Introduction
Ovarian cancer is the most lethal gynecological
malignancy. Approximately 80–90% of ovarian cancers originate from
the ovarian surface epithelium. The etiology of ovarian epithelial
cancer (OEC) is not yet fully clarified; currently, the
gonadotropin theory of ovarian cancer proposes that elevated serum
gonadotropins, follicle-stimulating hormone (FSH) and luteinizing
hormone (LH) contribute significantly to the development of ovarian
cancer. Wang et al reported that vascular endothelial growth
factor (VEGF) plays a role in the development of ovarian cancer,
and that elevated gonadotropin levels, as found in menopausal women
and most ovarian cancer patients after surgery, could accelerate
tumor growth and tumor recurrence by inducing expression of VEGF in
OECs (1). However, the detailed
molecular mechanisms by which FSH leads to expression of VEGF in
OET remain unclear.
Increasing evidence supports the hypothesis that
reactive oxygen species (ROS) are involved in the expression and
regulation of VEGF and angiogenesis (2–6), and
conversely, multiple ROS-mediated cellular functions can be induced
by growth factors and hormones (7–10). It
has been reported that estrogen-induced ROS-mediated signaling is
involved in the development of breast cancer (11). In addition, LH-induced ROS
generation contributes to ovulation (12). However, it is not clear whether FSH
can induce ROS generation and in turn contribute to FSH-induced
VEGF expression.
Cells have developed a variety of protective
mechanisms in response to oxidative stress to escape ROS-mediated
damage. Activated Nrf2 binds to the antioxidant-response element
(ARE) leading to the upregulation of a large number of antioxidant
genes (13–18). In a previous study, we observed that
Nrf2 was overexpressed in ovarian epithelial carcinoma, and
confirmed that FSH could induce the expression of Nrf2 in OEC
cells, suggesting that activation of Nrf2 signaling contributes to
the development of OEC (34). Kim
et al reported that Nrf2 blockade suppresses angiogenesis in
colon cancer by inhibiting hypoxia-induced activation of
hypoxia-inducible factor 1α (HIF1α) (19), implying that HIF1α signaling is
regulated by Nrf2, and suggesting that both HIF1α and Nrf2
signaling may regulate the expression of VEGF and cancer
angiogenesis.
Aberrant activation of HIF1α signaling induces
expression of VEGF in cancer (20–22).
High levels of HIF1α expression are observed in several types of
cancer and correlate with a poor prognosis. We previously confirmed
that FSH induced the expression of HIF1α in ovarian cancer cells
(23) and Lee et al revealed
that lysophosphatidic acid (LPA) induced the binding of HIF1α to
the VEGF promoter in cancer cells (24). However, it remains to be clarified
how FSH-induced HIF1α activation and VEGF expression occurs in OEC
cells. Therefore, in this study, we investigated whether ROS
regulate FSH-induced expression of VEGF via Nrf2 and HIF1α
signaling in OEC cells.
Materials and methods
Reagents and antibodies
Human FSH, dichlorofluorescein (DCF) and N-acetyl
cysteine (NAC) were obtained from Sigma-Aldrich (St. Louis, MO,
USA). Lipofectamine 2000, DMEM/F12 medium and fetal bovine serum
were purchased from Invitrogen (Carlsbad, CA, USA). Anti-Nrf2,
GAPDH and HIF1α primary antibodies were purchased from Abcam
(Cambridge, UK).
Cell lines and cell culture
Human ES2 (clear cell adenocarcinoma) and Hey
(papillary cystadenocarcinoma) cell lines were obtained from the
American Culture Collection (Manassas, VA, USA) and were cultured
in 1:1 DMEM/F12 supplemented with 100 U/ml penicillin, 100 μg/ml
streptomycin and 10% fetal bovine serum at 37°C in a humidified
incubator containing 95% room air and 5% CO2.
ROS detection
ES2 and Hey cells were seeded in 6-well plates and
incubated for 24 h. The culture medium was replaced with OPTI-MEM
and the cells were incubated for 24 h. The cells were then treated
with 40 mIU/ml FSH for 20 min or 6 h, incubated with 10 μg/ml DCF
for 30 min, and washed three times with PBS, fixed and imaged using
a fluorescence microscope.
Western blotting
Western blotting was performed in a routine manner.
Briefly, 60 μg protein samples was loaded on 10% SDS-PAGE gels,
transferred to polyvinylidene fluoride (PVDF) membranes, incubated
with specific primary antibodies at 4°C overnight, and incubated
with the appropriate secondary antibody for 1 h at room
temperature. The bands were visualized using the ECL Plus system
(Amersham, GE Healthcare; Chalfont St. Giles, UK).
ELISA assay
To investigate the effect of NAC on FSH-induced VEGF
expression, Hey cells were pretreated with different concentrations
of NAC (10, 50, 100 μM) for 30 min, and then treated with 40 mIU/ml
FSH for 48 h and the cell media were collected. To determine the
effect of blocking ROS signaling and knockdown of Nrf2 or
HIF1α on FSH-induced VEGF expression, Hey cells were treated
with NAC in the presence or absence of siNrf2 or siHIF1α for the
indicated times, and then the cell media were harvested. VEGF
protein concentration was measured using an ELISA (R&D Systems,
Minneapolis, MN, USA) according to the manufacturer’s
instructions.
