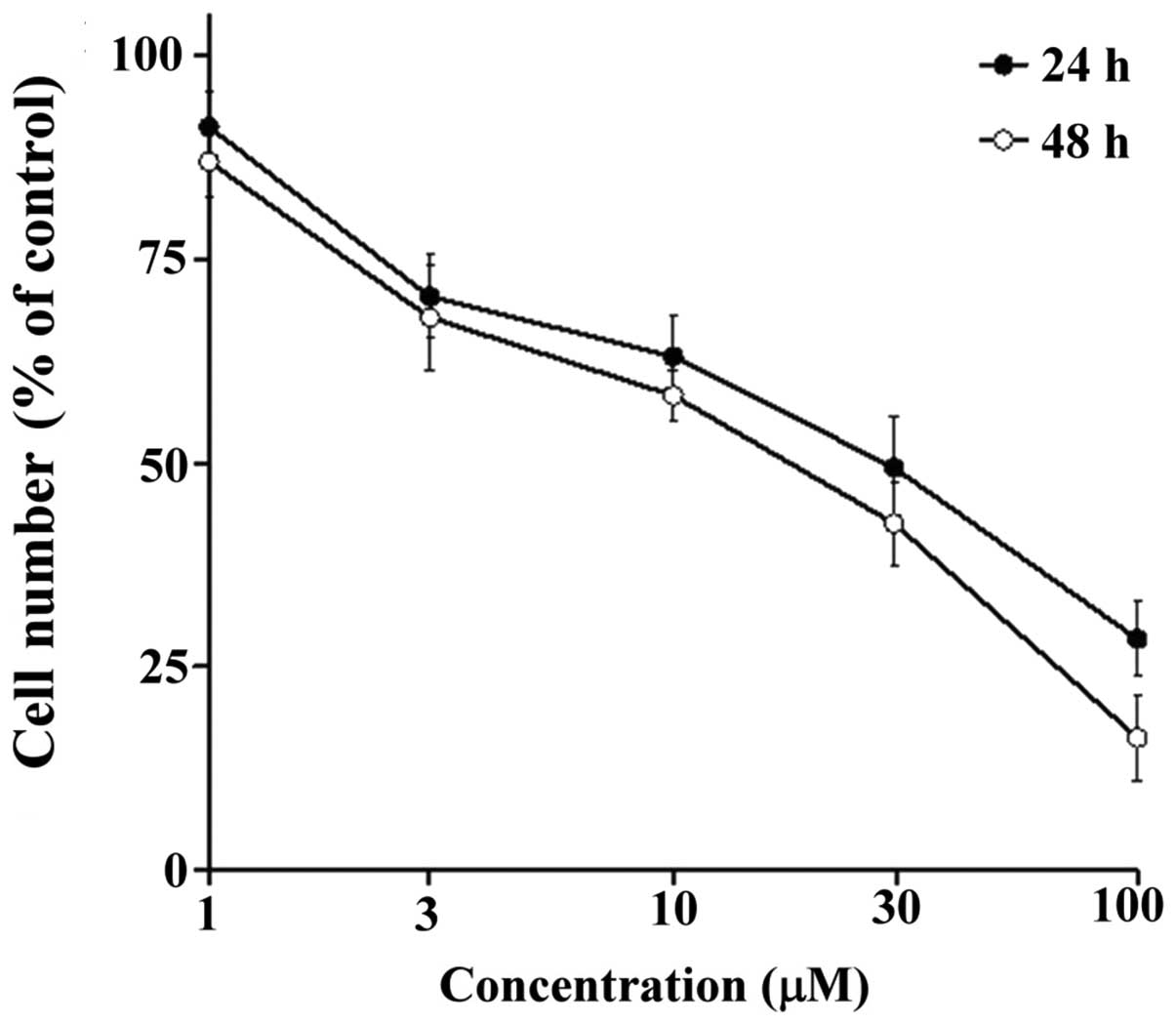
concentrations (1–100 μM) for 48 h. As shown in Figs. 2 and 3, HYL-6d exhibited different degrees of
dose- and time-dependent cytotoxic activities. HYL-6d appeared to
be the most potent to inhibit MCF-7 breast cancer cells with an
IC50 of 20–30 μM at 48 h.
Effects of HYL-6d on cell cycle
progression and regulatory proteins
To determine whether HYL-6d-induced cytotoxicity was
associated with a disturbance of cell cycle regulation, the
HYL-6d-treated MCF-7 cells were analyzed by flow cytometry. As
illustrated in Table I, treatment
of MCF-7 cells with HYL-6d resulted in a significant increase in
the percentage of cells in the S phase, accompanied by a decrease
in the percentage of cells in the G2/M phase.
HYL-6d-induced arrest in the S phase peaked at 48 h, with ~47.5% of
the cells in the S phase arrested, compared to 31.8% in
controls.
 | Table ICell cycle analysis of HYL-6d-treated
MCF-7 cells. |
Table I
Cell cycle analysis of HYL-6d-treated
MCF-7 cells.
