Introduction
The development of cervical carcinomas closely
correlates with the presence of certain high-risk human
papillomavirus (HR-HPV) types, such as HPV-16 and HPV-18. HPV-16 is
the most common HPV type detected in cervical cancer, accounting
for 50% of cancers and high-grade squamous intraepithelial lesions
(1). Carcinogenesis relies
primarily on the expression of two virally encoded oncoproteins, E6
and E7. These act synergistically to immortalize and transform the
infected cells partly through their ability to degrade p53 and Rb,
respectively (2–4).
The HR-HPV E6 protein has been demonstrated to lead
to the ubiquitin-mediated degradation of p53 by direct interaction
with the cellular E3 ubiquitin ligase, E6AP (5). The specific action of E6 on p53 is
functionally equivalent to p53 inactivation through mutation, which
indicates that the HR-HPV E6/p53 complex represents one of the most
important events in cervical carcinogenesis, given the interruption
of the cell cycle control points and inhibition of apoptosis. The
degradation of p53 induced by E6-AP is a significant effect of
HR-HPV and results in the malignant transformation of cervical
epithelial cells together with the inactivation of p53.
Genetic studies have shown natural amino acid
variants within the HPV-16 E6 oncoprotein (6). Variation within the E6 gene
leading to such changes in amino acids can alter the biological and
immunogenic properties of the encoded proteins (7,8).
Several studies have shown the existence of a link between E6
variants and the elevated risk of cervical intraepithelial
neoplasia and invasive cervical cancer (9,10).
However, little is known about the consequence of
sequence variants with respect to the function of E6. A previous
study analyzed a few of the HPV-16 E6 variants and showed that
amino acid changes can alter their ability to abrogate
serum/calcium-dependent differentiation leading to p53 degradation
in vitro(11). Other studies
have reported that the European variant, L83V, is associated with
an increased risk of developing invasive cervical carcinoma in the
Swedish population (10), and
enhances mitogen-activated protein kinase (MAPK) signaling and
cooperative transformation with deregulated Notch1 signaling
(12). Previous studies by us, as
well as others have indicated that E6 D25E, the most prevalent
variant type in Asian populations including Chinese (13), Japanese (14) and Korean populations (15), may have a unique oncogenic role
through different genes assoicated with the regulation of apoptosis
or the cell cycle, such as AIFM2 and RPL23(16). In the present study, we performed a
functional analysis of naturally occurring E6 variants (D25E and
L83V) to investigate the role of E6 polymorphisms in the
development of cervical cancer. The E6 variants were evaluated for
their ability to induce p53 degradation and inhibit p53
transactivation by comparing them with the reference HPV-16 E6
protein.
Materials and methods
Cell culture
The human cervical carcinoma cell lines, C33A, SiHa
and HeLa, were obtained from the American Type Culture Collection
(Manassas, VA, USA). The C33A and HeLa cells were cultured in MEM
Alpha medium (Gibco; Life Technologies, Carlsbad, CA, USA). The
SiHa cells were cultured in RPMI-1640 medium (Gibco) supplemented
with 10% fetal bovine serum (Gibco) and antibiotics (100 U/ml of
penicillin and 100 μg/ml of streptomycin), at 37°C under humidified
5% CO2 in air.
Gene construction of expression vectors
and transfection
Prototype E6 was cloned into the eukaryotic
expression vector, pFN21A HaloTag® CMV Flexi®
Vector (Promega, Madison, WI, USA) containing an N-terminal HaloTag
as described in the manufacturer’s instructions. E6 was amplified
by polymerase chain reaction (PCR) with primers including the
restriction site for SgfI or PmeI (underlined):
5′-CGAAGCGATCGCCATGCACCAAAAGAGAACTGC-3′
and 5′-CATCGTTTAAACTTACAGCGGGTTTCTCTAC-3′
from a previously constructed cell line [Jang et al(16)]. The E6 D25E and L83V variants were
acquired in the E6 prototype construct using the
QuikChange® site-directed mutagenesis kit (Stratagene,
La Jolla, CA, USA), according to the manufacturer’s instructions.
The primers used for the E6 D25E and L83V variants were as follows.
For E6 D25E, the primers were 5′-ACAACTATACATGAGATAATATTAG-3′ and
5′-CTAATATTATCTCATGTATAGTTGTTTG-3′; for E6 L83V, the primers were
5′-GACATTATTGTTATAGTGTGTATGGAACAACATTAG-3′ and
5′-GTAATGTTGTTCCATACACACTATAACAATAATGTC-3′. All vectors were
analyzed by nucleotide sequencing. Confirmed clones were
transfected into each cell line using FuGene X-treme GENE HP DNA
transfection reagent (Roche Applied Science, Pleasanton, CA, USA)
according to the manufacturer’s instructions.