Chromatin immunoprecipitation assay
A human HIF-1α chromatin immunoprecipitation (ChIP)
assay was performed using a kit purchased from R&D Systems,
according to the protocol recommended by the manufacturer. Hey
cells were treated as indicated in the figure legends, and then the
human VEGF promoter was amplified using the primers: forward,
5′-CCTCAGTTCCCTGGCAACATCTG-3′ and reverse,
5′-GAAGAATTTGGCACCAAGTTTGT-3′. The amplification products were
examined on 2% agarose gels using ethidium bromide staining.
RNA interference
Small interfering RNAs (siRNAs) against
HIF-1α and Nrf2 were designed and synthesized by
Dharmacon Thermo Scientific (Waltham, MA, USA). Hey cells were
seeded in 6-well plates, cultured to 50% confluence and then serum
starved for 24 h. The cells were transiently transfected with siRNA
using DharmaFECT transfection reagents according to the
manufacturer’s instructions, treated with 40 mIU/ml FSH or NAC for
48 h, and the protein expression levels of downstream target genes
were determined by western blotting or using an ELISA.
Statistical analysis
Data are presented as the mean ± standard deviation
(SD). Statistical significance was assessed using the Student’s
t-test or one-way ANOVA with SPSS 11.5 software (SPSS, Chicago, IL,
USA); P-values <0.05 were considered significant.
Results
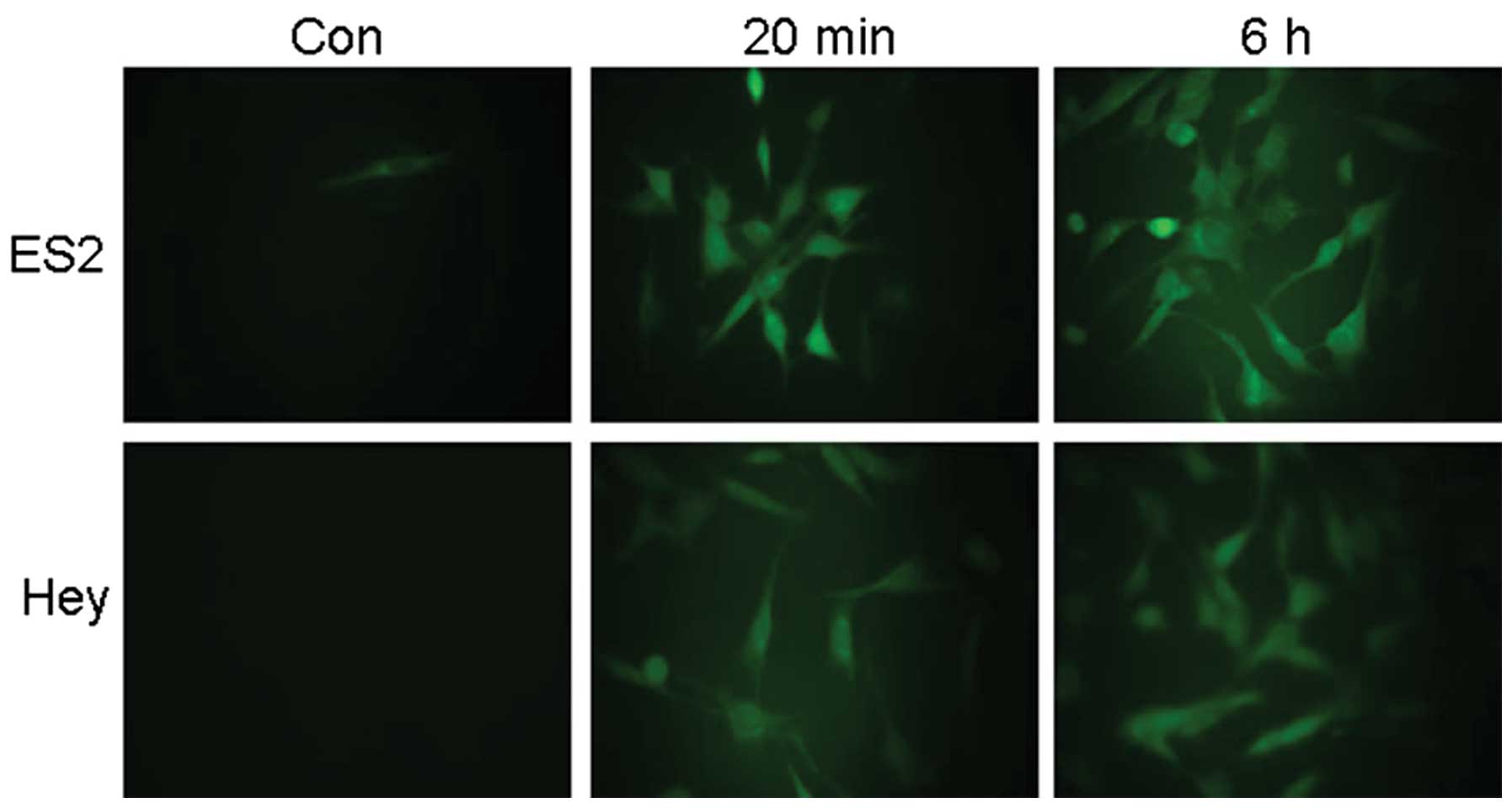
FSH stimulates ROS generation in ovarian
epithelial cancer cells
Increasing evidence supports the importance of ROS
as secondary messengers in a variety of cellular functions
(25–30). For example, ROS, particularly
H2O2, can mimic LH-induced ovulation
(12). To investigate the potential
involvement of Nrf2 in FSH-induced VEGF expression, Hey and ES2
ovarian epithelial cancer cells were treated with 40 mIU/ml FSH for
20 min or 6 h. As shown in Fig. 1,
FSH potently induced ROS production in both Hey and ES2 cell lines.