| | Treatment (% of the
cell no.) |
|---|
| |
|
|---|
| Time (h) | Cell cycle | Control | HYL-6d |
|---|
| 0 | G1 | 57.5±2.0 | |
| S | 34.0±1.5 | |
|
G2/M | 8.5±1.2 | |
| Apoptosis | 0.5±0.6 | |
| 12 | G1 | 59.0±2.0 | 58.5±0.9 |
| S | 28.4±2.7 | 39.4±1.2a |
|
G2/M | 12.6±1.3 | 2.1±1.1b |
| Apoptosis | 0.4±0.4 | 0.2±0.2 |
| 24 | G1 | 61.8±4.0 | 58.9±2.1 |
| S | 30.4±3.9 | 38.9±2.4 |
|
G2/M | 7.8±1.9 | 2.3±1.5a |
| Apoptosis | 0.2±0.2 | 1.2±1.2 |
| 36 | G1 | 57.9±3.8 | 57.5±2.2 |
| S | 33.5±2.4 | 39.5±3.0 |
|
G2/M | 8.6±1.8 | 3.0±2.0a |
| Apoptosis | 0.3±0.4 | 1.7±1.6 |
| 48 | G1 | 59.7±3.1 | 51.8±3.6 |
| S | 31.8±2.8 | 47.5±4.1b |
|
G2/M | 8.5±1.0 | 0.6±0.7 |
| Apoptosis | 0.7±0.9 | 25.1±5.3c |
Based on the fact that HYL-6d induced S phase arrest
in MCF-7 cells, we assessed the effect of HYL-6d in cell cycle
regulatory modules that play important roles in the S phase. The
cyclin A-CDK2 complex is the primary regulator of the S phase
progression and its expression serves as an index for the S phase
arrest (23). Cyclin D1 is one of
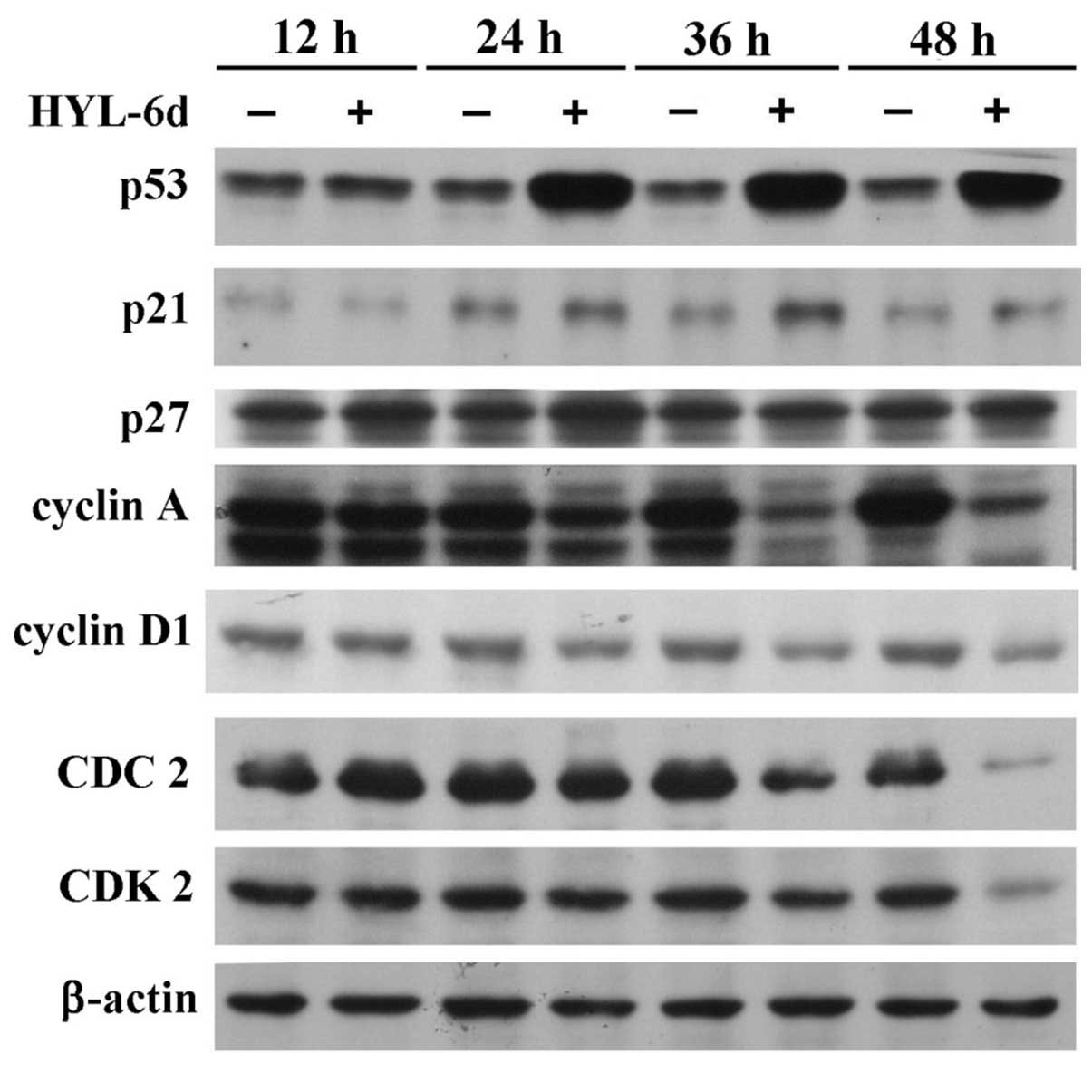
the most overexpressed oncogenes in breast cancer (6). As shown in Fig. 4, after 12 h of HYL-6d treatment in
MCF-7 cells, an increase of p53 protein up to 48 h was observed. By
contrast, cyclin D1, A and CDK2 levels were markedly decreased in
HYL-6d-treated MCF-7 cells. These results indicate that the
increase of p53, and the decrease of cyclin D1, A and CDK2, might
be involved in the S phase arrest caused by HYL-6d.