Western blot analysis
The C33A, HeLa and SiHa cells were transfected with
constructs containing the E6 prototype and variants. At 24 h after
transfection, cell extracts were obtained by lysis in a RIPA cell
lysis buffer (150 mM NaCl, 1% TritonX-100, 1% deoxycholic acid
sodium salt, 0.1% SDS, 50 mM Tris-HCl, pH 7.5 and 2 mM EDTA)
supplemented with complete protease inhibitor tablets (Roche
Applied Science). The extracts were then fractionated by 10% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred onto a polyvinylidene fluoride immunoblot membrane
(Millipore, Billerica, MA, USA). The blot was incubated
successively with the primary and the secondary antibodies, and the
resulting signal was detected using enhanced chemiluminescence
(Intron Biotechnology, Seoul, Korea). The antibodies used in this
study were as follows: anti-p53 (DO-1; Santa Cruz Biotechnology
Inc., Santa Cruz, CA, USA), anti-Halo (Promega), anti-p21 (Cell
Signaling Technology, Inc., Beverly, MA, USA) and anti-β-actin
(Cell Signaling Technology, Inc.). β-actin was used as the loading
control. To examine the effect of the proteasome inhibitor, MG132,
E6 protein-expressing HeLa cells were incubated with 10 μM MG132
for 2 h prior to western blot analysis.
Halo pull-down assay
The interaction between E6 variant proteins and p53
was identified using the HaloTag® Mammalian Pull-Down
System according to the manufacturer’s instructions (Promega).
Briefly, approximately 1–1.2×107 cells were washed with
phosphate-buffered saline (PBS) and lysed in 300 μl of mammalian
lysis buffer (GenDEPOT, Barker, TX, USA), containing protease
inhibitor (Roche Applied Science). Aliquots of 300 μl of clear cell
lysate were diluted with 700 μl of 1X TBS (50 mM Tris, pH 7.4 and
150 mM NaCl). Diluted cell extracts were incubated with
equilibrated HaloLink™ resin (Promega) at 4°C for 3 h. The beads
were washed three times with 1 ml of Promega resin
equilibration/wash buffer (with protease inhibitor) and washed
resins were resuspended in 35 μl of SDS sample buffer and boiled
for 5 min. The precipitated complexes were analyzed by western blot
analysis using an anti-p53 antibody.
Quantitative real-time (RT) PCR
cDNA was synthesized from 5 μg of total RNA using an
Omniscript RT kit (Qiagen, Hilden, Germany). We used 1 μl cDNA for
quantitive RT-PCR amplification using a SYBR Supermix kit (Bio-Rad
Laboratories, Richmond, CA, USA). Samples were subjected to 45
cycles of 95°C for 20 sec and 60°C for 1 min. PCR efficiency was
determined by running serial dilutions of template cDNA and melting
curve data were collected to assure PCR specificity. Each cDNA
sample was analyzed in triplicate and the corresponding non-RT mRNA
sample was included as the negative control. A β-actin primer was
included in every plate as the internal loading control. The
following primers were used for quantitative RT-PCR of the p21 and
β-actin genes: p21 forward, 5′-GCGGAACAAGGAGTCAGACA-3′ and reverse,
5′-GGAAGGTGTTTGGGGTCAGA-3′; β-actin forward,
5′-ATCTGGCACCACACCTTCTA-3′ and reverse,
5′-GGATAGCACAGCCTGGATAC-3′.
Cell cycle analysis using flow
cytometry
Cell cycle analysis was performed by flow cytometry
with propidium iodide (PI) staining. In brief, HeLa cells
(1.2×106 cells/10 cm2 dish) were transfected
with the empty vector, and with the Halo-E6, Halo-D25E and
Halo-L83V constructs. After 24 h, the harvested cells were fixed in
cold 75% ethanol at 4°C for 2 h and washed twice with PBS. The
cells were stained with 0.5 ml of 20 mg/ml PI containing 0.1 mg/ml
RNase in PBS for 30 min at room temperature. DNA contents in 10,000
cells were analyzed with ModFit LT software (Verity Software House,
Topsham, ME, USA) on a flow cytometer by gating on an area versus
width dot plot to exclude cell debris and aggregates.
Statistical analysis
Data of activity in the various functional assays
are presented in the figures as the means ± standard deviation
(SD). Data were subjected to a one-way ANOVA. A value of P<0.05
was considered to indicate a statistically significant
difference.