These observations suggest that ROS play a role in FSH-induced
cellular function.
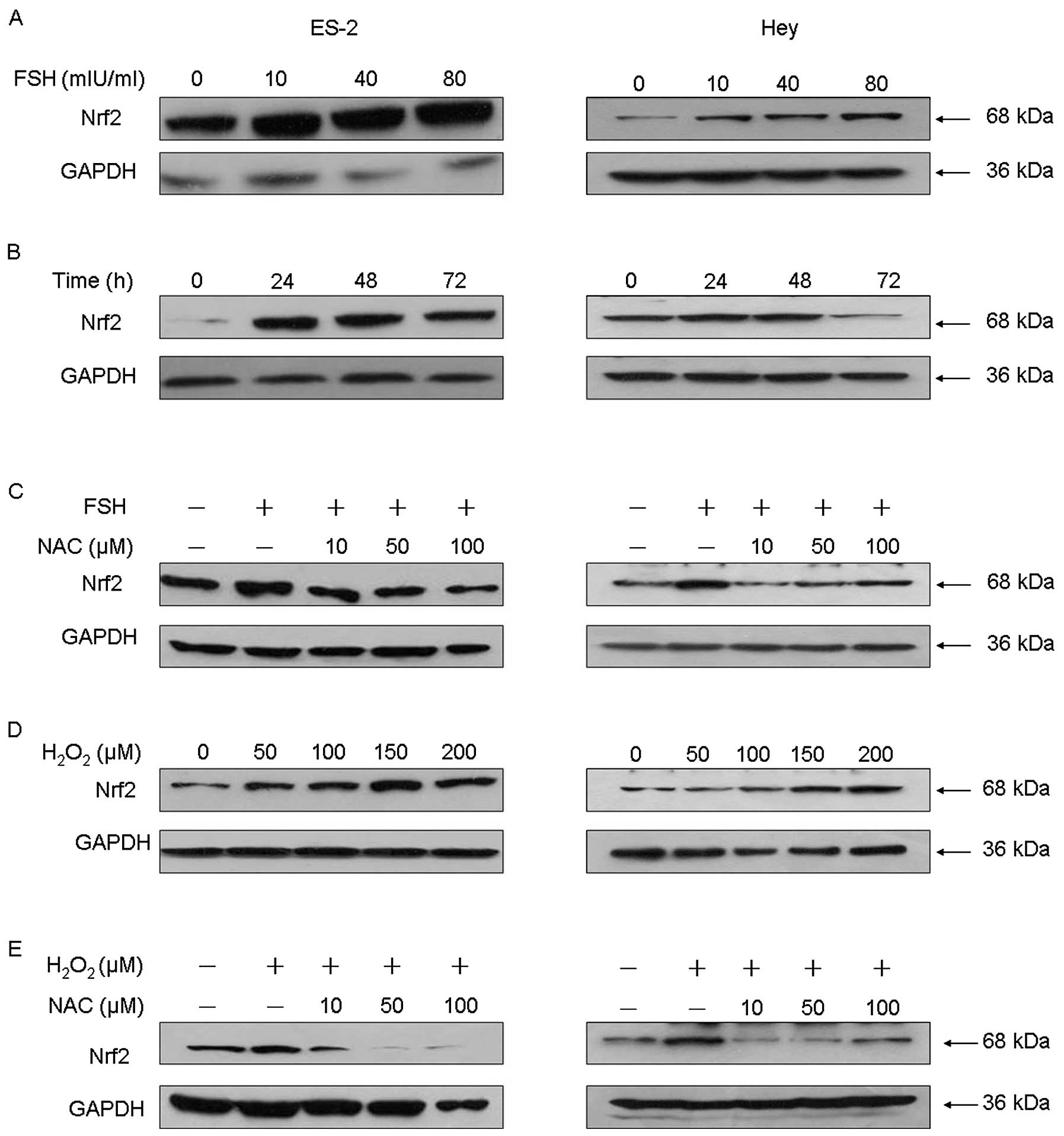
ROS generation is required for
FSH-induced Nrf2 signaling
Nrf2 is one of the most important cellular defense
mechanisms against oxidative stress. Our observation of increased
ROS production in FSH-treated cells prompted us to investigate
whether Nrf2 is also involved in FSH-induced ovarian cellular
function. As expected, FSH treatment induced Nrf2 expression in a
dose-dependent manner (Fig. 2A).
The peak Nrf2 expression level was observed in Hey and ES2 cells
exposed to 40 mIU/ml FSH for 48 h (Fig.
2B).
To confirm that ROS production is required for
FSH-induced Nrf2 signaling, we pretreated ovarian cancer cells with
the broad-range ROS scavenger N-acetyl cysteine (NAC) at various
concentrations for 30 min, and then treated the cells with 40
mIU/ml FSH for 48 h. As shown in Fig.
2C, NAC significantly and dose-dependently attenuated the
ability of FSH to induce Nrf2 protein expression. As expected,
H2O2 treatment also effectively induced Nrf2
expression in a dose-dependent manner (Fig. 2D), mimicking the effect of FSH; this
effect was also inhibited by pretreatment with NAC (Fig. 2E). Collectively, these results
suggest that ROS, especially H2O2, are
required for FSH-induced activation of Nrf2 signaling.