Effects of HYL-6d on cell apoptosis
HYL-6d-treated cells also accumulated in the
sub-G1 phase. Accumulation in the sub-G1 was
time-dependent and indicative of apoptosis, consistent with the
induction of cell death. To determine if HYL-6d-induced cell death
was mediated by apoptosis, the nuclear morphology of dying cells
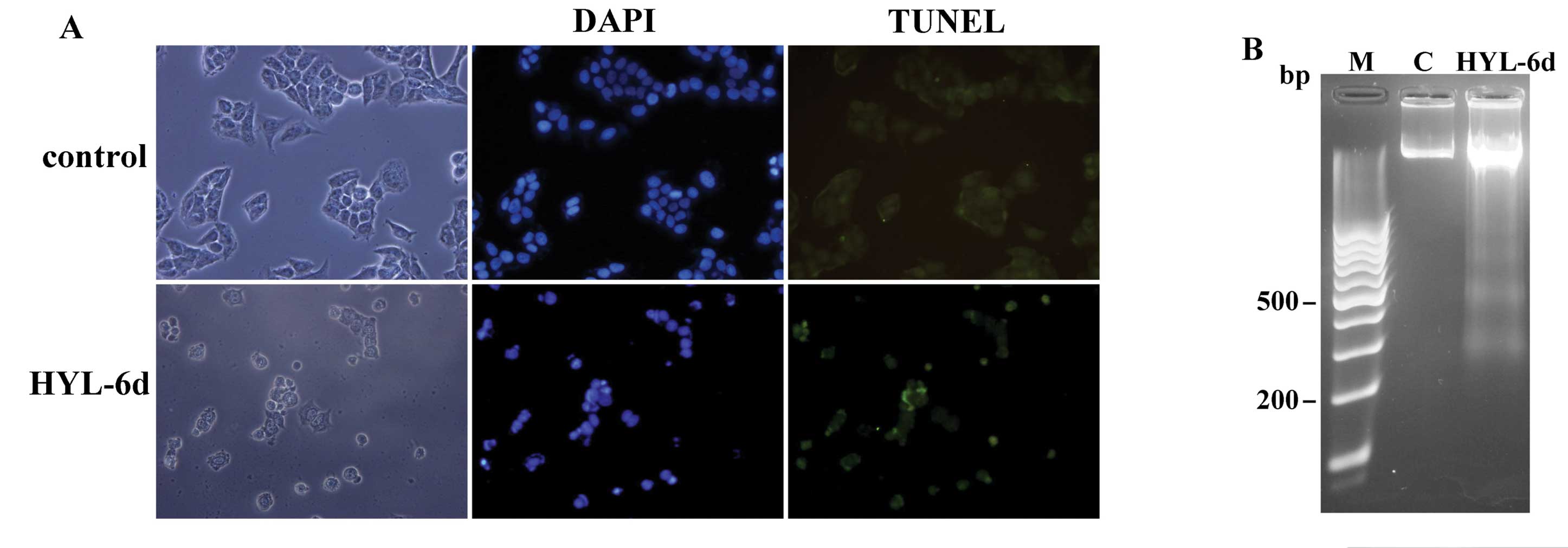
was examined using the fluorescent DNA-binding agent DAPI. As shown
in Fig. 5A, HYL-6d-treated MCF-7
cells displayed typical apoptotic morphological features, including
condensed and fragmented nuclei. Homogeneous nuclear chromatin was
evident in control cells (treated with 0.1% DMSO). To confirm the
morphological findings, in situ TUNEL assay and DNA
fragmentation analysis were performed. As shown in Fig. 5A, TUNEL positive cells (apoptotic
cells) and internucleosomal DNA fragments (Fig. 5B) were observed after HYL-6d
treatment. However, neither apoptotic cells nor DNA fragmentation
were observed in control cell lines within the 36-h culture
period.
Effects of HYL-6d on Bcl-2 family
proteins and caspase-9
Accumulating evidence indicates that Bcl-2-related
proteins play an important role in regulating apoptosis (24). To determine whether or not the
expression of these cell death-associated molecules are crucial for
HYL-6d-mediated apoptosis, MCF-7 cells were cultured in the
presence or absence of HYL-6d for 12, 24, 36 and 48 h. In these
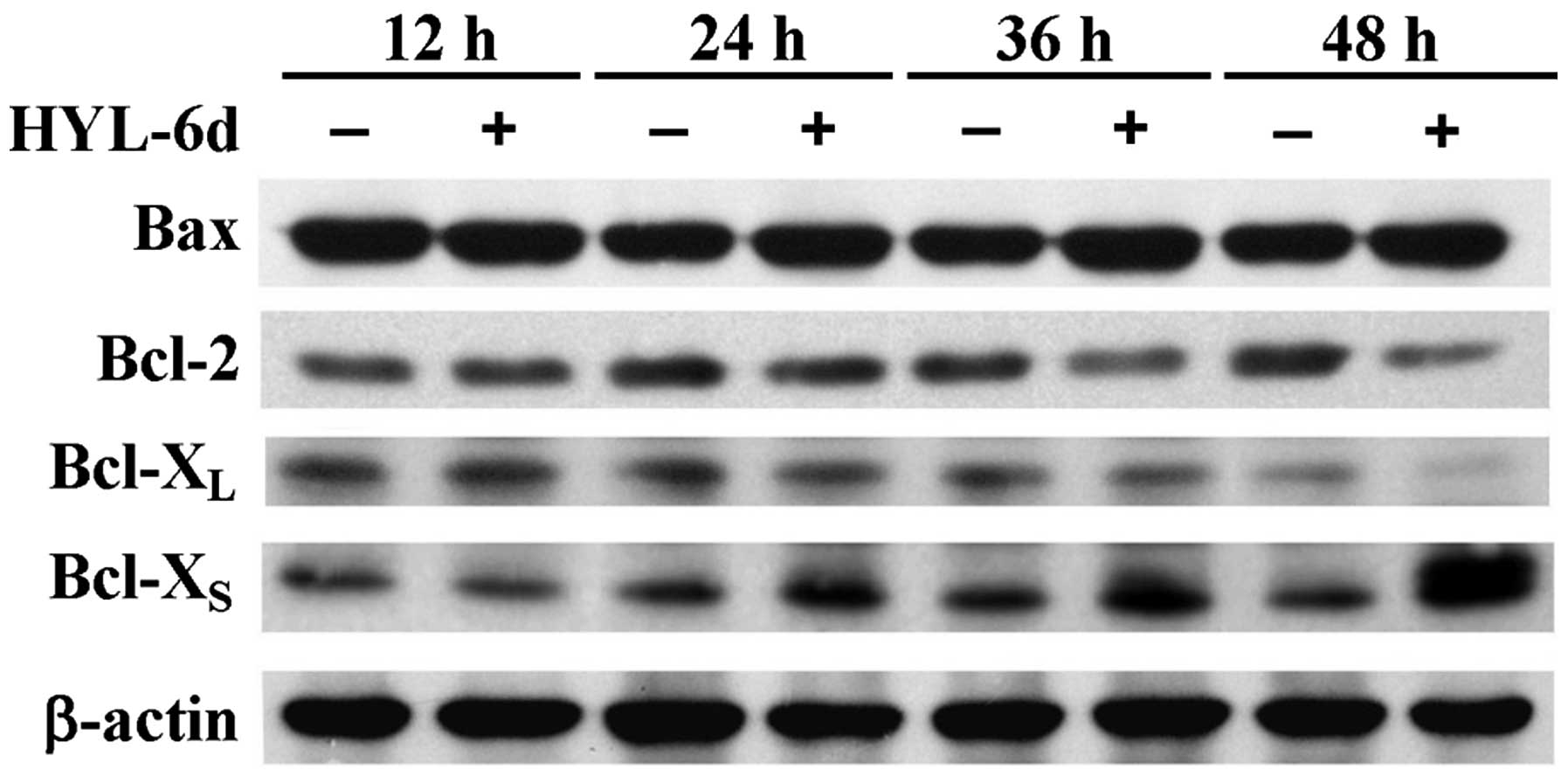
assays, significant increases of Bcl-XS and Bax protein
levels following a 48-h treatment of MCF-7 cells with HYL-6d were
found (Fig. 6). However, a decrease
of Bcl-2 protein was also observed after 48 h of HYL-6d treatment.