Results
E6 D25E and L83V variants reduce the
levels of p53 expression in several cervical carcinoma cell
lines
The targeting of p53 for degradation is believed to
be an essential event in HPV-mediated malignant cell transformation
(17). We investigated the ability
of the E6 variants to degrade p53 in transient expression assays
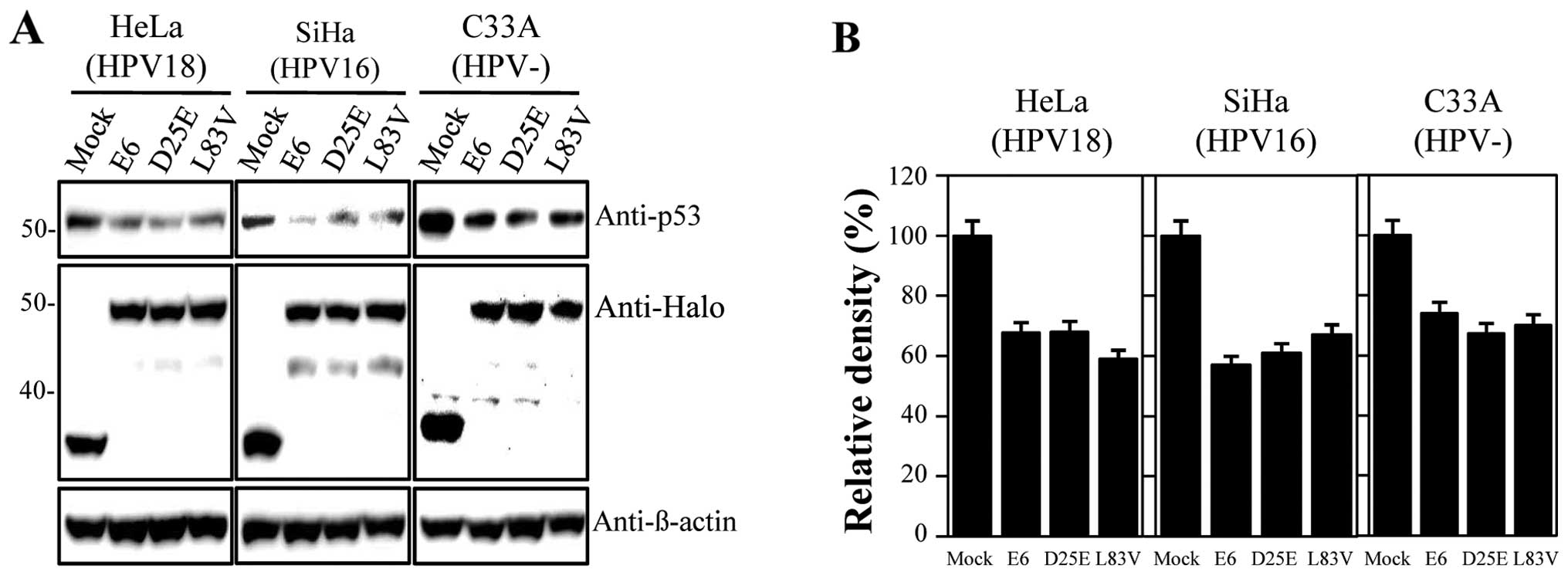
using Halo-tagged constructs expressing E6 proteins. The
constructed DNAs were confirmed through sequencing analysis (data
not shown). In our study, we used HPV-18-positive HeLa,
HPV-16-positive SiHa and HPV-negative C33A cervical carcinoma cell
lines, which were transfected with the empty vector, Halo-E6,
Halo-D25E and Halo-L83V constructs; prototype E6 and E6 variant
proteins were detected with the anti-Halo antibody. Levels of p53
were then measured by immunoblot analysis. The expression levels of
the E6 variant proteins were similar to those of the E6 prototype
(Fig. 1A). As indicated in Fig. 1, all examined proteins actively
promoted p53 degradation, and the level of p53 was similar in all
the tested cell lines compared with the empty vector-treated
samples. Degradation activities ranged between 30 and 40% of the
control levels (Fig. 1B) and did
not differ between the E6 prototype protein and variants. Thus, the
E6 D25E and L83V variants had similar abilities to degrade p53 as
the prototype protein. For further functional studies, we used HeLa
cells as there were no significant differences in the rate of
inactivation of p53 by E6 proteins among the cell lines
studied.