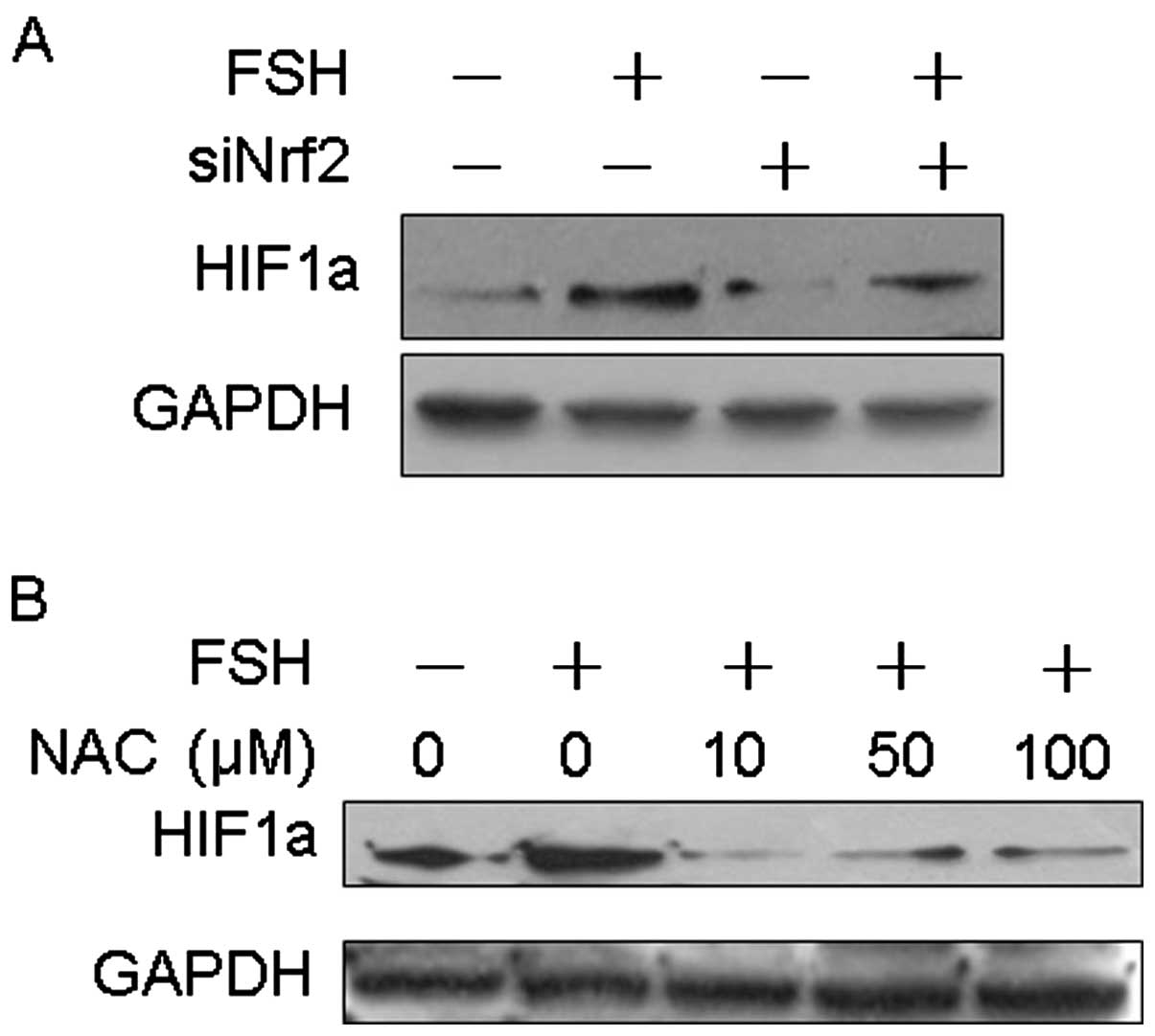
FSH-induced HIF1α expression requires ROS
generation and Nrf2 signaling activation
HIF1α accumulation is necessary for the induction of
VEGF expression, and our previous research demonstrated that HIF1α
is involved in FSH-induced VEGF expression. Therefore, we
hypothesized that ROS production and activation of Nrf2 are
necessary for FSH-induced HIF1α expression. To address this,
Nrf2 was knocked down using Nrf2-specific siRNA.
Knockdown of Nrf2 reduced the expression of HIF1α and
abolished FSH-induced HIF1α expression, compared to control
siRNA-transfected cells (Fig. 3A).
To further confirm the role of ROS as a mediator in FSH-induced
HIF1α expression, the cells were pretreated with various doses of
NAC prior to treatment with 40 mIU/ml FSH for 48 h. FSH-induced
HIF1α expression was considerably attenuated by NAC, suggesting
that ROS play a role in FSH-induced HIF1α expression (Fig. 3B).