Bcl-XL protein levels remained the same in these
experiments (Fig. 6). These results
suggest that HYL-6d-induced apoptosis might be mediated through the
downregulation of Bcl-2 anti-apoptotic proteins and upregulation of
Bcl-XS apoptotic proteins in MCF-7 cells.
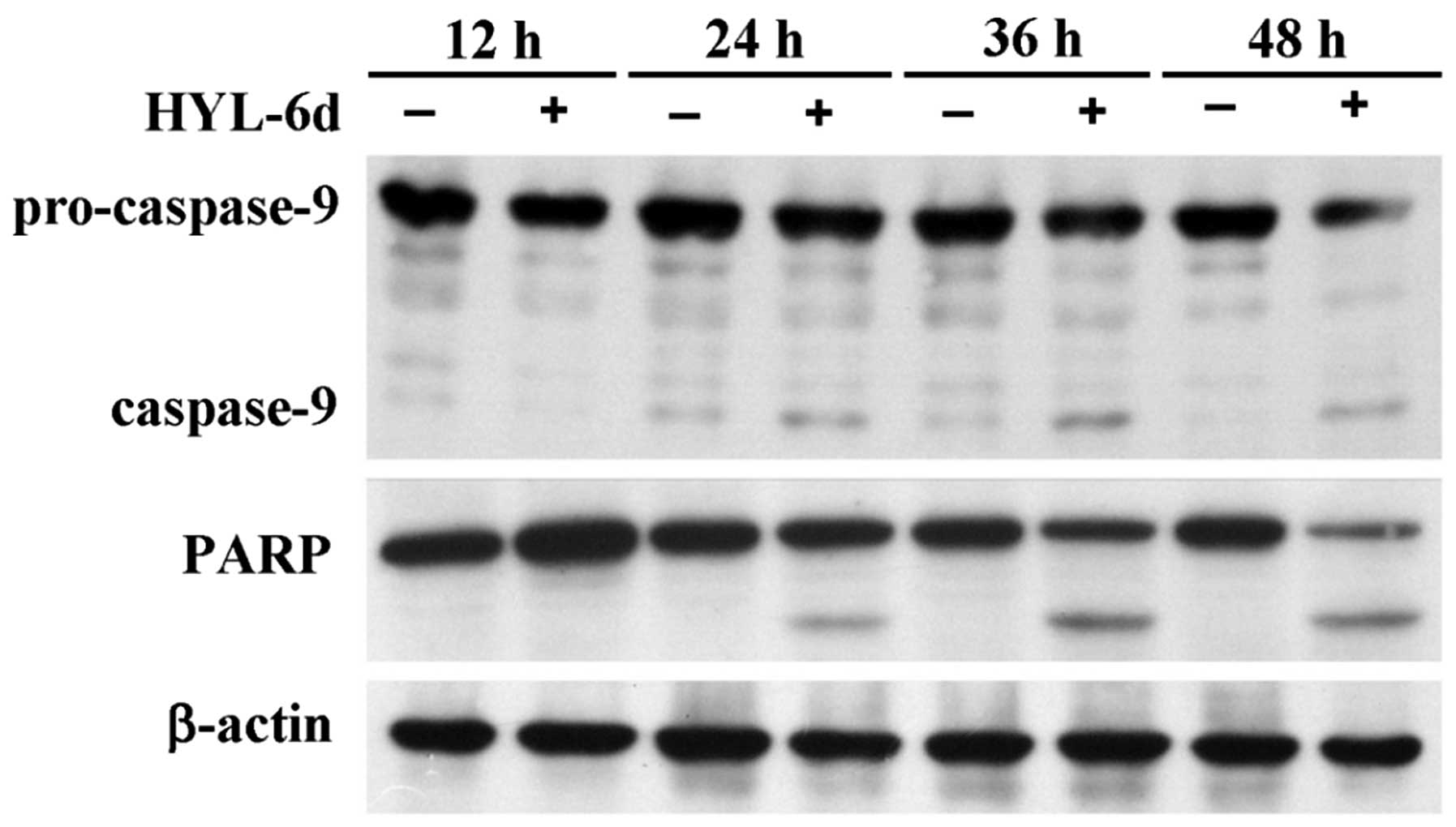
Activation of caspases during apoptosis is
correlated with the cleavage and activation/inactivation of a range
of critical cellular substrates, including activation of the DNA
repair enzyme Poly (ADP-ribos) polymerase (PARP). In this study,
the potential effects of HYL-6d on the cleavage of procaspase-9 and
PARP in MCF-7 cells were also investigated. As shown in Fig. 7, procaspase-9 and PARP showed a
time-dependent cleavage.