E6 D25E variant interaction with p53 in
vitro
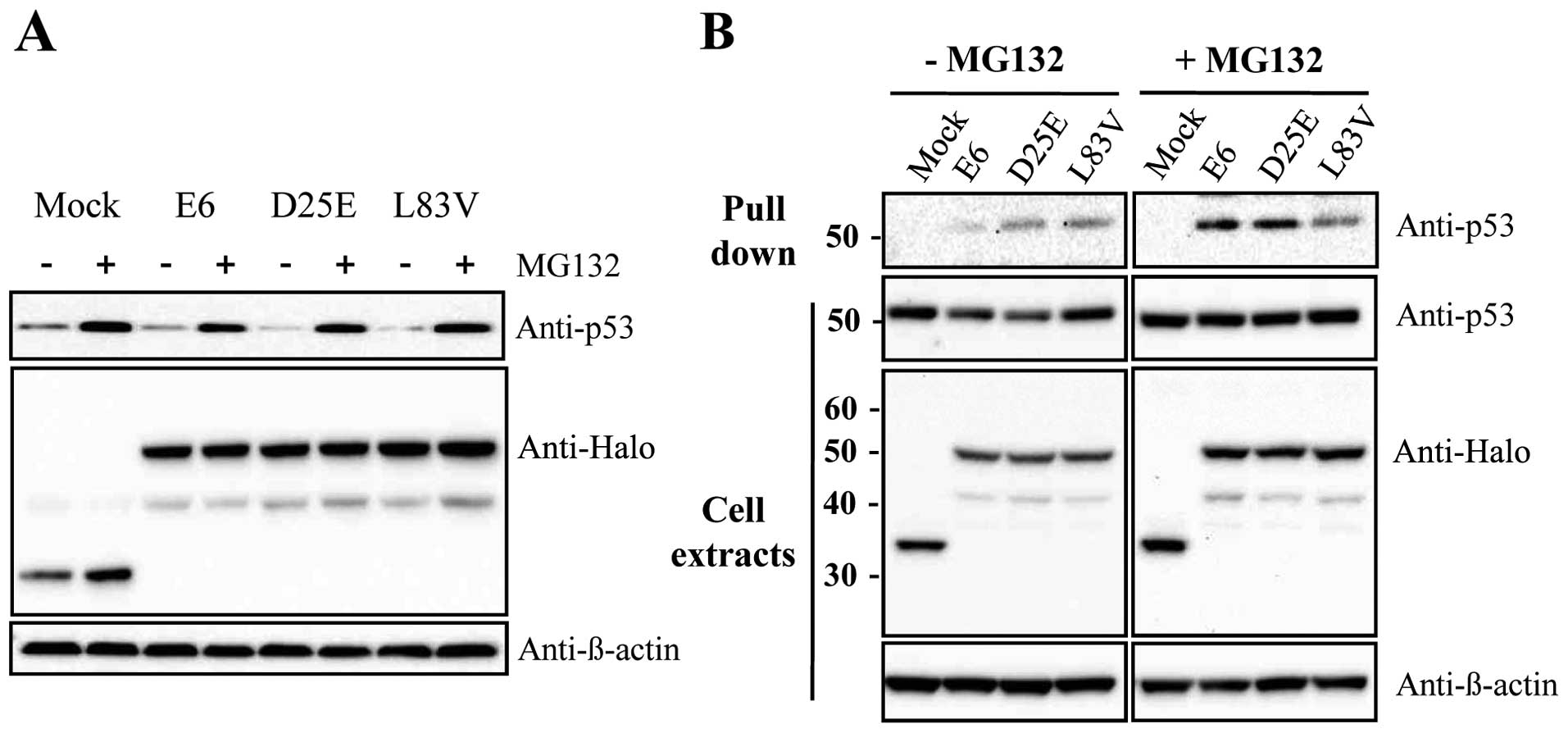
The E6 viral oncoprotein produced by HR-HPV fosters
ubiquitination and the proteasome-dependent degradation of p53
through protein-protein interactions. To examine whether E6
variants, such as the E6 prototype, can reduce p53 levels via a
proteasome-dependent pathway, the HeLa cells were transfected with
E6 variants or the E6 prototype protein as the control. At 24 h
after transfection, the cells were then treated with the proteasome
inhibitor, MG132, for 2 h before harvesting. As shown in Fig. 2A, the level of p53 was increased
with MG132 treatment. This suggests that the reduction in p53
levels by D25E and L83V was caused by proteasome-dependent
degradation. In addition, we examined whether these E6 variant
proteins can regulate p53 protein levels by direct interaction with
each other. Binding assays showed that D25E and L83V interact
directly with p53 in the presence or absence of MG132 (Fig. 2B). The interaction was increased
when the cells were treated with MG132, possibly due to the higher
concentration of existing p53 protein. Thus, both the E6 prototype
protein and its variants, D25E and L83V, affect p53 degradation by
binding to p53 directly.