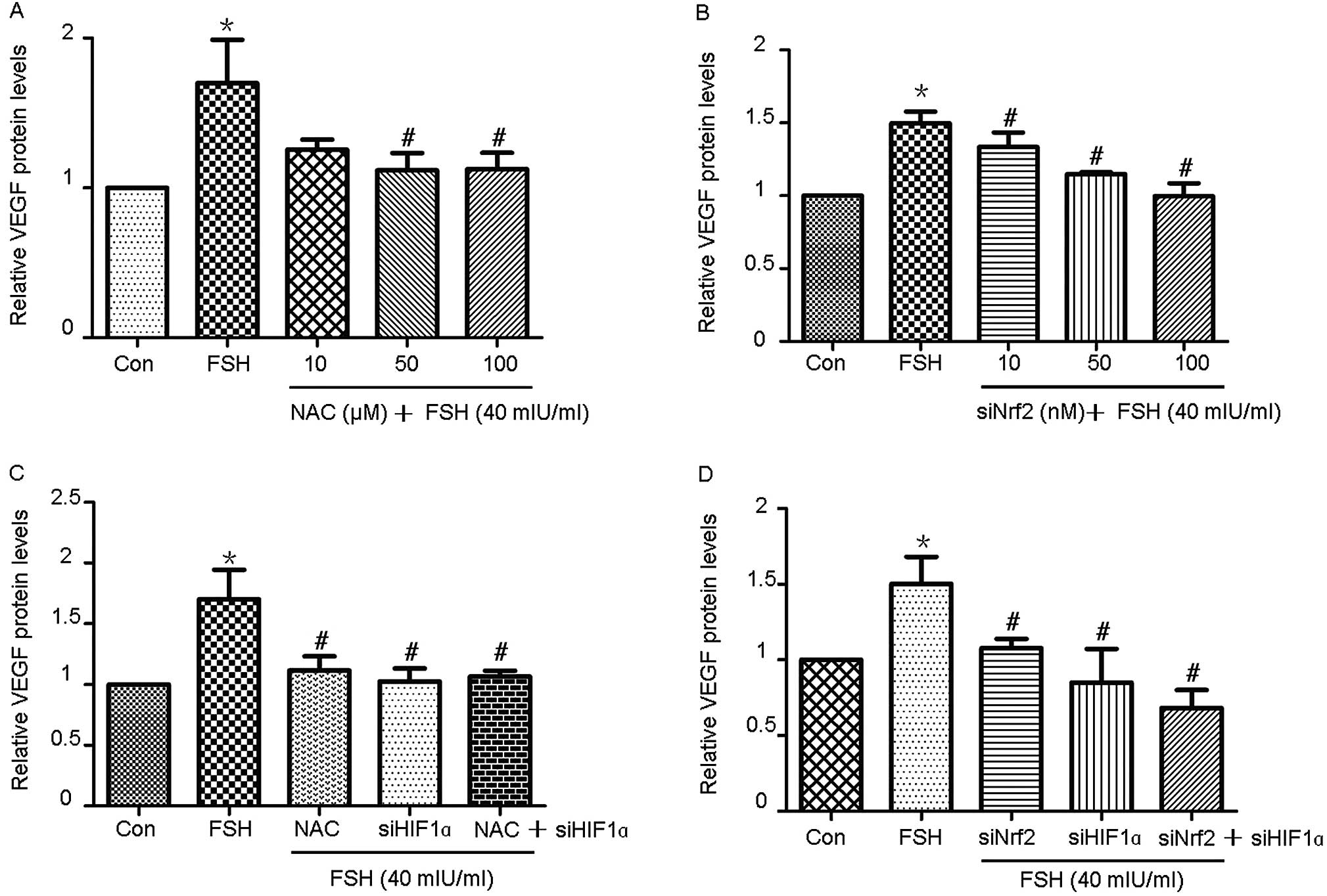
FSH induces VEGF expression through ROS
production and activation of Nrf2-HIF1α signaling
As demonstrated in Fig.
4A, treatment of Hey cells with FSH dramatically enhanced the
expression of VEGF; this effect was attenuated in a dose-dependent
manner by pretreatment of the cells with NAC. Furthermore,
siRNA-mediated knockdown of Nrf2 decreased FSH-induced VEGF
expression in a dose-dependent manner (Fig. 4B). ELISA assays were performed to
investigate the effect of combined NAC pretreatment and knockdown
of HIF1α and/or Nrf2 on FSH-induced VEGF expression.
NAC pretreatment, knockdown of HIF1α or knockdown of
Nrf2 alone potently attenuated FSH-induced VEGF expression.
Combined NAC pretreatment and knockdown of HIF1α did not
lead to a significant reduction in FSH-induced VEGF expression,
compared to pretreatment with NAC or knockdown of HIF1α
alone (Fig. 4C). However, double
knockdown of HIF1α and Nrf2 significantly inhibited
FSH-induced VEGF expression, compared to pretreatment with FSH,
knockdown of HIF1α or knockdown of Nrf2 alone
(Fig. 4D).