Effect of HYL-6d on VEGF- and
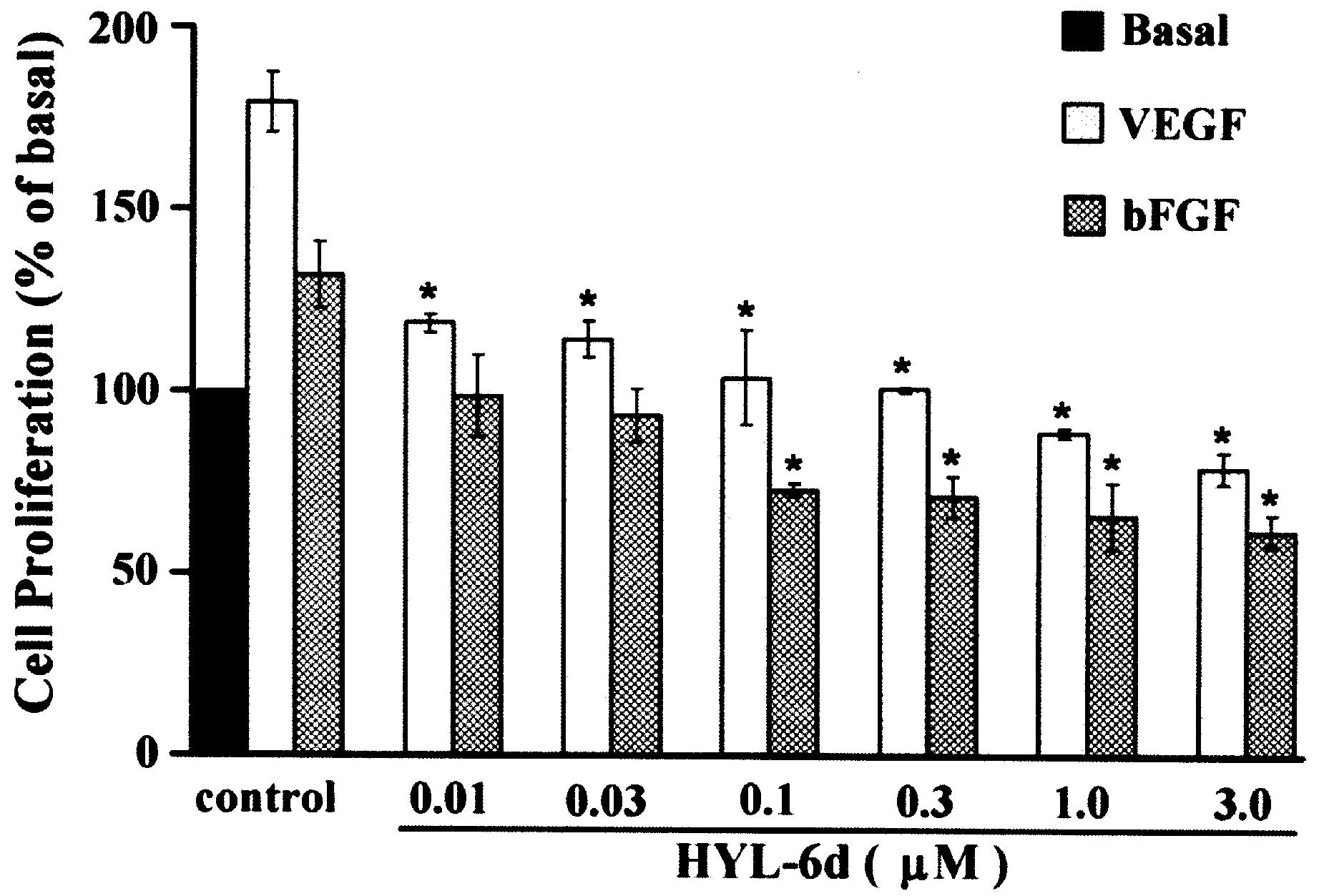
bFGF-induced cell proliferation of HUVECs
The ability of HYL-6d to influence the mitogen
activity of VEGF and bFGF was also determined using the BrdU cell
proliferation assay. In the presence of 10 ng/ml VEGF and bFGF,
different concentrations of HYL-6d (0.3, 3 and 30 μM) showed
inhibition of cell growth in a dose-dependent manner (Fig. 8). The data suggest that VEGF- and
bFGF-induced cell proliferation is inhibited by HYL-6d.
Effect of HYL-6d on VEGF- and
bFGF-induced cell migration and tube formation of HUVECs
As cell migration is necessary for endothelial cells
in angiogenesis and for cancer cells in tumor growth and metastasis
(10,25,26),
the potential regulatory role of HYL-6d on cell migration was also
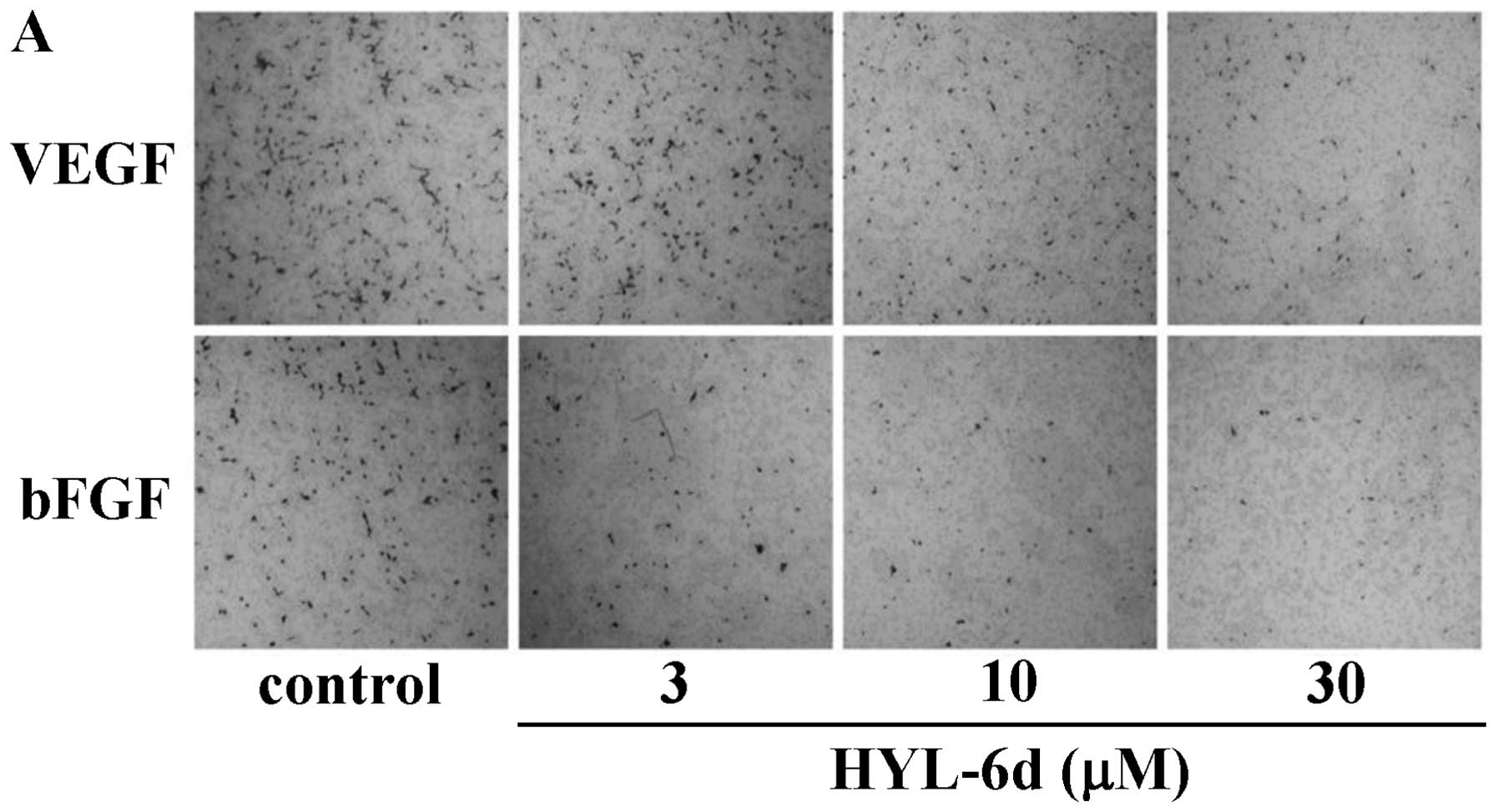
measured in this study. Treatment of HYL-6d (ranging from 3 to 30
μM) significantly inhibited VEGF- and bFGF-induced HUVEC cell
migration in a dose-dependent manner (Fig. 9). Angiogenesis is a complex process
involving several types of cells and tube formation of endothelial
cells is the key step (27). To
determine if HYL-6d-inhibited angiogenesis was mediated by its
effect on endothelial cell tube formation, assays were performed
using HUVECs (2×105 cells) in 500 μl M199/2% FBS treated
with different concentrations of HYL-6d and plated onto Matrigel
layers. Following 24 h of incubation, the ability of endothelial
cells to form tube-like structures was assessed by an inverted
photomicroscope (Fig. 10A and B).
Our results illustrated that HYL-6d blocks the formation of
capillary tubes in a concentration-dependent manner without
affecting HUVEC viability (Fig.
10C).
Discussion
Previous studies have shown that carbazole alkaloids
isolated from the alcohol extract of the Rutaceae root bark
display cytotoxic activity (28).
The antitumor activity of carbazole, and its ability to selectively
inhibit tumor growth, has been previously reported in human lung
cancer cells, colon cancer cells and monocytic leukemia cells
(15–17). In the present study, HYL-6d, a novel
synthetic carbazole derivative, was evaluated for its cytotoxic
activities in six human cancer cell lines. Our data revealed that
HYL-6d is the most effective antitumor agent in the human breast
cancer cell line MCF-7. In vitro studies have also
demonstrated that HYL-6d causes cell growth inhibition, cell cycle
arrest and induction of apoptosis in MCF-7 cells (Table I, Figs.
1 and 5). Moreover, HYL-6d also
displayed anti-angiogenic activity. Therefore, we concluded that