E6 D25E and L83V variants downregulate
the induction of p21 gene expression
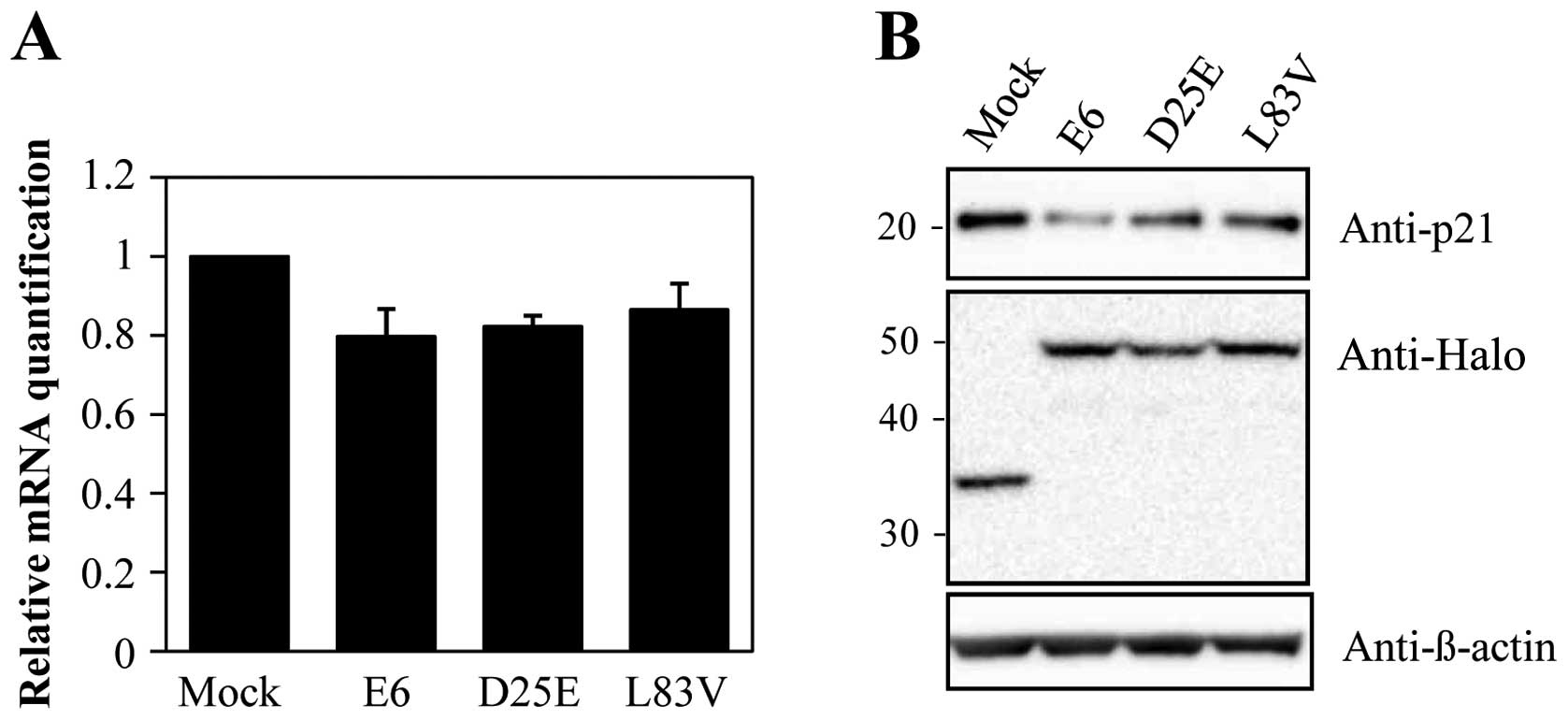
Abnormalities in the molecular pathways that mediate
the cell cycle have been implicated in p53-regulated pathways. A
well-known target of p53 is the p21 gene that causes cell cycle
arrest. Therefore, we evaluated the level of p21 to determine the
effect of degraded p53 in each cell line. As shown in Fig. 3, p21 mRNA and protein levels were
decreased in the E6 protein-expressing cell lines. These results
indicate that the downregulation of p53 by E6 proteins inhibits p21
expression. Additionally, the prototype E6 protein and its variants
inhibited p21 expression in a similar manner despite different
variant protein levels. These results suggest that the E6 protein
can accommodate amino acid changes without significantly perturbing
the activity of this protein in degrading p53 and overriding cell
growth arrest via p21.