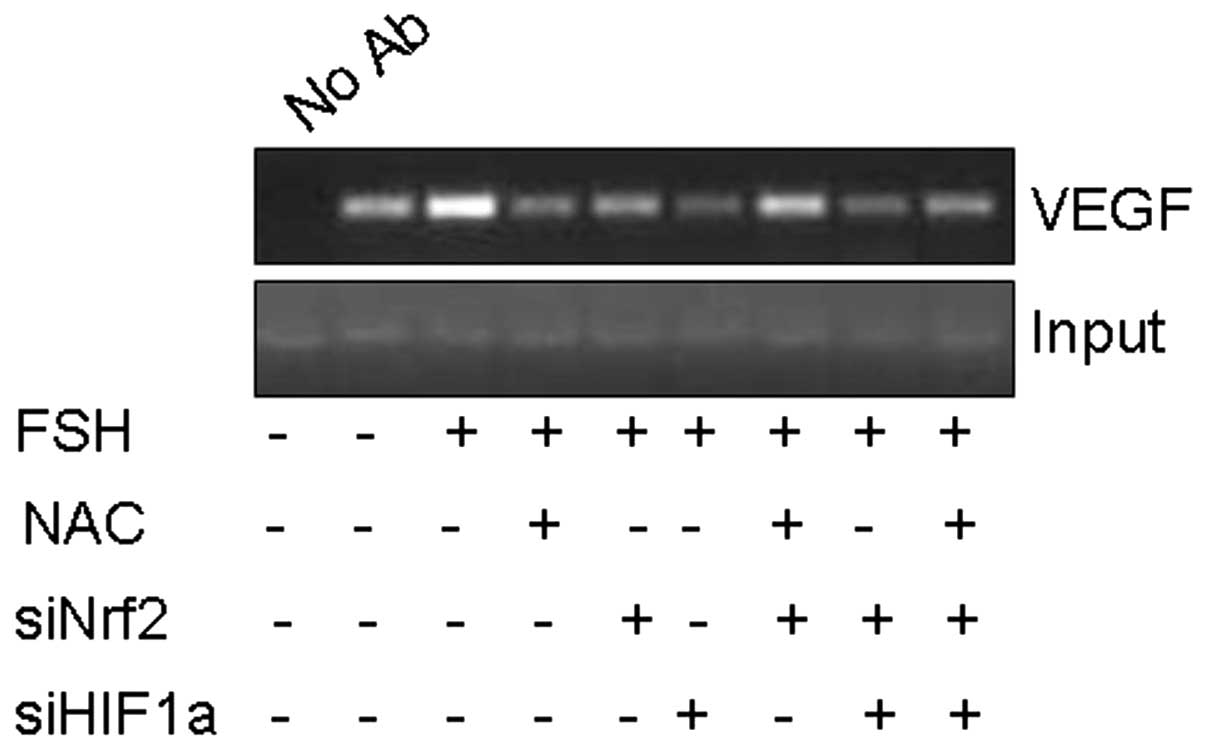
FSH enhances binding of HIF1α to the VEGF
promoter
To further investigate the role of HIF1α in
FSH-induced VEGF expression, we explored the ability of FSH to
affect the interaction of HIF1α with the native hypoxia response
element of the VEGF promoter using the chromatin
immunoprecipitation assay (ChIP). As shown in Fig. 5, HIF1α bound to the VEGF
promoter could be precipitated using an anti-HIF1α antibody.
Treatment of Hey cells with 40 mIU/ml FSH significantly enhanced
the ability of HIF1α to bind the VEGF promoter; however,
treatment with NAC, siNrf2 or siHIF1α alone or in combination
blocked FSH-induced binding of HIF1α to the VEGF
promoter.
Discussion
In the present study, our in vitro studies
and molecular analyses provided evidence that ROS are necessary for
FSH-induced VEGF expression in ovarian cancer, as ablation of ROS
or knockdown of Nrf2 attenuated FSH-induced VEGF expression.
Our data also demonstrated that the Nrf2 signaling pathway is
involved in FSH-induced cellular function, and that FSH enhances
the ability of HIF1α to bind the VEGF promoter. Increased
FSH levels are a significant risk factor for the development of
ovarian cancer. Our previous study demonstrated that activation of
the PI3K/AKT pathway mediated FSH-stimulated VEGF expression in
ovarian serous cystadenocarcinoma (23). That study also indicates that HIF1α
is involved in FSH-induced VEGF expression.
ROS production was observed in OEC cells treated
with FSH. ROS, such as hydrogen peroxide, the hydroxyl radical and
superoxide anion radical are produced in measurable quantities by
every aerobic system, and are considered to be toxic to living
cells in high concentrations. Oxyradicals can act as important
secondary messengers to regulate a variety of cellular functions.
For example, estrogen-induced ROS production contributes to the
development of breast cancer (11)
and ROS can mimic LH-induced ovulation (12). In agreement with these reports which
indicate that ROS mediated-signaling contributes to hormone-induced
cellular function, our results clearly demonstrate that ROS are
involved in FSH-induced VEGF expression. FSH-induced binding of
HIF1α to the VEGF promoter and FSH-induced VEGF expression
were attenuated by the antioxidant supplement NAC (Figs. 4A and 5), confirming that ROS are necessary for
FSH-induced VEGF expression.
The transcription factor Nrf2 regulates the cellular
antioxidant response which protects cells from various insults
(17) and facilitates cell survival
by inducing intracellular antioxidants, phase II detoxifying
enzymes and other molecules that detoxify xenobiotics and
neutralize ROS (31–33). In a previous study, we confirmed
that Nrf2 was overexpressed in ovarian cancer tissues (34). In agreement with our previous study
(34), the present study confirmed
that FSH upregulated the expression of Nrf2 in ovarian cancer cells
in a dose- and time-dependent manner (Fig. 2A and B). Moreover,
H2O2 treatment mimicked FSH-induced Nrf2
expression; this effect was abolished by NAC (Fig. 2C–E). Moreover, knockdown of
Nrf2 impaired FSH-induced VEGF expression and reduced the
ability of HIF1α to bind the VEGF promoter (Figs. 4B and 5). These data imply that Nrf2 plays a
critical role in ROS-mediated FSH-induced VEGF expression. Nrf2
normally exerts a protective role when oxyradicals are present,
which raises the question of how the Nrf2-mediated antioxidant
response fails to protect ovarian cells in patients with ovarian
cancer. We suggest that Nrf2 signaling can easily eliminate
FSH-induced ROS generation and prevent ROS-induced damage in the
early stages of ovarian epithelial cancer.