HYL-6d might be a potent carbazole derivative for breast cancer
therapy.
Alterations in the expression and activity of cell
cycle regulators have been associated with the occurrence of breast
cancer (29). In 2003, a synthetic
pyrrolo[3,4-c]carbazole was reported to induce cell cycle arrest in
G1 through cyclin D1/CDK4 inhibition in human lung
cancer cell lines (17).
Accordingly, HYL-6d is also able to induce cell cycle arrest.
Exposure of MCF-7 cells to 30 μM HYL-6d led to cell cycle arrest in
the S phase, and this arrest was accompanied by a marked decrease
in the number of cells in the G2/M phase (Table I). Several cell cycle regulators
including cyclins, CDK, CDKI and the tumor suppressor protein p53
were examined to determine their roles in HYL-6d-induced cell cycle
arrest. Using immunoblot analysis, we showed a marked increase of
p53 and p21 in MCF-7 cells treated with HYL-6d (Fig. 4). Furthermore, expression of cyclin
D1, A and CDK2 decreased in a time-dependent manner (Fig. 4). Collectively, these data indicated
that HYL-6d-induced S-phase arrest in MCF-7 cells was caused by the
inhibition of cyclins and CDK2, along with the induction of p21 and
p53.
Apoptosis plays an important role in cell
development and tissue homeostasis. The defects in cell death
during cell development might be associated with certain problems
such as autoimmune diseases and cancer, while excessive cell death
can lead to degenerative diseases in the nervous system (29). Induction of apoptosis is one of the
best strategies to treat cancer. Numerous current cytotoxic drugs
that mediate their effects through induction of apoptosis in cancer
cells have been reported (30). In
this study, we revealed that HYL-6d is another anticancer drug that
elicits apoptotic cell death as characterized by morphological
changes, chromatin condensation and internucleosomal DNA
fragmentation (Fig. 5). Bcl-2
family proteins, including the anti-apoptotic members Bcl-2 and
Bcl-XL and the pro-apoptotic members Bax and
Bcl-XS, are key regulators of apoptosis that act by
either inhibiting or promoting apoptosis (24). Among these Bcl-2 family proteins,
Bcl-X is an apoptotic effector (31), arising from an alternatively spliced
form of the Bcl-X transcript that generates either a long
Bcl-XL or short Bcl-XS form of the protein
(32). Bcl-XL is
associated with decreased apoptosis in cancer cells, increased risk
of metastasis and resistance to chemotherapeutic drugs (33,34).
By contrast, the overexpression of Bcl-XS can induce
apoptosis and sensitize cells to chemotherapeutic agents (35,36).