E6 D25E and L83V variants promote entry
into the S phase of the cell cycle in HeLa cells
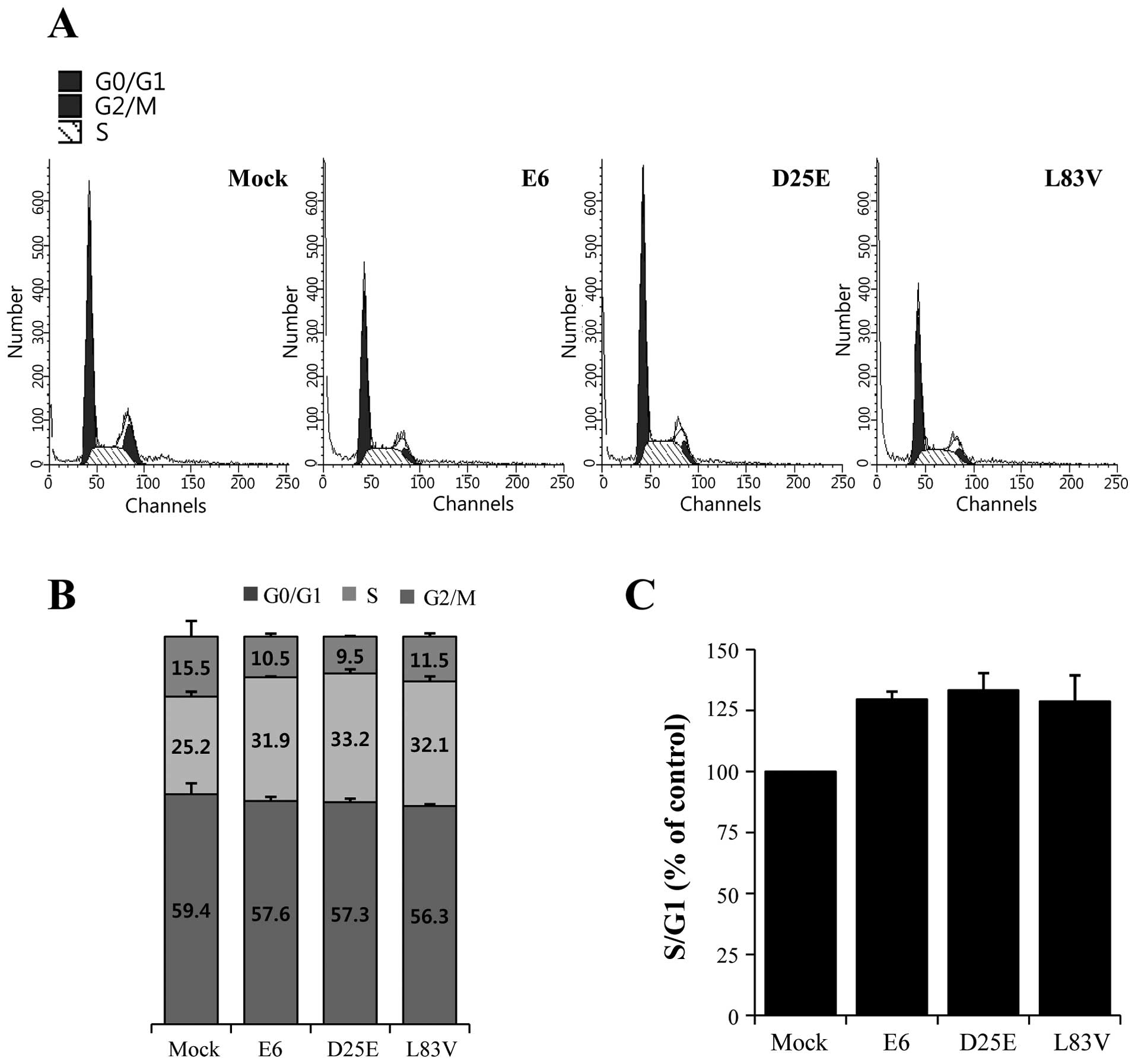
The ability of HR-HPV E6 to inhibit growth arrest
and to abrogate DNA damage response induced by p53 and the
downregulation of the p21 gene are crucial during HPV-associated
carcinogenesis (4,18). We examined the ability of the E6
variants to override growth arrest and determined the G1, S and
G2/M phases and the G1/S ratios in the E6 protein-expressing cell
lines (Fig. 4A) by ModFit LT
analysis using flow cytometry. HeLa cells were transfected with the
E6 prototype, D25E or L83V. The expression of the E6 protein
resulted in an increase in the percentage of cells at the S phase
entry stage (Fig. 4B), as evidenced
by an increase in the proportion of cells in the S phase with an
increase in the S/G1 ratio (Fig.
4C). The ratio was increased by >20% in the E6
variant-expressing cell lines. There were no differences among the
E6 protein variants, indicating that all overcame the growth arrest
induced by p21 repression. This showed that the natural E6
variants, D25E and L83V, contribute to cell growth and
proliferation through the reduction and downregulation of p53
expression.
Discussion
It has been proposed that intratypic variations of
HPV types can affect their carcinogenic potential (19). The causes for the pathogenic
differences between HPV-16 variants are not yet understood,
although in a limited number of studies, differences in the LCR,
E6, E2 and E7 sequences have been associated with altered
biological functions (19,20). The E6 L83V polymorphism has been
reported to be associated with an increased risk of cancer
progression in certain populations and has been detected at a high
frequency in all European populations (21–23).
This variant has been shown to enhance E6-mediated MAPK signaling
and differentially regulates tumorigenesis by Notch signaling and
oncogenic Ras expression (12).
These and perhaps other altered functions may underlie the
increased pathogenicity of L83V. However, there is no experimental
evidence of whether the HPV-16 E6 D25E variant protein contributes
to disease progression, as opposed to the prototype E6 protein. To
our knowledge, this is the first study to determine whether the
high prevalence of naturally occurring HPV-16 E6 variants in Asian
and European populations differ from prototype E6 in their ability
to regulate p53 expression. The D25E and L83V variants of HPV-16
E6, whose distribution in cervical precursor lesions and cancers
has been determined in Asian and European populations (10,15),
were compared for p53 degradation and changes in the cell cycle
dynamics via p21 downregulation in E6-expressing cell lines. There
were uniform patterns of activity with small differences among
variants in the assays tested.