According to the gonadotropin theory, persistent
stimulation with high concentrations of FSH contributes to the
progression of ovarian epithelial cancer. This stimulation may
induce persistent ROS generation; therefore, the Nrf2-mediated
protective mechanism may become saturated by excessive ROS,
resulting in the development of ovarian epithelial cancer. Our data
indicate that aberrant activation of Nrf2 in a highly oxidizing
environment may facilitate angiogenesis and tumor cell survival in
ovarian epithelial cancer.
In a previous study, we demonstrated that FSH
regulated the expression of HIF1α in a dose-dependent manner
(23). In the present study, we
showed that knockdown of Nrf2 impaired FSH-induced HIF1α
expression (Fig. 3A). In addition,
elimination of ROS using the antioxidant NAC also abolished
FSH-induced HIF1α expression (Fig.
3B). These data suggest that ROS and Nrf2 signaling are
involved in the regulation of HIF1α expression. Zhou et al
reported that HIF1α is indispensable during insulin-induced
VEGF transcriptional activation (35). Another study demonstrated that
Nrf2-deficient colon cancer cells failed to accumulate HIF1α
protein, which limited the expression of VEGF and other
HIF1α target genes (19). Our
research highlights Nrf2 as a potential candidate molecular target
for the control of tumor angiogenesis, as inhibition of Nrf2 may
block HIF1α signaling (19).
Blockage of ROS using NAC, knockdown of HIF1α or knockdown
of Nrf2 attenuated FSH-induced binding of HIF1α to the
VEGF promoter. However, knockdown of HIF1α combined
with NAC and/or knockdown of Nrf2 to a more significant
reduction in VEGF expression (Fig. 4C
and D), indicating that Nrf2/HIF1α signaling is involved in
FSH-induced VEGF expression. Most importantly, FSH induced the
binding of HIF1α to the VEGF promoter, which explains why
depletion of HIF1α abolished the expression of VEGF in our
previous study (23).
In summary, this study suggests that FSH induces ROS
generation, which activates Nrf2 signaling, which in turn triggers
HIF1α signaling and promotes the binding of HIF1α to the VEGF
promoter, which facilitates ovarian epithelial cancer progression.
Prevention of ROS accumulation and targeting of the Nrf2/HIF1α
signaling pathway may represent potential strategies to prevent the
development of ovarian epithelial cancer.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (NSFC nos. 81020108027,
30872755 and 81172478), and supported in part by grants (nos.
10JC1413100 and 09ZR1405000) from the Shanghai Science and
Technologic Committee.
References
|
1
|
Wang J, Luo F, Lu JJ, Chen PK, Liu P and
Zheng W: VEGF expression and enhanced production by gonadotropins
in ovarian epithelial tumors. Int J Cancer. 97:163–167. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee B, Kim KH, Jung HJ and Kwon HJ:
Matairesinol inhibits angiogenesis via suppression of mitochondrial
reactive oxygen species. Biochem Biophys Res Commun. 421:76–80.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sauer H and Wartenberg M: Reactive oxygen
species as signaling molecules in cardiovascular differentiation of
embryonic stem cells and tumor-induced angiogenesis. Antioxid Redox
Signal. 7:1423–1434. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ushio-Fukai M and Alexander RW: Reactive
oxygen species as mediators of angiogenesis signaling: role of
NAD(P)H oxidase. Mol Cell Biochem. 264:85–97. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ushio-Fukai M and Nakamura Y: Reactive
oxygen species and angiogenesis: NADPH oxidase as target for cancer
therapy. Cancer Lett. 266:37–52. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xia C, Meng Q, Liu LZ, Rojanasakul Y, Wang
XR and Jiang BH: Reactive oxygen species regulate angiogenesis and
tumor growth through vascular endothelial growth factor. Cancer
Res. 67:10823–10830. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Aljhni R, Ibrahim F, Guillaume YC and
Andre C: Reactive oxygen species and nitric oxide effect on the
steroid hormone binding with serum albumin. J Pharm Biomed Anal.
62:129–134. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tada H, Nakashima A, Awaya A, et al:
Effects of thymic hormone on reactive oxygen species-scavengers and
renal function in tacrolimus-induced nephrotoxicity. Life Sci.
70:1213–1223. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Choi JS, Paek AR, Kim SY and You HJ: GIPC
mediates the generation of reactive oxygen species and the
regulation of cancer cell proliferation by insulin-like growth
factor-1/IGF-1R signaling. Cancer Lett. 294:254–263. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu LZ, Hu XW, Xia C, et al: Reactive
oxygen species regulate epidermal growth factor-induced vascular
endothelial growth factor and hypoxia-inducible factor-1alpha
expression through activation of AKT and P70S6K1 in human ovarian
cancer cells. Free Radic Biol Med. 41:1521–1533. 2006. View Article : Google Scholar
|
|
11
|
Okoh V, Deoraj A and Roy D:
Estrogen-induced reactive oxygen species-mediated signalings
contribute to breast cancer. Biochim Biophys Acta. 1815:115–133.