In this study, our data showed that the expression levels of the
Bcl-2 and Bcl-XL proteins decreased, while
Bcl-XS expression increased, after 48 h of HYL-6d
treatment (Fig. 6). These
expression changes were accompanied by the caspase activation
(Fig. 7). Bcl-2, Bcl-XL,
Bcl-XS and Bax are located on the outer mitochondrial
membrane, leading to the release of pro-apoptotic factors from the
mitochondrial inner-membrane space into the cytosol (37,38).
These pro-apoptotic proteins lead to the subsequent activation of
caspase-9 and the executioner caspases, an event associated with
the induction of programmed cell death. These observations are
consistent with our findings that elevation of Bcl-XS
protein levels in HYL-6d-treated MCF-7 cells might alter the
permeability of the mitochondrial membrane, facilitating the
passage of cytochrome c, triggering cleavage, and activation
of downstream caspases and the onset of apoptosis.
The ability of tumor cells to produce various
cytokines, chemokines, angiogenic and growth factors is crucial for
tumor cell proliferation and the formation of stroma and blood
vessel networks that provide oxygen and nutrients to support
progressive tumor growth (39). The
level of angiogenic activity in breast cancer is a determinant of
disease progression and survival (40), suggesting that inhibition of blood
vessel formation could be a therapeutic target in this solid tumor
(41). Invasive breast cancer
generally expresses certain angiogenic factors, including VEGF and
bFGF (42). VEGF and bFGF stimulate
migration and proliferation of endothelial cells and formation of
blood vessels (43). Therefore, a
drug which targets angiogenesis has therapeutic value for cancer
treatment. In fact, there are several chemotherapeutic agents used
routinely in breast cancer treatment that are known to have
anti-angiogenic activity (44). In
this study, we demonstrated that HYL-6d significantly inhibited
angiogenic processes including proliferation and migration in VEGF-
or bFGF-stimulated HUVECs under pathological angiogenic conditions
(Figs. 8 and 10A). Stimulation of angiogenic activator
VEGF or bFGF can promote differentiation of HUVECS to form
capillary-like tubes (45), and
HYL-6d can effectively suppress this angiogenesis tube formation in
human endothelial cells (Figs. 9
and 10). These data indicate that
HYL-6d is a promising drug for the treatment of cancer by
inhibiting angiogenesis.
In conclusion, our data revealed that HYL-6d, a
novel carbazole derivative, possesses potent activity against
various human cancer cell lines. HYL-6d treatment results in an
arrest in the S phase and triggers apoptosis in MCF-7 cells through
a p53-dependent pathway. Furthermore, HYL-6d exerts its
anti-angiogenic activity by inhibiting HUVEC proliferation,
migration, and tube formation induced by VEGF or bFGF in
vitro. Thus, we have demonstrated that HYL-6d is a potent
chemotherapy agent with anti-angiogenic activity and promotes
apoptosis in MCF-7 cells.
Acknowledgements
This study was supported by grants from the National
Science Council of the Republic of China (NSC98-2628-B-039-018-MY3)
and China Medical University (CMU 95-249 and CMU 95-063).
References
|
1
|
Annual Reports of the Department of
Health, the Executive Yuan, Republic of China (Taiwan), 2011.
|
|
2
|
Lin A and Rugo HS: The role of trastuzumab
in early stage breast cancer: current data and treatment
recommendations. Curr Treat Options Oncol. 8:47–60. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schlotter CM, Vogt U, Allgayer H and
Brandt B: Molecular targeted therapies for breast cancer treatment.
Breast Cancer Res. 10:211–223. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Normanno N, Morabito A, De Luca A, et al:
Target-based therapies in breast cancer: current status and future
perspectives. Endocr Relat Cancer. 16:675–702. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mauri D, Polyzos NP, Salanti G, Pavlidis N
and Ioannidis JP: Multiple-treatments meta-analysis of chemotherapy
and targeted therapies in advanced breast cancer. J Natl Cancer
Inst. 100:1780–1791. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sutherland RL and Musgrove EA: Cyclins and
breast cancer. J Mammary Gland Biol Neoplasia. 9:95–104. 2004.
View Article : Google Scholar
|
|
7
|
Fulda S, Meyer E, Friesen C, Susin SA,
Kroemer G and Debatin KM: Cell type specific involvement of death
receptor and mitochondrial pathways in drug-induced apoptosis.
Oncogene. 20:1063–1075. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gasco M, Shami S and Crook T: The p53
pathway in breast cancer. Breast Cancer Res. 4:70–76. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brenner D and Mak TW: Mitochondrial cell
death effectors. Curr Opin Cell Biol. 21:871–877. 2009. View Article : Google Scholar
|
|
10
|
Folkman J: What is the evidence that
tumors are angiogenesis dependent? J Natl Cancer Inst. 82:4–6.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ma J and Waxman DJ: Combination of
antiangiogenesis with chemotherapy for more effective cancer
treatment. Mol Cancer Ther. 7:3670–3684. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Miller KD: Recent translational research:
antiangiogenic therapy for breast cancer - where do we stand?
Breast Cancer Res. 6:128–132. 2004. View
Article : Google Scholar
|
|
13
|
Nakamura K, Sugumi H, Yamaguchi A, et al:
Antitumor activity of ER-37328, a novel carbazole topoisomerase II
inhibitor. Mol Cancer Ther. 1:169–175. 2002.PubMed/NCBI
|
|
14
|
Kamata J, Okada T, Kotake Y, et al:
Synthesis and evaluation of novel pyrimido-acridone, -phenoxadine,
and -carbazole as topoisomerase II inhibitors. Chem Pharm Bull
(Tokyo). 52:1071–1081. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Riou JF, Guittat L, Mailliet P, et al:
Cell senescence and telomere shortening induced by a new series of
specific G-quadruplex DNA ligands. Proc Natl Acad Sci USA.
99:2672–2677. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chang CC, Kuo IC, Lin JJ, et al: A novel
carbazole derivative, BMVC: a potential antitumor agent and
fluorescence marker of cancer cells. Chem Biodivers. 1:1377–1384.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Engler TA, Furness K, Malhotra S, et al:
Novel, potent and selective cyclin D1/CDK4 inhibitors:
indolo[6,7-a]pyrrolo[3,4-c]carbazoles. Bioorg Med Chem Lett.