E6-mediated p53 degradation has been considered a
hallmark function of oncogenic HPV types, although E6 is a highly
multifunctional protein (24,25).
E6 proteins from HPV-16 are able to bind to p53 and this binding
promotes the degradation of p53 via the ubiquitin pathway (26). The degradation of p53 and the
induction of p53-mediated growth arrest associated with DNA damage
caused by E6 are believed to contribute to the accumulation of
genetic changes associated with cervical carcinogenesis (25). The variants examined in the present
study contained amino acid changes in positions 25 and 83 but
retained the prototype level of p53 degradation activity. Thus,
similar to the prototype E6 protein, the D25E and L83V variant
proteins are also able to suppress the elevation in levels of p53
protein and to override p53-mediated growth arrest through E6-p53
interaction. The binding of the prototype E6 and the two variants
with p53 occurred at similar levels. These results support the
notion that the HPV-16 E6 protein can accommodate some
non-conservative changes between natural variants for its
interaction with p53/E6, without significant changes in its ability
to target p53 for degradation. As with p53 degradation, the
variants examined in this study showed similar activities in p21
expression. The p21 tumor suppressor protein is a universal
inhibitor for cyclin-cyclin dependent kinase complexes and DNA
replication that induces cell cycle arrest at the G1/S checkpoint.
The expression of p21 is regulated transcriptionally via the p53
protein (27,28). The repression of p21 by p53
degradation through HPV-16 E6 proteins may result in the
stimulation of cell growth. To examine this possibility, we stably
transfected several HeLa cell lines with constructs containing E6
proteins and measured their cell cycle profiles. As expected, the
expression of the p21 protein was downregulated and the proportion
of cells in the S phase was increased in each E6-expressing cell
line. However, the results were similar between the E6 prototype
protein and its variants. These results are consistent with those
from a previous study (28),
indicating that HPV-16 E6 variant proteins repress the
transcription of p21 by the degradation of the p53 protein via
protein-protein interaction(s) and reduce the level of p21 protein
through transcriptional repression among different E6
protein-expressing cell lines.
The similarities in the modulation of DNA damage
responses between the E6 prototype and its variants, L83V and D25E,
both in terms of suppression of p53 accumulation and overcoming
growth arrest through p21 downregulation, strongly suggest that
these functions of E6 cannot be compromised to initiate
carcinogenesis.
In conclusion, using functional assays, we show that
naturally occurring amino acid variations in the E6 protein affect
pathogenesis by p53-associated proteins in human cervical cancer
cell lines. The ability of the proteins to induce p53 degradation
and modulate cell cycle profiles via p21 repression in various
HPV-16 E6 variant protein-expressing HeLa cells was similar to the
prototype E6 protein. Thus, the inactivation of p53 by E6 variants
may contribute to immortalization by preventing the cells from
arresting in response to genomic instability, in a similar manner
to the E6 prototype protein. To the best of our knowledge, this is
the first study to examine whether the high prevalence of naturally
occurring HPV-16 E6 variants in Asian and European populations
differ from the prototype E6 protein in their ability to regulate
p53 expression. The data presented in this study may open the way
for future mechanistic studies. Naturally occurring variants may
display biological differences other than those described in this
study, which could contribute to their pathogenicity. Further
structural and biochemical analyses with E6 variants are warranted
to improve our understanding of their biological functions and
epidemiology, and of how they modulate the progression of
carcinogenesis.
Acknowledgements
This study was supported by a grant from the health
Promotion against HIV/AIDS and STD (4800-4842-302) and from the
Pathogenic Proteome Management Program (4800-4847-300) of the
National Institute of Health, Ministry of Health and Welfare,
Republic of Korea.
References
|
1
|
Bosch FX, Manos MM, Muñoz N, et al:
Prevalence of human papillomavirus DNA in cervical cancer: a
worldwide perspective. International biological study on cervical
cancer (IBSCC) study group. J Natl Cancer Inst. 87:796–802. 1995.
View Article : Google Scholar
|
|
2
|
Liu Y, Chen JJ, Gao Q, et al: Multiple
functions of human papillomavirus type 16 E6 contribute to the
immortalization of mammary epithelial cells. J Virol. 73:7297–7307.
1999.PubMed/NCBI
|
|
3
|
Lehoux M, D’Abramo CM and Archambault J:
Molecular mechanisms of human papillomavirus-induced
carcinogenesis. Public Health Genomics. 12:268–280. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Moody CA and Laimins LA: Human
papillomavirus oncoproteins: pathways to transformation. Nat Rev
Cancer. 10:550–560. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huibregtse JM, Scheffner M and Howley PM:
A cellular protein mediates association of p53 with the E6
oncoprotein of human papillomavirus types 16 or 18. EMBO J.