2011.PubMed/NCBI
|
|
12
|
Shkolnik K, Tadmor A, Ben-Dor S, Nevo N,
Galiani D and Dekel N: Reactive oxygen species are indispensable in
ovulation. Proc Natl Acad Sci USA. 108:1462–1467. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen W, Sun Z, Wang XJ, et al: Direct
interaction between Nrf2 and p21(Cip1/WAF1) upregulates the
Nrf2-mediated antioxidant response. Mol Cell. 34:663–673. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sun Z, Chin YE and Zhang DD: Acetylation
of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2
during the antioxidant response. Mol Cell Biol. 29:2658–2672. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sun Z, Wu T, Zhao F, Lau A, Birch CM and
Zhang DD: KPNA6 (Importin {alpha}7)-mediated nuclear import of
Keap1 represses the Nrf2-dependent antioxidant response. Mol Cell
Biol. 31:1800–1811. 2012.
|
|
16
|
Villeneuve NF, Lau A and Zhang DD:
Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin
proteasome system: an insight into cullin-ring ubiquitin ligases.
Antioxid Redox Signal. 13:1699–1712. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang XJ, Sun Z, Chen W, Eblin KE, Gandolfi
JA and Zhang DD: Nrf2 protects human bladder urothelial cells from
arsenite and monomethylarsonous acid toxicity. Toxicol Appl
Pharmacol. 225:206–213. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang XJ, Sun Z, Villeneuve NF, et al: Nrf2
enhances resistance of cancer cells to chemotherapeutic drugs, the
dark side of Nrf2. Carcinogenesis. 29:1235–1243. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim TH, Hur EG, Kang SJ, et al: NRF2
blockade suppresses colon tumor angiogenesis by inhibiting
hypoxia-induced activation of HIF-1α. Cancer Res. 71:2260–2275.
2010.PubMed/NCBI
|
|
20
|
Cascio S, D’Andrea A, Ferla R, et al:
miR-20b modulates VEGF expression by targeting HIF-1 alpha and
STAT3 in MCF-7 breast cancer cells. J Cell Physiol. 224:242–249.
2010.PubMed/NCBI
|
|
21
|
Horiuchi A, Imai T, Shimizu M, et al:
Hypoxia-induced changes in the expression of VEGF, HIF-1 alpha and
cell cycle-related molecules in ovarian cancer cells. Anticancer
Res. 22:2697–2702. 2002.PubMed/NCBI
|
|
22
|
Wong C, Wellman TL and Lounsbury KM: VEGF
and HIF-1alpha expression are increased in advanced stages of
epithelial ovarian cancer. Gynecol Oncol. 91:513–517. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huang Y, Hua K, Zhou X, et al: Activation
of the PI3K/AKT pathway mediates FSH-stimulated VEGF expression in
ovarian serous cystadenocarcinoma. Cell Res. 18:780–791. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee J, Park SY, Lee EK, et al: Activation
of hypoxia-inducible factor-1alpha is necessary for
lysophosphatidic acid-induced vascular endothelial growth factor
expression. Clin Cancer Res. 12:6351–6358. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Finkel T: Oxidant signals and oxidative
stress. Curr Opin Cell Biol. 15:247–254. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Finkel T: Redox-dependent signal
transduction. FEBS Lett. 476:52–54. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Griendling KK and Ushio-Fukai M: Reactive
oxygen species as mediators of angiotensin II signaling. Regul
Pept. 91:21–27. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dimmeler S and Zeiher AM: Reactive oxygen
species and vascular cell apoptosis in response to angiotensin II
and pro-atherosclerotic factors. Regul Pept. 90:19–25. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Frank GD, Eguchi S, Yamakawa T, Tanaka S,
Inagami T and Motley ED: Involvement of reactive oxygen species in
the activation of tyrosine kinase and extracellular
signal-regulated kinase by angiotensin II. Endocrinology.
141:3120–3126. 2000.PubMed/NCBI
|
|
30
|
Hannken T, Schroeder R, Zahner G, Stahl RA
and Wolf G: Reactive oxygen species stimulate p44/42
mitogen-activated protein kinase and induce p27(Kip1): role in
angiotensin II-mediated hypertrophy of proximal tubular cells. J Am
Soc Nephrol. 11:1387–1397. 2000.PubMed/NCBI
|
|
31
|
Itoh K, Ishii T, Wakabayashi N and
Yamamoto M: Regulatory mechanisms of cellular response to oxidative
stress. Free Radic Res. 31:319–324. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kensler TW, Wakabayashi N and Biswal S:
Cell survival responses to environmental stresses via the
Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 47:89–116.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang DD: Mechanistic studies of the
Nrf2-Keap1 signaling pathway. Drug Metab Rev. 38:769–789. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liao H, Zhou Q, Zhang Z, et al: NRF2 is
overexpressed in ovarian epithelial carcinoma and is regulated by
gonadotrophin and sex-steroid hormones. Oncol Rep. 27:1918–1924.
2012.PubMed/NCBI
|
|
35
|
Zhou Q, Liu LZ, Fu B, et al: Reactive
oxygen species regulate insulin-induced VEGF and HIF-1alpha
expression through the activation of p70S6K1 in human prostate
cancer cells. Carcinogenesis. 28:28–37. 2007. View Article : Google Scholar : PubMed/NCBI
|