13:2261–2267. 2003.PubMed/NCBI
|
|
18
|
Hsu MJ, Chao Y, Chang YH, et al: Cell
apoptosis induced by a synthetic carbazole compound LCY-2-CHO is
mediated through activation of caspase and mitochondrial pathways.
Biochem Pharmacol. 70:102–112. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Arbiser JL, Govindarajan B, Battle TE, et
al: Carbazole is a naturally occurring inhibitor of angiogenesis
and inflammation isolated from antipsoriatic coal tar. J Invest
Dermatol. 126:1396–1402. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kongkathip B, Kongkathip N,
Sunthitikawinsakul A, Napaswat C and Yoosook C: Anti-HIV-1
constituents from Clausena excavata: Part II. Carbazoles and
a pyranocoumarin. Phytother Res. 19:728–731. 2005.PubMed/NCBI
|
|
21
|
Mathé G, Triana K, Pontiggia P, Blanquet
D, Hallard M and Morette C: Data of pre-clinical and early clinical
trials of acriflavine and hydroxy-methyl-ellipticine reviewed,
enriched by the experience of their use for 18 months to 6 years in
combinations with other HIV1 virostatics. Biomed Pharmacother.
52:391–396. 1998.PubMed/NCBI
|
|
22
|
Golob T, Biberger C, Walter GE and Angerer
E: Antiestrogenic activities of
3,8-dihydroxy-6,11-dihydrobenzo[a]carbazoles with sulfur-containing
side chains. Arch Pharm (Weinheim). 333:305–311. 2000.PubMed/NCBI
|
|
23
|
Chen T and Wong YS: Selenocystine induces
S-phase arrest and apoptosis in human breast adenocarcinoma MCF-7
cells by modulating ERK and Akt phosphorylation. J Agric Food Chem.
56:10574–10581. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Patel MP, Masood A, Patel PS and
Chanan-Khan AA: Targeting the Bcl-2. Curr Opin Oncol. 21:516–523.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Folkman J: Angiogenesis: an organizing
principle for drug discovery? Nat Rev Drug Discov. 6:273–286. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Baluk P, Hashizume H and McDonald DM:
Cellular abnormalities of blood vessels as targets in cancer. Curr
Opin Genet Dev. 15:102–111. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen H, Campbell RA, Chang Y, et al:
Pleiotrophin produced by multiple myeloma induces
transdifferentiation of monocytes into vascular endothelial cells:
a novel mechanism of tumor-induced vasculogenesis. Blood.
113:1992–2002. 2009. View Article : Google Scholar
|
|
28
|
Asche C and Demeunynck M: Antitumor
carbazoles. Anticancer Agents Med Chem. 7:247–267. 2007. View Article : Google Scholar
|
|
29
|
Joslyn KB and Anthony L: Control of
mitochondrial apoptosis by the Bcl-2 family. J Cell Sci.
122:437–441. 2009. View Article : Google Scholar
|
|
30
|
Lowe SW and Lin AW: Apoptosis in cancer.
Carcinogenesis. 21:485–495. 2000. View Article : Google Scholar
|
|
31
|
Akgul C, Moulding DA and Edwards SW:
Alternative splicing of Bcl-2-related genes: functional
consequences and potential therapeutic applications. Cell Mol Life
Sci. 61:2189–2199. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Garneau D, Revil T, Fisette JF and Chabot
B: Heterogeneous nuclear ribonucleoprotein F/H proteins modulate
the alternative splicing of the apoptotic mediator Bcl-x. J Biol
Chem. 280:22641–22650. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen N, Chen X, Huang R, et al: BCL-xL is
a target gene regulated by hypoxia-inducible factor-1{alpha}. J
Biol Chem. 284:10004–10012. 2009.
|
|
34
|
Olopade OI, Adeyanju MO, Safa AR, Hagos F,
Mick R, Thompson CB and Recant WM: Overexpression of BCL-x protein
in primary breast cancer is associated with high tumor grade and
nodal metastases. Cancer J Sci Am. 3:230–237. 1997.PubMed/NCBI
|
|
35
|
Clarke MF, Apel IJ, Benedict MA, et al: A
recombinant bcl-xs adenovirus selectively induces apoptosis in
cancer cells but not in normal bone marrow cells. Proc Natl Acad
Sci USA. 92:11024–11028. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sumantran VN, Ealovega MW, Nuñez G, Clarke
MF and Wicha MS: Overexpression of Bcl-XS sensitizes MCF-7 cells to
chemotherapy-induced apoptosis. Cancer Res. 55:2507–2510.
1995.PubMed/NCBI
|
|
37
|
Heermeier K, Benedict M, Li M, Furth P,
Nuñez G and Hennighausen L: Bax and Bcl-xs are induced at the onset
of apoptosis in involuting mammary epithelial cells. Mech Dev.
56:197–207. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tsujimoto Y and Shimizu S: Role of the
mitochondrial membrane permeability transition in cell death.
Apoptosis. 12:835–840. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Robinson SC and Coussens LM: Soluble
mediators of inflammation during tumor development. Adv Cancer Res.
93:59–87. 2005.
|
|
40
|
Bando H: Vascular endothelial growth
factor and bevacitumab in breast cancer. Breast Cancer. 14:163–173.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sledge GW Jr: VEGF-targeting therapy for
breast cancer. J Mammary Gland Biol Neoplasia. 10:319–323. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fox SB, Generali DG and Harris AL: Breast
tumour angiogenesis. Breast Cancer Res. 9:2162007. View Article : Google Scholar
|
|
43
|
Levina V, Su Y, Nolen B, Liu X, Gordin Y,
Lee M, Lokshin A and Gorelik E: Chemotherapeutic drugs and human
tumor cells cytokine network. Int J Cancer. 123:2031–2040. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Miller KD, Sweeney CJ and Sledge GW Jr:
Redefining the target: chemotherapeutics as antiangiogenics. J Clin
Oncol. 19:1195–1206. 2001.PubMed/NCBI
|
|
45
|
Kubota Y, Kleinman HK, Martin GR and
Lawley TJ: Role of laminin and basement membrane in the
morphological differentiation of human endothelial cells into
capillary-like structures. J Cell Biol. 107:1589–1598. 1988.
View Article : Google Scholar : PubMed/NCBI
|