10:4129–4135. 1991.PubMed/NCBI
|
|
6
|
Huertas-Salgado A, Martín-Gámez DC, Moreno
P, et al: E6 molecular variants of human papillomavirus (HPV) type
16: an updated and unified criterion for clustering and
nomenclature. Virology. 410:201–215. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ellis JR, Etherington I, Galloway D, et
al: Antibody responses to HPV16 virus-like particles in women with
cervical intraepithelial neoplasia infected with a variant HPV16.
Lancet. 34:1069–1070. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zehbe J, Mytilineos I, Wikstrom R, et al:
Association between human papillomavirus 16 E6 variants and human
leukocyte antigen class I polymorphism in cervical cancer of
Swedish women. Hum Immunol. 64:538–542. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Andersson S, Alemi M, Rylander E, et al:
Uneven distribution of HPV 16 E6 prototype and variant (L83V)
oncoprotein in cervical neoplastic lesions. Br J Cancer.
83:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zehbe I, Wilander E, Delius H, et al:
Human papillomavirus 16 E6 variants are more prevalent in invasive
cervical carcinoma than the prototype. Cancer Res. 58:829–833.
1998.PubMed/NCBI
|
|
11
|
Stoppler MC, Ching K, Stoppler H, et al:
Natural variants of the human papillomavirus type 16 E6 protein
differ in their abilities to alter keratinocyte differentiation and
to induce p53 degradation. J Virol. 70:6987–6993. 1996.PubMed/NCBI
|
|
12
|
Chakrabarti O, Veeraraghavalu K,
Tergaonkar V, et al: Human papillomavirus type 16 E6 amino acid 83
variants enhance E6-mediated MAPK signaling and differentially
regulate tumorigenesis by notch signaling and oncogenic Ras. J
Virol. 78:5934–5945. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cai HB, Chen CC and Ding XH: Human
papillomavirus type 16 E6 gene variations in Chinese population.
Eur J Surg Oncol. 36:160–163. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Matsumoto K, Yoshikawa H, Nakagawa S, et
al: Enhanced oncogenicity of human papillomvairus type 16 (HPV16)
variants in Japanese population. Cancer Lett. 56:159–165. 2000.
View Article : Google Scholar
|
|
15
|
Kang S, Jeon YT, Kim JW, et al:
Polymorphism in the E6 gene of human papillomavirus type 16 in the
cervical tissues of Korean women. Int J Gynecol Cancer. 15:107–112.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jang M, Rhee JE, Jang DH and Kim SS: Gene
expression profiles are altered in human papillomavirus-16 E6
D25E-expressing cell lines. Virol J. 8:4532011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thomas M, Pim D and Banks L: The role of
the E6-p53 interaction in the molecular pathogenesis of HPV.
Oncogene. 18:7690–7700. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kessis TD, Slebos RJ, Nelson WG, et al:
Human papillomavirus 16 E6 expression disrupts the p53-mediated
cellular response to DNA damage. Proc Natl Acad Sci USA.
90:3988–3992. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bernard HU, Calleja-Macias IE and Dunn ST:
Genome variation of human papillomavirus types: phylogenetic and
medical implications. Int J Cancer. 118:1071–1076. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hildesheim A and Wang SS: Host and viral
genetics and risk of cervical cancer: a review. Virus Res.
89:229–240. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zehbe I, Voglino G, Delius H, et al: Risk
of cervical cancer and geographical variations of human
papillomavirus 16 E6 polymorphisms. Lancet. 352:1441–1442. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kämmer C, Tommasino M, Syrjänen S, et al:
Variants of the long control region and the E6 oncogene in European
human papillomavirus type 16 isolates: implications for cervical
disease. Br J Cancer. 86:269–273. 2002.PubMed/NCBI
|
|
23
|
Grodzki M, Besson G, Clavel C, et al:
Increased risk for cervical disease progression of French women
infected with the human papillomavirus type 16 E6-350G variant.
Cancer Epidemiol Biomarkers Prev. 15:820–822. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
McLaughlin-Drubin ME and Munger K:
Oncogenic activities of human papillomaviruses. Virus Res.
143:195–208. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Howie HL, Katzenellenbogen RA and Galloway
DA: Papillomavirus E6 proteins. Virology. 384:324–334. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Scheffner M, Werness BA, Huibregtse JM,
Levine AJ and Howley PM: The E6 oncoprotein encoded by human
papillomavirus types 16 and 18 promotes the degradation of p53.
Cell. 63:1129–1136. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rowland BD and Peeper DS: KLF4, p21 and
context-dependent opposing forces in cancer. Nat Rev Cancer.
6:11–23. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Munger K and Howley PM: Human
papillomavirus immortalization and transformation functions. Virus
Res. 89:213–228. 2002. View Article : Google Scholar : PubMed/NCBI
|